

Abstract
Histone3-lysine79 (H3K79) methyltransferase DOT1L has been found to be a drug target for acute leukemia with MLL (mixed lineage leukemia) gene translocations. A total of 55 adenosine-containing compounds were designed and synthesized, among which several potent DOT1L inhibitors were identified with Ki values as low as 0.5 nM. These compounds also show high selectivity (>4,500-fold) over three other histone methyltransferases. Structure activity relationships (SAR) of these compounds for their inhibitory activities against DOT1L are discussed. Potent DOT1L inhibitors exhibit selective activity against the proliferation of MLL-translocated leukemia cell lines MV4;11 and THP1 with EC50 values of 4–11 μM. Isothermal titration calorimetry studies showed two representative inhibitors bind with a high affinity to the DOT1L:nucleosome complex, and only compete with the enzyme cofactor SAM (S-adenosyl-L-methionine), but not the substrate nucleosome.
INTRODUCTION
Post-translational modifications of a lysine residue of histone play important roles in gene regulation.1 These chemical modifications include acetylation and methylation, which change the electrostatic and/or steric properties of histone and thereby control the accessibility of genes wrapped around the histone core. Histone methyltransferase (HMT) consists of a family of >50 enzymes that methylate the sidechain amino (or guanidine) group of a lysine (or arginine) residue of histone or other proteins.2,3 As schematically illustrated in Figure 1, all HMTs use S-adenosyl-L-methionine (SAM) as the methyl donor (enzyme cofactor), producing methylated substrates as well as S-adenosyl-L-homocysteine (SAH). Aberrant histone methylations have been linked to many types of cancer,4 and there is therefore great interest in developing inhibitors of these enzymes.5 However, only a limited number of compounds have been reported to be histone methyltransferase inhibitors.5–8
Figure 1.

DOT1L catalyzed reaction and inhibitors.
DOT1L is a histone lysine methyltransferase that specifically methylates the residue Lys79 of histone 3 (H3K79).9,10 The catalytic domain of human DOT1L is highly conserved from yeasts to mammals. There are two features that make DOT1L a unique HMT. First, H3K79 is located in the ordered histone octamer core structure, while the methylation sites of all other HMTs are in the unordered tail of a histone protein. Second, DOT1L is the only histone lysine methyltransferase that belongs to class I methyltransferases. All other histone lysine methyltransferases are class V methyltransferases with a conserved SET domain. Of particular interest is that DOT1L has been proposed to be a target for acute leukemia with MLL (mixed lineage leukemia) gene translocations.11–13 Compared to other leukemias, this subtype of leukemia has a poor prognosis with a 5-year survival of <40%.14 Studies showed that several predominant fusion partners of onco-MLL genes, including AF4, AF9, AF10 and ENL, can recruit DOT1L, which methylates H3K79, causes overexpression of leukemia relevant genes (such as Hoxa9 and Meis1) that eventually leads to leukemia initiation. In addition, H3K79 hypermethylation was also found to be a hallmark of MLL-rearranged leukemia. A potent inhibitor EPZ004777 (1, Figure 1) was recently reported, which can selectively inhibit the proliferation of MLL-rearranged leukemia cell lines and prolong the life spans of experimental animals in a mouse model of human MLL leukemia.7a This further pharmacologically validated DOT1L as an anti-leukemia target. However, due to its very short half-life in plasma, 1 has a low plasma concentration of ~0.5 μM even when supplied continuous with an implanted pump. Clearly, more stable inhibitors are needed.
Our early work also disclosed a structure-based approach to the identification of several potent DOT1L inhibitors, such as 2–4 (Figure 1), showing introducing an N6-substituent on the adenine ring can provide high selectivity for DOT1L.7b Likely due to containing a reactive N-2-iodoethyl group (a precursor of aziridinium), these DOT1L selective inhibitors do not have specific activity in killing MLL-rearranged leukemia cells. In a parallel effort to find competitive DOT1L inhibitors, we also identified urea-containing compounds that possess strong inhibitory activity against DOT1L. Here, we report the synthesis, structure activity relationships (SAR) and biological activities of 55 compounds targeting DOT1L. In addition, isothermal titration calorimetry (ITC) was used to investigate the binding affinities of selected inhibitors to DOT1L.
RESULTS AND DISCUSSION
Inhibitor design and SARs for DOT1L inhibition
The poor pharmacokinetics of 1 is likely due to its 7-deaza-adenosine moiety, which can be recognized by many human enzymes, such as adenosine deaminase, hydrolase, or nucleosidase, leading to a rapid degradation. Other compounds containing adenosine or an adenosine-like group suffer from a similar metabolic instability.15,16 In our early work,7b we found DOT1L, but not other HMTs, can well tolerate an additional substituent as big as a benzyl group at the N6 position of adenine ring and the crystal structure of the DOT1L:2 complex shows the N6-methyl group is located in a mainly hydrophobic pocket, surrounded by Phe223, Leu224, Val249, Lys187 and Pro133 shown in Figure 2. Therefore, we first explore the structure activity relationship (SAR) at the N6 position of adenine, in an effort to find potent DOT1L inhibitors having a substituted adenosine moiety that could possess improved metabolic stability.
Figure 2.

Active site of the DOT1L:2 structure (PDB code: 3SR4), with compound 2 shown as a ball and stick model and hydrogen bonds as dotted lines.
First, we synthesized a series of SAH derivatives 2, 5–12 containing nine N6-substituents. The structures and inhibitory activities of these compounds are shown in Table 1, together with those of SAH, the common product of HMT catalyzed reactions and a non-selective HMT inhibitor. Compound 2 with an N6-methyl is still a potent DOT1L inhibitor with a Ki value of 290 nM. Although it is slightly less active than SAH (Ki = 160 nM), its methyl group provides excellent selectivity for DOT1L, with only weak or no activity against other HMTs.7b Compounds 5–8 with an allyl, cyclopropyl, isopropyl and n-butyl group, respectively, have considerably reduced activity with Ki values of 2.0–3.8 μM, as compared to 2. Compounds 9–11 bearing a branched butyl or pentyl chain are even less active (Ki = 6.5–8.9 μM). In addition, compound 12 having an N6-benzyl group has the second best inhibitory activity in this series with a Ki of 1.1 μM. These results show the small methyl group in 2 provides the best activity as well as selectivity.
Table 1.
Structures of inhibitors and their Ki values against DOT1L.
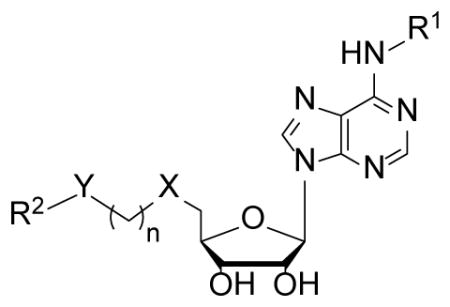
| ||
|---|---|---|
| compound | R- | Ki (μM) |
| SAH | -H | 0.16 |
| 2 | -CH3 | 0.29 |
| 5 | -allyl | 2.0 |
| 6 | -c-Pr | 2.0 |
| 7 | -i-Pr | 3.8 |
| 8 | -n-Bu | 2.8 |
| 9 | -C(CH3)3 | 7.8 |
| 10 | -CH2CH2CH(CH3)2 | 6.5 |
| 11 | -CH2C(CH3)3 | 8.9 |
| 12 | -CH2Ph | 1.1 |
Next, we wanted to replace the highly polar, charged amino acid moiety (at physiological pH) of compound 2 and SAH, since it renders these compounds limited bioavailability. Although the amino acid group forms five H-bonds with DOT1L, the pocket also contains several hydrophobic residues including Phe239, Val169, Thr139 and Tyr 136 (Figure 2), suggesting a more lipophilic sidechain might also be favorable. To this end, 15 compounds with a general structure in Chart 1 were synthesized and tested for their inhibition against DOT1L and the results are also shown in Chart 1. In general, these compounds are inactive or only weakly active. Compound 13 (Ki = ~100 μM) is decarboxy-SAH, but it is >600× less active than SAH. Compound 14 with a shorter linker has a Ki value of 25 μM, being slightly more active than 13. Changing -S- to -NH- resulted in an inactive compound 15. Compounds 16–18 having a carboxyl, hydroxyl or propargyl group possess no activity against DOT1L. Compounds 19–25 containing a phenyl or hetercyclic aromatic ring are also inactive. However, compounds 26 and 27 with a carbamate group, which are actually the precursors for making compounds 13 and 15, were found to be weak inhibitors of DOT1L with Ki values of 50 and 46 μM. These two compounds are of interest, since they possess greatly improved lipophilicity as well as unexpected, higher activity than 13 and 15.
Chart 1.

Structures and activities of compounds 13–27.
In order to find compounds with improved potency, three related series of compounds based on the structures of 26 and 27 were synthesized, including carbamates, amides and ureas, and the results are summarized in Table 2. Compound 28, a thioether analog of 27, possesses essentially the same activity (Ki = 47 μM), while changing the benzyl group to an ethyl for compound 29 makes it inactive, showing an aromatic ring is favored for the R2 group (in the general structure shown in Table 2). Introducing an additional isopropyl substituent into the 5′-N atom for compound 30 (Ki = 22 μM) resulted in a ~2-fold activity increase, as compared to its parent compound 27. We next sought whether other functional groups can provide improved inhibitory potency. Analogous amide compounds 31–33 were found to be inactive. A reversed amide compound 34 (with the same skeleton length) as well as a reversed carbamate compound 35, however, exhibit ~2× improved inhibition against DOT1L with Ki values of 22 and 16 μM, as compared to 26–28. Urea compound 36 was found to have a greatly enhanced activity with a Ki value of 1.9 μM. Compounds 37–40 containing a different R2 group were therefore synthesized to optimize this position. Although 37–39 having 2-furanyl-methyl, propyl, and cyclohexyl group are less active than 36, compound 40 with a Ki of 0.55 μM exhibits ~3× more activity, showing a phenyl group here is more favorable than an alkyl or aryl-substituted alkyl group. In addition, these results demonstrate both -NH- moieties of the urea group are very important, with each one offering ~25–>50-fold activity improvements, as compared to less favored -O- and -CH2- groups (e.g., compound 36 vs. 28, as well as compound 40 vs. 34/35). These two -NH- could serve as H-bond donors to increase the binding affinity to DOT1L. Next, compounds 41 and 42 having a 2 and 4 methylene linker, respectively, were prepared and found to possess less activity (Ki = 18 and 1.1 μM) than compound 40 with a 3 methylene linker. This shows a three -CH2- linker between the urea and the 5′-position represents the optimal length for the inhibition.
Table 2.
Structures of compounds 26–58 and their Ki values against DOT1L.

| ||||||
|---|---|---|---|---|---|---|
| Compound | R2 | Y | n | X | R1 | Ki (μM) |
| 26 | t-Bu |
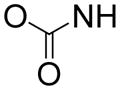
|
3 | S | H | 50 |
| 27 | PhCH2 | 3 | NH | H | 46 | |
| 28 | PhCH2 | 3 | S | H | 47 | |
| 29 | Et | 3 | S | H | >100 | |
| 30 | PhCH2 | 3 | (i-Pr)N | H | 22 | |
|
| ||||||
| 31 | PhCH2 |
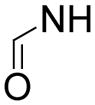
|
3 | S | H | >100 |
| 32 | Ph | 3 | S | H | >100 | |
| 33 | 2-naphthyl | 3 | S | H | >100 | |
|
| ||||||
| 34 | -Ph |
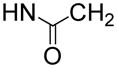
|
3 | S | H | 22 |
| 35 | -Ph |
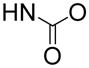
|
3 | S | H | 16 |
|
| ||||||
| 36 | PhCH2 |
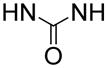
|
3 | S | H | 1.9 |
| 37 | 2-furanyl-CH2 | 3 | S | H | 5.5 | |
| 38 | n-Pr | 3 | S | H | 17 | |
| 39 | c-hexyl | 3 | S | H | 4.5 | |
| 40 | Ph | 3 | S | H | 0.55 | |
| 41 | Ph | 2 | S | H | 18 | |
| 42 | Ph | 4 | S | H | 1.1 | |
| 43 | 4-i-Pr-Ph | 3 | S | H | 0.35 | |
| 44 | 4-t-Bu-Ph | 3 | S | Me | 0.070 | |
| 45 | 4-Cl-Ph | 3 | S | H | 0.082 | |
| 46 | 4-Br-Ph | 3 | S | H | 0.19 | |
| 47 | 4-I-Ph | 3 | S | H | 0.27 | |
| 48 | 4-CF3-Ph | 3 | S | H | 0.058 | |
| 49 | 4-OCF3-Ph | 3 | S | H | 0.14 | |
| 50 | 2-Et-Ph | 3 | S | H | 0.69 | |
| 51 | 3-Et-Ph | 3 | S | H | 0.13 | |
| 52 | 2,4-di-F-Ph | 3 | S | H | 1.0 | |
| 53 | 3,5-di-CF3-Ph | 3 | S | H | 0.080 | |
| 54 | 1-naphthyl | 3 | S | H | 2.7 | |
| 55 | 4-t-Bu-Ph | 3 | (i-Pr)N | H | 0.00046 | |
| 56 | 4-t-Bu-Ph | 3 | (i-Pr)N | Me | 0.00076 | |
| 57 | 4-t-Bu-Ph | 3 | (i-Pr)N | allyl | 0.012 | |
| 58 | 4-t-Bu-Ph | 3 | (i-Pr)N | CH2Ph | 0.022 | |
Compounds 43–54 that are derivatives of 40 were synthesized, in an effort to find an appropriate substituent on the phenyl ring. As can be seen in Table 2, all of the 4-substituents in 43–49 (Ki = 58–350 nM), including isopropyl, tert-butyl, -Cl, -Br, -I, -CF3 and -OCF3, provide improved activity, as compared to the parent compound 40 (Ki = 550 nM). Among these, bulky tert-butyl (in 44) and electron-withdrawing -CF3 group (in 48) render >8-fold potency enhancement. For halogen substituents (in compounds 45–47), a trend of -Cl > -Br > -I was observed. Compound 50 with a 2-ethyl substituent possesses a slightly decreased activity (Ki = 690 nM), while 51 having a 3-ethyl exhibits a 4× stronger inhibition (Ki = 130 nM). 2,4-difluoro-substituened compound 52 is less active than 40, while another disubstituted compound 53 with 3,5-di-CF3 represents one of the best inhibitors with a Ki value of 80 nM. Compound 54 with a 1-naphthyl R2 group (i.e., 2,3-disubstituted phenyl) is considerably less active (Ki = 2700 nM) than compound 40.
We next synthesized compound 55 by replacing the -S- of 44 with a -N(i-Pr)- linkage, which in compound 30 shows improved activity. Compound 55 was found to be an extremely potent DOT1L inhibitor with a Ki value of 0.46 nM. The large activity boost (as compared to that of 44 with a Ki of 70 nM) might be due to coordinated protein conformational changes induced by co-binding of the N-(4-tert-butylphenyl)urea sidechain and the -N(i-Pr)- group of 55. Introducing a N6-methyl group led to compound 56, which possesses a similar inhibitory activity against DOT1L (Ki = 0.76 nM), while compounds 57 and 58 having N6-allyl and benzyl groups were found to be less active with Ki values of 12 and 22 nM, respectively.
Synthesis and activity testing of compounds 2, 5–58 targeting DOT1L provide several novel observations for structure activity relationship (SAR). First, a series of urea-containing adenosine derivatives were found to be potent DOT1L inhibitors with Ki values as low as 0.46 nM, with each of the two -NH- moieties of the urea offering 25–>50-fold activity enhancements. Second, 3- or 4-substituted phenyl is a preferred R2 group. Third, introducing a third substituent at the 5′-position, such as an isopropyl, can further improve the inhibitory activity. Fourth, a three-methylene linker between the urea and the 5′-position is the optimal length for DOT1L inhibition. Fifth, an N6-substituent on the adenine ring in general decreases the activity, while a small substituent at this position, such as methyl (as found in compound 56 with a Ki of 0.76 nM), could help provide desired selectivity as well as stability, without much activity loss.
Biological activity of selected DOT1L inhibitors
From the above SAR studies, we have obtained several highly potent DOT1L inhibitors having Ki values as low as 0.46 nM. We next investigated whether these compounds have enzyme activity against other HMTs. Selectivity is of importance for developing clinically useful DOT1L inhibitors that target the SAM binding site, since all HMTs use a similar mechanism of catalysis. Our previous structure-guided design showed a N6 substituent, such as the methyl in compound 2, provides excellent selectivity for DOT1L (Table 3).7b Potent DOT1L inhibitors 55–58 were chosen to be tested against 3 representative HMTs, i.e., PRMT1, CARM1 (or called PRMT4) and SUV39H1. PRMT1 and CARM1 are histone/protein arginine methyltransferases, which also belong to class I methyltransferases and share certain similar structural features as DOT1L. SUV39H1 is a typical SET domain histone lysine methyltransferase, belonging to the class V methyltransferases having a distinct structure. As can be seen in Table 3, drastically different from non-selective inhibitor SAH, compounds 55–58 exhibit essentially no activity against all of these three HMTs, showing excellent selectivity (>4,500-fold).
Table 3.
Ki values (μM) of DOT1L inhibitors on other HMTs.
| compound | DOT1L | PRMT1 | CARM1 | SUV39H1 |
|---|---|---|---|---|
| SAH | 0.16 | 0.40 | 0.86 | 4.9 |
| 2 | 0.29 | 22.7 | >100 | >100 |
| 55 | 0.00046 | >100 | >100 | >100 |
| 56 | 0.00076 | >100 | >100 | >100 |
| 57 | 0.012 | >100 | >100 | >100 |
| 58 | 0.022 | >100 | >100 | >100 |
Next, we tested the activities of compounds 55–58 against the proliferation of three human leukemia cell lines, MV4;11, THP1 and NB4. MV4;11 and THP1 are MLL gene translocated leukemia cells, harboring MLL-AF4 and MLL-AF9 oncogenes, respectively, while NB4 possesses a wild-type MLL. Figure 3 shows time-dependent activities of two representative compounds 55 and 56 on MV4;11 cells at 4 different concentrations (i.e., 1, 3, 10, 30 μM). These two potent DOT1L inhibitors work very slowly in blocking the proliferation of the MLL-translocated cells. For example, at 10 μM, they had negligible activity against the cell growth even on Day 10. However, 55 and 56 (at 10 μM) were able to exhibit 61.7% and 43.4% cell growth inhibition on Day 15 as well as more pronounced activities (91.7% and 77.6% inhibition) on Day 20. Other doses of these two compounds showed a similar tendency of activity. In addition, as shown in Table 4, compounds 55 and 56 exhibit good cell growth inhibition against MLL-translocated cells MV4;11 and THP1 with EC50 values of 4.4–11.0 μM, but are almost devoid of activity (EC50 > 77 μM) in a 20-day treatment against NB4 leukemia cells bearing a wild-type MLL gene. This indicates these two DOT1L inhibitors act in a mechanism drastically different from that of traditional chemotherapeutic agents, such as cisplatin, which is able to kill all of these leukemia cells within a short period of time (standard 2-day incubation) with EC50 values of 0.59–3.0 μM (Table 4). Previous biological studies7a demonstrate the slow action of DOT1L inhibitors is likely due to a relatively long time required for cellular events that lead to cell growth inhibition, including blocked H3K79 methylation, followed by decreased levels of mRNA expression for methyl-H3K79 targeted genes, as well as ultimately the depletion of the gene products (proteins) key to the cell proliferation. As can be seen in Table 4, two other potent DOT1L inhibitors 57 and 58 also exhibit good activity against MV4;11 and THP1 with EC50 values of 5.9–11.2 μM, although they (especially for 58) also have activity on NB4, showing a decreased selectivity.
Figure 3.

Time-dependent activity against MLL leukemia cell line MV4;11.
Table 4.
EC50 values (μM) for leukemia cells.
| compound | MV4;11 | THP1 | NB4 |
|---|---|---|---|
| 55a | 4.4 | 8.1 | 101 |
| 56a | 5.6 | 11.0 | 77.0 |
| 57a | 11.2 | 10.0 | 31.3 |
| 58a | 5.9 | 11.1 | 9.5 |
| cisplatinb | 0.59 | 2.5 | 3.0 |
with a 20-day treatment;
with a 2-day treatment.
Molecular modeling and isothermal titration calorimetry (ITC) studies
Our early X-ray crystallographic study revealed the binding site of the N6-subtituent is actually a mainly hydrophobic pocket that directly exposes to the solvent (Figure 2).7b This could explain although this group provides high selectivity for DOT1L, it does not increase the binding affinity (Table 1). However, the big, hydrophobic urea-containing sidechains render both greatly enhanced activity and selectivity. The next question of interest is where these urea sidechains bind in DOT1L. X-ray crystallography is the method of choice to determine this. However, we have not obtained quality single crystals of DOT1L in complex with any one of these urea-containing inhibitors. We explored molecular modeling (docking) to predict the binding site of these groups, using the crystal structure of the DOT1L:SAM complex10 as the docking template. As representatively shown in Supporting Information Figure S1 for compound 36, all of these urea-containing groups are invariably predicted by the docking program Glide17 in Schrödinger18 (version: 2010) to extend into the nucleosome (substrate) binding pocket of DOT1L through the “lysine binding channel”, which holds the tightly clustered -CH2CH2CH2- linkers of 10 docking structures of 36 (Figure S1a). The bulky and lipophilic aromatic group is predicted to be located in a mainly hydrophobic pocket on the nucleosome binding surface and the urea moiety to form a H-bond with the protein (Figure S1b). These results seem to be reasonable, since the amino acid binding pocket in the DOT1L:SAM structures has no room to hold such long and bulky sidechains.
The docking studies, if real, indicate these urea-containing compounds are bisubstrate-type inhibitors, which occupy the binding sites of SAM and nucleosome. These inhibitors should therefore compete with both SAM and the substrate nucleosome. Structurally similar inhibitor 1 was reported to be competitive with SAM.7a A kinetic study from the same group also showed the Km value for nucleosome is 8.6 nM,19 but did not report whether 1 competes with nucleosome on binding to DOT1L. It is known that the binding of nucleosome to DOT1L involves very large surface areas including those from histone proteins (H3, H4, H2A and H2B) and the DNA around the histone core.10 However, the Kd (dissociation constant) value of nucleosome has not been measured.
We used isothermal titration calorimetry (ITC) to determine the binding affinities of inhibitors 55, 56, SAH as well as the substrate nucleosome. First, each of these compounds was titrated individually into DOT1L in a 50 mM HEPES buffer (pH = 7.0) with 150 mM NaCl. As summarized in Table 5 and representatively shown in Figure 4, the Kd values for SAH, 55, 56, and nucleosome were determined to be 360, 170, 150 and 14 nM, respectively. The high-affinity binding of nucleosome to DOT1L (Kd = 14 nM) is consistent with their large surface interactions as well as the reported Km value. The binding affinity of SAH is also comparable to its Ki value (160 nM, Table 1) as determined by the enzyme inhibition assay. However, the measured Kd values of 55 and 56 are >200× larger than their Ki values of 0.46 and 0.76 nM. Next, we investigated the binding affinities of these ligands to the DOT1L:nucleosome (1:1 molar ratio, 5 μM) complex, which better mimics the enzyme inhibition assay given the tight binding of nucleosome to DOT1L. Compounds SAH, 55 and 56 were found to have considerably higher affinities to the DOT1L:nucleosome complex with Kd values of 150, 66 and 86 nM, respectively (Table 5). The enhanced affinity for SAH can be conceivable since SAH and nucleosome have different binding sites in DOT1L. However, for compounds 55 and 56, these experimental results are inconsistent with our docking studies, which suggest these two molecules are competitive with nucleosome, and (if real) should exhibit greatly reduced binding affinities to DOT1L in the presence of 5 μM of nucleosome (≫Kd of 14 nM). The ITC experiments therefore showed that these two inhibitors do not compete with nucleosome. One possibility is due to the flexibility of DOT1L, which could render appropriate conformational changes at the active site of DOT1L upon the binding of inhibitor 55/56. Such induced fit could allow co-binding of 55 (or 56) and nucleosome. In addition, we used ITC to determine whether 55 is competitive with SAH. Compound 55 was titrated into the DOT1L:nucleosome complex in the presence of increasing concentrations of SAH. As shown in Table 5 and Figure S2, the Kd values of 55 measured in the presence of 1, 2.5, 5, 10 and 20 μM of SAH increase in a linear fashion (r2 = 0.97), showing a competitive mode of action.
Table 5.
Kd values (μM) for ligand binding to DOT1L.
| SAH | 55 | 56 | nucleosome | ||
|---|---|---|---|---|---|
| DOT1L | 0.36 | 0.17 | 0.15 | 0.014 | |
| DOT1L:nucleosome | 0.15 | 0.066 | 0.086 | NTa | |
| DOT1L: nucleosome with | 1 μM SAH | NTa | 0.073 | NTa | NTa |
| 2.5 μM SAH | NTa | 0.13 | NTa | NTa | |
| 5 μM SAH | NTa | 0.20 | NTa | NTa | |
| 10 μM SAH | NTa | 0.24 | NTa | NTa | |
| 20 μM SAH | NTa | 0.55 | NTa | NTa | |
NT: not tested.
Figure 4.

Representative ITC results and fitting curves for inhibitor binding to DOT1L.
The large difference between the Kd and Ki values of 55 and 56 measured by ITC and enzyme inhibition assay, respectively, could also be due to the protein conformational changes induced by these ligands. The rapidly formed initial EI (enzyme:inhibitor) complex could undergo a time-dependent conversion to a more tightly bound complex EI*.20 For the enzyme inhibition assays, there was a pre-incubation of an inhibitor with DOT1L:nucleosome, which allows the formation of EI*. However, there was no pre-incubation for the ITC experiments.
Chemistry
The general methods for synthesizing compounds 2, 5–58 are shown in Scheme 1. N6-substituted adenosine 59 were prepared from inosine by tri-acetate protection, conversion to 6-chloro by treatment with SOCl2, deprotection, and heating with a primary amine. Compounds 2, 5–12 were synthesized from 59 according to our previous method.7b Compounds 13–25 were made by heating a thiol or amine with 5′-adenosyl chloride 60 readily prepared from adenosine. Other 5′-S containing compounds 26, 28, 29, 31–54 were prepared from 2′, 3′-acetonide protected adenosine by a Mitsunobu reaction with thioacetic acid, followed by in situ hydrolysis of the thioester product with NaOMe and a one-pot alkylation with a bromide in MeOH. A Mitsunobu reaction of 2′,3′-acetonide protected adenosine with phthalimide, followed by NH2NH2 treatment, gives rise to adenosine derivative 61 having a 5′-NH2, which was alkylated with Z-protected 3-iodopropylamine to give compound 27. To make N-isopropyl containing compounds 55 - 58, compound 61 was subjected successively to a reductive amination with acetone and NaCNBH3 to add an N-isopropyl group, a Michael addition with methyl acrylate, and a reduction with LiAlH4 to give compound 62 with a terminal -OH. The -OH was then converted, by a Mitsunobu reaction followed by NH2NH2 treatment, to an -NH2, which was further reacted with an isocyanate, affording, after depretection of 2′, 3′-acetonide, compounds 55–58.
Scheme 1a.

aReagents and conditions: (i) Ac2O, CH3CN, Et3N, reflux; (ii) SOCl2, DMF, CHCl3, reflux; (iii) NH3, MeOH; (iv) R1NH2, EtOH, 80 °C, 2h; (v) 2 equiv. SOCl2, acetone, pyridine, overnight, then aq. NH3; (vi) RH, reflux; (vii) acetone, SOCl2, CH(OMe)3; (viii) thioacetic acid, PPh3, diisopropyl azodicarboxylate, THF; (ix) NaOMe, then RBr, MeOH; (x) HCl, dioxane/H2O; (xi) phthalimide, PPh3, diisopropyl azodicarboxylate, THF; (xii) NH2NH2, EtOH, 80 °C; (xiii) RBr or RI, diisopropylethylamine, DMF; (ixv) acetone, AcOH, NaCNBH3, MeOH; (xv) methyl acrylate, MeOH; (xvi) LiAlH4, THF; (xvii) RNCO, CH2Cl2.
CONCLUSION
This work provides the synthesis, structure activity relationship and isothermal titration calorimetry (ITC) studies of a series of inhibitors of human histone methyltransferase DOT1L, a novel target for acute leukemia with MLL gene translocations. First, a total of 55 adenosine-containing compounds were designed and synthesized, among which several highly potent DOT1L inhibitors were identified with Ki values as low as 0.5 nM. Second, structure activity relationship analysis of these compounds shows that 1) replacing the amino acid moiety of SAH with an N-phenyl-substituted urea functional group leads to a series of potent and selective DOT1L inhibitors; 2) replacing the -S- as found in SAH to an -N(isopropyl)- group offers additional activity enhancement; 3) the optimal linker between the urea and the 5′-groups is -CH2CH2CH2-; and 4) a small substituent (e.g., methyl) at the N6-position of adenine ring renders high selectivity without much activity loss. Third, several representative DOT1L inhibitors demonstrate selective activity against the proliferation of MLL-rearranged leukemia cells with the EC50 values of 4–11 μM. However, it takes a relatively long time (> 10 days) for these compounds to exert growth arrest, showing a different mechanism of action from traditional chemotherapeutic drugs. Finally, ITC experiments showed urea-containing inhibitors 55 and 56 are able to bind with a high affinity (Kd: 66 and 86 nM) to the DOT1L:nucleosome complex, and only compete with SAM/SAH, but not the substrate nucleosome, on binding to DOT1L.
EXPERIMENTAL SECTION
All reagents were purchased from Alfa Aesar (Ward Hill, MA) or Aldrich (Milwaukee, WI). Compounds were characterized by 1H NMR on a Varian (Palo Alto, CA) 400-MR spectrometer. The purities of all compounds were determined by a Shimadzu Prominence HPLC with a Zorbax C18 or C8 column (4.6 × 250 mm) monitored by UV absorbance at 254 nm, or 1H (at 400 MHz) absolute spin-count quantitative NMR analysis using imidazole as an internal standard. The purities of all compounds were found to be >95%.
Synthesis and characterization of compounds 5–58 can be found in Supporting Information Experimental Section.
Enzyme inhibition
Expression, purification and inhibition of recombinant human DOT1L (catalytic domain 1–472) were performed according to our previous published methods.7b In brief, compounds with concentrations ranging from 1 nM to 100 μM were incubated with DOT1L (100 nM), 1.5 μM oligo-nucleosome in 20 μL of 20 mM Tris buffer (containing 1 mM EDTA, 0.5 mM DTT and 50 μg/mL BSA, pH = 8.0) for 10 min. 0.76 μM (equaling to the Km) of 3H-SAM (10 Ci/mM; Perkin-Elmer) was added to initiate the reaction. After 30 min at 30 °C, the reaction was stopped by adding SAH to a final concentration of 100 μM. 15 μL of reaction mixture was then transferred to a small piece of P81 filter paper (Whatman) that binds histone H3 protein, washed three times with 50 mM NaHCO3, dried, and transferred into a scintillation vial containing 2 mL of scintillation cocktail. Radioactivity on the filter paper was measured with a Beckman LS-6500 scintillation counter. IC50 values were obtained by using a standard sigmoidal dose response curve fitting in Prism (version 5.0, GraphPad Software, Inc., La Jolla, CA). The reported IC50s were the mean values from at least three experiments. Ki values for less potent inhibitors (IC50 > 0.8 μM) were calculated using the Cheng-Prusoff equation Ki = IC50/(1+[SAM]/Km). For more potent inhibitors (IC50 ≤ 0.8 μM), Ki values were calculated using the Morrison tight binding model fitting in Prism 5.0.
Enzyme inhibition assays for PRMT1, CARM1 and SUV39H1 were performed as described previously.7b
Proliferation inhibition assays
MV4;11, THP1 and NB4 cell lines were obtained from ATCC (American Type Culture Collection, Manassas, VA). We used an XTT cell viability assay kit available from Roche to assess the viability of suspension leukemia cells. In brief, 5 × 105 cells/well in 96-well plates were treated with an increasing concentration (0.1–100 μM) of a compound in RPMI1640 medium supplemented with 10% FBS (fetal bovine serum), penicillin (100 U/mL) and streptomycin (100 μg/mL). After a given period of time, cell viability were evaluated with the XTT assay and monitored at 490 nm with a Beckman DTX-880 microplate reader. All the experiments were done in triplicate. The EC50 values were calculated by using a standard sigmoidal dose response curve fitting in Prism 5.0.
Molecular modeling
Docking studies were performed using the program Glide17 as previously reported.21 The crystal structures of the DOT1L:SAM complex10 was prepared using the module “protein preparation wizard” in Maestro22 in Schrödinger18 with default protein parameters: water molecules were removed, hydrogens added and inhibitors extracted. H-bonds were then optimized and the protein was energy-minimized using OPLS_2005 force field. A receptor grid, which is large enough to contain the whole active site, was generated using Glide without constraints. Compounds were built, minimized using OPLS_2005 force field in Maestro and then docked into the prepared protein structure using Glide (docking parameters: standard-precision and dock flexibly).
Isothermal titration calorimetry
Using a previously reported method,23 ITC experiments were performed with a Nano ITC LV (190 μL) instrument from TA Instruments (New Castle, DE) at 279 K, at which DOT1L is more stable during the experiments (lasting for ~2h). A 190 μL solution containing DOT1L (5 μM) (or a 1:1 DOT1L:nucleosome complex (5 μM) containing an increasing concentration of SAH from 0–20 μM) in 50 mM HEPES buffer (pH = 7.0) with 150 mM NaCl was added into the sample cell. A ligand (40–60 μM) solution in the same buffer (50 μL) was transferred into the syringe of the instrument. Upon reaching temperature equilibrium, 25 injections (2 μL each) of the ligand were used for the titration. ITC data were imported into NanoAnalyze software (TA Instruments, New Castle, DE). The titration baseline was corrected and Kd values for the binding of the ligand were obtained by fitting into the independent model in the software. All experiments were repeated for at least one time to ensure the reported data are reliable.
Supplementary Material
Acknowledgments
This work was supported by a grant (RP110050) from Cancer Prevention and Research Institute of Texas (CPRIT) to Y.S. J.L.A. was supported by a training fellowship from the Keck Center of the Gulf Coast Consortia, on the Pharmacological Sciences Training Program, National Institute of General Medical Sciences (NIGMS) T32GM089657.
ABBREVATIONS
- HMT
Histone methyltransfers
- H3K79
Histone3-lysine79
- ITC
isothermal titration calorimetry
- MLL
mixed lineage leukemia
- SAM
S-adenosyl-L-methionine
- SAH
S-adenosyl-L-homocysteine
- SAR
structure activity relationship
Footnotes
Supporting Information Available. Figures S1, S2, and Experimental Section showing docking results, linear correlation of Kd values of 55 with the competing SAH concentrations, and detailed compound synthesis and characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.For a recent review, see Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005.
- 2.Cheng X, Collins RE, Zhang X. Structural and sequence motifs of protein (histone) methylation enzymes. Annu Rev Biophys Biomol Struct. 2005;34:267–294. doi: 10.1146/annurev.biophys.34.040204.144452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schubert HL, Blumenthal RM, Cheng X. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem Sci. 2003;28:329–335. doi: 10.1016/S0968-0004(03)00090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov. 2009;8:724–732. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- 6.For reviews of HMT inhibitors, see Ref. 5 and Cole PA. Chemical probes for histone-modifying enzymes. Nat Chem Biol. 2008;4:590–597. doi: 10.1038/nchembio.111.
- 7.For DOT1L inhibitors, see Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, Jin L, Kuntz KW, Chesworth R, Moyer MP, Bernt KM, Tseng JC, Kung AL, Armstrong SA, Copeland RA, Richon VM, Pollock RM. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009.Yao Y, Chen P, Diao J, Cheng G, Deng L, Anglin JL, Prasad BV, Song Y. Selective inhibitors of histone methyltransferase DOT1L: design, synthesis, and crystallographic studies. J Am Chem Soc. 2011;133:16746–16749. doi: 10.1021/ja206312b.
- 8.For inhibitors of other HMTs, see Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3–9. Nat Chem Biol. 2005;1:143–145. doi: 10.1038/nchembio721.Liu F, Barsyte-Lovejoy D, Allali-Hassani A, He Y, Herold JM, Chen X, Yates CM, Frye SV, Brown PJ, Huang J, Vedadi M, Arrowsmith CH, Jin J. Optimization of cellular activity of G9a inhibitors 7-aminoalkoxy-quinazolines. J Med Chem. 2011;54:6139–6150. doi: 10.1021/jm200903z.Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, Wigle TJ, Dimaggio PA, Wasney GA, Siarheyeva A, Dong A, Tempel W, Wang SC, Chen X, Chau I, Mangano TJ, Huang XP, Simpson CD, Pattenden SG, Norris JL, Kireev DB, Tripathy A, Edwards A, Roth BL, Janzen WP, Garcia BA, Petronis A, Ellis J, Brown PJ, Frye SV, Arrowsmith CH, Jin J. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol. 2011;7:566–574. doi: 10.1038/nchembio.599.Liu F, Chen X, Allali-Hassani A, Quinn AM, Wigle TJ, Wasney GA, Dong A, Senisterra G, Chau I, Siarheyeva A, Norris JL, Kireev DB, Jadhav A, Herold JM, Janzen WP, Arrowsmith CH, Frye SV, Brown PJ, Simeonov A, Vedadi M, Jin J. Protein lysine methyltransferase G9a inhibitors: design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines. J Med Chem. 2010;53:5844–5857. doi: 10.1021/jm100478y.Liu F, Chen X, Allali-Hassani A, Quinn AM, Wasney GA, Dong A, Barsyte D, Kozieradzki I, Senisterra G, Chau I, Siarheyeva A, Kireev DB, Jadhav A, Herold JM, Frye SV, Arrowsmith CH, Brown PJ, Simeonov A, Vedadi M, Jin J. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a. J Med Chem. 2009;52:7950–7953. doi: 10.1021/jm901543m.Kubicek S, O’Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, Rea S, Mechtler K, Kowalski JA, Homon CA, Kelly TA, Jenuwein T. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017.Allan M, Manku S, Therrien E, Nguyen N, Styhler S, Robert MF, Goulet AC, Petschner AJ, Rahil G, Robert Macleod A, Deziel R, Besterman JM, Nguyen H, Wahhab A. N-Benzyl-1-heteroaryl-3-(trifluoromethyl)-1H-pyrazole-5-carboxamides as inhibitors of co-activator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett. 2009;19:1218–1223. doi: 10.1016/j.bmcl.2008.12.075.Huynh T, Chen Z, Pang S, Geng J, Bandiera T, Bindi S, Vianello P, Roletto F, Thieffine S, Galvani A, Vaccaro W, Poss MA, Trainor GL, Lorenzi MV, Gottardis M, Jayaraman L, Purandare AV. Optimization of pyrazole inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1) Bioorg Med Chem Lett. 2009;19:2924–2927. doi: 10.1016/j.bmcl.2009.04.075.Purandare AV, Chen Z, Huynh T, Pang S, Geng J, Vaccaro W, Poss MA, Oconnell J, Nowak K, Jayaraman L. Pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett. 2008;18:4438–4441. doi: 10.1016/j.bmcl.2008.06.026.
- 9.Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, Zhang Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol. 2002;12:1052–1058. doi: 10.1016/s0960-9822(02)00901-6. [DOI] [PubMed] [Google Scholar]
- 10.Min J, Feng Q, Li Z, Zhang Y, Xu RM. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell. 2003;112:711–723. doi: 10.1016/s0092-8674(03)00114-4. [DOI] [PubMed] [Google Scholar]
- 11.Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, Zhang Y. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 12.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 13.Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU, Xia X, Jesneck J, Bracken AP, Silverman LB, Kutok JL, Kung AL, Armstrong SA. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14:355–368. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hilden JM, Dinndorf PA, Meerbaum SO, Sather H, Villaluna D, Heerema NA, McGlennen R, Smith FO, Woods WG, Salzer WL, Johnstone HS, Dreyer Z, Reaman GH. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the Children’s Oncology Group. Blood. 2006;108:441–451. doi: 10.1182/blood-2005-07-3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coulombe RA, Sharma RP, Huggins JW. Pharmacokinetics of the antiviral agent 3-deazaneplanocin A. Eur J Drug Metab Pharmacokinet. 1995;20:197–202. doi: 10.1007/BF03189670. [DOI] [PubMed] [Google Scholar]
- 16.Obara T, Shuto S, Saito Y, Snoech R, Andrei G, Balzarini J, De Clercq E, Matsuda A. New Neplanocin analogues. 7. Synthesis and antiviral activity of 2-halo derivatives of Neplanocin A. J Med Chem. 1996;39:3847–3852. doi: 10.1021/jm960145+. [DOI] [PubMed] [Google Scholar]
- 17.Glide, version 5.5. Schrödinger, LLC; New York, NY: 2010. [Google Scholar]
- 18.Schrödinger Suite, version 2010. Schrödinger, LLC; New York, NY: 2010. [Google Scholar]
- 19.Richon VM, Johnston D, Sneeringer CJ, Jin L, Majer CR, Elliston K, Jerva LF, Scott MP, Copeland RA. Chemogenetic analysis of human protein methyltransferases. Chem Biol Drug Des. 2011;78:199–210. doi: 10.1111/j.1747-0285.2011.01135.x. [DOI] [PubMed] [Google Scholar]
- 20.Koppisch AT, Fox DT, Blagg BS, Poulter CD. E. coli MEP synthase: steady-state kinetic analysis and substrate binding. Biochemistry. 2002;41:236–243. doi: 10.1021/bi0118207. [DOI] [PubMed] [Google Scholar]
- 21.Deng L, Endo K, Kato M, Cheng G, Yajima S, Song Y. Structures of 1-deoxy-D-xylulose-5-phosphate reductoisomerase/lipophilic phosphonate complexes. ACS Med Chem Lett. 2011;2:165–170. doi: 10.1021/ml100243r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maestro, version 9.1. Schrödinger, LLC; New York, NY: 2010. [Google Scholar]
- 23.Cai G, Deng L, Fryszczyn BG, Brown NG, Liu Z, Jiang H, Palzkill T, Song Y. Thermodynamic investigation of inhibitor binding to 1-deoxy-D-xylulose-5-phosphate reductoisomerase. ACS Med Chem Lett. 2012;3:496–500. doi: 10.1021/ml300071w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.