

Abstract
Reverse transcription and integration are the defining features of the Retroviridae; the common name “retrovirus” derives from the fact that these viruses use a virally encoded enzyme, reverse transcriptase (RT), to convert their RNA genomes into DNA. Reverse transcription is an essential step in retroviral replication. This article presents an overview of reverse transcription, briefly describes the structure and function of RT, provides an introduction to some of the cellular and viral factors that can affect reverse transcription, and discusses fidelity and recombination, two processes in which reverse transcription plays an important role. In keeping with the theme of the collection, the emphasis is on HIV-1 and HIV-1 RT.
More than half of the drugs currently approved to treat HIV-1 infections are reverse transcriptase inhibitors. Therefore, the continued characterization of HIV-1 reverse transcriptase and the process of reverse transcription is vital.
It has been 40 years since the discovery of reverse transcriptase (RT) was announced by Howard Temin and David Baltimore, who independently showed that retroviral virions contain an enzymatic activity that can copy RNA into DNA (Baltimore 1970; Mizutani et al. 1970). These experiments provided the crucial proof of Temin’s provirus hypothesis that retroviral infections persist because the RNA genome found in the virions is converted into DNA (Temin 1964). The sequences of the genomes of eukaryotes show how pervasive reverse transcription is in nature; not only do these genomes contain large numbers of endogenous retroviruses, but also a variety of retroposons and reverse-transcribed elements. The discovery in the early 1980s, that AIDS is caused by a human retrovirus, HIV-1, invigorated retroviral research and focused attention on the viral enzymes, which have become the primary target of anti-AIDS drugs. Not surprisingly, the focus of RT research shifted from the RTs of the murine leukemia viruses (MLV) and the avian myeloblastosis virus to HIV-1 RT. The first approved anti-HIV drug, AZT, targets RT, and of the 26 drugs currently approved to treat HIV-1 infections, 14 are RT inhibitors. In addition, RTs (primarily recombinant MLV RTs) have become extremely valuable tools that are widely used in research, in clinical/diagnostic tests, and in biotechnology. We provide here a relatively brief description of the process of reverse transcription, the structure and biochemical functions of RT, some information about how other viral and cellular factors influence reverse transcription, and briefly consider how the reverse transcription process affects both the mutations that arise during the retroviral life cycle and recombination. The focus will be HIV-1 and HIV-1 RT; however, in some cases, we will draw on insights and include information obtained with other retroviruses and other RTs. Although the issues of the inhibition of HIV-1 RT by anti-RT drugs and the mechanisms of drug resistance are of considerable importance, these issues will not be addressed in detail here; the reader is directed to Arts and Hazuda (2011). Given that the literature on RT and reverse transcription is both vast and complex, and the space allowed for this article is limited, we have had to make some difficult choices in what to present, and what to omit, both in terms of the material and the references. For the omissions, we apologize.
THE PROCESS OF REVERSE TRANSCRIPTION
When a mature HIV-1 virion infects a susceptible target cell, interactions of the envelope glycoprotein with the coreceptors on the surface of the cell brings about a fusion of the membranes of the host cell and the virion (Wilen et al. 2011). This fusion introduces the contents of the virion into the cytoplasm of the cell, setting the stage for reverse transcription. There are complexities to the early events that accompany reverse transcription in an infected cell, not all of which are well understood, which will be considered later in this article. We will begin by discussing the mechanics of the conversion of the single-stranded RNA genome found in the virion into the linear double-stranded DNA that is the substrate for the integration process. The synthesis of this linear DNA is a reasonably well-understood process; additional details and references can be found in the books Retroviruses (Telesnitsky and Goff 1997) and Reverse Transcriptase (Skalka and Goff 1993). In orthoretroviruses, including HIV-1, reverse transcription takes place in newly infected cells. There is some debate in the literature about whether reverse transcription is initiated in producer cells. Primer tagging experiments suggest that most HIV-1 virions initiate reverse transcription in newly infected cells (Whitcomb et al. 1990); however, there are claims that a small number of nucleotides may be incorporated before the virions initiate infection of target cells (Lori et al. 1992; Trono 1992; Zhu and Cunningham 1993; Huang et al. 1997). Either way, the vast majority of the viral DNA is synthesized in newly infected cells. This is a lifestyle choice; spumaretroviruses and the more distantly related hepadna viruses carry out extensive reverse transcription in producer cells (Summers and Mason 1982; Yu et al. 1996, 1999). Although there are viral and cellular factors that assist in the process of reverse transcription (these will be discussed later) the two enzymatic activities that are necessary and sufficient to carry out reverse transcription are present in RT. These are a DNA polymerase that can copy either a RNA or a DNA template, and an RNase H that degrades RNA if, and only if, it is part of an RNA–DNA duplex.
Like many other DNA polymerases, RT needs both a primer and a template. Genomic RNA is plus-stranded (the genome and the messages are copied from the same DNA strand), and the primer for the synthesis of the first DNA strand (the minus strand) is a host tRNA whose 3′ end is base paired to a complementary sequence near the 5′ end of the viral RNA called the primer binding site (pbs). Different retroviruses use different host tRNAs as primers. HIV-1 uses Lys3. It would appear, based on in vitro experiments, that the addition of the first few nucleotides is slow and difficult. DNA synthesis speeds up considerably once the first five to six deoxyribonucleotides have been added to the 3′ end of the tRNA primer (Isel et al. 1996; Lanchy et al. 1998). In HIV-1, the pbs is approximately 180 nucleotides from the 5′ end of genomic RNA. DNA synthesis creates an RNA–DNA duplex, which is a substrate for RNase H. There are perhaps 50 RTs in an HIV-1 virion; it is unclear whether the same RT that synthesizes the DNA plays a significant role in degrading the RNA. This is not a requirement—retroviruses can replicate (at a considerably reduced efficiency) with a mixture of RTs, some of which have only polymerase activity and some that have only RNase H activity (Telesnitsky and Goff 1993; Julias et al. 2001). Moreover, in in vitro assays, little or no RNase H cleavage is detected while RT is actively synthesizing DNA; instead, cleavages occur at sites where DNA synthesis pauses (Driscoll et al. 2001; Purohit et al. 2007). Whatever the exact mechanism, RNase H degradation removes the 5′ end of the viral RNA, exposing the newly synthesized minus-strand DNA (see Fig. 1).
Figure 1.

Conversion of the single-stranded RNA genome of a retrovirus into double-stranded DNA. (A) The RNA genome of a retrovirus (light blue) with a tRNA primer base paired near the 5′ end. (B) RT has initiated reverse transcription, generating minus-strand DNA (dark blue), and the RNase H activity of RT has degraded the RNA template (dashed line). (C) Minus-strand transfer has occurred between the R sequences at both ends of the genome (see text), allowing minus-strand DNA synthesis to continue (D), accompanied by RNA degradation. A purine-rich sequence (ppt), adjacent to U3, is resistant to RNase H cleavage and serves as the primer for the synthesis of plus-strand DNA (E). Plus-strand synthesis continues until the first 18 nucleotides of the tRNA are copied, allowing RNase H cleavage to remove the tRNA primer. Most retroviruses remove the entire tRNA; the RNase H of HIV-1 RT leaves the rA from the 3′ end of the tRNA attached to minus-strand DNA. Removal of the tRNA primer sets the stage for the second (plus-strand) transfer (F); extension of the plus and minus strands leads to the synthesis of the complete double-stranded linear viral DNA (G).
The ends of the viral RNA are direct repeats, called R. These repeats act as a bridge that allows the newly synthesized minus-strand DNA to be transferred to the 3′ end of the viral RNA. Retroviruses package two copies of the viral RNA genome; the first (or minus-strand) transfer can involve the R sequence at the 3′ ends of either of the two RNAs (Panganiban and Fiore 1988; Hu and Temin 1990b; van Wamel and Berkhout 1998; Yu et al. 1998). After this transfer, minus-strand synthesis can continue along the length of the genome. As DNA synthesis proceeds, so does RNase H degradation. However, there is a purine-rich sequence in the RNA genome, called the polypurine tract, or ppt, that is resistant to RNase H cleavage and serves as the primer for the initiation of the second (or plus) strand DNA. All retroviruses have at least one ppt. HIV-1 has two, one near the 3′ end of the RNA, the other (the central ppt) near the middle of the genome. The 3′ ppt is essential for viral replication, the central ppt probably increases the ability of the virus to complete plus-strand DNA synthesis, but is not essential (Charneau et al. 1992; Hungnes et al. 1992). When RT generates the plus-stand DNA that is initiated from the 3′ ppt, it not only copies the minus-strand DNA, but also the first 18 nucleotides of the Lys3 tRNA primer. Experiments performed with avian sarcoma-leukosis virus (ASLV) suggest that the ppt-primed plus-strand DNA synthesis stops when it encounters a modified A that RT cannot copy (Swanstrom et al. 1981). It is reasonable to expect that the same mechanism defines the portion of the HIV tRNA primer that is copied. Once the tRNA has been copied into DNA, it becomes a substrate for RNase H. Most retroviruses remove the entire tRNA; however, HIV-1 RT is the exception. It cleaves the tRNA one nucleotide from the 3′ end, leaving a single A ribonucleotide at the 5′ end of the minus strand (the specificity of RNase H cleavage is discussed at the end of this section) (Whitcomb et al. 1990; Pullen et al. 1992; Smith and Roth 1992).
In theory, minus-strand DNA synthesis can proceed along the entire length of the RNA genome; however, the genomic RNAs found in virions are often nicked. The fact that there is a second copy of the RNA genome allows minus-strand DNA synthesis to transfer to the second RNA template, thus bypassing the nick in the original template. This template switching ability contributes to efficient recombination, a topic that is considered later in this article. When minus-strand DNA synthesis nears the 5′ end of the genomic RNA, the pbs is copied, setting the stage for the second, or plus-strand transfer. The 3′ end of the plus-strand DNA contains 18 nucleotides copied from the tRNA primer, which are complementary to 18 nucleotides at the 3′ end of the minus-strand DNA that were copied from the pbs. These two complementary sequences anneal, and DNA synthesis extends both the minus and plus strands to the ends of both templates. The synthesis of plus-strand DNA does not have to be continuous; it is clear that, in ASLV, the plus strand is made in segments (Kung et al. 1981; Hsu and Taylor 1982). It has been reported that HIV-1 plus-strand DNA is also synthesized from multiple initiation sites (Miller et al. 1995; Klarmann et al. 1997; Thomas et al. 2007); however, that raises a question about the role played by the second ppt: If plus-strand DNA is made in segments, what advantage does the second ppt give HIV-1?
The reverse transcription process creates a DNA product that is longer than the RNA genome from which it is derived: both ends of the DNA contain sequences from each end of the RNA (U3 from the 3′ end and U5 from the 5′ end). Thus, each end of the viral DNA has the same sequence, U3-R-U5; these are the long terminal repeats (LTRs) that will, after integration, be the ends of the provirus. It is important to remember that the sequences at the ends of the full-length linear viral DNA are defined, on the U5 end, by the RNase H cleavage that removes the tRNA primer, and on the U3 end, by the cleavages that generate and remove the ppt primer. Despite the fact that RNase H does not have any specific sequence recognition motifs, it cleaves these substrates with single nucleotide specificity, a specificity that appears to be based on the structures of the nucleic acid substrates when they are in a complex with RT (Pullen et al. 1993; Julias et al. 2002; Rausch et al. 2002; Dash et al. 2004; Yi-Brunozzi and Le Grice 2005). The specificity of RNase H cleavage is important because the ends of the linear viral DNA are the substrates for integration. Although DNA substrates whose ends differ modestly from the consensus sequence can be used for retroviral DNA integration, the consensus sequence is the preferred substrate (Colicelli and Goff 1985, 1988; Esposito and Craigie 1998; Oh et al. 2008).
THE GENESIS, STRUCTURE, AND ENZYMATIC FUNCTIONS OF HIV-1 RT
HIV-1 RT is produced from a Gag-Pol polyprotein by cleavage with the viral protease (PR). HIV-1 Gag-Pol is produced by a frameshift readthrough event in the p6 coding region that occurs about 5% of the time Gag is translated from an unspliced full-length viral RNA transcript. The Gag portion of Gag-Pol allows it to associate with Gag during virion assembly, ensuring that the Pol portion of Gag-Pol, which includes PR, RT, and integrase, is inside the assembled virion.
The mature form of HIV-1 RT is a heterodimer that is composed of two related subunits: the larger, p66, is 560 amino acids long; the smaller, p51, contains the first 440 amino acids of p66 (Lightfoote et al. 1986). The p66 subunit consists of two domains: polymerase and RNase H; in the mature HIV-1 RT heterodimer, p66 contains the active sites for the two enzymatic activities of RT (see Movie 1). The polymerase domain has been compared to a human right hand and is composed of the fingers, palm, thumb, and connection subdomains (see Fig. 2) (Kohlstaedt et al. 1992). The p51 subunit corresponds closely, but not exactly, to the polymerase domain of p66, and contains the same four subdomains. However, the relative arrangement of the subdomains differs in the two subunits. The p66 domain plays the catalytic role, whereas the p51 subunit plays a structural role (Movie 1) (Kohlstaedt et al. 1992; Jacobo-Molina et al. 1993).
Figure 2.

Structure of a ternary complex of HIV-1 RT, double-stranded DNA, and an incoming dNTP. HIV-1 RT is composed of two subunits, p51 and p66. P51 is shown in gray. The RNase H domain of p66 is gold, and the four subdomains of the polymerase domain of p66 are color-coded: fingers, blue; palm, red; thumb, green; and connection, yellow. The template strand of the DNA is brown, and the primer strand is purple. The incoming dNTP is light blue. (Figure courtesy of K. Das and E. Arnold.)
Movie 1.
The active site of reverse transcriptase.
We are fortunate to have crystal structures that correspond to HIV-1 RT in multiple states that are important intermediates in the reverse transcription process (see Fig. 3 and Movie 2). Some of the structures also tell us a great deal about how anti-RT drugs work, and how resistance mutations allow RT to evade the currently approved drugs (Arts and Hazuda 2011). Considering the structures in a way that corresponds to steps in reverse transcription, unliganded RT has the thumb subdomain of the p66 subunit folded over into the nucleic acid binding cleft, in a position such that the tip of thumb nearly touches the fingers (Esnouf et al. 1995; Rodgers et al. 1995; Hsiou et al. 1996). Because the thumb is in the closed configuration in unliganded RT, the thumb must move away from the fingers to create the nucleic acid binding cleft for the template primer. The nucleic acid binding cleft has a structure that allows it to bind a double-stranded nucleic acid (Jacobo-Molina et al. 1993). There are modest interactions of the single-stranded 5′ extension of the template with RT; this helps to position the end of the primer at the polymerase active site, which is composed of three aspartatic acid residues (D110, D185, and D186), that help position the two divalent metal ions (Mg2+ during viral replication) required for polymerization (Movie 2).
Figure 3.

Structural changes in RT that occur during polymerization. In unliganded RT (A), the thumb is in the closed configuration. Binding a double-stranded nucleic acid substrate (B) is accompanied by movement of the thumb (upper left, A,B) that creates the nucleic acid binding site. Binding of the incoming dNTP (C) is accompanied by a movement of the fingers that closes the β3-β4 loop down onto the incoming dNTP (lower left, B,C). These movements correspond to steps in DNA synthesis (bottom). (Figure courtesy of K. Kirby and S. Sarafianos.)
Movie 2.
Conformational changes in the reverse transcriptase complex on binding the nucleic acid substrate and dNTP.
We do not have any structures of RT with an RNA–RNA duplex (which would correspond to minus-strand initiation); however, there is one structure with an RNA–DNA duplex and several that contain DNA–DNA duplexes (Jacobo-Molina et al. 1993; Huang et al. 1998; Sarafianos et al. 2001, 2002; Tuske et al. 2004; Das et al. 2009; Lansdon et al. 2010; Tu et al. 2010). The sequence of the RNA in the RNA–DNA duplex structure was based on the ppt, which was chosen for this structural analysis because it is poorly cleaved by RNase H. The RNA–DNA duplex is bent approximately 40° near where it passes under the thumb. A similar bend is seen in the DNA–DNA structures (discussed below). However, the surprising thing about the RNA–DNA duplex is that it contains two unpaired and two mispaired bases that take the duplex out of, then back into, proper register (Sarafianos et al. 2001). We only have one RNA–DNA structure, so it is possible that this misalignment could be specific to the ppt, and, if it is, the misalignment could play a role in the resistance of the ppt to RNase H cleavage. Although the RNA strand is the template strand in this structure, the RNase H active site amino acids D443, E478, D498, and D549, which help position the two divalent metal ions (again, Mg2+ in vivo) do not make close contact with the RNA strand in the structure, which helps account for the inability of RNase H to cleave the ppt. The polymerase and RNase H active sites are 17 to 18 base pairs apart along the nucleic acid, depending on the nucleic acid substrate (Jacobo-Molina et al. 1993; Sarafianos et al. 2001). Despite the fact that the RNA template does not make close contact with the RNase H active site in the one structure we have of RT bound to an RNA–DNA duplex, the two active sites are positioned so that they should be able to simultaneously engage a nucleic acid substrate. There are several motifs that play important roles in holding and properly positioning the nucleic acid relative to the two active sites. The RNase H primer grip, which is near the RNase H active site, plays a role in positioning an RNA–DNA duplex for proper (specific) cleavage (Julias et al. 2002; Rausch et al. 2002). The primer grip and template grip, which are nearer the polymerase active site, help position nucleic acid duplexes at both the polymerase and RNase H active sites (Ghosh et al. 1996, 1997; Powell et al. 1997, 1999; Gao et al. 1998).
The complexes of RT bound to a DNA–DNA duplex are globally quite similar to the complex with the RNA–DNA duplex but with some interesting differences. None of the DNA–DNA duplexes contain unpaired bases, possibly because none of the sequences of the DNA–DNA duplexes in the RT structures have the sequence of the ppt. The DNA–DNA duplex follows a similar bent trajectory as the RNA–DNA duplex (Jacobo-Molina et al. 1993; Sarafianos et al. 2001). In the DNA–DNA duplex, the portion of the double-stranded DNA near the polymerase active site is A form. Where the DNA bends, near the thumb of p66, there is a transition, which occurs over a stretch of four base pairs, from an A-form to a B-form duplex, and the DNA beyond the thumb is B form. In contrast, the RNA–DNA duplex is neither entirely A form nor B form, being somewhat intermediate between the two (this is common for RNA–DNA duplexes; these structures have been called H form), although the region near the polymerase active site is more similar to A form and the region beyond the thumb more similar to B form.
There are also structures of HIV-1 RT with both a bound DNA–DNA duplex and an incoming dNTP or the triphosphate of a nucleoside analog (Huang et al. 1998; Tuske et al. 2004; Das et al. 2009; Lansdon et al. 2010). Overall, the structures of these ternary complexes are similar to the corresponding structures of HIV-1 RT bound to a DNA–DNA duplex, with one important difference: When there is a bound dNTP, a portion of the p66 fingers (the β3-β4 loop) closes down on the incoming triphosphate, forming part of the dNTP-binding pocket. A similar movement of the fingers has been seen with other polymerases (bacterial DNA polymerases, T7 RNA polymerase, and some viral RNA-dependent RNA polymerases) when an incoming nucleoside triphosphate is bound. In the structures of HIV-1 RT in a complex with a DNA–DNA duplex and an incoming dNTP, the last nucleotide at the 3′ end of the primer strand is a dideoxy; this prevents incorporation of the incoming dNTP. The dNTP is bound at the active site, which is also called the N, or nucleoside-binding site. The end of the primer is at the P, or priming site. During normal polymerization, the incorporation of the incoming dNTP links the α-phosphate of the dNTP to the 3′OH of the deoxyribose of the nucleotide at the 3′ end of the primer, releasing pyrophosphate. At this point, the end of the primer is still in the N site, and there are structures that correspond to this state. For polymerization to continue, the nucleic acid substrate must move (translocate) relative to RT, moving the end of the primer to the P site, so that the next incoming dNTP can bind. Release of the pyrophosphate appears to be accompanied by an opening of the fingers and it has been suggested that a movement of the conserved YMDD loop that contains two of the active site aspartates (D185 and D186) acts as a springboard, affecting translocation. In this model, the binding of the incoming dNTP causes a downward movement that loads the springboard; thus the incorporation of the nucleotide and the release of the pyrophosphate are the ultimate source of the energy that drives translocation (Sarafianos et al. 2002). Once the end of the primer is in the P site, another incoming dNTP can bind, and polymerization can continue.
The availability of the various different RT structures has guided and informed the analysis of the biochemical properties of RT. This combination of biochemical analysis and structural insights have made it possible to gain a good understanding of the roles played by structural elements like the primer grip, the template grip, the RNase H primer grip, and, in some cases, of the roles played by individual amino acids. In many cases, ideas that originated by looking at one or more of the RT structures was tested by reverse genetics: Mutations were made in specific amino acids to determine their effects on the in vitro properties of recombinant HIV-1 RT, on the replication of an HIV-1-based vector, or both. This literature is too large and complex to review here; however, a few simple ideas are worth presenting. For the most part, mutations in the structures that appear to be important in binding the substrates have phenotypes that match what the structure shows. This brings up an important point: Any mutation that causes a change in the polymerase domain of HIV-1 RT makes two changes in the mature HIV-1 RT heterodimer, one in p51 and one in p66. It is possible to express forms of recombinant RT that have changes in only one subunit. Despite the fact that both the polymerase and the RNase H active sites are in p66, in some cases the change in p51, which interacts extensively with p66 and helps form the nucleic acid binding cleft, can contribute to the behavior of the recombinant enzyme. Despite this complexity, individual mutations (e.g., active site mutations) can selectively affect one of the two activities of RT, polymerase or RNase H. However, there are a number of mutations, such as mutations that change the binding of the nucleic acid substrate, that affect both polymerase and RNase H. For example, there are mutations in the polymerase domain that affect not only polymerase activity and the fidelity of DNA synthesis, but also RNase H cleavage (Palaniappan et al. 1997; Gao et al. 1998; Powell et al. 1999; Sevilya et al. 2001, 2003). Mutations in the RNase H primer grip can affect the specificity of RNase H cleavage, but can also have some effect on the initiation of viral DNA synthesis (Julias et al. 2002; Rausch et al. 2002). Mutations in and around the polymerase active site can profoundly affect dNTP selection and polymerization. Not surprisingly, some, but not all, of the mutations that cause resistance to nucleoside analogs are near the polymerase active site (Tantillo et al. 1994; Sarafianos et al. 2009). Mutations that do not directly impact the enzymatic activities of RT can still have important effects on reverse transcription and the viral life cycle. For example, there are mutations in HIV-1 RT that affect the stability of the heterodimer. There are also mutations that permit the degradation of RT in virions by the viral PR. Some of these mutants have been shown to have a temperature-sensitive phenotype. It would appear that the mutations allow RT to partially unfold, making it susceptible to cleavage by PR. In some cases, the mutant virions contain no detectable RT. As expected, virions in which RT is extensively degraded have little or no infectivity (Huang et al. 2003; Takehisa et al. 2007; Dunn et al. 2009; Wang et al. 2010).
REVERSE TRANSCRIPTION IN INFECTED CELLS
Although only purified RT is required to carry out DNA synthesis from an RNA template in vitro, reverse transcription in target cells is a complex process that is intimately interconnected with other early events in the viral life cycle. The reverse transcription complex (RTC), in which DNA synthesis occurs in infected cells, contains multiple proteins. At some point, late in the reverse transcription process, the RTC transitions into a preintegration complex (PIC), and the PIC is transported into the nucleus. Although the ends of the DNA are completed in the cytoplasm, the plus-strand DNA of HIV-1 comprises at least two segments before integration. Viral DNA synthesis is a highly regulated event. Mutations in other viral genes, such as CA, can have a profound effect on reverse transcription in vivo (Forshey et al. 2002).
Multiple viral proteins, including MA, CA, NC, IN, and Vpr, have been reported to be present in the RTC (Fassati and Goff 2001; Nermut and Fassati 2003; Iordanskiy et al. 2006). The role of mature MA protein in the RTC/PIC is unclear. There were claims that MA directs the nuclear import of PICs and allows HIV-1 to infect nondividing cells; however, more recent data suggest that this hypothesis is incorrect (Gallay et al. 1995a,b; Freed et al. 1997). The mature CA protein most likely provides the overall structure of the RTC. CA may play a role in the transport of the RTC and the nuclear import of the RTC/PIC, allowing HIV-1 to infect nondividing cells (Yamashita and Emerman 2004; Dismuke and Aiken 2006; Qi et al. 2008; Lee et al. 2010). NC has nucleic acid chaperone activity; it affects the reverse transcription process, both in terms of helping RT through regions of secondary structure and facilitating strand transfer (Feng et al. 1996; Zhang et al. 2002; Buckman et al. 2003; Golinelli and Hughes 2003; Houzet et al. 2008; Thomas and Gorelick 2008; Thomas et al. 2008). Vpr is present in the RTC and it has been suggested that Vpr interacts with the host enzyme uracil DNA glycosylase/uracil N-glycosylase (UNG2), a factor that could modify newly synthesized viral DNA (Selig et al. 1997; Mansky et al. 2000; Chen et al. 2004; Schrofelbauer et al. 2005). The precise role and effects of Vpr/UNG2 is not yet clear. Some lentiviruses encode a deoxyuridine triphosphatase; however, this accessory gene is not found in HIV-1 (Elder et al. 1992; Wagaman et al. 1993; Lerner et al. 1995). Other HIV-1 gene products such as Vif and Tat have been shown to affect DNA synthesis in vitro; however, their roles in in vivo DNA synthesis, if any, are unclear (Harrich et al. 1997; Kameoka et al. 2002; Liang and Wainberg 2002; Apolloni et al. 2007; Henriet et al. 2007; Carr et al. 2008). Conversely, some, but not all, IN mutations have a profound negative effect on reverse transcription in an infected cell; however, these same IN mutations do not affect the activity of RT in viral lysates, and IN has not been shown to enhance the activity of purified RT in vitro, which suggest that IN might have an indirect role in the structure of the RTC in vivo (Engelman et al. 1995; Masuda et al. 1995; Leavitt et al. 1996; Wu et al. 1999). Certain host restriction factors, such as APOBEC3G and APOBEC3F, can be incorporated into virions and become part of the RTCs where they can cause mutations during DNA synthesis (Bishop et al. 2004b; Zheng et al. 2004); these factors are discussed briefly later in this article, and in more detail by Malim and Bieniasz (2011).
The structure of the RTC is not known. It is clear that there are changes in the structure of the core found in a mature virion (collectively called “uncoating”), which convert the core into a complex that can efficiently carry out reverse transcription. One hypothesis is that as uncoating is a continuous process, the structure of RTC changes as DNA synthesis proceeds, eventually transforming the RTC into a PIC (Forshey et al. 2002; Dismuke and Aiken 2006), which can be isolated from the cytoplasm and is capable of integrating viral DNA into a DNA target in vitro (Craigie and Bushman 2011). An alternative hypothesis is that the RTCs have a structure similar to that of the virus core, within which DNA synthesis occurs. In this proposal, the “core-like” structures are transported to the nuclear pore and converted into PICs before they enter the nucleus (Arhel et al. 2007). The RTC is a target for host restriction factors such as TRIM5α, TRIMCyp, and Fv-1; these host restriction factors interact with hexameric CA protein in the RTC, interfering with uncoating in a way that blocks reverse transcription or some later step that is essential for nuclear import and integration (Frankel et al. 1989; Best et al. 1996; Nisole et al. 2004; Sayah et al. 2004; Stremlau et al. 2004, 2006; Wu et al. 2006; Brennan et al. 2008; Newman et al. 2008; Virgen et al. 2008; Wilson et al. 2008).
Reverse transcription is initiated shortly after virus entry; viral DNA can be detected within hours of infection (Butler et al. 2001; Julias et al. 2001; Thomas et al. 2006; Mbisa et al. 2007). The rate of HIV-1 DNA synthesis had been measured in 293 T cells and activated primary human CD4+ T cells: minus-strand DNA is synthesized at a rate of ∼70 nucleotides per minute (Thomas et al. 2007). Plus-strand DNA synthesis is rapid, which agrees with the proposals that multiple initiation sites are used (Miller et al. 1995; Klarmann et al. 1997; Thomas et al. 2007). The minus-strand and plus-strand transfer reactions were first studied in vitro, in experiments performed with MLV and ASLV. The in vitro strand transfer reactions are slow, and the transfer intermediates, minus- and plus-strand strong-stop DNA, are easy to detect. These transfers occur more rapidly in an infected cell, where it has been estimated for HIV-1 that the transfers take ∼4 and ∼9 mins, respectively (Thomas et al. 2007). The rate of DNA synthesis, however, can vary depending on the nature of the target cell. For example, the rate of synthesis is expected to be slow in quiescent cells where the dNTP levels are low; it has been shown that DNA synthesis can stall in resting T cells (Zack et al. 1992). Additionally, the rate of DNA synthesis can be affected by mutations in viral genes, including RT.
DNA synthesis is often used to monitor the early stages of virus infection. The progression of reverse transcription can be determined using real-time polymerase chain reaction (PCR) and primer sets that anneal to various regions of the viral genomes (Butler et al. 2001; Julias et al. 2001; Mbisa et al. 2009). Generally, primer sets that anneal to R-U5 are used to measure the initiation of DNA synthesis, U3-R for minus-strand DNA transfer, Gag for the extension of the minus-strand DNA, and U5-5′UTR for plus-strand DNA transfer. Additionally, primers that anneal to U5-U3 can be used to measure 2-LTR circles, which are often used as a surrogate for nuclear import; lastly, primers that anneal to the LTR and human repetitive element Alu have been used to detect viral DNAs integrated in the human genome (proviruses).
GENETIC CONSEQUENCES OF REVERSE TRANSCRIPTION: MUTATION AND RECOMBINATION
Mutations and Fidelity
HIV-1 sequences vary considerably, not only between individuals, but also within an infected patient. The large variation seen in individual patients is somewhat surprising given that most patients are infected with a single virus (Keele et al. 2008); this means that the diversity of viral sequences seen in most patients arises after the patients are infected. Although it is the large numbers of infected cells and the rapid turnover of these infected cells that are the major reasons why the virus diverges so rapidly, it is the mutations that arise during the viral life cycle that are the ultimate source of viral diversity (Coffin 1995). The ability of the virus to diverge rapidly plays an important role in its ability to stay ahead of the immune system in an infected individual, and plays a key role in the ability of the virus to become resistant to all the known anti-AIDs drugs. The diversity of known viral strains also makes the daunting task of developing an effective vaccine even more difficult. As will be discussed in the next section, the fact that the virus can, and does, recombine efficiently complicates attempts to deal with these problems.
What is the cause of the mutations? A casual reading of the HIV-1 literature would suggest that mutations are caused by errors made by RT, which has no proofreading function. However, the data that speak directly to this question are quite limited. In theory, there are three ways in which mutations could arise during the HIV-1 replication cycle. Reverse transcription is one possibility. However, the RNA genome is synthesized by the host DNA-dependent RNA polymerase II (RNA pol II), another enzyme that lacks a proofreading function, and the contribution of RNA pol II to the mutation rate has not been determined. In addition, if a cell that harbors a provirus replicates, it is possible that the host DNA replication machinery could generate a mutation in the provirus. However, for exogenous retroviruses like HIV-1, in which there is a rapid turnover of infected cells, the contribution of the host DNA polymerases (which have elaborate proofreading functions) to the mutation rate is negligible. That leaves RT and RNA pol II. Unfortunately it has not been possible to separate their contributions to the overall viral mutation rate (Kim et al. 1996; O’Neil et al. 2002). It is possible to run RT fidelity assays in vitro, using purified RT, but, as will be discussed in more detail later, the in vitro data obtained with purified RT do not match the fidelity data obtained when an HIV-1 vector is used to infect cultured cells. If we could identify the errors made by RNA pol II, the errors made by RT could be identified by subtraction, but as yet there is no good way to determine which errors are made by pol II.
It has been suggested that the fidelity of HIV-1 RT is particularly low, and this accounts for the observed sequence variation. This is incorrect. The mutation rate for HIV-1 replication, which represents the combined error rate for RT and RNA pol II, is approximately 2 × 10−5 per nucleotide per replication cycle, a rate that is similar of other retroviruses (Pathak and Temin 1990a,b; Mansky and Temin 1994, 1995; Kim et al. 1996; Julias and Pathak 1998; Halvas et al. 2000; Abram et al. 2010). As has already been mentioned, the rapid variation in HIV-1 in patients is primarily because of the rapid turnover of a relatively large population of infected cells. Other retroviruses, which have a mutation rate similar to that of HIV-1, have a lower variation because the viruses replicate less rapidly in their infected hosts. In some cases (e.g., the ASLV and MLV viruses) the natural host (in this example, chickens and mice) carries closely related endogenous viruses whose proteins are expressed early enough in development to be recognized as self. This means that the exogenous viruses are sheltered, to some degree, from the host’s immune system. Because the degree of protection afforded by the proteins of endogenous viruses depends on the similarity of the proteins encoded by the exogenous viruses, the immune selection tends to restrict the overall variation of these exogenous viruses. Given that the overall mutation rate for HIV-1 and other retroviruses is similar, we can infer that the error rates of their RTs are similar. It is likely that RT makes a significant contribution to the overall error rate because, in a system in which the genetic information copied by RT is supplied by RNA pol II, there can be no selection for an RT that has a fidelity higher than the enzyme that provides the template RNA copies. This idea is supported by the analysis of mutations in the LTR that must be owing to the activity of RT (Kim et al. 1996; O’Neil et al. 2002). These data leave open the possibility that RT may have a lower fidelity than RNA pol II. However, even if we assume that RT makes the majority of the errors, its fidelity could be no lower than the error rate for the viral replication cycle (2 × 10−5). Thus, the fidelity of RT in an infected cell is at least 10 times higher than most groups have reported based on assays that involve using the purified enzyme in vitro.
Although it is convenient to calculate a specific number for the overall mutation rate, providing a single number is somewhat misleading. Errors do not arise uniformly throughout the sequence. Errors arise more frequently at some positions than at others; sites where errors occur frequently are called “hotspots.” In theory, it should be possible to use the in vitro assays to understand why RT preferentially makes mistakes at certain sites. Some of the in vitro fidelity assays have been performed with a substrate (the α-complementing fragment of Lac Z) similar to the one used in the single-cycle cell culture assays. Unfortunately, none of the in vitro assays produced a pattern of hotspots that was similar to what has been seen with a viral vector in cultured cells (Mansky and Temin 1995; Abram et al. 2010). To make matters worse, the pattern of hotspots reported from the various labs that did the in vitro experiments are all different. There are several possible explanations: (1) As has already been discussed, there are a number of ancillary viral and host proteins that contribute to the efficiency of the reverse transcription process; it is possible that some of these factors also contribute to fidelity. (2) The various groups used different assay conditions, and the purified RTs used in the assays are not identical. (3) Although it is likely that RT makes a real contribution to hotspots seen in the viral vector system, some of the hotspots could be caused by RNA pol II.
One of the important underlying issues is the emergence of mutations that allow HIV-1 to evade anti-AIDS drugs and mutations that cause immune escape. It is clear that the virus almost always finds a way to evade the host’s immune system and, unless the therapy completely blocks viral replication, the virus also finds ways to evade all the known anti-HIV drugs. However, the virus can use different mutations for immune escape and drug resistance. There are several possible explanations. One interesting possibility is that differences in the sequences of the various RTs could change their ability to make specific errors (mutations). As a result, these RTs (e.g., a drug-resistant RT) might, when carrying out viral DNA synthesis, generate a different spectrum of errors (mutations), thereby altering the spectrum of variants that eventually emerge in response to immunological (or drug) selection.
In addition to the errors made by RT and RNA pol II, the reverse transcription process can be affected by cellular factors, in particular, the APOBEC proteins. The APOBECs are covered in greater detail by Malim and Bieniasz (2011); however, it is important to remember that they affect the fidelity and efficiency of reverse transcription. Broadly speaking, the APOBECs are cytidine deaminases; those that affect retroviruses use a single-stranded DNA substrate (Harris et al. 2003; Mangeat et al. 2003; Bishop et al. 2004b). Although APOBEC3G is the best studied of the human APOBEC proteins, there are several human APOBECs that can affect HIV replication in cultured cells; these could have effects on HIV-1 replication in patients (Bishop et al. 2004a; Yu et al. 2004; Holmes et al. 2007). The APOBECs that affect HIV replication are packaged into virions and modify minus-strand DNA after the RNA has been degraded, but before the plus strand has been synthesized. At this stage the APOBECs can convert some of the Cs in the minus strand to Us. When the virus replicates, the C-to-U mutations in minus-strand DNA lead to the conversion of Gs in the RNA genome to As. The APOBECs are part of the host’s innate defense against retroviruses, and, as might have been expected, HIV-1 has a counter, the Vif protein, which interacts with host machinery to cause the degradation of APOBEC (Mariani et al. 2003; Yu et al. 2003; Liu et al. 2004; Sawyer et al. 2004; Schrofelbauer et al. 2004; Luo et al. 2005; Fang and Landau 2007; Russell and Pathak 2007). The impact of APOBEC on HIV replication is much greater if the virus lacks a functional Vif. Moreover, the ability of the APOBECs to block HIV-1 replication does not appear to be entirely attributable to their cytidine deaminase activity; it appears that the APOBECs can have other negative effects on reverse transcription and integration, although the exact nature of these effects is not yet clear (Bishop et al. 2006; Holmes et al. 2007; Mbisa et al. 2007, 2010). There are also hints that enzymes that act as RNA adenine deaminases (ADARs) can cause mutations in HIV-1, at least in cultured cells (Abram et al. 2010) and mutations that appear to have been caused by ADARs have been reported when HIV replication is challenged with antisense RNAs (Lu et al. 2004; Mukherjee et al. 2011). However, despite the fact that viruses isolated from patients almost always have an intact Vif-coding region, it is easy to find, among the HIV-1 sequences from patients, G-to-A hypermutations that appear to be the result of APOBEC activity. A search of the same sequence databases showed no obvious indication of ADAR-induced hypermutations (Abram et al. 2010).
RECOMBINATION
The recombination rate for retroviruses is higher than for most other viruses and the recombination rate for HIV-1 is higher than other retroviruses such as MLV and spleen necrosis viruses (Hu and Temin 1990a; Anderson et al. 1998; Onafuwa et al. 2003; Rhodes et al. 2003, 2005). Mapping of HIV-1 genomes by direct sequencing shows that there is frequent recombination during DNA synthesis (Robertson et al. 1995; Jetzt et al. 2000; Zhuang et al. 2002; Dykes et al. 2004; Levy et al. 2004; Chin et al. 2008; Galli et al. 2010). During minus-strand DNA synthesis, RT can switch between the two copackaged RNAs, using portions of each RNA as a template to generate a chimeric DNA containing sequences from each of the two genomic RNAs. Template switching can occur between two copackaged RNAs with identical sequences; however, only virions that package two genetically different RNAs can generate a recombinant with a genotype distinct from that of the parents (Hu and Temin 1990a). Multiple steps are required for the generation of a novel recombinant; first, the virus producer cell needs to be infected by more than one virus, the RNAs from the two proviruses have to be copackaged into the same virion, and template switching has to occur during reverse transcription to generate a chimeric DNA copy, which needs to integrate into the genome of the target cell. Lastly, this recombinant provirus needs to be able to generate replication-competent virus for the impact of the recombination event to be observed. For these reasons, factors that affect any of these steps can influence recombination. Currently, little is known about how frequently target cells in patients are infected by more than one HIV-1 (double infection). In culture, double infection occurs more frequently than expected from random events, in both T-cell lines and primary CD4+ T cells (Dang et al. 2004). This result is at least partly attributable to the fact that some cells have more receptors/coreceptors, and are, therefore, more susceptible to HIV-1 infection (Chen et al. 2005). Double infection is increased when HIV-1 is transmitted via cell-mediated events because multiple viruses are passed from the donor cell to the target cells (Dang et al. 2004).
During HIV-1 assembly, Gag packages full-length genomic RNA in a dimer form (for details, see Sundquist and Kräusslich 2011). Hence, RNA partner selection occurs before the encapsidation of the RNA genomes; a major determinant for RNA partner selection is the dimerization initiation signal (DIS) located in the loop of stem loop 1 in the 5′ untranslated region of HIV-1 RNA (Chin et al. 2005; Moore et al. 2007; Chen et al. 2009). Most subtype B and D variants have GCGCGC in their DIS, whereas most subtype A, C, F, and G variants have GTGCAC, although other sequences have been found (St Louis et al. 1998; Hussein et al. 2010). It is thought that the palindromic nature of the DIS promotes an intermolecular base pairing of the two RNAs that initiates RNA dimerization. Other sequences in the viral genome can also affect the frequencies of RNA heterodimerization, albeit with a milder effect than the DIS (Chin et al. 2007).
HIV-1 recombination rates have been measured using marker genes; these results indicated that recombination rates increase proportionally with the distances that separate the two alleles when the distance between the markers is less than 0.6 kb; the maximum possible recombination rate is reached when the two alleles are separated by 1.3 kb (Rhodes et al. 2003, 2005). Although recombination has been shown to occur throughout the HIV-1 genome, RNA structure may affect the frequency of recombination of certain regions (Galetto et al. 2004). Sequence homology can affect both the recombination rate and the distribution of the crossover junctions (Baird et al. 2006); for example, there is more frequent recombination between two copackaged HIV-1 RNAs from the same subtype than there is when the two copackaged HIV-1 RNAs are from different subtypes (Galli et al. 2010).
It has been proposed that RT switches to the copackaged RNA copy where there is a break in the RNA template; this is known as the copy-choice recombination model (Fig. 4A) (Coffin 1979). The original copy-choice model has been revised and renamed the dynamic copy-choice model (Hwang et al. 2001), which proposes that a balance between polymerase activity and RNase H activity of RT determines the stability of the association between the nascent DNA and the RNA template, and that a perturbation of this balance affects the recombination rate (Fig. 4B). If the balance is shifted toward greater RNase H activity relative to the polymerase activity, there is less extensive base-pairing between nascent DNA and RNA template, which promotes dissociation of the DNA–RNA complex and template switching (Hwang et al. 2001). Indeed, RT mutants that have decreased RNase H activity relative to their polymerase activity show a reduction in template switching (Nikolenko et al. 2005, 2007). It has also been proposed that, for retroviruses in which plus-strand DNA synthesis is initiated at multiple sites, plus-strand DNA fragments can be annealed to a second minus-strand DNA synthesized using the copackaged RNA as a template. After host DNA repair, a recombinant can be generated (Junghans et al. 1982). This model, which was originally proposed for recombination in avian retroviruses, requires that considerable portions of two minus-strand DNAs be synthesized. However, extensive minus-strand recombination will lead to the synthesis of a single minus-strand DNA. Currently, there is little data to suggest that plus-strand recombination occurs at a significant level during HIV-1 replication.
Figure 4.
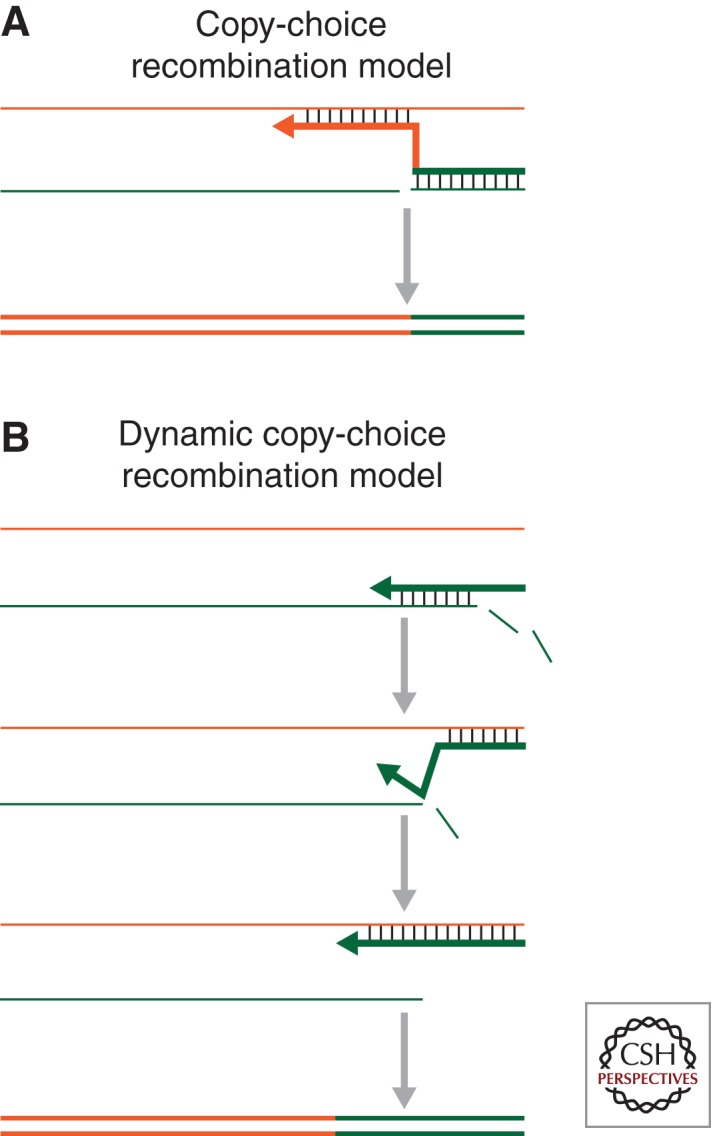
Recombination models. (A) Copy-choice model, (B) dynamic copy-choice model. The top of A shows two RNA strands (thin orange and green lines). Minus-strand DNA synthesis uses the green RNA strand as a template; however, the green strand is nicked, causing DNA synthesis to switch to the orange RNA strand. This switch leads to the generation of a double-stranded DNA that is composed of sequences from both the green and the orange RNAs. In B, minus-strand DNA synthesis also uses the green RNA strand as a template. When the viral DNA is synthesized, RNase H degrades the green RNA strand, and the DNA that was copied from the green RNA strand can hybridize to the orange RNA (middle panels). This facilitates a transfer of the growing minus-strand DNA from the green to the orange strand, which results in the synthesis of a double-stranded DNA with sequences from both of the parental RNAs.
Last, for viral recombinants to establish themselves, they have to be able to replicate efficiently. Most template switching events use the complementarity between the nascent DNA and the acceptor template, and the recombination junctions are generally precise (Zhuang et al. 2002; Chin et al. 2008). However, because the resulting recombinants contain portions of the genomes of each parent, these sequences may or may not work together efficiently (Baird et al. 2006; Simon-Loriere et al. 2009; Galli et al. 2010). This issue is more pronounced when the two parental viruses are separated by a greater genetic distance (e.g., when the parental viruses are from different subtypes). For this reason, many newly generated intersubtype recombinants are eliminated by purifying selection during virus replication (Galli et al. 2010). The generation of a successful intersubtype recombinant faces multiple challenges: possible decreased efficiencies of RNA copackaging, relative inefficient template switching, and the impact of a decrease of replication fitness on the resulting recombinants. However, a conservative estimate suggests that >20% of the currently circulating HIV-1 variants are intersubtype recombinants (Hemelaar et al. 2006). This shows that recombination is a major force in the evolution of the HIV-1 population.
Frequent HIV-1 recombination reassorts existing mutations and increases genetic diversity in the viral population, thereby allowing the emergence of the variants that are best suited for any given environment. Recombination can combine drug-resistance mutations to produce multidrug-resistant variants (Kellam and Larder 1995; Moutouh et al. 1996); similarly, recombination can also produce variants that can evade multiple challenges from the host’s immune system (Streeck et al. 2008). Recombination can generate chimeras between two viruses from the same subtype, from different subtypes, or even from different groups. Therefore, recombination not only can affect the evolution of a viral population within an infected individual but can also affect HIV diversity worldwide. Given that different virus strains express different epitopes and vary in their susceptibility to antivirals, the increased diversity caused by recombination makes it more difficult to develop effective vaccines and antiviral regimens.
ANTI-RT COMPOUNDS AND RESISTANCE
Currently there are two types of antivirals targeting the reverse transcription process: nucleoside reverse transcriptase inhibitors (NRTIs), and nonnucleoside reverse transcriptase inhibitors (NNRTIs). NRTIs are given as prodrugs; after they are taken up by cells, and phosphorylated by the host cell enzymes, NRTIs can be incorporated into viral DNA by RT. NRTIs lack a 3′ hydroxyl group, thus, their incorporation blocks viral DNA synthesis. The common NRTI resistance mutations cause resistance by two general mechanisms: (1) mutations that reduce the incorporation of the NRTITP relative to the normal dNTPs, or (2) mutations that lead to a selective excision of the incorporated NRTIs by RT, unblocking the viral DNA. NNRTIs bind to RT and block the chemical step of DNA synthesis (see Movie 3); resistance mutations generally change the NNRTI-binding pocket in ways that make the binding of the NNRTIs less favorable. This topic will be described in more detail by Arts and Hazuda (2011).
Movie 3.
The binding pocket for nonnucleoside reverse transcriptase inhibitors.
CONCLUDING REMARKS
Reverse transcription and integration are the hallmarks of retroviruses; in this article, we provide an overview of RT and the reverse transcription process. The crystal structures of the HIV-1 RT, particularly those that reveal the structure of complexes with bound nucleic acids and incoming dNTPs, have allowed us to understand in molecular detail how the enzyme works. Structures of RT with bound anti-RT drugs have allowed us to better understand drug action and drug resistance. Complementary biochemical and genetic experiments have helped complete the picture. However, there is still much to be learned about the interactions of the various viral and cellular components that facilitate viral DNA synthesis in an infected cell and how the viral core is first converted into an RTC and then into a PIC. Reverse transcription has important genetic consequences—mutation and recombination provide the basis for the genetic diversity of HIV-1, which allows the emergence of viral strains that can escape the host’s immune response and/or become resistant to drug treatment. Modern highly active antiretroviral therapy (HAART) therapy rests on the development and use of anti-AIDS drugs that target RT. However, the prevalence of drug-resistant HIV-1 strains makes it necessary to continue our efforts to develop more and better antiviral drugs. A better understanding of the complexities of the replication process, RT, and the reverse transcription pathway, can help us develop better ways to combat HIV-1.
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
We thank K. Das, E. Arnold, K. Kirby, S. Sarafianos and A. Kane for help with the figures and K. Kirby and S. Sarafianos for help with movies.
Footnotes
Editors: Frederic D. Bushman, Gary J. Nabel, and Ronald Swanstrom
Additional Perspectives on HIV available at www.perspectivesinmedicine.org
REFERENCES
*Reference is also in this collection.
- Abram ME, Ferris AL, Shao W, Alvord WG, Hughes SH 2010. The nature, position and frequency of mutations made in a single-cycle of HIV-1 replication. J Virol 84: 9864–9878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JA, Bowman EH, Hu WS 1998. Retroviral recombination rates do not increase linearly with marker distance and are limited by the size of the recombining subpopulation. J Virol 72: 1195–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apolloni A, Meredith LW, Suhrbier A, Kiernan R, Harrich D 2007. The HIV-1 Tat protein stimulates reverse transcription in vitro. Curr HIV Res 5: 473–483 [DOI] [PubMed] [Google Scholar]
- Arhel NJ, Souquere-Besse S, Munier S, Souque P, Guadagnini S, Rutherford S, Prevost MC, Allen TD, Charneau P 2007. HIV-1 DNA flap formation promotes uncoating of the pre-integration complex at the nuclear pore. EMBO J 26: 3025–3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Arts EJ, Hazuda DJ 2011. HIV-1 antiretroviral drug therapy. Cold Spring Harb Perspect Med 10.1101/cshperspect.a007161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird HA, Gao Y, Galetto R, Lalonde M, Anthony RM, Giacomoni V, Abreha M, Destefano JJ, Negroni M, Arts EJ 2006. Influence of sequence identity and unique breakpoints on the frequency of intersubtype HIV-1 recombination. Retrovirology 3: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltimore D 1970. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature 226: 1209–1211 [DOI] [PubMed] [Google Scholar]
- Best S, Le Tissier P, Towers G, Stoye JP 1996. Positional cloning of the mouse retrovirus restriction gene Fv1. Nature 382: 826–829 [DOI] [PubMed] [Google Scholar]
- Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH 2004a. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol 14: 1392–1396 [DOI] [PubMed] [Google Scholar]
- Bishop KN, Holmes RK, Sheehy AM, Malim MH 2004b. APOBEC-mediated editing of viral RNA. Science 305: 645. [DOI] [PubMed] [Google Scholar]
- Bishop KN, Holmes RK, Malim MH 2006. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J Virol 80: 8450–8458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan G, Kozyrev Y, Hu SL 2008. TRIMCyp expression in Old World primates Macaca nemestrina and Macaca fascicularis. Proc Natl Acad Sci 105: 3569–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckman JS, Bosche WJ, Gorelick RJ 2003. Human immunodeficiency virus type 1 nucleocapsid Zn2+ fingers are required for efficient reverse transcription, initial integration processes, and protection of newly synthesized viral DNA. J Virol 77: 1469–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler SL, Hansen MS, Bushman FD 2001. A quantitative assay for HIV DNA integration in vivo. Nat Med 7: 631–634 [DOI] [PubMed] [Google Scholar]
- Carr JM, Coolen C, Davis AJ, Burrell CJ, Li P 2008. Human immunodeficiency virus 1 (HIV-1) virion infectivity factor (Vif) is part of reverse transcription complexes and acts as an accessory factor for reverse transcription. Virology 372: 147–156 [DOI] [PubMed] [Google Scholar]
- Charneau P, Alizon M, Clavel F 1992. A second origin of DNA plus-strand synthesis is required for optimal human immunodeficiency virus replication. J Virol 66: 2814–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Le Rouzic E, Kearney JA, Mansky LM, Benichou S 2004. Vpr-mediated incorporation of UNG2 into HIV-1 particles is required to modulate the virus mutation rate and for replication in macrophages. J Biol Chem 279: 28419–28425 [DOI] [PubMed] [Google Scholar]
- Chen J, Dang Q, Unutmaz D, Pathak VK, Maldarelli F, Powell D, Hu WS 2005. Mechanisms of nonrandom human immunodeficiency virus type 1 infection and double infection: Preference in virus entry is important but is not the sole factor. J Virol 79: 4140–4149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Nikolaitchik O, Singh J, Wright A, Bencsics CE, Coffin JM, Ni N, Lockett S, Pathak VK, Hu WS 2009. High efficiency of HIV-1 genomic RNA packaging and heterozygote formation revealed by single virion analysis. Proc Natl Acad Sci 106: 13535–13540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin MP, Rhodes TD, Chen J, Fu W, Hu WS 2005. Identification of a major restriction in HIV-1 intersubtype recombination. Proc Natl Acad Sci 102: 9002–9007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin MP, Chen J, Nikolaitchik OA, Hu WS 2007. Molecular determinants of HIV-1 intersubtype recombination potential. Virology 363: 437–446 [DOI] [PubMed] [Google Scholar]
- Chin MP, Lee SK, Chen J, Nikolaitchik OA, Powell DA, Fivash MJ Jr, Hu WS 2008. Long-range recombination gradient between HIV-1 subtypes B and C variants caused by sequence differences in the dimerization initiation signal region. J Mol Biol 377: 1324–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffin JM 1979. Structure, replication, and recombination of retrovirus genomes: Some unifying hypotheses. J Gen Virol 42: 1–26 [DOI] [PubMed] [Google Scholar]
- Coffin JM 1995. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 267: 483–489 [DOI] [PubMed] [Google Scholar]
- Colicelli J, Goff SP 1985. Mutants and pseudorevertants of Moloney murine leukemia virus with alterations at the integration site. Cell 42: 573–580 [DOI] [PubMed] [Google Scholar]
- Colicelli J, Goff SP 1988. Sequence and spacing requirements of a retrovirus integration site. J Mol Biol 199: 47–59 [DOI] [PubMed] [Google Scholar]
- *.Craigie R, Bushman FD 2011. HIV DNA integration. Cold Spring Harb Perspect Med 10.1101/cshperspect.a006890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang Q, Chen J, Unutmaz D, Coffin JM, Pathak VK, Powell D, KewalRamani VN, Maldarelli F, Hu WS 2004. Nonrandom HIV-1 infection and double infection via direct and cell-mediated pathways. Proc Natl Acad Sci 101: 632–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das K, Bandwar RP, White KL, Feng JY, Sarafianos SG, Tuske S, Tu X, Clark AD Jr, Boyer PL, Hou X, et al. 2009. Structural basis for the role of the K65R mutation in HIV-1 reverse transcriptase polymerization, excision antagonism, and tenofovir resistance. J Biol Chem 284: 35092–35100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash C, Rausch JW, Le Grice SF 2004. Using pyrrolo-deoxycytosine to probe RNA/DNA hybrids containing the human immunodeficiency virus type-1 3′ polypurine tract. Nucleic Acids Res 32: 1539–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dismuke DJ, Aiken C 2006. Evidence for a functional link between uncoating of the human immunodeficiency virus type 1 core and nuclear import of the viral preintegration complex. J Virol 80: 3712–3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll MD, Golinelli MP, Hughes SH 2001. In vitro analysis of human immunodeficiency virus type 1 minus-strand strong-stop DNA synthesis and genomic RNA processing. J Virol 75: 672–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn LL, McWilliams MJ, Das K, Arnold E, Hughes SH 2009. Mutations in the thumb allow human immunodeficiency virus type 1 reverse transcriptase to be cleaved by protease in virions. J Virol 83: 12336–12344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykes C, Balakrishnan M, Planelles V, Zhu Y, Bambara RA, Demeter LM 2004. Identification of a preferred region for recombination and mutation in HIV-1 gag. Virology 326: 262–279 [DOI] [PubMed] [Google Scholar]
- Elder JH, Lerner DL, Hasselkus-Light CS, Fontenot DJ, Hunter E, Luciw PA, Montelaro RC, Phillips TR 1992. Distinct subsets of retroviruses encode dUTPase. J Virol 66: 1791–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A, Englund G, Orenstein JM, Martin MA, Craigie R 1995. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J Virol 69: 2729–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnouf R, Ren J, Ross C, Jones Y, Stammers D, Stuart D 1995. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat Struct Biol 2: 303–308 [DOI] [PubMed] [Google Scholar]
- Esposito D, Craigie R 1998. Sequence specificity of viral end DNA binding by HIV-1 integrase reveals critical regions for protein-DNA interaction. EMBO J 17: 5832–5843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Landau NR 2007. Analysis of Vif-induced APOBEC3G degradation using an α-complementation assay. Virology 359: 162–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassati A, Goff SP 2001. Characterization of intracellular reverse transcription complexes of human immunodeficiency virus type 1. J Virol 75: 3626–3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng YX, Copeland TD, Henderson LE, Gorelick RJ, Bosche WJ, Levin JG, Rein A 1996. HIV-1 nucleocapsid protein induces “maturation” of dimeric retroviral RNA in vitro. Proc Natl Acad Sci 93: 7577–7581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forshey BM, von Schwedler U, Sundquist WI, Aiken C 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol 76: 5667–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel WN, Stoye JP, Taylor BA, Coffin JM 1989. Genetic analysis of endogenous xenotropic murine leukemia viruses: Association with two common mouse mutations and the viral restriction locus Fv-1. J Virol 63: 1763–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO, Englund G, Maldarelli F, Martin MA 1997. Phosphorylation of residue 131 of HIV-1 matrix is not required for macrophage infection. Cell 88: 171–173; discussion 173–174 [DOI] [PubMed] [Google Scholar]
- Galetto R, Moumen A, Giacomoni V, Veron M, Charneau P, Negroni M 2004. The structure of HIV-1 genomic RNA in the gp120 gene determines a recombination hot spot in vivo. J Biol Chem 279: 36625–36632 [DOI] [PubMed] [Google Scholar]
- Gallay P, Swingler S, Aiken C, Trono D 1995a. HIV-1 infection of nondividing cells: C-terminal tyrosine phosphorylation of the viral matrix protein is a key regulator. Cell 80: 379–388 [DOI] [PubMed] [Google Scholar]
- Gallay P, Swingler S, Song J, Bushman F, Trono D 1995b. HIV nuclear import is governed by the phosphotyrosine-mediated binding of matrix to the core domain of integrase. Cell 83: 569–576 [DOI] [PubMed] [Google Scholar]
- Galli A, Kearney M, Nikolaitchik OA, Yu S, Chin MP, Maldarelli F, Coffin JM, Pathak VK, Hu WS 2010. Patterns of human immunodeficiency virus type 1 recombination ex vivo provide evidence for coadaptation of distant sites, resulting in purifying selection for intersubtype recombinants during replication. J Virol 84: 7651–7661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HQ, Boyer PL, Arnold E, Hughes SH 1998. Effects of mutations in the polymerase domain on the polymerase, RNase H and strand transfer activities of human immunodeficiency virus type 1 reverse transcriptase. J Mol Biol 277: 559–572 [DOI] [PubMed] [Google Scholar]
- Ghosh M, Jacques PS, Rodgers DW, Ottman M, Darlix JL, Le Grice SF 1996. Alterations to the primer grip of pp66 HIV-1 reverse transcriptase and their consequences for template-primer utilization. Biochemistry 35: 8553–8562 [DOI] [PubMed] [Google Scholar]
- Ghosh M, Williams J, Powell MD, Levin JG, Le Grice SF 1997. Mutating a conserved motif of the HIV-1 reverse transcriptase palm subdomain alters primer utilization. Biochemistry 36: 5758–5768 [DOI] [PubMed] [Google Scholar]
- Golinelli MP, Hughes SH 2003. Secondary structure in the nucleic acid affects the rate of HIV-1 nucleocapsid-mediated strand annealing. Biochemistry 42: 8153–8162 [DOI] [PubMed] [Google Scholar]
- Halvas EK, Svarovskaia ES, Pathak VK 2000. Role of murine leukemia virus reverse transcriptase deoxyribonucleoside triphosphate-binding site in retroviral replication and in vivo fidelity. J Virol 74: 10349–10358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrich D, Ulich C, Garcia-Martinez LF, Gaynor RB 1997. Tat is required for efficient HIV-1 reverse transcription. EMBO J 16: 1224–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113: 803–809 [DOI] [PubMed] [Google Scholar]
- Hemelaar J, Gouws E, Ghys PD, Osmanov S 2006. Global and regional distribution of HIV-1 genetic subtypes and recombinants in 2004. AIDS 20: W13–W23 [DOI] [PubMed] [Google Scholar]
- Henriet S, Sinck L, Bec G, Gorelick RJ, Marquet R, Paillart JC 2007. Vif is a RNA chaperone that could temporally regulate RNA dimerization and the early steps of HIV-1 reverse transcription. Nucleic Acids Res 35: 5141–5153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes RK, Koning FA, Bishop KN, Malim MH 2007. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J Biol Chem 282: 2587–2595 [DOI] [PubMed] [Google Scholar]
- Houzet L, Morichaud Z, Didierlaurent L, Muriaux D, Darlix JL, Mougel M 2008. Nucleocapsid mutations turn HIV-1 into a DNA-containing virus. Nucleic Acids Res 36: 2311–2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiou Y, Ding J, Das K, Clark AD Jr, Hughes SH, Arnold E 1996. Structure of unliganded HIV-1 reverse transcriptase at 2.7 A resolution: Implications of conformational changes for polymerization and inhibition mechanisms. Structure 4: 853–860 [DOI] [PubMed] [Google Scholar]
- Hsu TW, Taylor JM 1982. Single-stranded regions on unintegrated avian retrovirus DNA. J Virol 44: 47–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WS, Temin HM 1990a. Genetic consequences of packaging two RNA genomes in one retroviral particle: Pseudodiploidy and high rate of genetic recombination. Proc Natl Acad Sci 87: 1556–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WS, Temin HM 1990b. Retroviral recombination and reverse transcription. Science 250: 1227–1233 [DOI] [PubMed] [Google Scholar]
- Huang Y, Wang J, Shalom A, Li Z, Khorchid A, Wainberg MA, Kleiman L 1997. Primer tRNA3Lys on the viral genome exists in unextended and two-base extended forms within mature human immunodeficiency virus type 1. J Virol 71: 726–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Chopra R, Verdine GL, Harrison SC 1998. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: Implications for drug resistance. Science 282: 1669–1675 [DOI] [PubMed] [Google Scholar]
- Huang W, Gamarnik A, Limoli K, Petropoulos CJ, Whitcomb JM 2003. Amino acid substitutions at position 190 of human immunodeficiency virus type 1 reverse transcriptase increase susceptibility to delavirdine and impair virus replication. J Virol 77: 1512–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hungnes O, Tjotta E, Grinde B 1992. Mutations in the central polypurine tract of HIV-1 result in delayed replication. Virology 190: 440–442 [DOI] [PubMed] [Google Scholar]
- Hussein IT, Ni N, Galli A, Chen J, Moore MD, Hu WS 2010. Delineation of the preferences and requirements of the human immunodeficiency virus type 1 dimerization initiation signal by using an in vivo cell-based selection approach. J Virol 84: 6866–6875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang CK, Svarovskaia ES, Pathak VK 2001. Dynamic copy choice: Steady state between murine leukemia virus polymerase and polymerase-dependent RNase H activity determines frequency of in vivo template switching. Proc Natl Acad Sci 98: 12209–12214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iordanskiy S, Berro R, Altieri M, Kashanchi F, Bukrinsky M 2006. Intracytoplasmic maturation of the human immunodeficiency virus type 1 reverse transcription complexes determines their capacity to integrate into chromatin. Retrovirology 3: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isel C, Lanchy JM, Le Grice SF, Ehresmann C, Ehresmann B, Marquet R 1996. Specific initiation and switch to elongation of human immunodeficiency virus type 1 reverse transcription require the post-transcriptional modifications of primer tRNA3Lys. EMBO J 15: 917–924 [PMC free article] [PubMed] [Google Scholar]
- Jacobo-Molina A, Ding J, Nanni RG, Clark AD Jr, Lu X, Tantillo C, Williams RL, Kamer G, Ferris AL, Clark P, et al. 1993. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 A resolution shows bent DNA. Proc Natl Acad Sci 90: 6320–6324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetzt AE, Yu H, Klarmann GJ, Ron Y, Preston BD, Dougherty JP 2000. High rate of recombination throughout the human immunodeficiency virus type 1 genome. J Virol 74: 1234–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julias JG, Pathak VK 1998. Deoxyribonucleoside triphosphate pool imbalances in vivo are associated with an increased retroviral mutation rate. J Virol 72: 7941–7949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julias JG, Ferris AL, Boyer PL, Hughes SH 2001. Replication of phenotypically mixed human immunodeficiency virus type 1 virions containing catalytically active and catalytically inactive reverse transcriptase. J Virol 75: 6537–6546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julias JG, McWilliams MJ, Sarafianos SG, Arnold E, Hughes SH 2002. Mutations in the RNase H domain of HIV-1 reverse transcriptase affect the initiation of DNA synthesis and the specificity of RNase H cleavage in vivo. Proc Natl Acad Sci 99: 9515–9520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junghans RP, Boone LR, Skalka AM 1982. Retroviral DNA H structures: Displacement-assimilation model of recombination. Cell 30: 53–62 [DOI] [PubMed] [Google Scholar]
- Kameoka M, Morgan M, Binette M, Russell RS, Rong L, Guo X, Mouland A, Kleiman L, Liang C, Wainberg MA 2002. The Tat protein of human immunodeficiency virus type 1 (HIV-1) can promote placement of tRNA primer onto viral RNA and suppress later DNA polymerization in HIV-1 reverse transcription. J Virol 76: 3637–3645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, et al. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci 105: 7552–7557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellam P, Larder BA 1995. Retroviral recombination can lead to linkage of reverse transcriptase mutations that confer increased zidovudine resistance. J Virol 69: 669–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Mudry RA Jr, Rexrode CA 2nd, Pathak VK 1996. Retroviral mutation rates and A-to-G hypermutations during different stages of retroviral replication. J Virol 70: 7594–7602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarmann GJ, Yu H, Chen X, Dougherty JP, Preston BD 1997. Discontinuous plus-strand DNA synthesis in human immunodeficiency virus type 1-infected cells and in a partially reconstituted cell-free system. J Virol 71: 9259–9269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA 1992. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 256: 1783–1790 [DOI] [PubMed] [Google Scholar]
- Kung HJ, Fung YK, Majors JE, Bishop JM, Varmus HE 1981. Synthesis of plus strands of retroviral DNA in cells infected with avian sarcoma virus and mouse mammary tumor virus. J Virol 37: 127–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanchy JM, Keith G, Le Grice SF, Ehresmann B, Ehresmann C, Marquet R 1998. Contacts between reverse transcriptase and the primer strand govern the transition from initiation to elongation of HIV-1 reverse transcription. J Biol Chem 273: 24425–24432 [DOI] [PubMed] [Google Scholar]
- Lansdon EB, Samuel D, Lagpacan L, Brendza KM, White KL, Hung M, Liu X, Boojamra CG, Mackman RL, Cihlar T, et al. 2010. Visualizing the molecular interactions of a nucleotide analog, GS-9148, with HIV-1 reverse transcriptase-DNA complex. J Mol Biol 397: 967–978 [DOI] [PubMed] [Google Scholar]
- Leavitt AD, Robles G, Alesandro N, Varmus HE 1996. Human immunodeficiency virus type 1 integrase mutants retain in vitro integrase activity yet fail to integrate viral DNA efficiently during infection. J Virol 70: 721–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, et al. 2010. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7: 221–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner DL, Wagaman PC, Phillips TR, Prospero-Garcia O, Henriksen SJ, Fox HS, Bloom FE, Elder JH 1995. Increased mutation frequency of feline immunodeficiency virus lacking functional deoxyuridine-triphosphatase. Proc Natl Acad Sci 92: 7480–7484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DN, Aldrovandi GM, Kutsch O, Shaw GM 2004. Dynamics of HIV-1 recombination in its natural target cells. Proc Natl Acad Sci 101: 4204–4209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Wainberg MA 2002. The role of Tat in HIV-1 replication: An activator and/or a suppressor? AIDS Rev 4: 41–49 [PubMed] [Google Scholar]
- Lightfoote MM, Coligan JE, Folks TM, Fauci AS, Martin MA, Venkatesan S 1986. Structural characterization of reverse transcriptase and endonuclease polypeptides of the acquired immunodeficiency syndrome retrovirus. J Virol 60: 771–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Yu X, Luo K, Yu Y, Yu XF 2004. Influence of primate lentiviral Vif and proteasome inhibitors on human immunodeficiency virus type 1 virion packaging of APOBEC3G. J Virol 78: 2072–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lori F, di Marzo Veronese F, de Vico AL, Lusso P, Reitz MS Jr, Gallo RC 1992. Viral DNA carried by human immunodeficiency virus type 1 virions. J Virol 66: 5067–5074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Yu Q, Binder GK, Chen Z, Slepushkina T, Rossi J, Dropulic B 2004. Antisense-mediated inhibition of human immunodeficiency virus (HIV) replication by use of an HIV type 1-based vector results in severely attenuated mutants incapable of developing resistance. J Virol 78: 7079–7088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo K, Xiao Z, Ehrlich E, Yu Y, Liu B, Zheng S, Yu XF 2005. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc Natl Acad Sci 102: 11444–11449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Malim MH, Bieniasz PD 2011. HIV restriction factors and mechanisms of evasion. Cold Spring Harb Perspect Med 10.1101/cshperspect.a006940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424: 99–103 [DOI] [PubMed] [Google Scholar]
- Mansky LM, Temin HM 1994. Lower mutation rate of bovine leukemia virus relative to that of spleen necrosis virus. J Virol 68: 494–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansky LM, Temin HM 1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol 69: 5087–5094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansky LM, Preveral S, Selig L, Benarous R, Benichou S 2000. The interaction of vpr with uracil DNA glycosylase modulates the human immunodeficiency virus type 1 in vivo mutation rate. J Virol 74: 7039–7047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR 2003. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114: 21–31 [DOI] [PubMed] [Google Scholar]
- Masuda T, Planelles V, Krogstad P, Chen IS 1995. Genetic analysis of human immunodeficiency virus type 1 integrase and the U3 att site: Unusual phenotype of mutants in the zinc finger-like domain. J Virol 69: 6687–6696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbisa JL, Barr R, Thomas JA, Vandegraaff N, Dorweiler IJ, Svarovskaia ES, Brown WL, Mansky LM, Gorelick RJ, Harris RS, et al. 2007. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J Virol 81: 7099–7110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbisa JL, Delviks-Frankenberry KA, Thomas JA, Gorelick RJ, Pathak VK 2009. Real-time PCR analysis of HIV-1 replication post-entry events. Methods Mol Biol 485: 55–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbisa JL, Bu W, Pathak VK 2010. APOBEC3F and APOBEC3G inhibit HIV-1 DNA integration by different mechanisms. J Virol 84: 5250–5259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MD, Wang B, Bushman FD 1995. Human immunodeficiency virus type 1 preintegration complexes containing discontinuous plus strands are competent to integrate in vitro. J Virol 69: 3938–3944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani S, Boettiger D, Temin HM 1970. A DNA-depenent DNA polymerase and a DNA endonuclease in virions of Rous sarcoma virus. Nature 228: 424–427 [DOI] [PubMed] [Google Scholar]
- Moore MD, Fu W, Nikolaitchik O, Chen J, Ptak RG, Hu WS 2007. Dimer initiation signal of human immunodeficiency virus type 1: Its role in partner selection during RNA copackaging and its effects on recombination. J Virol 81: 4002–4011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutouh L, Corbeil J, Richman DD 1996. Recombination leads to the rapid emergence of HIV-1 dually resistant mutants under selective drug pressure. Proc Natl Acad Sci 93: 6106–6111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee R, Plesa G, Sherrill-Mix S, Richardson MW, Riley JL, Bushman FD HIV sequence variation associated with env antisense adoptive T-cell therapy in the hNSG mouse model. Mol Ther 18: 803–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nermut MV, Fassati A 2003. Structural analyses of purified human immunodeficiency virus type 1 intracellular reverse transcription complexes. J Virol 77: 8196–8206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman RM, Hall L, Kirmaier A, Pozzi LA, Pery E, Farzan M, O’Neil SP, Johnson W 2008. Evolution of a TRIM5-CypA splice isoform in old world monkeys. PLoS Pathog 4: e1000003 10.1371/journal.ppat.1000003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolenko GN, Palmer S, Maldarelli F, Mellors JW, Coffin JM, Pathak VK 2005. Mechanism for nucleoside analog-mediated abrogation of HIV-1 replication: Balance between RNase H activity and nucleotide excision. Proc Natl Acad Sci 102: 2093–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolenko GN, Delviks-Frankenberry KA, Palmer S, Maldarelli F, Fivash MJ Jr, Coffin JM, Pathak VK 2007. Mutations in the connection domain of HIV-1 reverse transcriptase increase 3′-azido-3′-deoxythymidine resistance. Proc Natl Acad Sci 104: 317–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisole S, Lynch C, Stoye JP, Yap MW 2004. A Trim5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc Natl Acad Sci 101: 13324–13328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Chang KW, Wierzchoslawski R, Alvord WG, Hughes SH 2008. Rous sarcoma virus (RSV) integration in vivo: A CA dinucleotide is not required in U3, and RSV linear DNA does not autointegrate. J Virol 82: 503–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onafuwa A, An W, Robson ND, Telesnitsky A 2003. Human immunodeficiency virus type 1 genetic recombination is more frequent than that of Moloney murine leukemia virus despite similar template switching rates. J Virol 77: 4577–4587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil PK, Sun G, Yu H, Ron Y, Dougherty JP, Preston BD 2002. Mutational analysis of HIV-1 long terminal repeats to explore the relative contribution of reverse transcriptase and RNA polymerase II to viral mutagenesis. J Biol Chem 277: 38053–38061 [DOI] [PubMed] [Google Scholar]
- Palaniappan C, Wisniewski M, Jacques PS, Le Grice SF, Fay PJ, Bambara RA 1997. Mutations within the primer grip region of HIV-1 reverse transcriptase result in loss of RNase H function. J Biol Chem 272: 11157–11164 [DOI] [PubMed] [Google Scholar]
- Panganiban AT, Fiore D 1988. Ordered interstrand and intrastrand DNA transfer during reverse transcription. Science 241: 1064–1069 [DOI] [PubMed] [Google Scholar]
- Pathak VK, Temin HM 1990a. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: Deletions and deletions with insertions. Proc Natl Acad Sci 87: 6024–6028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak VK, Temin HM 1990b. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: Substitutions, frameshifts, and hypermutations. Proc Natl Acad Sci 87: 6019–6023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell MD, Ghosh M, Jacques PS, Howard KJ, Le Grice SF, Levin JG 1997. Alanine-scanning mutations in the “primer grip” of pp66 HIV-1 reverse transcriptase result in selective loss of RNA priming activity. J Biol Chem 272: 13262–13269 [DOI] [PubMed] [Google Scholar]
- Powell MD, Beard WA, Bebenek K, Howard KJ, Le Grice SF, Darden TA, Kunkel TA, Wilson SH, Levin JG 1999. Residues in the αH and αI helices of the HIV-1 reverse transcriptase thumb subdomain required for the specificity of RNase H-catalyzed removal of the polypurine tract primer. J Biol Chem 274: 19885–19893 [DOI] [PubMed] [Google Scholar]
- Pullen KA, Ishimoto LK, Champoux JJ 1992. Incomplete removal of the RNA primer for minus-strand DNA synthesis by human immunodeficiency virus type 1 reverse transcriptase. J Virol 66: 367–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen KA, Rattray AJ, Champoux JJ 1993. The sequence features important for plus strand priming by human immunodeficiency virus type 1 reverse transcriptase. J Biol Chem 268: 6221–6227 [PubMed] [Google Scholar]
- Purohit V, Roques BP, Kim B, Bambara RA 2007. Mechanisms that prevent template inactivation by HIV-1 reverse transcriptase RNase H cleavages. J Biol Chem 282: 12598–12609 [DOI] [PubMed] [Google Scholar]
- Qi M, Yang R, Aiken C 2008. Cyclophilin A-dependent restriction of human immunodeficiency virus type 1 capsid mutants for infection of nondividing cells. J Virol 82: 12001–12008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch JW, Lener D, Miller JT, Julias JG, Hughes SH, Le Grice SF 2002. Altering the RNase H primer grip of human immunodeficiency virus reverse transcriptase modifies cleavage specificity. Biochemistry 41: 4856–4865 [DOI] [PubMed] [Google Scholar]
- Rhodes T, Wargo H, Hu WS 2003. High rates of human immunodeficiency virus type 1 recombination: Near-random segregation of markers one kilobase apart in one round of viral replication. J Virol 77: 11193–11200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes TD, Nikolaitchik O, Chen J, Powell D, Hu WS 2005. Genetic recombination of human immunodeficiency virus type 1 in one round of viral replication: Effects of genetic distance, target cells, accessory genes, and lack of high negative interference in crossover events. J Virol 79: 1666–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson DL, Sharp PM, McCutchan FE, Hahn BH 1995. Recombination in HIV-1. Nature 374: 124–126 [DOI] [PubMed] [Google Scholar]
- Rodgers DW, Gamblin SJ, Harris BA, Ray S, Culp JS, Hellmig B, Woolf DJ, Debouck C, Harrison SC 1995. The structure of unliganded reverse transcriptase from the human immunodeficiency virus type 1. Proc Natl Acad Sci 92: 1222–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RA, Pathak VK 2007. Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J Virol 81: 8201–8210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafianos SG, Das K, Tantillo C, Clark AD Jr, Ding J, Whitcomb JM, Boyer PL, Hughes SH, Arnold E 2001. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA: DNA. EMBO J 20: 1449–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafianos SG, Clark AD Jr, Das K, Tuske S, Birktoft JJ, Ilankumaran P, Ramesha AR, Sayer JM, Jerina DM, Boyer PL, et al. 2002. Structures of HIV-1 reverse transcriptase with pre- and post-translocation AZTMP-terminated DNA. EMBO J 21: 6614–6624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafianos SG, Marchand B, Das K, Himmel DM, Parniak MA, Hughes SH, Arnold E 2009. Structure and function of HIV-1 reverse transcriptase: Molecular mechanisms of polymerization and inhibition. J Mol Biol 385: 693–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Emerman M, Malik HS 2004. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol 2: pE275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayah DM, Sokolskaja E, Berthoux L, Luban J 2004. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430: 569–573 [DOI] [PubMed] [Google Scholar]
- Schrofelbauer B, Chen D, Landau NR 2004. A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif). Proc Natl Acad Sci 101: 3927–3932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrofelbauer B, Yu Q, Zeitlin SG, Landau NR 2005. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J Virol 79: 10978–10987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selig L, Benichou S, Rogel ME, Wu LI, Vodicka MA, Sire J, Benarous R, Emerman M 1997. Uracil DNA glycosylase specifically interacts with Vpr of both human immunodeficiency virus type 1 and simian immunodeficiency virus of sooty mangabeys, but binding does not correlate with cell cycle arrest. J Virol 71: 4842–4846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevilya Z, Loya S, Hughes SH, Hizi A 2001. The ribonuclease H activity of the reverse transcriptases of human immunodeficiency viruses type 1 and type 2 is affected by the thumb subdomain of the small protein subunits. J Mol Biol 311: 957–971 [DOI] [PubMed] [Google Scholar]
- Sevilya Z, Loya S, Adir N, Hizi A 2003. The ribonuclease H activity of the reverse transcriptases of human immunodeficiency viruses type 1 and type 2 is modulated by residue 294 of the small subunit. Nucleic Acids Res 31: 1481–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon-Loriere E, Galetto R, Hamoudi M, Archer J, Lefeuvre P, Martin DP, Robertson DL, Negroni M 2009. Molecular mechanisms of recombination restriction in the envelope gene of the human immunodeficiency virus. PLoS Pathog 5: e1000418 10.1371/journal.ppat.1000418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalka AM, Goff SP, ed. 1993. Reverse transcriptase. Cold Spring Harbor Laboratory Press, Plainview, NY: [PubMed] [Google Scholar]
- Smith JS, Roth MJ 1992. Specificity of human immunodeficiency virus-1 reverse transcriptase-associated ribonuclease H in removal of the minus-strand primer, tRNA(Lys3). J Biol Chem 267: 15071–15079 [PubMed] [Google Scholar]
- St Louis DC, Gotte D, Sanders-Buell E, Ritchey DW, Salminen MO, Carr JK, McCutchan FE 1998. Infectious molecular clones with the nonhomologous dimer initiation sequences found in different subtypes of human immunodeficiency virus type 1 can recombine and initiate a spreading infection in vitro. J Virol 72: 3991–3998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streeck H, Li B, Poon AF, Schneidewind A, Gladden AD, Power KA, Daskalakis D, Bazner S, Zuniga R, Brander C, et al. 2008. Immune-driven recombination and loss of control after HIV superinfection. J Exp Med 205: 1789–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J 2004. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature 427: 848–853 [DOI] [PubMed] [Google Scholar]
- Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, Sodroski J 2006. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5α restriction factor. Proc Natl Acad Sci 103: 5514–5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers J, Mason WS 1982. Replication of the genome of a hepatitis B–like virus by reverse transcription of an RNA intermediate. Cell 29: 403–415 [DOI] [PubMed] [Google Scholar]
- *.Sundquist WI, Kräusslich H-G 2011. HIV assembly, budding, and maturation. Cold Spring Harb Perspect Med 10.1101/cshperspect.a006924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanstrom R, Varmus HE, Bishop JM 1981. The terminal redundancy of the retrovirus genome facilitates chain elongation by reverse transcriptase. J Biol Chem 256: 1115–1121 [PubMed] [Google Scholar]
- Takehisa J, Kraus MH, Decker JM, Li Y, Keele BF, Bibollet-Ruche F, Zammit KP, Weng Z, Santiago ML, Kamenya S, et al. 2007. Generation of infectious molecular clones of simian immunodeficiency virus from fecal consensus sequences of wild chimpanzees. J Virol 81: 7463–7475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantillo C, Ding J, Jacobo-Molina A, Nanni RG, Boyer PL, Hughes SH, Pauwels R, Andries K, Janssen PA, Arnold E 1994. Locations of anti-AIDS drug binding sites and resistance mutations in the three-dimensional structure of HIV-1 reverse transcriptase. Implications for mechanisms of drug inhibition and resistance. J Mol Biol 243: 369–387 [DOI] [PubMed] [Google Scholar]
- Telesnitsky A, Goff SP 1993. Two defective forms of reverse transcriptase can complement to restore retroviral infectivity. EMBO J 12: 4433–4438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telesnitsky A, Goff G.P 1997. Reverse transcriptase and the generation of retroviral DNA. In Retroviruses (ed. Coffin JM, Hughes SH, Varmus HE), pp. 121–160 Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- Temin HM 1964. Homology between RNA from Rous sarcoma virus and DNA from Rous sarcoma virus-infected cells. Proc Natl Acad Sci 52: 323–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JA, Gorelick RJ 2008. Nucleocapsid protein function in early infection processes. Virus Res 134: 39–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JA, Gagliardi TD, Alvord WG, Lubomirski M, Bosche WJ, Gorelick RJ 2006. Human immunodeficiency virus type 1 nucleocapsid zinc-finger mutations cause defects in reverse transcription and integration. Virology 353: 41–51 [DOI] [PubMed] [Google Scholar]
- Thomas DC, Voronin YA, Nikolenko GN, Chen J, Hu WS, Pathak VK 2007. Determination of the ex vivo rates of human immunodeficiency virus type 1 reverse transcription by using novel strand-specific amplification analysis. J Virol 81: 4798–4807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JA, Bosche WJ, Shatzer TL, Johnson DG, Gorelick RJ 2008. Mutations in human immunodeficiency virus type 1 nucleocapsid protein zinc fingers cause premature reverse transcription. J Virol 82: 9318–9328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trono D 1992. Partial reverse transcripts in virions from human immunodeficiency and murine leukemia viruses. J Virol 66: 4893–4900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu X, Das K, Han Q, Bauman JD, Clark AD Jr, Hou X, Frenkel YV, Gaffney BL, Jones RA, Boyer PL, et al. 2010. Structural basis of HIV-1 resistance to AZT by excision. Nat Struct Mol Biol 17: 1202–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuske S, Sarafianos SG, Clark AD Jr, Ding J, Naeger LK, White KL, Miller MD, Gibbs CS, Boyer PL, Clark P, et al. 2004. Structures of HIV-1 RT-DNA complexes before and after incorporation of the anti-AIDS drug tenofovir. Nat Struct Mol Biol 11: 469–474 [DOI] [PubMed] [Google Scholar]
- van Wamel JL, Berkhout B 1998. The first strand transfer during HIV-1 reverse transcription can occur either intramolecularly or intermolecularly. Virology 244: 245–251 [DOI] [PubMed] [Google Scholar]
- Virgen CA, Kratovac Z, Bieniasz PD, Hatziioannou T 2008. Independent genesis of chimeric TRIM5-cyclophilin proteins in two primate species. Proc Natl Acad Sci 105: 3563–3568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagaman PC, Hasselkus-Light CS, Henson M, Lerner DL, Phillips TR, Elder JH 1993. Molecular cloning and characterization of deoxyuridine triphosphatase from feline immunodeficiency virus (FIV). Virology 196: 451–457 [DOI] [PubMed] [Google Scholar]
- Wang J, Bambara RA, Demeter LM, Dykes C 2010. Reduced fitness in cell culture of HIV-1 with nonnucleoside reverse transcriptase inhibitor-resistant mutations correlates with relative levels of reverse transcriptase content and RNase H activity in virions. J Virol 84: 9377–9389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitcomb JM, Kumar R, Hughes SH 1990. Sequence of the circle junction of human immunodeficiency virus type 1: Implications for reverse transcription and integration. J Virol 64: 4903–4906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Wilen CB, Tilton JC, Doms RW 2011. HIV: Cell binding and entry. Cold Spring Harb Perspect Med 10.1101/cshperspect.a006866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SJ, Webb BL, Ylinen LM, Verschoor E, Heeney JL, Towers GJ 2008. Independent evolution of an antiviral TRIMCyp in rhesus macaques. Proc Natl Acad Sci 105: 3557–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Liu H, Xiao H, Conway JA, Hehl E, Kalpana GV, Prasad V, Kappes JC 1999. Human immunodeficiency virus type 1 integrase protein promotes reverse transcription through specific interactions with the nucleoprotein reverse transcription complex. J Virol 73: 2126–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Anderson JL, Campbell EM, Joseph AM, Hope TJ 2006. Proteasome inhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infection. Proc Natl Acad Sci 103: 7465–7470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Emerman M 2004. Capsid is a dominant determinant of retrovirus infectivity in nondividing cells. J Virol 78: 5670–5678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi-Brunozzi HY, Le Grice SF 2005. Investigating HIV-1 polypurine tract geometry via targeted insertion of abasic lesions in the (−)-DNA template and (+)-RNA primer. J Biol Chem 280: 20154–20162 [DOI] [PubMed] [Google Scholar]
- Yu SF, Baldwin DN, Gwynn SR, Yendapalli S, Linial ML 1996. Human foamy virus replication: A pathway distinct from that of retroviruses and hepadnaviruses. Science 271: 1579–1582 [DOI] [PubMed] [Google Scholar]
- Yu H, Jetzt AE, Ron Y, Preston BD, Dougherty JP 1998. The nature of human immunodeficiency virus type 1 strand transfers. J Biol Chem 273: 28384–28391 [DOI] [PubMed] [Google Scholar]
- Yu SF, Sullivan MD, Linial ML 1999. Evidence that the human foamy virus genome is DNA. J Virol 73: 1565–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302: 1056–1060 [DOI] [PubMed] [Google Scholar]
- Yu Q, Chen D, Konig R, Mariani R, Unutmaz D, Landau NR 2004. APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J Biol Chem 279: 53379–53386 [DOI] [PubMed] [Google Scholar]
- Zack JA, Haislip AM, Krogstad P, Chen IS 1992. Incompletely reverse-transcribed human immunodeficiency virus type 1 genomes in quiescent cells can function as intermediates in the retroviral life cycle. J Virol 66: 1717–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WH, Hwang CK, Hu WS, Gorelick RJ, Pathak VK 2002. Zinc finger domain of murine leukemia virus nucleocapsid protein enhances the rate of viral DNA synthesis in vivo. J Virol 76: 7473–7484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YH, Irwin D, Kurosu T, Tokunaga K, Sata T, Peterlin BM 2004. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J Virol 78: 6073–6076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Cunningham JM 1993. Minus-strand DNA is present within murine type C ecotropic retroviruses prior to infection. J Virol 67: 2385–2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang J, Jetzt AE, Sun G, Yu H, Klarmann G, Ron Y, Preston BD, Dougherty JP 2002. Human immunodeficiency virus type 1 recombination: Rate, fidelity, and putative hot spots. J Virol 76: 11273–11282 [DOI] [PMC free article] [PubMed] [Google Scholar]