

Abstract
The synthetic organoselenium agent 1,4- phenylenebis(methylene)selenocyanate (p-XSC) and its glutathione (GSH) conjugate (p-XSeSG), are potent chemopreventive agents in several preclinical models. p-XSC is also an effective inducer of GSH in mouse lung. Our objectives were to test the hypothesis that GSH induction by p-XSC occurs through upregulation of the rate-limiting GSH biosynthetic enzyme glutamate cysteine ligase (GCL), through activation of antioxidant response elements (ARE) in GCL genes via activation of nuclear factor-erythroid 2-related factor 2 (Nrf2). p-XSC feeding (10 ppm Se) increased GSH (230%) and upregulated the catalytic subunit of GCL (GCLc) (55%), extracellular related kinase (ERK) (220%) and nuclear Nrf2 (610%) in lung but not liver after 14 days in the rat (P<0.05). Similarly, p-XSeSG feeding (10 ppm) induced lung GCLc (88%) and GSH (200%) (P<0.05), while the naturally-occurring selenomethionine had no effect. Both p-XSC and p-XSeSG activated a luciferase reporter in HepG2 ARE reporter cells up to 3-fold for p-XSC and ≥5-fold for p-XSeSG. Luciferase activation by p-XSeSG was associated with enhanced levels of GSH, GCLc and nuclear Nrf2, which were significantly reduced by co-incubation with short interfering RNA targeting Nrf2 (siNrf2). The dependence of GCL induction on Nrf2 was confirmed in Nrf2 deficient mouse embryonic fibroblasts (MEF) where p-XSeSG induced GCL subunits in wildtype, but not Nrf2 deficient cells (p<0.05). These results indicate that p-XSC may act through the Nrf2 pathway in vivo, and that p-XSeSG is the putative metabolite responsible for such activation, thus offering p-XSeSG as a less toxic, yet highly efficacious inducer of GSH.
Keywords: Organoselenium, p-XSC, p-XSeSG, glutathione, γ-GCL, Nrf2, MEF, antioxidant response element (ARE), HepG2-ARE, Lung cancer, Fisher 344 rat
Introduction
Lung cancer is the second most common cancer and is the leading cause of cancer deaths in the world. Because of difficulties associated with smoking cessation, early detection, and chemotherapeutic effectiveness, research into chemoprevention has become a priority. Selenium, in both organic and inorganic forms, has played a major role in the field of chemoprevention, particularly after the reporting of an almost 50% reduction in morbidity and mortality by major cancers following dietary supplementation with selenized brewer’s yeast [1]. Various synthetic selenium compounds have been developed with goals of lowering toxicity while enhancing efficacy when compared to other historic chemopreventive agents such as selenite, selenate, or selenomethionine [2]. The organoselenium agents 1,4-phenylenebis(methylene)selenocyanate (p-XSC) and its putative metabolic glutathione conjugate, p-XSeSG have proven to be effective in preventing a variety of carcinogen-induced cancers in several animal models, including lung cancer [3-5].
We reported that p-XSC-mediated protection against lung tumors induced by the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in A/J mice was associated with increased levels of lung GSH [5]. Also, selenized yeast supplementation enhanced levels of blood GSH in a clinical study of healthy adult men [6]. Glutathione plays a number of critical roles in protection against carcinogenesis through its activities as the most important and abundant endogenous antioxidant and in the detoxification of exogenous and endogenous carcinogens [7, 8]. Thus, the maintenance of cellular GSH and/or induction of supra-normal levels are likely part of an agent’s chemopreventive potential.
Both catalytic and modulatory subunits of the rate-limiting enzyme for GSH synthesis, glutamate cysteine ligase (GCLc and GCLm, respectively), are regulated in part by the presence of antioxidant response elements (ARE) in upstream promoter regions of each gene [9]. Enhanced nuclear translocation and subsequent binding of the nuclear factor-erythroid 2-related factor 2 (Nrf2) transcription factor to ARE-containing promoters activates a variety of chemoprotective Phase II detoxification genes, including many in the GSH biosynthetic and homeostasis pathways [10].
Nrf2 is normally sequestered in the cytoplasm by the actin-bound protein Keap1, a substrate adaptor for an E3 ubiquitin ligase, which targets Nrf2 for rapid turnover [11]. Keap1 contains multiple reactive cysteine residues that, when modified directly or indirectly by a variety of inducers, reduce its affinity for Nrf2 and promote the translocation of Nrf2 to the nucleus [12]. The synthetic organoselenium compound ebselen has been shown in a cell culture model to directly modify Keap1 [13]. Phosphorylation of Nrf2, mediated by extracellular signal-regulated kinase and c-Jun-NH2-kinase pathways, has also been shown to disrupt its association with Keap1 [14].
The primary objective of the studies reported here was to determine if Nrf2 activation of ARE elements in GSH biosynthetic genes is responsible for GSH enhancement by p-XSC and its metabolite p-XSeSG in vitro and in vivo in rat lung.
Materials and Methods
Reagents
Organoselenium compounds p-XSC and p-XSeSG and selenomethionine were synthesized as described previously [15]. L-selenomethionine was obtained commercially (Sigma, St. Louis). Antibodies (Nrf2, GCLc, GCLm, p-ERK, Actin, Lamin A) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Animals and diet preparation
Pathogen-free male Fisher 344 rats 4 weeks of age were purchased from Charles River Breeding Laboratory (Kingston, NY). After a 1 wk. quarantine, rats were weighed and placed into 4 treatment groups (Control, p-XSC, p-XSeSG and selenomethionine) of 6 rats each such that average weights per group were similar. All rats were fed a semipurified diet (AIN-93G) containing 0.1 ppm of selenium as sodium selenite throughout the study. At 6 weeks of age, diets of rats in the 3 selenium groups were supplemented with 10 ppm p-XSC, 10 ppm p-XSeSG or 10 ppm selenomethionine, respectively. Levels of selenium (10 ppm) for incorporation into the diet and time period for GSH induction were based on our previous studies [5][16]. The organoselenium compounds were blended into the diets by Harlan Teklad (Ijamsville, MD), and stored at 4° C. Rats were fed either the control or supplemented diets for 2 weeks and were reweighed weekly. After the second week animals were sacrificed as described below. The 2 week time period was based on preliminary studies (data not shown). All procedures involving animals were conducted in accordance with guidelines of the Institutional Animal Care and Use Committee of the Pennsylvania State University, as promulgated by the National Institutes of Health.
Necropsy and tissue harvesting and processing
Animals were euthanized by CO2 inhalation between 9:00 and 10:00 am to avoid the confounding effects of circadian fluctuations. Whole blood was collected into EDTA tubes by cardiac puncture and frozen at -80°C for analysis of GSH. Lungs and livers were harvested, rinsed in saline and blotted dry, then divided into portions and snap-frozen in liquid nitrogen. For Western blot analysis, nuclear and cytoplasmic extracts were obtained from frozen tissue using reagents supplied by Marligen (Madison, WI) according to the manufacturer’s protocol. Briefly, 0.15 g of frozen tissue was placed into 1 ml of ice-cold cytoplasm extract buffer containing EDTA and protease inhibitors, then homogenized using an all-glass Ten Broeck homogenizer and centrifuged at 800g for 5 min at 0-4°C. The resulting supernatant and nuclear pellets were stored at -80°C until analysis.
Determination of total selenium in lung
Lung tissues were homogenized in 1.15% KCl (0.1gm/ml) using a glass hand homogenizer and digested in a MARS Xpress microwave digestion system (CEM Corp., Mathews, NC) equipped with 55 ml Teflon PFA vessels and a turntable. The digestion was conducted in 50% nitric acid and then diluted to 20% before selenium analysis by Atomic Absorption Spectroscopy. We used an AAnalyst 600 instrument from PerkinElmer with Graphite Furnace for total selenium analysis by measuring the absorbance peak area at 196 nm for each sample. Palladium matrix modifier was added along with each sample to the furnace. Reference standard solutions of selenium dioxide at various concentrations range were used to construct a standard curve for comparison with all assays. Analysis was performed in duplicate for each sample and the average value was recorded. For each group at least six samples were analyzed and the results were expressed as mean ± SE.
Blood and Tissue Glutathione
For GSH analysis, portions of lung (left lung) and liver (right lobe) (~0.1g) were homogenized in 3 ml of 5% (w/v) metaphosphoric acid (MPA) using an all-glass Ten Broeck homogenizer at 0-4°C and whole blood (0.1 ml) was hemolyzed in 0.4 ml of 5% MPA. Precipitated protein was removed by centrifugation at 14,000g for 2 min and acid-soluble fractions were removed and stored at -80°C until analysis for free glutathione by the enzymatic recycling method using Elman’s reagent [17] with modifications [18]. Tissue GSH levels were normalized to tissue weight, while GSH in blood samples was normalized to hemoglobin levels.
Cell Culture
HepG2 human hepatoma cells stably transfected with ARE-luciferase plasmid were obtained courtesy of Dr. Muriel Cuendet, Purdue University [19]. Cells were maintained in F-12 media (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, antibiotics, MEM amino acid solution, and insulin (1.45 μg/ml). Wildtype and Nrf2 deficient (Nrf2 -/-) mouse embryonic fibroblasts (MEF) were obtained courtesy of Drs. Nobunao Wakabayashi and Thomas Kensler, Johns Hopkins University [20]. Cells were maintained in Iscov’s MDM (Gibco12440, Coon Rapids, MN) supplemented with 10% fetal bovine serum, and antibiotics.
Viability Assay
Cell viability was assessed using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) reduction assay, according to the manufacturer’s instructions (CellTiter 96™, Promega, Madison WI). Absorbance at 490 nm was measured using a Bio-Tek Synergy HT plate reader (Bio-Tek Instruments, Winooski, VT).
Luciferase Assay
Treated HepG2 ARE reporter cells were washed in PBS, lysed, and luciferase activity of resulting extracts was measured according to the Luciferase Assay System protocol (Promega, Madison WI) using a single tube luminometer.
Glutathione Assay
Aliquots of cells were washed in PBS and lysed in 5% MPA. Precipitated protein was removed by centrifugation and acid soluble GSH was measured as described above, with levels were normalized to protein levels as determined by BCA assay.
Cellular Extracts and Western Blot Analysis
Cellular extracts were obtained using NE-PER® Nuclear and Cytoplasmic Extraction Reagents according to the manufacturer’s protocol (Pierce, Rockford, IL). Western blotting for nuclear Nrf2 and lamin A, or cytoplasmic GCLc, GCLm, actin, or GAPDH was performed according to manufacturer’s instructions using the Criterion™ Cell system (Bio-Rad, Hercules, CA). The protein content of cellular extracts was determined by BCA assay according to the manufacturer’s protocol (Pierce Protein Research Products, Rockford, IL). Absorbance was measured using a Bio-Tek Synergy HT plate reader (Bio-Tek Instruments, Winooski, VT). Samples were normalized in Laemli’s buffer. Cytoplasmic or nuclear protein (50 or 20 μg per lane, respectively) were loaded in 4-20% gradient polyacrylamide gels (TRIS-HCL) immediately following boiling for 5 minutes in Laemli’s buffer. Resolved protein was then transferred to nitrocellulose membranes using a CAPS discontinuous buffer system and Trans-Blot® SD transfer cell according to manufacturer’s instructions (Bio-Rad, Hercules, CA). Membranes were blocked for 1 hour at room temperature in 5% non-fat dry milk/TBST, and probed overnight at 4° C with primary antibody diluted in TBST at concentrations recommended by the manufacturer (Santa Cruz Biotechnology, Santa Cruz, CA). Membranes were incubated with HRP-conjugated secondary antibodies (Pierce Protein Research Products, Thermo Scientific, Rockford, IL), diluted 1:1000 in TBST, for 1 hour at room temperature and developed using SuperSignal West Dura Extended Duration Substrate reagents according to the manufacturer’s protocol (Pierce Protein Research Products, Thermo Scientific, Rockford, IL), before exposure. Membranes were re-probed for actin (cytoplasmic marker) or Lamin A (nuclear marker) to insure equal loading and purity of cytoplasmic or nuclear extracts. Bands were quantitated with the BioSpectrum AC Imaging System (UVP BioImaging Systems, Upland, CA), and data was normalized to corresponding actin or Lamin A values.
RNA Interference
HepG2 ARE reporter cells were grown to 60% confluence in 6 cm dishes then transfected with non-targeting siRNA-Control (siControl) or siRNA-Nrf2 (siNrf2, sc-37030) using manufacturer-recommended reagents and protocols (Santa Cruz Biotechnology, Santa Cruz, CA). Thirty hours after siRNA transfection, siControl and siNrf2 cells were exposed to vehicle control, 10 μM p-XSC, or 100 μM p-XSeSG added to the cell media. Following 18 hours of exposure to the chemoprevention agent, cells were assayed for viability and luciferase activity and cytoplasmic or nuclear extracts were subjected to western blotting.
Statistical Analysis
Data are reported as mean ± standard deviation. Significance was assessed using either the Student’s t-test or ANOVA where appropriate. Differences between data sets were considered statistically significant if p < 0.05.
Results
Effect of p-XSC, p-XSeSG and selenomethionine administration in vivo
In this experiment, in addition to p-XSC and p-XSeSG, selenomethionine was tested since it has been show to lack activity in the SELECT trial as well as numerous animal models.[21, 22] Following dietary administration of these compounds for 2 weeks, rats gained an average of 37.5, 35.0, 36.0 and 31.0 g per week in control, p-XSC, p-XSeSG and selenomethionine groups, respectively (Table 1). Body weights did not differ significantly between controls and organoselenium diets at any time point. These data suggest that the organoselenium compounds used were not toxic within the 2-week study period at the concentrations used.
Table 1.
Effects of Dietary Organoselenium Compounds on Bodyweights
| Diet | Bodyweights (g)
a
|
||
|---|---|---|---|
| Baseline | 1 week | 2 weeks | |
| Control | 105 ± 7.79 | 138 ± 10.6 | 180 ± 13.4 |
| p-XSC | 105 ± 11.1 | 137 ± 15.9 | 175 ± 21.4 |
| p-XSeSG | 104 ± 8.81 | 135 ± 11.1 | 176 ± 13.8 |
| Selenomethionine | 105 ± 7.83 | 128 ± 12.7 | 167 ± 17.8 |
Values are mean ± SE (n=6).
The effects of feeding organoselenium on lung selenium levels were determined (Figure 1). Lung selenium levels were enhanced by all three organoselenium compounds with selenomethionine being most effective (11.2-fold, P<0.005) followed by p-XSeSG (4.8-fold, P<0.0001) and p-XSC (3.6-fold, P<0.0001).
Figure 1.
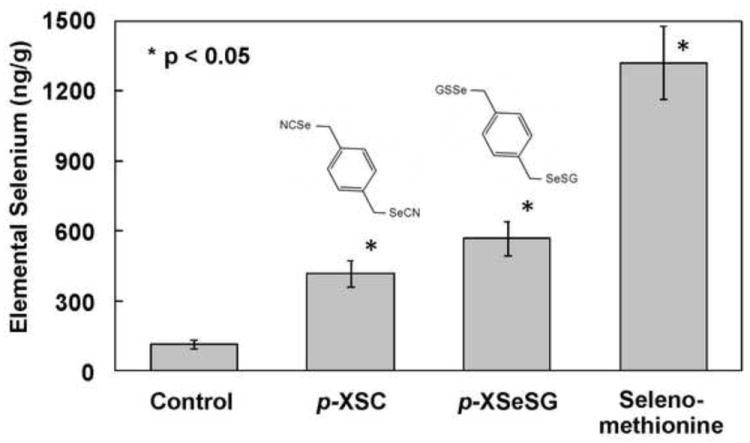
Effect of feeding organoselenium agents on selenium content in the lung. Male F344 rats were fed control diet (AIN-93A) or diets supplemented with p-XSC, p-XSeSG or selenium methionine (10 ppm as selenium) for 2 weeks. Lung selenium levels were determined by atomic absorption as described in text. Values are mean ± SD (n=6).
To investigate the effects of organoselenium compounds on GSH levels, GSH was measured in whole blood and lung and liver homogenates (Table 2). Feeding p-XSC resulted in significant increases in GSH levels of 37% (P=0.01) and 230% (P=0.0005) over controls in whole blood and lung, respectively. Feeding p-XSeSG also resulted in significant increases in GSH levels of 24% (P=0.04) and 200% (P=0.0002) over controls in whole blood and lung, respectively. Selenomethionine resulted in a significant 34% induction in whole blood GSH (p = 0.003), but no significant change in lung GSH was observed. No significant changes were detected in liver GSH by any of the organoselenium compounds tested.
Table 2.
Impact of organoselenium compounds on glutathione levels and GCL expression in rat lung, liver and blood.
| Diet | Glutathione a | GCLc a, b | GCLm a, b | ||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Whole Blood (μmol/g hemoglobin) | Lung (μmol/g tissue) | Liver (μmol/g tissue) | Lung | Liver | Lung | Liver | |
| Control | 8.17 ± 1.24 | 1.95 ± 0.13 | 8.68 ± 1.88 | 1.00 ± 0.12 | 1.00 ± 0.23 | 1.00 ± 0.22 | 1.00 ± 0.32 |
| p-XSC | 11.2 ± 1.76 c | 4.39 ± 0.74 c | 8.48 ± 0.96 | 1.55 ± 0.29 c | 1.08 ± 0.14 | 1.09 ± 0.08 | 0.94 ± 0.64 |
| p-XSeSG | 10.1 ± 1.75 c | 3.73 ± 0.44 c | 9.12 ± 0.82 | 1.88 ± 0.55 c | 1.06 ± 0.15 | 1.19 ± 0.23 | 1.60 ± 0.71 |
| Selenomethionine | 11.1 ± 0.89 c | 2.32 ± 0.27 | 10.4 ± 1.32 | 1.53 ± 0.31 | 1.05 ± 0.29 | 1.00 ± 0.19 | 1.21 ± 0.95 |
Values are mean ± SE (n=6)
Expression relative to control group
Significantly different from control group, P<0.05
To determine if the rate-limiting enzyme responsible for de novo GSH synthesis, GCL, was induced by organoselenium compounds, protein levels of GCLm and GCLc were compared by western blotting of cytoplasmic lung and liver extracts (Table 2). In the lung, protein levels of GCLc, were significantly induced 55% by p-XSC (P<0.05), and 88% by p-XSeSG (P<0.001). While mean lung GCLc levels in selenomethionine-fed rats were 53% higher than controls, this difference was not significant (P=0.06). No significant differences in relative GCLm protein expression was observed by any organoselenium compound in the lung. Likewise, in the liver, no significant difference in relative protein expression of either GCLc or GCLm was observed by any organoselenium compound.
To investigate the effects of organoselenium compounds on nuclear Nrf2 levels, western blotting was applied to nuclear extracts of lung and liver homogenates from rats fed control, p-XSC, p-XSeSG, or selenomethionine containing diets. Since Nrf2 normally resides in the cytoplasm, it was important to demonstrate that nuclear extracts were not contaminated by cytoplasmic protein. Therefore, western blots of nuclear and cytoplasmic samples were simultaneously probed for the nuclear marker, Lamin A and the cytoplasmic marker, GAPDH (Figure 2A). No cross contamination was detected, even at saturating exposure levels, suggesting high efficacy of the nuclear and cytoplasmic extraction process. Results of nuclear Nrf2 analyses are shown in Figure 2B. Feeding of p-XSC resulted in a significant 6.1-fold increase in nuclear Nrf2 in the lung relative to controls (P=0.0002). No significant changes in lung nuclear Nrf2 were observed by p-XSeSG or selenomethionine. Likewise, in the liver, no significant effects on relative nuclear Nrf2 levels were observed by any organoselenium compound.
Figure 2.
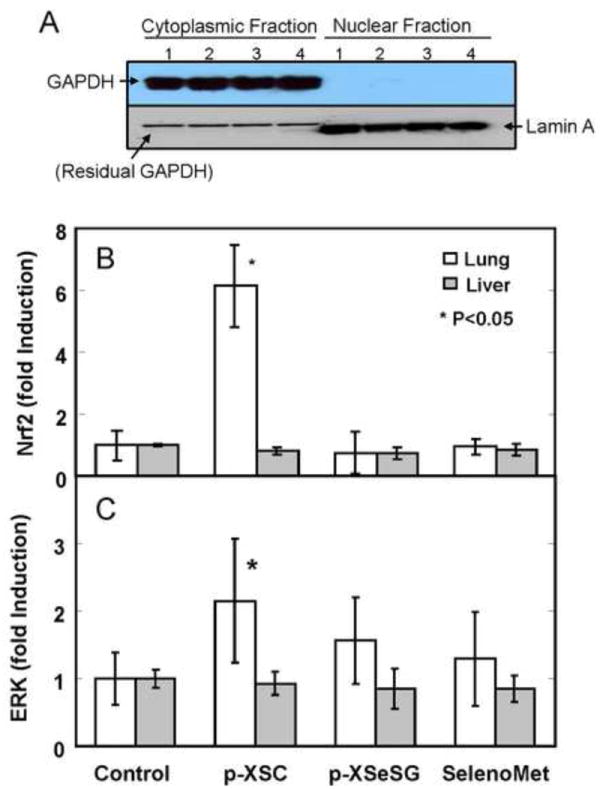
Analysis of nuclear Nrf2 and p-ERK in lung and liver of rats fed organoselenium compounds. (A) Purity of cytoplasmic and nuclear extracts from rat lung were assessed by analyzing levels of GAPDH (cytoplasmic marker) and Lamin A (nuclear marker) of cytoplasmic fractions (lanes 1-4) and nuclear fractions (lanes 5-8) from 4 separate samples of rat lung. Samples were processed and analyzed by Western blot analysis as described in text. Resulting membranes were probed initially with GAPDH antibody (top panel) and later with Lamin A antibody (bottom panel). Relative Nuclear Nrf2 (B) and p-ERK (C) expression in lung and liver of rats fed organoselenium compounds were analyzed. Male F344 rats were fed control diet (AIN-93A) or diets supplemented with p-XSC, p-XSeSG or selenium methionine (10 ppm as selenium) for 2 weeks. Protein expression of nuclear Nrf2 (A) and p-ERK (B), normalized to nuclear Lamin A, in lung and liver was determined by western blotting. Values are mean ± SD (n=6).
Since the ERK pathway is involved in Nrf2 stabilization in vitro [14], we investigated the in vivo effects of organoselenium compounds on nuclear p-ERK levels by western blotting of nuclear extracts of lung and liver homogenates from rats fed control, p-XSC, p-XSeSG, or selenomethionine containing diets (Figure 2C). Feeding of p-XSC resulted in a significant 2.2-fold increase in nuclear p-ERK in the lung relative to controls (p = 0.04) despite a relatively high degree of variation in the p-XSC group. In contrast, no significant changes in lung nuclear p-ERK were observed by p-XSeSG or selenomethionine. Furthermore, no significant induction of nuclear p-ERK was observed in the liver by any organoselenium compound.
Effect of p-XSC and p-XSeSG on viability of HepG2 ARE reporter cell viability
To determine if p-XSC and p-XSeSG activate the ARE pathway in vitro, HepG2-ARE-luciferase reporter cells were employed. Initially, we examined the effect of these agents on cell viability. Results of preliminary time course experiments indicated that a 24-hour exposure time was appropriate to minimize toxic effects by p-XSC, while still allowing observation of luciferase induction as a function of drug concentration. With increasing concentration of either p-XSC or p-XSeSG, viability was reduced, however at equimolar concentrations, p-XSC reduced viability to a greater extent than p-XSeSG for every concentration tested (P<0.05) (Figure 3A). At concentrations of 100 μM, approximately 80% of p-XSeSG treated cells remained viable while cells exposed to this same concentration of p-XSC exhibited a relative viability of 0%, suggesting that all cells had been killed. The concentration of p-XSC resulting in 50% viability was approximately 25 μM.
Figure 3.
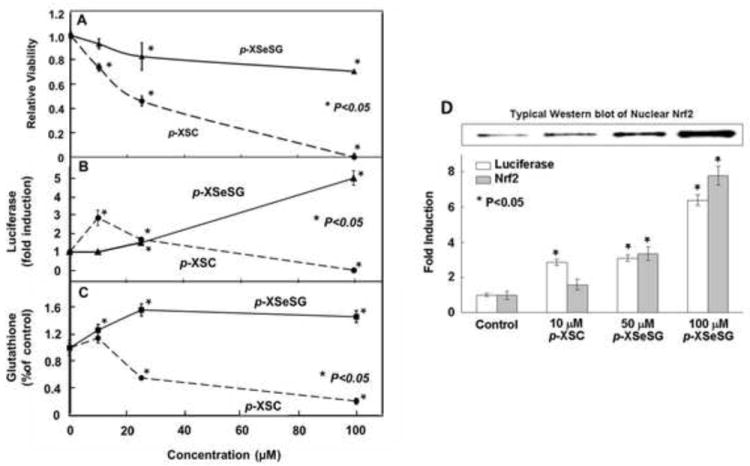
Effect of p-XSC or p-XSeSG on HepG2 ARE-luciferase cell viability, luciferase activity and Nrf2 and GSH levels. Cells at 50% confluence were provided fresh media containing vehicle control or 10, 25 or 100 μM of p-XSC or p-XSeSG. After 24 hours, cells were (A) assayed for viability by MTS absorbance at 490 nm or lysed and assayed for luciferase activity using Promega’s Luciferase Assay System (B) or extracted with 5% metaphosphoric acid and assayed for GSH was measured (C). Data are normalized to control values. (D) For assessment of nuclear Nrf2, cells treated for 24 hr with either p-XSC or p-XSeSG, were washed with PBS, trypsinized and a portion was lysed for analysis of luciferase activity. Nuclear extracts were obtained from remaining cells and subjected to western blotting for Nrf2 and normalized to Lamin A. All data are presented as mean ± SD (n=3).
Effect of p-XSC and p-XSeSG on the activation of ARE-luciferase reporter and the levels of intracellular GSH and nuclear Nrf2 in HepG2 cells
To determine if p-XSC and p-XSeSG could activate Nrf2 and enhance GSH levels in an ARE luciferase reporter cell line, 24-hr time-course studies of luciferase activation and viability were performed after exposure of HepG2 ARE-luciferase reporter cells to various concentrations of each selenium agent. Both p-XSC and p-XSeSG activated the ARE-luciferase reporter in HepG2 cells; fold induction as a function of agent concentration after 24-hour exposures are shown in Figure 3B. A peak induction of 3-fold occurred at a p-XSC concentration of 10 μM with no increases observed at higher concentrations (P<0.05). In contrast, 10 μM p-XSeSG resulted in no significant induction. However, increasing the concentration of p-XSeSG resulted in a higher luciferase activity than was attainable by p-XSC. Above 10 μM, p-XSeSG caused a dose-dependent induction and the highest concentration tested (100 μM) yielded a 5-fold luciferase activity over vehicle-treated control cells (P<0.05).
Because the genes for the rate-limiting enzyme of glutathione synthesis GCL are regulated in part by AREs, it was hypothesized that in HepG2 ARE-luciferase cells GSH induction would occur concurrently with reporter activation. Relative GSH levels in HepG2 ARE-luciferase cells when exposed for 24 hours to varying concentrations of p-XSC or p-XSeSG are shown in Figure 3C. Treatment with p-XSeSG resulted in significant increases in GSH relative to vehicle treated control cells at all concentrations tested. Elevations of 20%, 55%, and 50% were observed at concentrations of 10, 25, and 100 μM, respectively (P<0.05). However, no significant increases in GSH levels were observed by p-XSC treatment at any concentration tested and levels were depleted at the 2 highest doses.
To determine if Nrf2 is responsible for the observed inductions in luciferase expression by p-XSC and p-XSeSG, nuclear extracts from treated HepG2 reporter cells were subjected to Western blot analysis for Nrf2. In Figure 3D a typical blot is shown along with a bar graph showing nuclear Nrf2 quantitation in tandem with luciferase activation after treatment with either selenium agent at 24 hr. Overall, increased nuclear Nrf2 protein levels were generally associated with luciferase activation over all concentrations of organoselenium compounds. Nuclear Nrf2 protein levels were significantly induced in a dose-dependent manner by p-XSeSG (P<0.05). With p-XSC, an increase in luciferase expression was associated with an increase in nuclear Nrf2 levels, however, this difference was not statistically significant (P=0.12).
Dependency of p-XSC and p-XSeSG induction of the ARE-luciferase reporter on Nrf2 activation in HepG2 cells
To examine if increased nuclear Nrf2 is responsible for luciferase induction by organoselenium compounds, short interfering RNA targeting Nrf2 (siNrf2) or non-targeting control RNA (siControl) were transfected into HepG2 ARE-luciferase cells prior to an 24-hour treatment by vehicle control, 10 μM p-XSC or 100 μM p-XSeSG and luciferase induction, nuclear Nrf2 protein and GSH levels were measured (Figure 4). Western blot analysis revealed that nuclear Nrf2 protein levels were successfully reduced by siNrf2 after treatment with either p-XSC or p-XSeSG with an average Nrf2 knockdown of approximately 50%. For both p-XSC and p-XSeSG, luciferase activity was also lowered by siNrf2 treatment. While, glutathione levels were unchanged by siNrf2 in vehicle treated control cells, the approximately 8-fold increase observed in GSH by 100 μM p-XSeSG in siControl cells was reduced to a 3-fold increase after incubation by siNrf2. p-XSC (10 μM) resulted in small and non-significant increases which were marginally reduced by siNrf2.
Figure 4.
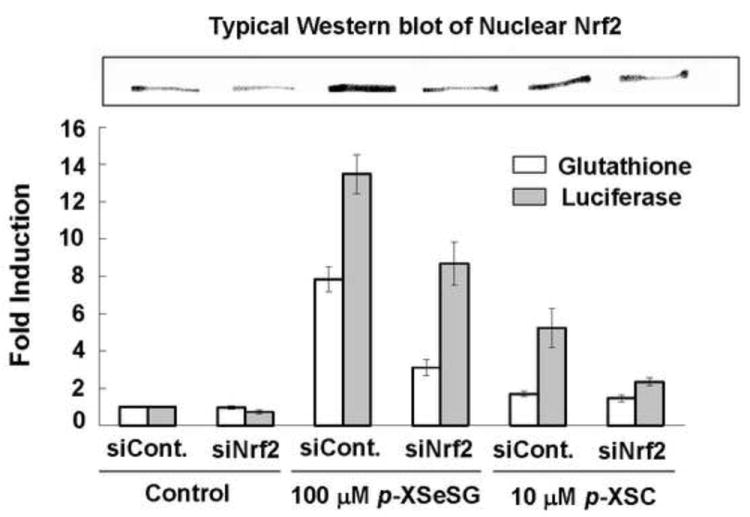
Effects of siNrf2 on Luciferase Activity, GSH and Nuclear Nrf2 in p-XSC or p-XSeSG Treated HepG2 ARE Cells. Cells were grown to 60% confluence then transfected with non-targeting siRNA-Control (siControl) or siRNA-Nrf2 (siNrf2). Thirty hours after siRNA transfection, siControl and siNrf2 cells were exposed to vehicle control, 10 μM p-XSC, or 100 μM p-XSeSG in fresh media. After 18 hours of drug exposure, cell extracts were assayed for luciferase activity and GSH levels and nuclear extracts were analyzed for Nrf2 by Western blotting using Lamin A as a control marker. Data are presented as mean ± SD (n=3).
Dependency of p-XSC and p-XSeSG induction of GSH on Nrf2 activation in mouse embryonic fibroblasts (MEF)
To quantify the dependence of GSH induction by p-XSeSG on the Nrf2 transcription factor, wildtype or Nrf2-deficient (Nrf2 -/-) MEF were employed. The degree of toxicity to MEF cells was assessed using the MTS assay after a 24-hour exposure to various concentrations of p-XSC and p-XSeSG, and results are plotted in Figure 5A. Cell viability was not affected by p-XSeSG, however p-XSC proved to be toxic to both wildtype and Nrf2 -/- cells, with relative viability being reduced to approximately 50% of vehicle treated control values at concentrations of 10 μM. Thus, additional experiments could not be performed with p-XSC, and only p-XSeSG was used in further experiments with MEF cells. Nuclear Nrf2 protein levels, as determined by western blotting of nuclear extracts, were significantly enhanced 2.3-fold (p=0.02) by a 24-hour exposure to 100 μM p-XSeSG in wildtype MEF cells (data not shown). In Nrf2 -/- cells, a similar induction of mutated (dysfunctional) Nrf2 protein was observed.
Figure 5.
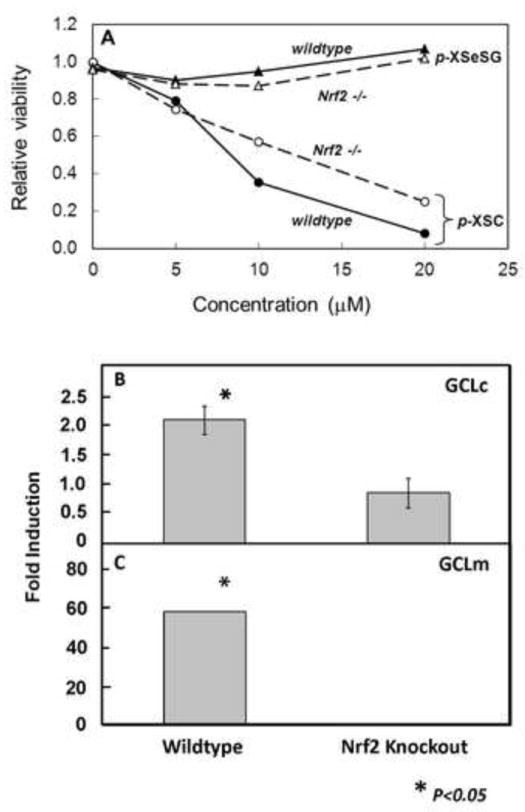
Effect of p-XSC and p-XSeSG on viability and Nrf2 and GCL induction in wildtype and Nrf2 (-/-) mouse embryonic fibroblasts. (A) Cells at 50% confluence were provided fresh media containing vehicle control or 5, 10 or 20 μM of p-XSC or p-XSeSG. After 24 hours, cells were assayed for viability by MTS absorbance at 490 nm. Cells at 50% confluence were provided fresh media containing vehicle control or 100 μM p-XSeSG. After 24 hours, cells were washed with PBS, trypsinized and collected. Cytoplasmic extracts were subjected to Western blotting for GCLm (B) and GCLc (C). All data are normalized to control values and are presented as mean ± SD (n=3).
It was hypothesized that the p-XSeSG-induced Nrf2 in wildtype MEF cells would result in enhanced protein expression of GCLc and/or GCLm, and that this would not be seen in Nrf2 -/- cells. Results of western blotting of cytoplasmic extracts for these subunits are shown in Figures 5B & 5C. The catalytic and modulatory subunits of GCL were significantly enhanced 2-fold (P<0.02) and 60-fold (P<0.05), respectively in wildtype, but not Nrf2 -/-, MEF cells after a 24-hour exposure to 100 μM p-XSeSG.
Discussion
We observed that p-XSC is a potent inducer of GSH in rat lung, but not liver, and that the mechanism involves induction of GCL, the rate-limiting enzyme for GSH biosynthesis. Further, we demonstrate that that the induction of both GCL subunits GCLc and GCLm is likely mediated through the activation of the Nrf2/ARE pathway. Since p-ERK can stabilize Nrf2 and promote its nuclear translocation,[14] its observed induction by p-XSC in the rat lung may indicate a role for this pathway in the enhancement of nuclear Nrf2 levels. We previously observed that p-XSC feeding results in elevated GSH in lungs of A/J mice, but this is tempered by the fact that this strain is highly susceptible to spontaneous lung tumors [5]. The present finding that p-XSC induces GSH in the lung of the Fisher rat, a species without susceptibility to lung tumors, thus generalizes the in vivo potential of p-XSC as a chemopreventive and GSH inducing agent.
Feeding of selenomethionine resulted in no significant change in lung GSH, GCLc, nuclear Nrf2 or p-ERK. Differential effects between p-XSC, p-XSeSG and selenomethionine do not appear to be driven by differences in delivery of the agent or its metabolites to the target tissue as the resulting increases in lung selenium levels in selenomethionine fed rats were 2-3-fold greater than for either p-XSC or p-XSeSG. These findings highlight the importance of the form of selenium, rather than simply the absolute levels of elemental selenium reaching the target tissue. Previous studies have shown that selenomethionine lacks chemopreventive activity in laboratory animals and is weakly active at toxic non-physiological selenium levels [22-24].
To examine the dependence of GCL induction on Nrf2 we utilized cell lines previously developed to screen for inducers of Phase II chemoprotective genes [19, 20]. We show that the chemopreventive organoselenium compounds p-XSC and p-XSeSG induced a HepG2-ARE luciferase reporter, suggesting they act through similar mechanisms and that Nrf2 is involved in the process. Previous studies have demonstrated that selenium-containing agents, including p-XSC and related compounds, are effective inducers of Phase II detoxification enzymes [27][28]. Our present results suggest that induction of the Nrf2-ARE pathway may also be responsible for the effects of p-XSC on these important detoxification pathways.
The GSH conjugate of p-XSC, p-XSeSG has been less studied than p-XSC, but nevertheless has been shown to be an effective chemopreventive agent against both mammary and colon cancer in vivo [4, 25]. For example, in a rat model, p-XSeSG was found to inhibit COX activity and to be effective against AOM-induced colon cancer with low toxicity [4]. Here we show, for the first time that p-XSeSG induces lung GSH in vivo, and to a similar extent as p-XSC. Furthermore, like p-XSC, GCLc was significantly induced in the rat lung by p-XSeSG. These data suggest that p-XSC and p-XSeSG may act via similar pathways. A peak induction of 3-fold occurred at a p-XSC concentration of 10 μM, whereas higher doses were required for p-XSeSG.
In cells in culture, we demonstrated that Nrf2 is involved in p-XSeSG-mediated activation of in vitro GSH synthesis, and that p-XSeSG is a more potent and less toxic inducer of the ARE-regulated genes, GCLc and GCLm, than is p-XSC. The addition of p-XSeSG to cells in culture resulted in significantly elevated levels of nuclear Nrf2 protein in both HepG2 and MEF cells. Furthermore, luciferase activity in HepG2 ARE reporter cells is induced to levels that correlate well with nuclear Nrf2 levels, thus providing strong evidence that Nrf2 is the transcription factor responsible for induction of the luciferase reporter, and therefore is available for activation of other ARE regulated genes such as the subunits of GCL. Since in both cell culture systems, p-XSeSG, the putative major in vivo metabolite of p-XSC, was more effective at inducing GCLc and GSH levels and the efficacy of p-XSC was limited by its toxic effects on cell survival, it is likely that GSH induction by p-XSC is mediated by p-XSeSG.
It has been established that the genes responsible for de novo glutathione synthesis are regulated in part by the ARE pathway [9]. Indeed, we find that glutathione levels are significantly enhanced by p-XSeSG treatment in HepG2 cells, and demonstrated that both catalytic and modulatory subunits of the rate limiting enzyme of glutathione synthesis, GCLc and GCLm, are significantly enhanced by p-XSeSG in wildtype MEF cells. That nuclear Nrf2, GCL subunit protein expression and cellular GSH were concurrently induced upon p-XSeSG treatment does not prove causality. However, the effects of specific experimental manipulation of Nrf2, either by short interfering RNA in HepG2 cells or through the use of Nrf2-deficient MEF cells, provide powerful evidence of such a relationship. It was hypothesized that depletion of available Nrf2 in HepG2 reporter cells would result in proportionally lowered ARE-luciferase activity and GSH levels. Short interfering RNA specifically targeting Nrf2 in HepG2 cells successfully resulted in an average 50% knockdown of nuclear Nrf2 protein levels, and that this brought about similar reductions in p-XSeSG-mediated luciferase activation and GSH levels.
While the experiments employing partial knockdown of Nrf2 by RNA interference support the hypothesis that Nrf2 is involved in p-XSeSG-mediated GSH synthesis, such experiments do not fully prove dependence. However, complete reliance of this process upon Nrf2 is supported by experiments with Nrf2-deficient MEF cells. Neither subunit of GCL was induced by p-XSeSG treatment of Nrf2 -/- cells despite elevated, albeit dysfunctional, nuclear Nrf2 protein levels and marked and significant enhancements of GCL subunits in their wildtype MEF counterpart.
It is important to address possible reasons for the differences observed between p-XSC and p-XSeSG-mediated responses in both HepG2 and MEF cells. Luciferase activity was induced by p-XSC in HepG2 ARE reporter cells, however concentration-response characteristics differ from those observed by p-XSeSG. At 10uM, the concentration at which p-XSC exhibited maximal ARE induction, p-XSeSG had no effect. However, induction by 100uM p-XSeSG surpassed the peak induction attainable by p-XSC by almost 2-fold. Viability data indicates that p-XSeSG is less toxic to HepG2 cells than is p-XSC, and it is likely that toxicity of p-XSC may diminish its ability to induce the HepG2 ARE-luciferase reporter. The implications of these differential effects of p-XSC and p-XSeSG on cell viability to in vivo toxicity and efficacy are not known, and will likely depend on the metabolism and distribution of these agent as well as target cell type. Glutathione was not significantly induced by p-XSC at any concentration tested, although the concentration-response curve paralleled that of ARE-luciferase activation. It is likely that GSH depletion at toxic doses of p-XSC is responsible in part for limiting its ability to induce the ARE. p-XSC was also shown to reduce GSH to less than 50% of control values in human prostate cancer cells [26].
While p-XSeSG was effective at inducing Nrf2 in vitro, no increase in Nrf2 was observed by p-XSeSG in rat lung after 24 hr. As a glutathiolated product of p-XSC, p-XSeSG is likely to have a significantly shorter half-life and thus may cause an earlier and shorter-lived induction of Nrf-2 than for p-XSC, which may have been missed in our experimental design. Even a short pulse of Nrf2 induction following feeding of an inducer could result in a sustained induction of GCLc and GSH. Alternatively, it is possible that Nrf2-independent pathways are operative in p-XSeSG-mediated GSH induction in vitro.
It is of interest that the liver was not responsive to GSH-induction by either p-XSC or p-XSeSG in vivo. This is apparently due to the lack of an induction of GCLc or GCLm by the nrf2 pathway as neither nrf2 nor p-ERK were induced by supplementation with either compound. These findings are consistent with previous findings in rats where p-XSC feeding induced lung but not liver GST-α and GST-π activities after 1 week, however total GST activity (using CDNB as the substrate) was enhanced in both liver and lung [27]. In mice, GPX activity was induced by p-XSC feeding in lung but not in liver [28]. However, in that same study, similar increases in GST-π and GST-μ were observed in lung and liver. It is possible that the 2-week time point used in the present study was not optimal for observation of an effect in liver, however, other preliminary findings from our laboratory suggest that p-XSC is not effective at inducing GSH after 1 week and 1 month as well. The differing results of p-XSC on enzyme activities in liver may result from the complex nature mechanisms involved in their regulation.
In summary, we have shown that induction of the Nrf2 transcription factor is involved in the enhancement of lung GSH by p-XSC and that p-XSeSG may be the metabolite responsible for such activation. Thus, p-XSeSG may represent a less toxic, yet highly efficacious alternative for p-XSC as an inducer of lung GSH. Induction of GCLc, and possibly other Nrf2-mediated Phase II genes, may represent a general mechanism of chemoprevention by p-XSC and related organoselenium compounds. Induction of GSH with less toxic agents like p-XSeSG may prove useful for treatment of other conditions associated with GSH depletion such as chronic obstructive pulmonary disease (COPD), acute respiratory distress syndrome and cystic fibrosis [29].
Highlights.
-
>
The potent chemopreventive agent p-XSC selectively induces lung GSH levels
-
>
GSH enhancement involves induction of GCL through activation of nrf2
-
>
The GSH conjugate (p-XSeSG) may be the active p-XSC metabolite
-
>
p-XSeSG may represent a potent and less toxic alternative for glutathione induction
Acknowledgments
This work was supported in part by Penn State Hershey Cancer Institute seed funding.
Abbreviations
- p-XSC
1,4-phenylenebis(methylene)selenocyanate
- p-XSeSG
glutathione conjugate of p-XSC or N,N’-[1,4-phenylenebis[methyleneselenothio[(1R)-1-[[(carboxymethyl)amino]carbonyl]-2,1-ethanediyl]]]bis-L-glutamine
- GSH
glutathione
- GCL
γ-glutamyl cysteine ligase
- GCLc
catalytic subunit of γ-glutamyl cysteine ligase
- GCLm
modulatory subunit of γ-glutamyl cysteine ligase
- ARE
antioxidant response element
- Nrf2
nuclear factor-erythroid 2-related factor 2
- siNrf2
short interfering RNA targeting Nrf2
- MEF
mouse embryonic fibroblasts
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- siControl
non-targeting control RNA
- Nrf2 -/-
Nrf2-deficient
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Clark LC, Combs GF, Jr, Turnbull BW, Slate EH, Chalker DK, Chow J, Davis LS, Glover RA, Graham GF, Gross EG, Krongrad A, Lesher JL, Jr, Park HK, Sanders BB, Jr, Smith CL, Taylor JR. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. Jama. 1996;276:1957–1963. [PubMed] [Google Scholar]
- 2.El-Bayoumy K, Sinha R. Mechanisms of mammary cancer chemoprevention by organoselenium compounds. Mutation research. 2004;551:181–197. doi: 10.1016/j.mrfmmm.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 3.Tanaka T, Makita H, Kawabata K, Mori H, El-Bayoumy K. 1,4-phenylenebis(methylene)selenocyanate exerts exceptional chemopreventive activity in rat tongue carcinogenesis. Cancer Res. 1997;57:3644–3648. [PubMed] [Google Scholar]
- 4.Rao CV, Wang CQ, Simi B, Rodriguez JG, Cooma I, El-Bayoumy K, Reddy BS. Chemoprevention of colon cancer by a glutathione conjugate of 1,4-phenylenebis(methylene)selenocyanate, a novel organoselenium compound with low toxicity. Cancer Res. 2001;61:3647–3652. [PubMed] [Google Scholar]
- 5.Richie JP, Jr, Kleinman W, Desai DH, Das A, Amin SG, Pinto JT, El-Bayoumy K. The organoselenium compound 1,4-phenylenebis(methylene)selenocyanate inhibits 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced tumorgenesis and enhances glutathione-related antioxidant levels in A/J mouse lung. Chemico-biological interactions. 2006;161:93–103. doi: 10.1016/j.cbi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 6.El-Bayoumy K, Richie JP, Jr, Boyiri T, Komninou D, Prokopczyk B, Trushin N, Kleinman W, Cox J, Pittman B, Colosimo S. Influence of selenium-enriched yeast supplementation on biomarkers of oxidative damage and hormone status in healthy adult males: a clinical pilot study. Cancer Epidemiol Biomarkers Prev. 2002;11:1459–1465. [PubMed] [Google Scholar]
- 7.Townsend DM, Tew KD, Tapiero H. The importance of glutathione in human disease. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2003;57:145–155. doi: 10.1016/s0753-3322(03)00043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huber WW, Parzefall W. Thiols and the chemoprevention of cancer. Current opinion in pharmacology. 2007;7:404–409. doi: 10.1016/j.coph.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 9.Wild AC, Moinova HR, Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. The Journal of biological chemistry. 1999;274:33627–33636. doi: 10.1074/jbc.274.47.33627. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Gordon GB. A strategy for cancer prevention: stimulation of the Nrf2-ARE signaling pathway. Molecular cancer therapeutics. 2004;3:885–893. [PubMed] [Google Scholar]
- 11.Zhang DD, Lo SC, Sun Z, Habib GM, Lieberman MW, Hannink M. Ubiquitination of Keap1, a BTB-Kelch substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteasome-independent pathway. The Journal of biological chemistry. 2005;280:30091–30099. doi: 10.1074/jbc.M501279200. [DOI] [PubMed] [Google Scholar]
- 12.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Molecular and cellular biology. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakurai T, Kanayama M, Shibata T, Itoh K, Kobayashi A, Yamamoto M, Uchida K. Ebselen a seleno-organic antioxidant, as an electrophile. Chemical research in toxicology. 2006;19:1196–1204. doi: 10.1021/tx0601105. [DOI] [PubMed] [Google Scholar]
- 14.Xu C, Yuan X, Pan Z, Shen G, Kim JH, Yu S, Khor TO, Li W, Ma J, Kong AN. Mechanism of action of isothiocyanates: the induction of ARE-regulated genes is associated with activation of ERK and JNK and the phosphorylation and nuclear translocation of Nrf2. Molecular cancer therapeutics. 2006;5:1918–1926. doi: 10.1158/1535-7163.MCT-05-0497. [DOI] [PubMed] [Google Scholar]
- 15.Sohn OS, Desai DH, Das A, Rodriguez JG, Amin SG, El-Bayoumy K. Comparative excretion and tissue distribution of selenium in mice and rats following treatment with the chemopreventive agent 1,4-phenylenebis(methylene)selenocyanate. Chemico-biological interactions. 2005;151:193–202. doi: 10.1016/j.cbi.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 16.El-Bayoumy K, Narayanan BA, Desai DH, Narayanan NK, Pittman B, Amin SG, Schwartz J, Nixon DW. Elucidation of molecular targets of mammary cancer chemoprevention in the rat by organoselenium compounds using cDNA microarray. Carcinogenesis. 2003;24:1505–1514. doi: 10.1093/carcin/bgg103. [DOI] [PubMed] [Google Scholar]
- 17.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 18.Richie JP, Jr, Skowronski L, Abraham P, Leutzinger Y. Blood glutathione concentrations in a large-scale human study. Clin Chem. 1996;42:64–70. [PubMed] [Google Scholar]
- 19.Cuendet M, Oteham CP, Moon RC, Pezzuto JM. Quinone reductase induction as a biomarker for cancer chemoprevention. Journal of natural products. 2006;69:460–463. doi: 10.1021/np050362q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, Ford LG, Parnes HL, Minasian LM, Gaziano JM, Hartline JA, Parsons JK, Bearden JD, 3rd, Crawford ED, Goodman GE, Claudio J, Winquist E, Cook ED, Karp DD, Walther P, Lieber MM, Kristal AR, Darke AK, Arnold KB, Ganz PA, Santella RM, Albanes D, Taylor PR, Probstfield JL, Jagpal TJ, Crowley JJ, Meyskens FL, Jr, Baker LH, Coltman CA., Jr Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) Jama. 2009;301:39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El-Bayoumy K. The negative results of the SELECT study do not necessarily discredit the selenium-cancer prevention hypothesis. Nutrition and cancer. 2009;61:285–286. doi: 10.1080/01635580902892829. [DOI] [PubMed] [Google Scholar]
- 23.Facompre ND, El-Bayoumy K, Sun YW, Pinto JT, Sinha R. 1,4-phenylenebis(methylene)selenocyanate, but not selenomethionine, inhibits androgen receptor and Akt signaling in human prostate cancer cells. Cancer prevention research (Philadelphia, Pa) 3:975–984. doi: 10.1158/1940-6207.CAPR-10-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li GX, Lee HJ, Wang Z, Hu H, Liao JD, Watts JC, Combs GF, Jr, Lu J. Superior in vivo inhibitory efficacy of methylseleninic acid against human prostate cancer over selenomethionine or selenite. Carcinogenesis. 2008;29:1005–1012. doi: 10.1093/carcin/bgn007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El-Bayoumy K, Das A, Boyiri T, Desai D, Sinha R, Pittman B, Amin S. Comparative action of 1,4-phenylenebis(methylene)selenocyanate and its metabolites against 7,12-dimethylbenz[a]anthracene-DNA adduct formation in the rat and cell proliferation in rat mammary tumor cells. Chemico-biological interactions. 2003;146:179–190. doi: 10.1016/j.cbi.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 26.Pinto JT, Sinha R, Papp K, Facompre ND, Desai D, El-Bayoumy K. Differential effects of naturally occurring and synthetic organoselenium compounds on biomarkers in androgen responsive and androgen independent human prostate carcinoma cells. International journal of cancer. 2007;120:1410–1417. doi: 10.1002/ijc.22500. [DOI] [PubMed] [Google Scholar]
- 27.Sohn OS, Fiala ES, Upadhyaya P, Chae YH, El-Bayoumy K. Comparative effects of phenylenebis(methylene)selenocyanate isomers on xenobiotic metabolizing enzymes in organs of female CD rats. Carcinogenesis. 1999;20:615–621. doi: 10.1093/carcin/20.4.615. [DOI] [PubMed] [Google Scholar]
- 28.Prokopczyk B, Rosa JG, Desai D, Amin S, Sohn OS, Fiala ES, El-Bayoumy K. Chemoprevention of lung tumorigenesis induced by a mixture of benzo(a)pyrene and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone by the organoselenium compound 1,4-phenylenebis(methylene)selenocyanate. Cancer letters. 2000;161:35–46. doi: 10.1016/s0304-3835(00)00590-5. [DOI] [PubMed] [Google Scholar]
- 29.Rahman I, MacNee W. Oxidative stress and regulation of glutathione in lung inflammation. Eur Respir J. 2000;16:534–554. doi: 10.1034/j.1399-3003.2000.016003534.x. [DOI] [PubMed] [Google Scholar]