

Abstract
The signaling mechanisms that regulate CLC anion channels are poorly understood. Caenorhabditis elegans CLH-3b is a member of the CLC-1/2/Ka/Kb channel subfamily. CLH-3b is activated by meiotic cell-cycle progression and cell swelling. Inhibition is brought about by GCK-3 kinase-mediated phosphorylation of S742 and S747 located on a ∼176 amino acid disordered domain linking CBS1 and CBS2. Much of the inter-CBS linker is dispensable for channel regulation. However, deletion of a 14 amino acid activation domain encompassing S742 and S747 inhibits channel activity to the same extent as GCK-3. The crystal structure of CmCLC demonstrated that CBS2 interfaces extensively with an intracellular loop connecting membrane helices H and I, the C-terminus of helix D, and a short linker connecting helix R to CBS1. Point mutagenesis of this interface identified two highly conserved aromatic amino acid residues located in the H-I loop and the first α-helix (α1) of CBS2. Mutation of either residue to alanine rendered CLH-3b insensitive to GCK-3 inhibition. We suggest that the dephosphorylated activation domain normally interacts with CBS1 and/or CBS2, and that conformational information associated with this interaction is transduced through a conserved signal transduction module comprising the H-I loop and CBS2 α1.
Introduction
Members of the CLC superfamily of anion transport proteins are found in all major groups of life including archaebacteria. CLC genes encode anion channels and Cl−/H+ exchangers and are expressed in plasma and intracellular organelle membranes where they perform essential physiological functions including transepithelial transport, regulation of membrane potential, and regulation of cytoplasmic and intraorganelle Cl− and H+ concentrations (1,2).
CLC channels are homodimers. Each monomer forms an independently gated protopore that is opened and closed by a fast gating process. A common or slow gate opens and closes both protopores simultaneously. A CLC monomer comprises 18 α-helical domains (designated A–R). Helices B through R span or are embedded in the membrane (1–5).
Eukaryotic CLC proteins have extensive cytoplasmic C-termini that contain two cystathionine-β-synthase (CBS) domains (6–9). The CBS domain is a highly conserved 50–60 amino acid motif found in numerous diverse proteins (10). Mutations in CBS domains give rise to several diseases (10) and alter CLC gating (6,11–17) indicating that they play a critical role in protein structure/function.
The structural bases of CLC gating and ion transport have been studied extensively. However, little is known about the cell signaling and biophysical mechanisms that regulate CLC activity. We have addressed this important problem using the nematode Caenorhabditis elegans. The C. elegans clh-3 gene encodes two CLC anion channel splice variants, CLH-3a and CLH-3b, which are members of the CLC-1/2/Ka/Kb channel subfamily. CLH-3b is expressed in the worm oocyte and is activated by meiotic cell cycle progression and cell swelling (18). Activation requires dephosphorylation mediated by type 1 phosphatases (19). Phosphorylation-dependent channel inhibition is mediated by the Ste20 kinase GCK-3 (20–22). GCK-3 is a homolog of the SPAK and OSR1 kinases, which play key roles in both cellular and systemic ion and water homeostasis (23).
CBS1 and CBS2 of CLH-3b are connected by a ∼176 amino acid linker. The last 101 amino acids comprise a regulatory domain. GCK-3 binds to the regulatory domain via a SPAK interaction motif (21). Inhibition of CLH-3b by GCK-3 requires concomitant phosphorylation of S742 and S747 (20).
Multiple CLC proteins are regulated by phosphorylation as well as by intracellular nucleotide binding. How are C-terminus signaling events transduced into changes in CLC activity? The crystal structure of the eukaryotic Cl−/H+ exchanger CmCLC from the thermophilic red alga Cyanidioschyzon merolae (5) provides possible insights. CBS2 of CmCLC interfaces extensively with a short intracellular loop connecting membrane helices H and I, the C-terminus of membrane helix D and a short linker connecting membrane helix R to CBS1. The interface has a shape complementarity index, which is a measure of how well two protein domains fit together, similar to the interface between an antibody and antigen. This suggests that the interaction is highly specific and functionally significant and therefore may provide a signal transduction pathway for regulating CLC function.
The goal of the current study was to identify CLH-3b structural components important in phosphorylation-dependent regulation. We demonstrate that much of the ∼176 amino acid inter-CBS linker is dispensable for channel regulation. However, deletion of a 14 amino acid domain encompassing the two regulatory phosphorylation sites inhibits channel activity to the same extent as GCK-3. Mutation of a conserved tyrosine residue in the intracellular H-I loop or a conserved histidine residue located in the first α-helix of CBS2 prevents GCK-3-mediated channel inhibition. We suggest that the 14 amino acid domain in the inter-CBS linker of CLH-3b interacts with one or both CBS domains when it is dephosphorylated, and that phosphorylation disrupts this interaction leading to conformational changes that inhibit channel activity. We also suggest that the H-I loop/CBS2 interface serves to transduce cytoplasmic C-terminus conformational information to channel membrane domains. Our studies provide novel, to our knowledge, molecular insights into long-range intraprotein signaling in CLC channels.
Material and Methods
Transfection and whole cell patch-clamp recording
Human embryonic kidney (HEK293) cells were cultured and patch clamped as described previously (24). Cells were transfected using FuGENE 6 or X-tremeGENE HP (Roche Diagnostics, Indianapolis, IN) with 1.0 μg GFP, 1.5–3 μg CLH-3b, and 1.5–3 μg GCK-3 ligated into pcDNA3.1. Point and deletion mutations were generated using QuikChange Lightning Site-Directed and Multi Site-Directed mutagenesis kits (Agilent Technologies, Santa Clara, CA). All mutations were confirmed by DNA sequencing. Experimental protocols were performed on at least two independently transfected groups of cells.
Transfected cells were identified by GFP fluorescence and patch clamped using a bath solution containing 90 mM NMDG-Cl, 5 mM MgSO4, 1 mM CaCl2, 12 mM Hepes free acid titrated to pH 7.0 with CsOH, 8 mM Tris, 5 mM glucose, 95 mM sucrose, and 2 mM glutamine (pH 7.4, 310 mOsm), and a pipette solution containing 116 mM NMDG-Cl, 2 mM MgSO4, 20 mM Hepes, 6 mM CsOH, 1 mM EGTA, 2 mM ATP, 0.5 mM GTP, and 10 mM sucrose (pH 7.2, 275 mOsm). Cells were swollen by exposure to a hypotonic (250 mOsm) bath solution that contained no added sucrose.
Patch electrodes were pulled from 1.5 mm outer diameter silanized borosilicate microhematocrit tubes; electrode resistance ranged from 4 to 8 MΩ. Currents were measured with an Axopatch 200B (Axon Instruments, Foster City, CA) patch clamp amplifier. Electrical connections to the patch clamp amplifier were made using Ag/AgCl wires and 3 M KCl/agar bridges. Data acquisition and analysis were performed using pClamp 8 software (Axon Instruments).
Quantification of CLH-3b current properties
Whole cell currents were elicited by stepping membrane potential from a holding voltage of 0 mV to test voltages of −140 to +60 mV in 20 mV increments. Test voltages were maintained for 1 s and cells were then returned to the holding voltage of 0 mV for 1 s. CLH-3b is strongly inwardly rectifying and does not exhibit tail currents due to rapid inactivation at positive potentials. Current-to-voltage plots were therefore used to estimate channel activation voltage (i.e., the voltage at which current activation is first detected) (21). A line was first drawn by linear regression analysis of currents measured between 0 and 60 mV where channel activity is low. A second line was then drawn by linear regression analysis of currents measured between the first test voltage at which inward current was detected and a second voltage 20 mV more negative. The point at which these two lines intersect is defined as the activation voltage.
The kinetics of hyperpolarization-induced activation of CLH-3b are described by either mono- or biexponential fits describing slow or fast and slow time constants, respectively. The nature of the fit is dictated by channel phosphorylation (21). To simplify presentation and interpretation of activation kinetics under different experimental conditions, time constants are not used. Instead, the time required for whole cell current to reach 50% activation when membrane voltage is stepped from 0 to −100 mV for 1 s is quantified. This time is defined as the 50% rise time.
Production of recombinant inter-CBS linker protein
The inter-CBS linker (amino acids 619–793) was cloned into the bacterial expression vector pET47b(+) (Novagen, Darmstadt, Germany) resulting in a 197 amino acid (∼22 kD) construct that included an N-terminal His tag and an HRVC protease cleavage site. The linker construct was transformed into Rosetta 2(DE3) competent cells (Novagen) and expressed using the Overnight Express Autoinduction System 1 (Novagen). Bacterial cells were grown at 22°C to a final culture volume of 400 ml.
Approximately 1 g of bacterial pellet was lysed in 16 ml of Bugbuster Protein Extraction Reagent (Novagen) and inclusion bodies were purified according to the manufacturer’s instructions. Protein was bound to Ni-NTA His-Bind resin (Novagen) under denaturing conditions (100 mM sodium phosphate buffer, 8.0 M urea, 10 mM Tris, pH 8.0), washed 10 times with 1 ml of binding buffer and then dialyzed against 100 mM sodium phosphate buffer, 10 mM Tris, pH 8.0. The resin-bound protein was then washed 10 times with 1 ml of 20 mM imidazole in 50 mM phosphate buffer, 300 mM NaCl, pH 8.0, followed by 10 1 ml washes of 75 mM imidazole, and finally eluted with four 1 ml aliquots of 250 mM imidazole and two 1 ml aliquots of 300 mM imidazole. Each elution fraction was collected into a tube containing 1 ml of the elution buffer to keep the protein concentration low and prevent aggregation. Eluates were analyzed for purity by SDS-PAGE gel electrophoresis before they were pooled for the final dialysis into 200 mM NaCl, 50 mM Tris, 10% glycerol, pH 7.5. Protein concentrations were estimated using SDS-PAGE gel electrophoresis with 22 kD protein standards and by Coomassie Blue Plus (Thermo Fisher Scientific, Rockford, IL) protein assay. The protein was concentrated to 1 μg/μl before analysis using 4 ml capacity centrifugal filter units with a 10 kD molecular mass cutoff (Millipore, Cork, Ireland).
Circular dichroism (CD) measurements
CD spectra of the linker protein were recorded on a Jasco J-810 system (Jasco, Easton, MD) at room temperature in a 0.1 cm path length cell. Data were collected from 190 to 260 nm at a scanning speed of 50 nm/min. The raw CD signal in millidegrees was converted to molar ellipticity [θ] using the relationship:
where n is the number of amino acid residues, C is the protein concentration in mM, and l is the path length in cm.
Statistical analyses
Electrophysiological data are presented as mean ± SE. n represents the number of patch-clamped cells from which CLH-3b currents were recorded. Statistical significance was determined using Student’s t-test for unpaired means.
Results
The distal C-terminus of CLH-3b is not required for regulation by GCK-3 or cell swelling
Fig. 1 A shows a schematic diagram of the organization of CLC membrane-associated α-helices and the cytoplasmic C-terminus of CLH-3b. We first deleted the last 156 amino acids of CLH-3b downstream from CBS2 to determine if the distal C-terminus was required for channel regulation. Wild-type (WT) and Δ846-1001 mutant channels were coexpressed with kinase dead or WT GCK-3 and current properties were measured before and 1 min after cell swelling was induced by exposure to a 250 mOsm bath solution. GCK-3 reduced current amplitude, hyperpolarized channel activation voltage, and slowed voltage-dependent activation kinetics (Fig. 2). Cell swelling reversed these effects (Fig. 2). The GCK-3 and swelling-induced changes in the functional properties of the Δ846-1001 mutant were not significantly (P > 0.3) different from those of WT channels. We conclude therefore that the distal C-terminus of CLH-3b is not required for regulation of CLH-3b by either GCK-3 or cell volume changes.
Figure 1.
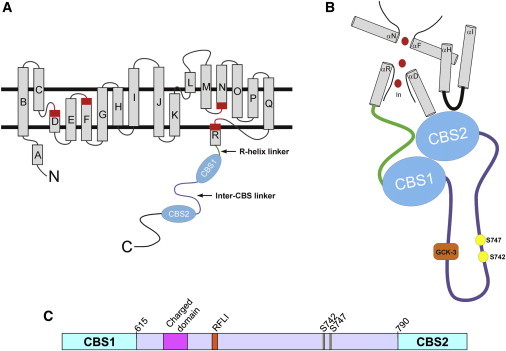
CLH-3b structural features. (A) Schematic diagram showing organization of the membrane-associated domain of a CLC protein monomer and cytoplasmic C-terminus of CLH-3b. Rectangles indicate α-helices. Regions of helices highlighted in red function in channel gating and to coordinate Cl− within the ion-conducting pore. R-helix is connected to CBS1 via a short cytoplasmic linker (green). (B) Model showing putative interface of CLH-3b intracellular domains with membrane-associated domains. Interface features are based on the crystal structure of CmCLC (5). CBS1 and CBS2 dimerize to form a Bateman module. Helices D, F, N, and R form the Cl− (red circles) conducting pore of a CLC monomer. CBS2 interacts with the R-helix linker (green), the C-terminus of the D-helix, and an intracellular loop (black) connecting helices H and I, which form part of the interface between two CLC monomers. The inter-CBS linker is shown in purple. (C) Diagram of inter-CBS linker domain. Linker is ∼176 amino acids long (amino acids 615–790) and contains a highly charged motif, a GCK-3-binding motif (RFLI), and two regulatory phosphorylation sites, S742 and S747.
Figure 2.
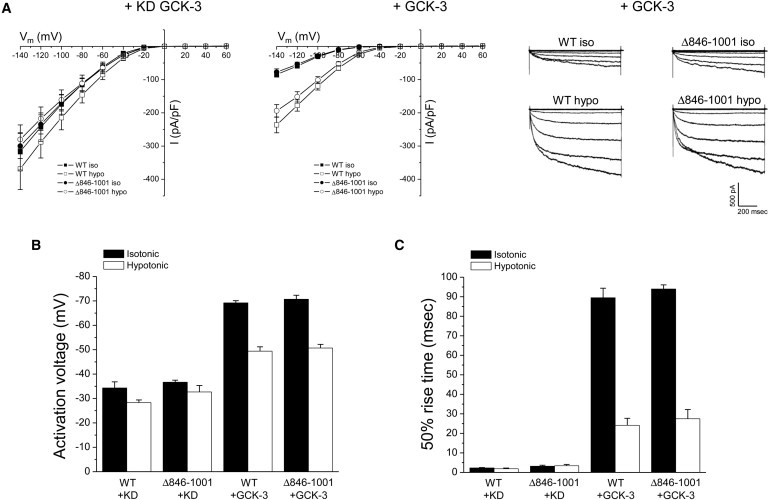
Effect of deletion of the distal C-terminus (Δ846-1001) on CLH-3b regulation. WT or Δ846-1001 mutant channels were coexpressed with kinase dead or WT GCK-3. Current properties were measured before and 1 min after cell swelling in 250 mOsm bath medium. Changes in WT and Δ846-1001 current amplitude (A), activation voltage (B), and 50% rise time (C) induced by GCK-3 and swelling were not significantly (P > 0.3) different. Representative whole cell current traces for WT and Δ846-1001 channels coexpressed with GCK-3 before and 1 min after induction of cell swelling are shown in the right panel of A. Values are mean ± SE (n = 4–5).
Structural properties of the inter-CBS linker domain
Fig. 1, B and C, show details of the ∼176 amino acid inter-CBS linker containing the GCK-3-binding motif (21) and two regulatory phosphorylation sites, S742 and S747 (20). Several protein structure analysis programs (e.g., DisEMBL Disopred, FoldIndex, Disprot, PONDR) predicted that the inter-CBS linker is largely disordered. Fig. 3 A shows PONDR analyses (25) for this domain. Two small regions had low PONDR scores suggesting that they are relatively structured compared to the rest of the protein. The first region includes the GCK-3-binding motif (RFLI; amino acids 668–671; 21).
Figure 3.
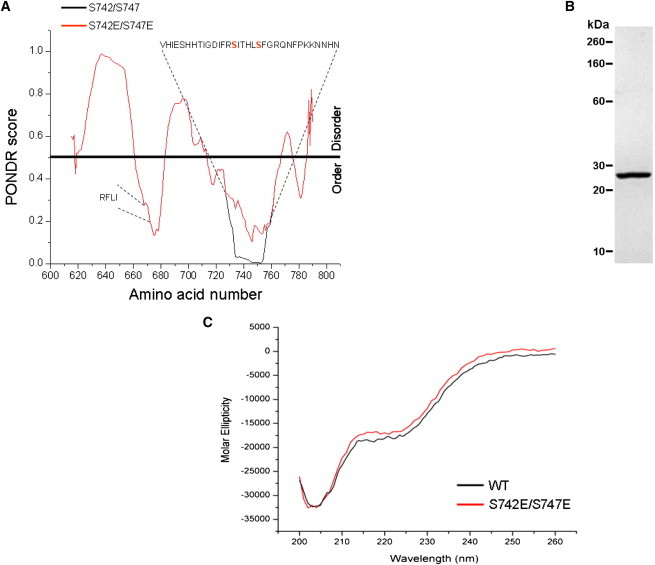
Biophysical properties of recombinant inter-CBS linker protein. (A) Prediction of disorder in the inter-CBS linker domain using PONDR (25). Predictions were generated for WT and S742E/S747E linkers. PONDR scores were identical for the two proteins except for amino acids 728–760, which are shown on the plot and contain the two regulatory phosphorylation sites (S742 and S747 shown in red). In the WT linker (black line), amino acids 734–753 have near-zero PONDR scores suggesting a high degree of order. Replacement of S742 and S747 with glutamate (red line) to mimic phosphorylation increases the PONDR scores in this region suggesting an increase in disorder. The GCK-3-binding motif (RFLI; amino acids 668–671) is shown. (B) SDS-PAGE gel of 1 μg of purified inter-CBS linker protein (amino acids 619–793). (C) Near ultraviolet CD spectra of WT and S742E/S747E mutant linkers. Spectra indicate that the WT and S742E/S747E mutant linkers contain little secondary structure.
Amino acids 734–753, which encompass the regulatory phosphorylation sites S742 and S747, had near-zero PONDR scores. When S742 and S747 were substituted with glutamate, which mimics the effects of phosphorylation on channel activity (20), the PONDR scores increased suggesting an increase in disorder.
We generated a recombinant protein encompassing amino acids 619–793 (Fig. 3 B) and used CD to directly assess the overall structural characteristics of the inter-CBS linker. Fig. 3 C shows CD spectra for a WT linker and a mutant linker in which S742 and S747 were mutated to glutamate. Glutamate substitution had no obvious effect on CD spectra indicating that phosphorylation is unlikely to cause large-scale secondary structure changes in the linker. However, phosphorylation could induce secondary structure changes in small domains within the linker that are not detectable by CD spectroscopy.
The overall properties of the spectra suggest that the linker contains little secondary structure. We used K2D3 (26) software to analyze the spectra. This software predicts protein secondary structure from a CD spectrum by comparing it to spectra theoretically derived from over 16,000 protein chains with known structure. The software predicted that ∼62% of the linker comprises random coil. However, it should be noted that the error associated with this estimate is likely to be high. The K2D3 software compares a queried CD spectrum with the most similar CD spectrum in its data set and calculates the distance between them over a given wavelength interval. The distances between highly similar spectra are small and the errors in structural predictions are therefore expected to be correspondingly small. For the CLH-3b inter-CBS linker, the distance between its CD spectrum and the most similar spectrum in the K2D3 data set was above the maximal value reported by the program. Because reference spectra are obtained from protein chains with well-defined secondary structure, this finding again suggests that the linker is largely disordered.
Identification of regions within the inter-CBS linker domain required for GCK-3 regulation
Deletion of linker amino acids 668–768, which includes the GCK-3-binding motif and S742 and S747, generates a mutant channel phenotype resembling that of WT CLH-3b inhibited by GCK-3 (11). This observation suggested that residues within the linker might interact with another portion of the channel, and that phosphorylation or deletion of this domain disrupts the interaction leading to inhibitory conformational changes. However, the specific amino acids involved in this putative interaction are unclear. Despite the widely accepted importance of disordered domains in protein regulation (27), it has been difficult to identify motifs that are required for their function and that mediate putative protein-protein interactions (28). We therefore generated a series of deletion mutations to assess the role of domains within the inter-CBS linker required for phosphorylation-dependent channel regulation.
A highly charged domain in the linker extends from amino acids 635–651 (Fig. 1 C). Large deletions in this domain (Δ628-647 and Δ648-667) or replacement of multiple charged amino acids with alanine (Ala-636–647) had little effect on channel regulation by GCK-3 (Table 1).
Table 1.
Functional properties of inter-CBS linker domain mutants
| Current @−140 mV (pA/pF) |
Activation voltage (mV) |
50% rise time (ms) |
||||
|---|---|---|---|---|---|---|
| Mutant | −GCK-3 | +GCK-3 | −GCK-3 | +GCK-3 | −GCK-3 | +GCK-3 |
| Δ628-647 | −360.4 ± 26.2 | −167.2 ± 27.6 | −40.0 ± 1.1 | −67.2 ± 2.1 | 4.1 ± 0.9 | 74.7 ± 14.8 |
| P < 0.0015 | P < 0.0001 | P < 0.02 | ||||
| Δ648-667 | −403.9 ± 39.5 | −108.5 ± 13.2 | −36.2 ± 0.8 | −60.5 ± 1.7 | 2.2 ± 0.2 | 32.6 ± 12.8 |
| P < 0.0004 | P < 0.0001 | P < 0.05 | ||||
| Ala-636-647 | −318.9 ± 48.8 | −122.4 ± 14.8 | −36.5 ± 0.8 | −58.5 ± 1.1 | 3.0 ± 0.2 | 59.1 ± 5.3 |
| P < 0.0001 | P < 0.0001 | P < 0.0001 | ||||
| Δ668-687 | −299.0 ± 42.4 | −406.8 ± 23.3 | −35.7 ± 0.6 | −35.4 ± 1.3 | 2.7 ± 0.2 | 2.7 ± 0.4 |
| P > 0.1 | P > 0.8 | P > 0.99 | ||||
| Δ688-709 | −362.3 ± 14.8 | −382.3 ± 52.3 | −33.8 ± 0.7 | −34.5 ± 0.4 | 1.8 ± 0.3 | 1.9 ± 0.1 |
| P > 0.7 | P > 0.4 | P > 0.8 | ||||
| Δ688-709/S742E/S747E | −223.1 ± 27.1∗ | −43.7 ± 1.1† | 11.2 ± 2.5‡ | |||
| Δ708-727 | −404.9 ± 43.8 | −211.6 ± 36.5 | −34.7 ± 0.4 | −57.2 ± 1.3 | 2.2 ± 0.2 | 29.2 ± 7.5 |
| P < 0.01 | P < 0.0001 | P < 0.01 | ||||
| Δ769-777 | −344.0 ± 34.9 | −66.7 ± 10.7 | −45.6 ± 1.1 | −65.5 ± 0.3 | 15.6 ± 2.7 | 79.4 ± 5.3 |
| P < 0.001 | P < 0.0001 | P < 0.0001 | ||||
| Δ728-737 | −154.2 ± 34.5 | −60.0 ± 12.8 | −48.2 ± 1.0 | −66.7 ± 0.8 | 15.1 ± 2.4 | 56.3 ± 10.2 |
| P < 0.04 | P < 0.0001 | P < 0.01 | ||||
| Δ752-759 | −134.3 ± 23.0 | −49.9 ± 14.5 | −43.5 ± 1.1 | −70.8 ± 1.6 | 7.8 ± 1.9 | 51.1 ± 7.5 |
| P < 0.02 | P < 0.0001 | P < 0.005 | ||||
| Δ760-768 | −195.0 ± 24.0 | −91.1 ± 6.0 | −45.2 ± 3.5 | −62.8 ± 2.9 | 6.2 ± 1.8 | 66.0 ± 5.6 |
| P < 0.0007 | P < 0.008 | P < 0.0002 | ||||
Values are mean ± SE (n = 3–12). Channels were coexpressed with either kinase dead GCK-3 (−GCK-3) or WT GCK-3 (+GCK-3).
∗P < 0.005, †P < 0.0001, and ‡P < 0.02, and compared to Δ688-709 mutant. P values shown for all other mutants were generated by comparing to the mutant channel expressed with kinase dead GCK-3.
Deletion of a region containing the GCK-3-binding motif (Δ668-687) and 22 amino acids immediately following this region (Δ688-709) prevented channel inhibition by the kinase (Table 1). To determine if the effect of the Δ688-709 deletion was due to disruption of GCK-3 binding and/or altered phosphorylation-dependent conformational changes, we replaced S742 and S747 with glutamate residues (i.e., Δ688-709/S742E/S747E), which fully mimics phosphorylation (20). The glutamate for serine substitutions in the Δ688-709 mutant significantly (P < 0.02) reduced current amplitude, hyperpolarized activation voltage, and slowed voltage-dependent activation kinetics (Table 1).
Δ708-727 and Δ769-777 mutant channels had WT properties and were inhibited by GCK-3 (Table 1). In contrast, Δ728-748 and Δ748-768 mutants coexpressed with kinase dead GCK-3 exhibited greatly reduced current amplitudes (Fig. 4, A and B), hyperpolarized activation voltages (Fig. 4 C), and greatly slowed activation kinetics (Fig. 4 D). These properties resemble those of WT CLH-3b coexpressed with and inhibited by GCK-3 (see Fig. 2). Coexpression of Δ748-768 or Δ728-748 with GCK-3 had no further inhibitory effect on channel properties (Fig. 4). Cell swelling did not activate Δ728-748 and Δ748-768 mutant channels coexpressed with the kinase (data not shown). These results show that the region between amino acids 728–768 mediates putative interchannel protein-protein interactions required for phosphorylation-dependent inhibition.
Figure 4.
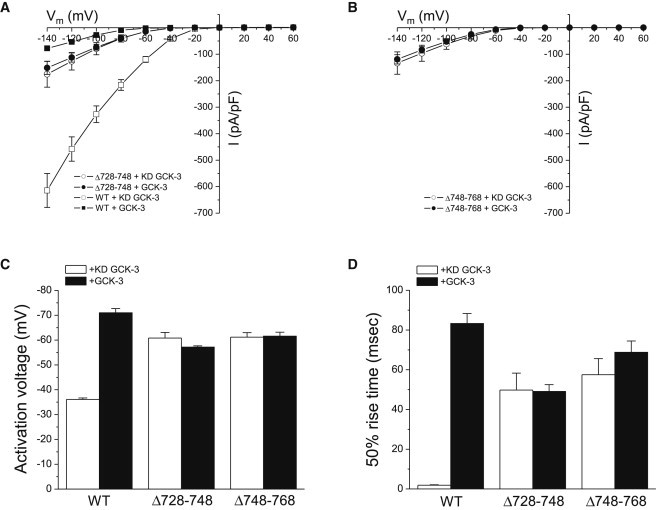
Effect of inter-CBS linker deletions Δ728-748 and Δ748-768 on channel functional properties. Mutant and WT channels were coexpressed with kinase dead or WT GCK-3. When coexpressed with kinase dead GCK-3, Δ728-748 and Δ748-768 current amplitudes (A and B), activation voltages (C) and 50% rise times (D) were similar to those of GCK-3 inhibited WT channels. Coexpression of the deletion mutants with GCK-3 had no effect on functional properties. Values are mean ± SE (n = 4–8).
Smaller deletions (Fig. 5 A) in the region containing amino acids 728–768 were generated to identify the minimal domain required for GCK-3-dependent channel inhibition. Δ728-737, Δ752-759, and Δ760-768 mutants exhibited largely normal kinase regulation (Table 1). In contrast, deletion of amino acids 738–751, which includes S742 and S747, resulted in a channel with functional properties similar to those of WT CLH-3b coexpressed with GCK-3 (Fig. 5, B–D). We conclude that amino acids 738–751 comprise an activation domain. Deletion or phosphorylation of this domain inhibits channel activity.
Figure 5.
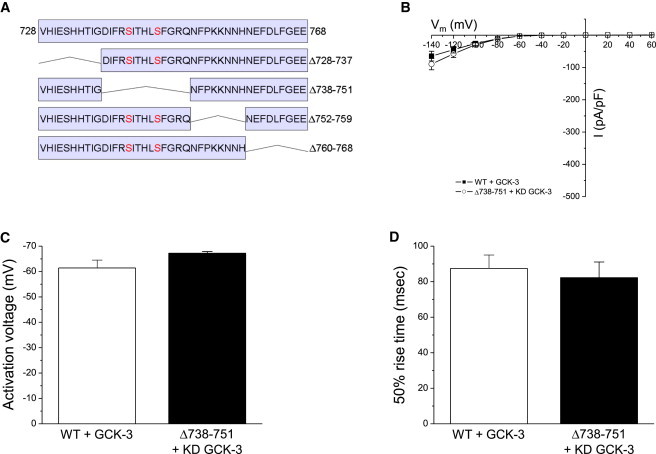
Effect of deletions in inter-CBS linker amino acids 728–768 on CLH-3b functional properties. (A) Sequence of amino acids 728–768 and schematic diagram showing location of 8–14 amino acid deletions. (B) Current amplitudes, (C) activation voltages, and (D) 50% rise times for WT CLH-3b coexpressed with functional GCK-3 and the Δ738-751 mutant coexpressed with kinase dead GCK-3. Functional properties of the WT and mutant channels were not significantly (P > 0.1) different. Values are mean ± SE (n = 6–11).
Conserved aromatic amino acid residues in CBS2 and the intracellular loop connecting membrane helices H and I are essential for GCK-3 regulation
The linker connecting the C-terminus of the R-helix to CBS1 of CmCLC (5) passes over and makes multiple close contacts with CBS2. CBS2 also interacts with the D-helix and a short intracellular loop connecting helices H and I (H-I loop). Helices D and R form part of the CLC ion-conducting pore (3,4) and the H and I helices contribute to the interface between CLC monomers (4) (Fig. 1, A and B). We used the CmCLC structure to design mutagenesis experiments that would test the role of the CBS2/membrane domain interface in phosphorylation-dependent regulation of CLH-3b. Fig. 6 A shows molecular details of this interface. CmCLC F273 is located on the H-I loop and faces the first α-helix, α1, of CBS2. The distance between F273 and α1 is ∼3.5 Å (5). F273 is conserved or exhibits conservative substitution in many CLCs proteins (Fig. 6 B). The equivalent amino acid in CLH-3b is Y232 (Fig. 6 B). We mutated Y232 and neighboring amino acids to assess the role of the H-I loop in signal transduction. Mutating Y232 to alanine completely prevented GCK-3-mediated channel inhibition (Fig. 7). In contrast, V231A and F233A mutant channels showed normal kinase regulation (Table 2).
Figure 6.
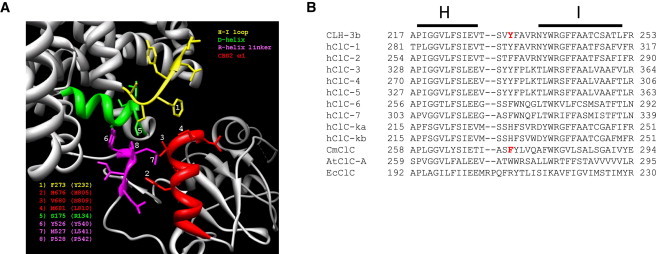
Structural and sequence features of CLC intracellular domains. (A) Ribbon diagram showing interface of CmCLC membrane and intracellular domains with CBS2. Intracellular loop connecting membrane helices H and I (yellow), membrane helix D (green), N-terminus of R-helix linker (purple), and α1 of CBS2 (red) are shown. Numbers indicate CmCLC amino acids that were used to guide point mutation studies in CLH-3b. The specific CmCLC amino acids are identified in the lower left corner and the equivalent residues mutated in CLH-3b are shown in parentheses. Figure was generated using UCSF Chimera software. (B) Alignments of CLH-3b membrane helices H and I and intracellular loop connecting them with various CLC proteins including CmCLC. Red shows Y232 in CLH-3b and F273 in CmCLC.
Figure 7.
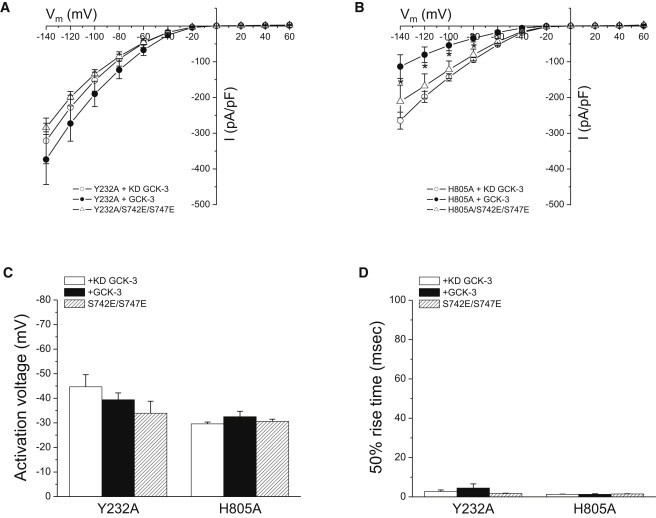
Effect of the H-I loop mutation Y232A and the CBS2 α1 H805A on CLH-3b regulation. Y232A and H805A mutant channels were expressed in the presence of kinase dead or functional GCK-3. Current amplitudes (A and B), (C) activation voltages, and (D) 50% rise times of Y232A and H805A mutants. Activation voltage and 50% rise times are plotted on the same scale as data in Fig. 2, B and C, to facilitate comparison with WT CLH-3b activity. Values are mean ± SE (n = 5–7). ∗P < 0.02 compared to H805A mutant expressed with functional GCK-3.
Table 2.
Functional properties of channels with mutations in the H-I loop, R-helix linker, D-helix, and α1 of CBS2
| Current @−140 mV (pA/pF) |
Activation voltage (mV) |
50% rise time (ms) |
||||
|---|---|---|---|---|---|---|
| Mutant | −GCK-3 | +GCK-3 | −GCK-3 | +GCK-3 | −GCK-3 | +GCK-3 |
| V231A | −392.2 ± 12.6 | −125.4 ± 43.5 | −36.3 ± 1.5 | −66.7 ± 1.1 | 4.0 ± 0.6 | 94.6 ± 6.2 |
| P < 0.004 | P < 0.0001 | P < 0.0007 | ||||
| F233A | −248.0 ± 36.0 | −133.9 ± 20.0 | −42.2 ± 2.8 | −69.1 ± 1.1 | 2.6 ± 0.7 | 12.3 ± 4.1 |
| P < 0.01 | P < 0.0001 | P < 0.03 | ||||
| Y540A | −126.9 ± 18.1 | −61.3 ± 7.9 | −48.6 ± 0.8 | −84.9 ± 3.5 | 4.0 ± 0.4 | 22.6 ± 2.7 |
| P < 0.006 | P < 0.0001 | P < 0.0001 | ||||
| L541A | −171.7 ± 21.2 | −83.1 ± 13.0 | −43.5 ± 1.4 | −84.0 ± 2.9 | 2.9 ± 0.4 | 9.2 ± 1.7 |
| P < 0.0012 | P < 0.0001 | P < 0.01 | ||||
| P542A | −215.3± 28.6 | −47.2 ± 16.2 | −36.6 ± 2.7 | −62.1 ± 2.9 | 1.7 ± 0.2 | 20.7 ± 3.4 |
| P < 0.0002 | P < 0.0001 | P < 0.0008 | ||||
| R134A | −158.3 ± 20.8 | −32.1 ± 7.2 | −29.4 ± 0.5 | −74.8 ± 2.3 | 1.5 ± 0.2 | 27.0 ± 8.0 |
| P < 0.0007 | P < 0.0001 | P < 0.013 | ||||
| S809A | −191.3 ± 67.3 | −51.1 ± 13.7 | −33.0 ± 1.1 | −65.4 ± 4.0 | 3.1 ± 0.5 | 36.4 ± 8.5 |
| P < 0.03 | P < 0.001 | P < 0.02 | ||||
| L810A | −290.8 ± 43.7 | −342.3 ± 63.1 | −31.6 ± 0.9 | −49.0 ± 1.1 | 5.3 ± 0.4 | 17.5 ± 2.0 |
| P > 0.5 | P < 0.0001 | P < 0.0001 | ||||
Values are mean ± SE (n = 3–10). Channels were coexpressed with either kinase dead GCK-3 (−GCK-3) or WT GCK-3 (+GCK-3). P values shown were generated by comparing to the mutant channel expressed with kinase dead GCK-3.
The close apposition of the H-I loop to α1 of CBS2 suggested that it may also play a role in phosphorylation-dependent signal transduction. We therefore mutated H805, S809, and L810 α1 to alanine (Fig. 6 A). The S809A mutant was inactivated in the presence of GCK-3 (Table 2). GCK-3 had no significant (P > 0.5) effect on current amplitude of the L810A mutant, but it did hyperpolarize activation voltage and slowed voltage-dependent activation kinetics (P < 0.0001; Table 2).
Activation voltage (Fig. 7 C) and 50% rise time (Fig. 7 D) for the H805A mutant were unaffected (P > 0.2) by GCK-3. However, current amplitude was significantly (P < 0.02) reduced at membrane voltages of −80 to −140 mV (Fig. 7 B). It is possible that GCK-3 may regulate CLH-3b current amplitude and voltage-dependent properties via distinct mechanisms that are differentially sensitive to the H805A mutation. Alternatively, reduced membrane expression of the phosphorylated H805A mutant channel could account for the apparent GCK-3-dependent reduction in current amplitude. We tested these possibilities by swelling cells coexpressing GCK-3 and the H805A mutant. Four minutes after cell swelling was induced, the mean ± SE relative whole cell current measured at −100 mV was 0.96 ± 0.03 (n = 4), which was not significantly (P > 0.3) different from 1. WT CLH-3b in contrast exhibits a three- to fourfold increase in current amplitude under similar conditions (24). We conclude therefore that the GCK-3 induced reduction in H805A current amplitude reflects reduced membrane expression rather than the existence of independent mechanisms for regulating channel conductance and voltage-dependent gating.
It is conceivable that the Y232A and H805A mutants cannot be phosphorylated by GCK-3. To test for that possibility, we bypassed the requirement for phosphorylation by mutating S742 and S747 to glutamate (20). Current amplitude, activation voltage, and 50% rise time for the triple mutant channels Y232A/S742E/S747E and H805A/S742E/S747E were not significantly (P > 0.2) different from those of the Y232A and H805A mutants expressed with or without functional GCK-3 (Fig. 7). Thus, we conclude that the Y232A and H805A mutant channels cannot be inactivated by either phosphorylation or mutation of regulatory phosphorylation sites to glutamate.
A short stretch of amino acids termed the R-helix linker connects the C-terminus of helix R to the N-terminus of CBS1. Several CLC crystal structures show that this linker is ordered (5,7–9). In the CmCLC crystal, amino acids Y526, M527, and P528 are located on the N-terminus of the R-helix linker and in close proximity to α1 of CBS2 (Fig. 6 A). These amino acids are highly conserved in eukaryotic CLCs and are typically considered to form the C-terminus of the R-helix itself (7–9,29,30). Given their conservation and apparent close association with CBS2, we mutated the equivalent amino acids, Y540, L541, and P542, to alanine. Y540A, L541A, and P542A mutants were responsive to GCK-3 (Table 2).
The CmCLC crystal structure revealed a close interaction between CBS2 and the C-terminus of membrane helix D (Fig. 6 A) (5). The most C-terminal amino acid in the D-helix of CLH-3b is R134. This amino acid aligns with CmCLC S175, which lies close to α1 of CBS2 (Fig. 6 A). Mutation of R134 to alanine had little effect on the responsiveness of CLH-3b to GCK-3 (Table 2).
Discussion
Regulatory phosphorylation sites have been identified in the cytoplasmic N- and C-termini of CLC-1, CLC-2, and CLC-3 (31–35). However, there is as yet no clear understanding of the physiological context under which phosphorylation-dependent regulation occurs, the signaling pathways that mediate changes in the protein phosphorylation state, and the molecular mechanisms by which phosphorylation induces changes in the activity of these CLC proteins. Detailed understanding of phosphorylation-dependent regulation of CLCs has been hampered by contradictory results due to species, splice variant, and/or experimental differences.
We have exploited our extensive understanding of CLH-3b function and regulation to gain insights into how phosphorylation of the cytoplasmic C-terminus regulates channel activity. Previous studies (11) as well as the current work (Fig. 4) show that deletion of a region within the inter-CBS linker encompassing the regulatory phosphorylation sites S742 and S747 inhibits CLH-3b. This indicates that the linker is required for maintaining CLH-3b in an active state. The minimal activation domain is ∼14 amino acids long and extends from D738 to Q751 (Fig. 5).
Deletion of the activation domain inactivates CLH-3b to the same extent as coexpression with GCK-3 (Figs. 4 and 5, B–D). The simplest interpretation of these data is that the activation domain normally interacts with another part of the channel and that phosphorylation or deletion disrupts that putative interaction leading to channel inactivation. The small size of the activation domain and the results of our deletion studies (Table 1) suggest that it is unlikely that it interacts with another part of the inter-CBS linker. Given that CBS2 of CmCLC interacts in a highly specific manner with membrane-associated domains of the channel (5), we postulate that the most likely site for the putative activation domain interaction is one or both CLH-3b CBS motifs.
CBS domains are found in numerous proteins from all kingdoms of life and mutations in these motifs give rise to diverse human diseases. The precise functions of CBS domains are not well understood, but they are clearly involved in protein regulation (10,36). For example, CBS domains bind adenosyl compounds such as ATP, ADP, and AMP, and this in turn modulates protein activity (37–39), including the activity of CLCs (9,15,37,40–42). The CBS domains of the MgtE Mg2+ transporter bind Mg2+ and likely function as part of an intracellular Mg2+ sensor that regulates transporter function (43). Recent studies on the bacterial osmoregulatory organic osmolyte transporter OpuA have shown that CBS motifs regulate the transporter on/off state by binding in an ionic strength-dependent manner to charged membrane domains (44).
CBS domains are ∼50–60 amino acids long and have a highly conserved secondary structure consisting of an N-terminal β-strand (β1) followed by an α-helix (α1), two β-strands (β2 and β3), and an α-helix (α2). They are typically present in proteins as two or four copies. Pairs of CBS motifs associate to form a dimeric structure known as a Bateman module or domain, and Bateman domains in turn can interact to form oligomeric structures (5,7,9,10).
What mechanisms could mediate the putative regulated interaction of the CLH-3b activation domain with CBS motifs? One possibility is suggested by recent studies on cystic fibrosis transmembrane regulator (CFTR). The channel is activated by phosphorylation of S768, which is located in the cytoplasmic regulatory (R) domain. Mutagenesis and sulhydryl cross-linking studies suggest that unphosphorylated S768 forms a hydrogen bond with a histidine residue located on a cytoplasmic loop of a membrane spanning domain. Phosphorylation disrupts the hydrogen bonding and activates the channel (45).
S742 and S747 could conceivably form hydrogen bonds with CBS domain amino acids when CLH-3b is active. Phosphorylation would disrupt these hydrogen bonds resulting in channel inhibition. However, CLH-3b remains fully activated when alanine, which is poorly reactive and does not form hydrogen bonds, is substituted for S742 and S747 (20). Similarly, channels in which S742 and S747 are replaced by phenylalanine, a bulky and strongly hydrophobic amino acid, remain fully active (H. Miyazaki and K. Strange, unpublished).
As shown in Fig. 3, the isolated CLH-3b inter-CBS linker appears to be largely unstructured, a finding consistent with structural studies of CLC-0 and CmCLC (5,8,46). Intrinsically disordered domains are common in proteins and are thought to play critical functional roles (27,28). It has been suggested that unstructured domains located within well-structured protein regions function to fish in or sample a large volume of the cytoplasm for binding partners such as kinases and phosphatases (27).
Phosphorylation sites occur predominately in disordered domains (27,28). There is a growing body of work in diverse proteins demonstrating that phosphorylation induces order-to-disorder transitions or the converse in intrinsically disordered domains, and that these transitions in turn modulate function (47–51). For example, recent work on CFTR has demonstrated that the R-domain is intrinsically disordered, but contains small helical regions. R-domain phosphorylation, which is required for channel activation (52,53), reduces the propensity of these regions to form helices and this in turn reduces the interaction of the R-domain with nucleotide-binding domain 1 (NBD1) (54).
Numerous structure prediction programs suggest that although largely disordered, the CLH-3b inter-CBS linker may also contain small, well-ordered helical regions. One of these predicted helical regions encompasses much of the activation domain (see Fig. 3 A). Interestingly, substitution of S742 and S747 with glutamate residues to mimic phosphorylation (20) increases the PONDR scores and hence predicted disorder in this region (Fig. 3 A). Although highly speculative at this stage, phosphorylation may induce an order-to-disorder transition in the activation domain similar to that proposed for the R-domain of CFTR (54). This in turn may disrupt interactions with CBS motifs leading to channel inactivation. Clearly, extensive biochemical and structural studies are required to determine if there is a regulated interaction between activation and CBS domains.
How could phosphorylation-dependent changes in CBS conformation be transduced into changes in CLH-3b channel activity? The CmCLC crystal structure demonstrated close and apparently highly specific interactions with the short loop connecting membrane helices H and I, the C-terminus of membrane helix D, and a short linker connecting membrane helix R to CBS1 (5). Using the CmCLC structure as a guide, we made mutations in each of these interacting domains. Mutations in the R-helix linker or the D-helix had no effect on the ability of GCK-3 to inhibit CLH-3b (Table 2) suggesting that these domains may not be important for signal transduction. In contrast, a single point mutation in the H-I loop, Y232A, completely prevented inhibition by GCK-3 or mutation of S742 and S747 to glutamate (Fig. 7, A, C, and D).
Y232 shows strong conservation or conservative substitution in CLH-3b, CmCLC, and various mammalian CLC proteins (Fig. 6 B). Of importance, the H and I helices form part of the subunit interface of the CLC homodimer (4). The subunit interface is the site of multiple disease causing mutations (55,56). Disease and site-directed interface mutations alter CLC biophysical properties as well as protein folding and trafficking. Studies on CLC-1 have demonstrated that the subunit interface plays an important role in common gating and may function in intersubunit communication thereby allowing the common gate to close both protopores simultaneously (57,58). The interface may also play a role in Ca2+-dependent regulation of CLC-K channels (59). Given the functional importance of the subunit interface then, it is logical to propose that it may also function as at least one site at which CBS domain conformational changes transmitted through the H-I loop are transduced into changes in channel activity. An attractive and testable hypothesis is that phosphorylation-dependent changes in the structural properties of the subunit interface regulate CLH-3b common gating.
A phenylalanine residue at position 318 in CLC-7 aligns with Y232 (Fig. 6 B). Mutation of F318 to leucine causes a dominant form of osteopetrosis (60). Interestingly, the F318L mutant expresses and traffics normally (60). This observation suggests that F318L disrupts CLC-7 ion transport or possibly its regulation by the accessory protein Ostm1.
In CmCLC, the H-I loop lies close to α1 of CBS2 (5). Consistent with this, we found that mutating H805 to alanine renders the channel insensitive to GCK-3 or the S742E/S747E double mutation (Fig. 7, B–D, and Results). We conclude that CBS2 α1 plays a central role in phosphorylation-dependent regulation of CLH-3b activity.
Of importance, H805 shows strong conservation in eukaryotic CLC proteins. H805 aligns with H736 in CLC-0, H838 in CLC-1, and H811 in CLC-2 (6). Mutating these histidine residues to alanine causes striking changes in the voltage-dependent gating properties of these three channels (6,13). Aromatic-aromatic amino acid interactions play critical roles in protein structure/function (61). We postulate that Y232 and H805 may interact directly and that this putative interaction represents a conserved signal transduction module in CLC proteins.
In conclusion, our studies provide the first, to our knowledge, detailed mechanistic insights into how structural information associated with C-terminus phosphorylation is transduced into changes in CLC channel activity. Although our studies do not rule out the possibility that other CLH-3b membrane-associated domains may also play a signaling role, we suggest that α1 of CBS2 and the H-I loop represent a critical phosphorylation-dependent signal transduction module. This module may also be important in regulation of CLC proteins by nucleotide binding (9,15,37,40–42) and accessory proteins (60,62–64).
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant R01 DK51610 to K.S.
Experiments described in this paper were proposed and designed by H.M., T.Y., A.P., R.M., S.K., A.H.B. and K.S. H.M., T.Y., A.P., R.M., and S.K performed experimental studies. All authors participated in the analysis and interpretation of the data, in the writing of the manuscript, and in the approval of the final version of the manuscript for publication.
References
- 1.Duran C., Thompson C.H., Hartzell H.C. Chloride channels: often enigmatic, rarely predictable. Annu. Rev. Physiol. 2010;72:95–121. doi: 10.1146/annurev-physiol-021909-135811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jentsch T.J. CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Crit. Rev. Biochem. Mol. Biol. 2008;43:3–36. doi: 10.1080/10409230701829110. [DOI] [PubMed] [Google Scholar]
- 3.Dutzler R., Campbell E.B., MacKinnon R. Gating the selectivity filter in ClC chloride channels. Science. 2003;300:108–112. doi: 10.1126/science.1082708. [DOI] [PubMed] [Google Scholar]
- 4.Dutzler R., Campbell E.B., MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- 5.Feng L., Campbell E.B., MacKinnon R. Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science. 2010;330:635–641. doi: 10.1126/science.1195230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Estévez R., Pusch M., Jentsch T.J. Functional and structural conservation of CBS domains from CLC chloride channels. J. Physiol. 2004;557:363–378. doi: 10.1113/jphysiol.2003.058453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Markovic S., Dutzler R. The structure of the cytoplasmic domain of the chloride channel ClC-Ka reveals a conserved interaction interface. Structure. 2007;15:715–725. doi: 10.1016/j.str.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 8.Meyer S., Dutzler R. Crystal structure of the cytoplasmic domain of the chloride channel ClC-0. Structure. 2006;14:299–307. doi: 10.1016/j.str.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 9.Meyer S., Savaresi S., Dutzler R. Nucleotide recognition by the cytoplasmic domain of the human chloride transporter ClC-5. Nat. Struct. Mol. Biol. 2007;14:60–67. doi: 10.1038/nsmb1188. [DOI] [PubMed] [Google Scholar]
- 10.Ignoul S., Eggermont J. CBS domains: structure, function, and pathology in human proteins. Am. J. Physiol. Cell Physiol. 2005;289:C1369–C1378. doi: 10.1152/ajpcell.00282.2005. [DOI] [PubMed] [Google Scholar]
- 11.He L., Denton J., Strange K. Carboxy terminus splice variation alters ClC channel gating and extracellular cysteine reactivity. Biophys. J. 2006;90:3570–3581. doi: 10.1529/biophysj.105.078295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denton J., Nehrke K., Strange K. Altered gating and regulation of a carboxy-terminal ClC channel mutant expressed in the Caenorhabditis elegans oocyte. Am. J. Physiol. Cell Physiol. 2006;290:C1109–C1118. doi: 10.1152/ajpcell.00423.2005. [DOI] [PubMed] [Google Scholar]
- 13.Yusef Y.R., Zúñiga L., Sepúlveda F.V. Removal of gating in voltage-dependent ClC-2 chloride channel by point mutations affecting the pore and C-terminus CBS-2 domain. J. Physiol. 2006;572:173–181. doi: 10.1113/jphysiol.2005.102392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennetts B., Rychkov G.Y., Cromer B.A. Cytoplasmic ATP-sensing domains regulate gating of skeletal muscle ClC-1 chloride channels. J. Biol. Chem. 2005;280:32452–32458. doi: 10.1074/jbc.M502890200. [DOI] [PubMed] [Google Scholar]
- 15.Bennetts B., Parker M.W., Cromer B.A. Inhibition of skeletal muscle ClC-1 chloride channels by low intracellular pH and ATP. J. Biol. Chem. 2007;282:32780–32791. doi: 10.1074/jbc.M703259200. [DOI] [PubMed] [Google Scholar]
- 16.Wu W., Rychkov G.Y., Bretag A.H. Functional complementation of truncated human skeletal-muscle chloride channel (hClC-1) using carboxyl tail fragments. Biochem. J. 2006;395:89–97. doi: 10.1042/BJ20050966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dave S., Sheehan J.H., Strange K. Unique gating properties of C. elegans ClC anion channel splice variants are determined by altered CBS domain conformation and the R-helix linker. Channels (Austin) 2010;4:289–301. doi: 10.4161/chan.4.4.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rutledge E., Bianchi L., Strange K. CLH-3, a ClC-2 anion channel ortholog activated during meiotic maturation in C. elegans oocytes. Curr. Biol. 2001;11:161–170. doi: 10.1016/s0960-9822(01)00051-3. [DOI] [PubMed] [Google Scholar]
- 19.Rutledge E., Denton J., Strange K. Cell cycle- and swelling-induced activation of a Caenorhabditis elegans ClC channel is mediated by CeGLC-7α/β phosphatases. J. Cell Biol. 2002;158:435–444. doi: 10.1083/jcb.200204142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Falin R.A., Morrison R., Strange K. Identification of regulatory phosphorylation sites in a cell volume- and Ste20 kinase-dependent ClC anion channel. J. Gen. Physiol. 2009;133:29–42. doi: 10.1085/jgp.200810080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Denton J., Nehrke K., Strange K. GCK-3, a newly identified Ste20 kinase, binds to and regulates the activity of a cell cycle-dependent ClC anion channel. J. Gen. Physiol. 2005;125:113–125. doi: 10.1085/jgp.200409215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Falin R.A., Miyazaki H., Strange K. C. elegans STK39/SPAK ortholog-mediated inhibition of ClC anion channel activity is regulated by WNK-independent ERK kinase signaling. Am. J. Physiol. Cell Physiol. 2011;300:C624–C635. doi: 10.1152/ajpcell.00343.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delpire E., Gagnon K.B. SPAK and OSR1: STE20 kinases involved in the regulation of ion homoeostasis and volume control in mammalian cells. Biochem. J. 2008;409:321–331. doi: 10.1042/BJ20071324. [DOI] [PubMed] [Google Scholar]
- 24.Miyazaki H., Strange K. Differential regulation of a CLC anion channel by SPAK kinase ortholog-mediated multisite phosphorylation. Am. J. Physiol. Cell Physiol. 2012;302:C1702–C1712. doi: 10.1152/ajpcell.00419.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Romero P., Obradovic Z., Dunker A.K. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 26.Louis-Jeune C., Andrade-Navarro M.A., Perez-Iratxeta C. Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins. 2011;80:374–381. doi: 10.1002/prot.23188. [DOI] [PubMed] [Google Scholar]
- 27.Gsponer J., Babu M.M. The rules of disorder or why disorder rules. Prog. Biophys. Mol. Biol. 2009;99:94–103. doi: 10.1016/j.pbiomolbio.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen Ba A.N., Yeh B.J., Moses A.M. Proteome-wide discovery of evolutionary conserved sequences in disordered regions. Sci. Signal. 2012;5:rs1. doi: 10.1126/scisignal.2002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Estévez R., Schroeder B.C., Pusch M. Conservation of chloride channel structure revealed by an inhibitor binding site in ClC-1. Neuron. 2003;38:47–59. doi: 10.1016/s0896-6273(03)00168-5. [DOI] [PubMed] [Google Scholar]
- 30.Engh A.M., Maduke M. Cysteine accessibility in ClC-0 supports conservation of the ClC intracellular vestibule. J. Gen. Physiol. 2005;125:601–617. doi: 10.1085/jgp.200509258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furukawa T., Ogura T., Inagaki N. Phosphorylation and functional regulation of ClC-2 chloride channels expressed in Xenopus oocytes by M cyclin-dependent protein kinase. J. Physiol. 2002;540:883–893. doi: 10.1113/jphysiol.2001.016188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsiao K.M., Huang R.Y., Lin M.J. Functional study of CLC-1 mutants expressed in Xenopus oocytes reveals that a C-terminal region Thr-891-Ser-892-Thr-893 is responsible for the effects of protein kinase C activator. Cell. Physiol. Biochem. 2010;25:687–694. doi: 10.1159/000315088. [DOI] [PubMed] [Google Scholar]
- 33.Cuppoletti J., Tewari K.P., Malinowska D.H. Sites of protein kinase A activation of the human ClC-2 Cl(-) channel. J. Biol. Chem. 2004;279:21849–21856. doi: 10.1074/jbc.M312567200. [DOI] [PubMed] [Google Scholar]
- 34.Duan D., Cowley S., Hume J.R. A serine residue in ClC-3 links phosphorylation-dephosphorylation to chloride channel regulation by cell volume. J. Gen. Physiol. 1999;113:57–70. doi: 10.1085/jgp.113.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robinson N.C., Huang P., Nelson D.J. Identification of an N-terminal amino acid of the CLC-3 chloride channel critical in phosphorylation-dependent activation of a CaMKII-activated chloride current. J. Physiol. 2004;556:353–368. doi: 10.1113/jphysiol.2003.058032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baykov A.A., Tuominen H.K., Lahti R. The CBS domain: a protein module with an emerging prominent role in regulation. ACS Chem. Biol. 2011;6:1156–1163. doi: 10.1021/cb200231c. [DOI] [PubMed] [Google Scholar]
- 37.Scott J.W., Hawley S.A., Hardie D.G. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Invest. 2004;113:274–284. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Townley R., Shapiro L. Crystal structures of the adenylate sensor from fission yeast AMP-activated protein kinase. Science. 2007;315:1726–1729. doi: 10.1126/science.1137503. [DOI] [PubMed] [Google Scholar]
- 39.Jin X., Townley R., Shapiro L. Structural insight into AMPK regulation: ADP comes into play. Structure. 2007;15:1285–1295. doi: 10.1016/j.str.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 40.De Angeli A., Moran O., Gambale F. ATP binding to the C-terminus of the Arabidopsis thaliana nitrate/proton antiporter, AtCLCa, regulates nitrate transport into plant vacuoles. J. Biol. Chem. 2009;284:26526–26532. doi: 10.1074/jbc.M109.005132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tseng P.Y., Yu W.P., Chen T.Y. Binding of ATP to the CBS domains in the C-terminal region of CLC-1. J. Gen. Physiol. 2011;137:357–368. doi: 10.1085/jgp.201010495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wellhauser L., Luna-Chavez C., Bear C.E. ATP binding induces conformational changes in the carboxy terminal region of ClC-5. J. Biol. Chem. 2011;268:6733–6741. doi: 10.1074/jbc.M110.175877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hattori M., Tanaka Y., Nureki O. Crystal structure of the MgtE Mg2+ transporter. Nature. 2007;448:1072–1075. doi: 10.1038/nature06093. [DOI] [PubMed] [Google Scholar]
- 44.Mahmood N.A., Biemans-Oldehinkel E., Poolman B. Engineering of ion sensing by the cystathionine beta-synthase module of the ABC transporter OpuA. J. Biol. Chem. 2009;284:14368–14376. doi: 10.1074/jbc.M901238200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang G. The inhibition mechanism of non-phosphorylated Ser768 in the regulatory domain of cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 2011;286:2171–2182. doi: 10.1074/jbc.M110.145540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alioth S., Meyer S., Pervushin K. The cytoplasmic domain of the chloride channel ClC-0: structural and dynamic characterization of flexible regions. J. Mol. Biol. 2007;369:1163–1169. doi: 10.1016/j.jmb.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 47.Karim C.B., Zhang Z., Thomas D.D. Phosphorylation-dependent conformational switch in spin-labeled phospholamban bound to SERCA. J. Mol. Biol. 2006;358:1032–1040. doi: 10.1016/j.jmb.2006.02.051. [DOI] [PubMed] [Google Scholar]
- 48.Metcalfe E.E., Traaseth N.J., Veglia G. Serine 16 phosphorylation induces an order-to-disorder transition in monomeric phospholamban. Biochemistry. 2005;44:4386–4396. doi: 10.1021/bi047571e. [DOI] [PubMed] [Google Scholar]
- 49.Wynn R.M., Kato M., Chuang D.T. Molecular mechanism for regulation of the human mitochondrial branched-chain alpha-ketoacid dehydrogenase complex by phosphorylation. Structure. 2004;12:2185–2196. doi: 10.1016/j.str.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 50.Sanchez E.J., Munske G.R., Kang C. Phosphorylation of human calsequestrin: implications for calcium regulation. Mol. Cell. Biochem. 2011;353:195–204. doi: 10.1007/s11010-011-0787-4. [DOI] [PubMed] [Google Scholar]
- 51.Nelson W.D., Blakely S.E., Thomas D.D. Site-directed spin labeling reveals a conformational switch in the phosphorylation domain of smooth muscle myosin. Proc. Natl. Acad. Sci. USA. 2005;102:4000–4005. doi: 10.1073/pnas.0401664102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alzamora R., King J.D., Jr., Hallows K.R. CFTR regulation by phosphorylation. Methods Mol. Biol. 2011;741:471–488. doi: 10.1007/978-1-61779-117-8_29. [DOI] [PubMed] [Google Scholar]
- 53.Hwang T.C., Sheppard D.N. Gating of the CFTR Cl- channel by ATP-driven nucleotide-binding domain dimerisation. J. Physiol. 2009;587:2151–2161. doi: 10.1113/jphysiol.2009.171595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baker J.M., Hudson R.P., Forman-Kay J.D. CFTR regulatory region interacts with NBD1 predominantly via multiple transient helices. Nat. Struct. Mol. Biol. 2007;14:738–745. doi: 10.1038/nsmb1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lourdel S., Grand T., Teulon J. ClC-5 mutations associated with Dent’s disease: a major role of the dimer interface. Pflugers Arch. 2012;463:247–256. doi: 10.1007/s00424-011-1052-0. [DOI] [PubMed] [Google Scholar]
- 56.Pusch M. Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Hum. Mutat. 2002;19:423–434. doi: 10.1002/humu.10063. [DOI] [PubMed] [Google Scholar]
- 57.Cederholm J.M., Rychkov G.Y., Bretag A.H. Inter-subunit communication and fast gate integrity are important for common gating in hClC-1. Int. J. Biochem. Cell Biol. 2010;42:1182–1188. doi: 10.1016/j.biocel.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 58.Duffield M., Rychkov G., Roberts M. Involvement of helices at the dimer interface in ClC-1 common gating. J. Gen. Physiol. 2003;121:149–161. doi: 10.1085/jgp.20028741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gradogna A., Babini E., Pusch M. A regulatory calcium-binding site at the subunit interface of CLC-K kidney chloride channels. J. Gen. Physiol. 2010;136:311–323. doi: 10.1085/jgp.201010455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leisle L., Ludwig C.F., Stauber T. ClC-7 is a slowly voltage-gated 2Cl(-)/1H(+)-exchanger and requires Ostm1 for transport activity. EMBO J. 2011;30:2140–2152. doi: 10.1038/emboj.2011.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burley S.K., Petsko G.A. Aromatic-aromatic interaction: a mechanism of protein structure stabilization. Science. 1985;229:23–28. doi: 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- 62.Jeworutzki E., López-Hernández T., Estévez R. GlialCAM, a protein defective in a leukodystrophy, serves as a ClC-2 Cl(-) channel auxiliary subunit. Neuron. 2012;73:951–961. doi: 10.1016/j.neuron.2011.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fischer M., Janssen A.G., Fahlke C. Barttin activates ClC-K channel function by modulating gating. J. Am. Soc. Nephrol. 2010;21:1281–1289. doi: 10.1681/ASN.2009121274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scholl U., Hebeisen S., Fahlke C. Barttin modulates trafficking and function of ClC-K channels. Proc. Natl. Acad. Sci. USA. 2006;103:11411–11416. doi: 10.1073/pnas.0601631103. [DOI] [PMC free article] [PubMed] [Google Scholar]