

Abstract
Objective:
Parkinson disease (PD), a devastating neurodegenerative disorder, affects motor abilities and cognition as well. It is not clear whether the proapoptotic protein, Bid, is involved in tumor necrosis factor death receptor I (TNFRI)–mediated destructive signal transduction pathways such as cell dysfunction or neurodegeneration in the temporal cortex of patients with PD.
Methods:
Molecular and biochemical approaches were used to dissect mitochondrial related components of the destructive signaling pathway in the temporal cortex from rapidly autopsied brains (postmortem interval mean 2.6 hours). Brains from patients with PD (n = 15) had an average age of 81.4 years, compared to the average age of 84.36 years in age-matched control patient brains (n = 15).
Results:
TNFRI and its adaptor protein, TRADD, were not only present in the cytoplasm of the temporal cortex, but were significantly elevated (42.3% and 136.1%, respectively) in PD brains compared to age-matched control brains. Bid in the PD temporal cortex could be further cleaved into tBid in the cytosol, which is translocated into the mitochondria, where cytochrome c is then released and caspase-3 is subsequently activated.
Conclusion:
Patients with PD have an activated Bid-mediated destructive signal pathway via TNFRI in the temporal cortex. Such deficits are pervasive, suggesting that they might contribute to cortex degeneration as PD manifests.
Parkinson disease (PD) is a progressive neurodegenerative disorder characterized by the loss of dopaminergic neurons in the substantia nigra. Apoptosis may be involved in dopaminergic cell death in sporadic and familial PD.1–7 However, whether other brain regions undergo the destructive signal pathways is still unknown. Cell apoptosis is regulated by a wide number of factors, such as tumor necrosis factor (TNF) and Fas ligand. Evidence implicates neuroinflammation from TNF as the fundamental piece in the pathogenesis of PD.8–12 Elevated levels of TNF in the CSF and postmortem brains of patients with PD suggest that this proinflammatory cytokine plays an important role in the pathophysiology of the disease. Elucidation of the mechanisms underlying neuroinflammation in PD may contribute to new and effective therapies, especially for cognitive decline of patients with PD. TNF receptor subtype I (TNFRI) contains a “death domain” (DD) that is associated with DD-containing adaptor proteins, including TNFRI-associated DD protein (TRADD), Fas-associated DD (FADD), or receptor-interacting protein. The detailed apoptotic pathway that may be mediated by TNFRI is still unknown in PD, although studies have shown elevated TNFRI levels in the substantia nigra of patients with PD.13
Bid is a proapoptotic protein of the Bcl-2 family that is crucial for death receptor–mediated apoptosis and is activated post-translationally via caspase-8-mediated cleavage into a truncated form, tBid.14–16 In the present study, we have identified an activated Bid-mediated destructive pathway in the temporal cortex of patients with PD, suggesting that they might contribute to cognitive decline manifestation.
METHODS
Standard protocol approvals, registrations, and patient consents.
All subjects or their legally authorized representatives sign an informed consent to research and autopsy; the research has been approved by the institutional review board. Brain tissue was obtained from the Banner Sun Health Research Institute brain and body donation program (BBDP), Sun City, AZ.17
Acquisition of samples.
The research subjects were recruited primarily from the Sun City retirement communities located in the northwest metropolitan region of Phoenix, AZ. These communities have a population of approximately 900,000, an average age of 72 years, and a minimum age of 55 years. The BBDP is unique for its rapid autopsy program (median postmortem interval is 2.8 hours), allowing the provision of unusually high- quality brain tissue. The study subjects consisted of 15 nonpathologic control (NPC) subjects (at age of 78 to 90 years) and 15 patients with PD (at age of 72 to 90 years, disease duration of 14.4 ± 8.4 years). The subject profiles are presented in table 1.
Table 1.
Human subject profiles from the patients with PD and control subjects
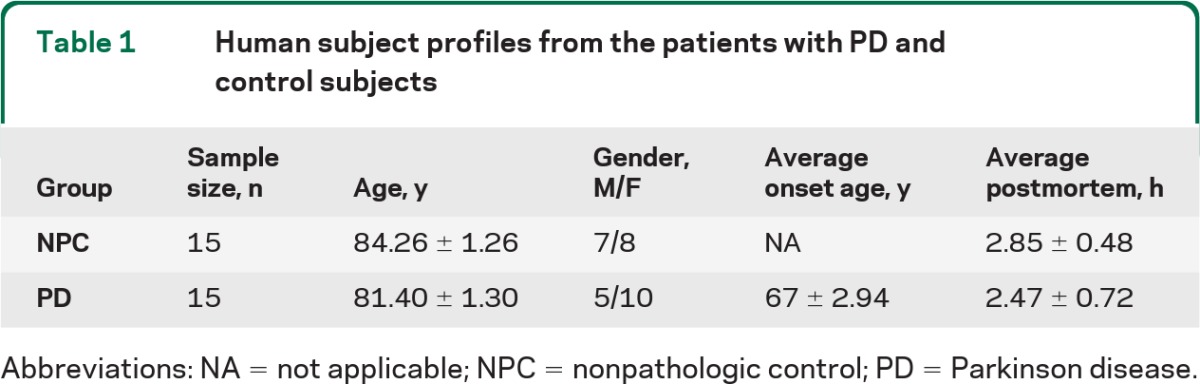
Abbreviations: NA = not applicable; NPC = nonpathologic control; PD = Parkinson disease.
Clinical diagnosis and pathologic confirmation.
Human subjects were specifically chosen with no dementia and PD in the temporal cortex who might be showing early changes but not be end-stage as might be seen in individuals with dementia. The criteria for Alzheimer disease (AD) are defined by NIA–Reagan “intermediate or high”18 and pathologic Consortium to Establish a Registry for Alzheimer's Disease neuritic plaque density19 as well as Braak staging.20 The detail information is shown in table e-1 on the Neurology® Web site at www.neurology.org. PD diagnosis was determined when there were at least 2 of the cardinal clinical signs of PD as well as pathologic evidence of Lewy bodies and pigmented neuron loss in the substantia nigra (SN). Scores for brain regions are based on Dementia with Lewy Body Consortium density estimates21 and Unified Staging System for Lewy body disorders.22 The detail information is shown in table e-2. Patients who met pathologic criteria for PD are levodopa responsive. NPCs were selected based on the absence of a clinical history of parkinsonism or dementia and the absence of Lewy bodies on neuropathologic examination.
Brain tissue preparation.
Protein was extracted from homogenates of frozen tissue from the gray matter of the temporal cortex in 15 PD cases and 15 controls. Approximately 500 mg of tissue was homogenized in phosphate-buffered saline containing 1% Triton X-100 and 0.1% sodium dodecyl sulfate (SDS) (pH 7.0) plus proteinase inhibitors, and it was centrifuged at 14,000 rpm for 30 minutes. The protein concentration in the supernatant was measured with a Bio-Rad protein assay kit. A total of 50 μg of protein was mixed with 1:1 of sample buffer (25 mM Tris, 250 mM Glycine, pH 7.0) and loaded on a 12% SDS polyacrylamide gel electrophoresis (SDS-PAGE) gel. The protein was then transferred to a PDVF membrane.
ELISA.
The content of TNFRI in the lysed extracts was determined using a commercially available ELISA kit for human TNFRI (R&D System). Dilution curve homogenate samples of the brain were parallel to the standard dilution curve. The assay followed the manufacturer's recommended procedures. The absorbance was measured at 450 nm with an automatic wavelength correction of 540 nm. The experiments were repeated 3 times.
Immunoprecipitation.
For the immunoprecipitation of TRADD, 500 μg of brain extracts in a 300 μL volume containing 20 μg anti-TRADD polyclonal antibody (catalog: ab18914, Abcam) was incubated at 4°C overnight. A total of 20 μL of Protein-G agarose (Pierce) was then mixed with the samples and incubated with orbitary rotation at 4°C for 4 hours. The beads were collected and resuspended in 50 μL of SDS-PAGE loading dye and heated at 95°C for 5 minutes. A total of 10 μL of each sample was separated by 12% SDS-PAGE gel and transferred to PDVF membranes.
Immunoblotting.
Western blots were probed with the following antibodies: anti-TRADD monoclonal antibody (1:500, catalog: sc-46653, Santa Cruz); anti-Bid polyclonal antibody (1:5,000, catalog: AF846, R&D Systems); anti-cytochrome c monoclonal antibody (1:5,000, clone: 7H8.2C12, catalog: ab13575, Abcam); and anti-caspase-3 polyclonal antibody (1:5,000, catalog: sc-7148, Santa Cruz). Blots were also probed with anti-β-actin monoclonal antibody (1:5,000, catalog: #1978, Sigma) as a loading control. For mitochondria samples, the blots were performed with anti-Cox IV monoclonal antibody (1:5,000, clone: 20E8C12, catalog: ab14744, Abcam) as a mitochondrial loading control. The protein levels were normalized to β-actin or Cox IV (for mitochondria samples) and indicated in arbitrary units.
Immunohistochemistry.
The immunohistostaining was performed as described previously.23,24 Fresh tissues of the temporal cortex were postfixed with 4% paraformaldehyde and sectioned to 30 μm. Pretreatment of sections was performed with 0.3% triton X-100 and blocked with 10% goat serum for 30 minutes. Primary antibodies were incubated with polyclonal rabbit anti-Bid (1:400, clone: Y8, catalog: ab32060, Abcam), mouse anti-NeuN (MAB377, Millipore, Billerica, MA), monoclonal mouse anti–glial fibrillary acid protein (GFAP, 1:5,000, clone: SMI22, catalog: SMI-22R, Covance), mouse anti-human CD45 (1:400, clone: F10-89-4, catalog: MCA87, Serotec) overnight at 4°C. Secondary antibodies of goat against mouse or rabbit immunoglobulin G with fluorescence 488 or 568 (1:1,000; Invitrogen, Carlsbad, CA) were incubated for 30 minutes. The images were taken with BX63 microscope (Olympus, Tokyo, Japan).
Mitochondria isolation.
Mitochondria were isolated from approximately 100 mg of gray matter tissue from the temporal cortex using a mitochondria isolation kit (BioChain Institute Inc, Hayward, CA). The isolation procedure followed the manufacturer's recommended procedures. Protein concentration was measured with Bio-Rad protein assay kit. Samples for separation on a 12% SDS-PAGE gel were prepared as described in the immunoprecipitation section.
Caspase-3 activity assay.
Caspase-3 activity was determined using a caspase-3 activity kit according to the manufacturer's protocol (Calbiochem). Briefly, the colorimetric assay was conducted with an equal amount of brain lysate combined with the fluorescent labeled DEVD substrate in a 96-well plate. The plate was incubated at 37°C for 2 hours and the fluorescence was measured at 400 nm and 505 nm. The amount of luminescence expressed as relative light units reflects caspase activity. These assays were performed in triplicate.
Statistical analysis.
Data were presented as mean ± SEM. Differences in means were determined by an unpaired t test. A probability value of p < 0.05 was considered statistically significant.
RESULTS
TNFRI signaling is elevated in PD brains.
Quantification of TNFRI protein levels in the temporal cortex of PD and age-matched NPC brains were determined by ELISA. As shown in figure 1A, the levels of TNFRI in PD brains (2.22 ± 0.24 ng/mg) was higher (p < 0.05) than that in NPC brains (1.56 ± 0.12 ng/mg). The activation of TNFRI recruited several adaptor proteins containing DDs. We examined the expression of TRADD, a molecule involved in a TNFRI-mediated destructive signal transduction pathway, in the temporal cortex using immunoprecipitation. We observed a band of TRADD around 34 kD (figure 1B). Density assay showed increased TRADD levels in PD brains (figure 1C, p < 0.01).
Figure 1. Tumor necrosis factor death receptor I (TNFRI) signaling is increased in Parkinson disease (PD) brains.

(A) ELISA showed that TNFRI levels are elevated in the temporal cortex of PD brains compared to the control brains. (B) Representative immunoprecipitation of tumor necrosis factor death receptor I–associated death domain protein (TRADD) was presented in the temporal cortex from parkinsonian brains and controls. (C) Spot density analysis of TRADD was normalized to corresponding β-actin level (ap < 0.05; bp < 0.01) compared to nonpathologic control (NPC).
Bid expression in neurons and activated astrocytes as well as cleaved Bid translocation to the mitochondria in PD brains.
To verify whether Bid expression is activated and in which cell type in brains, immunostaining was performed with the brain sections from the temporal cortex. We found Bid expression in most of the neurons (NeuN, a neuron marker, figure 2A) and some activated astrocytes (GFAP, an astrocyte marker, figure 2B) but not in activated microglia (CD45, an activated microglial marker, figure 2C). Both forms of Bid, full length Bid (∼22 kD) and tBid (∼14 kD), were detected in temporal cortex brain lysate and mitochondria extractions. The levels of full-length Bid in the cytosol were increased in PD brains compared to NPC brains. A dramatically increased level of tBid was detected in all PD brains, but only existed in a few NPC brains (figure 2, D and E). In the mitochondria, there was an increase in full-length Bid expression exclusively in PD group compared to NPC brains. Similar results were observed in tBid levels (figure 2, F and G); tBid was not found in mitochondria extractions from NPC brain samples.
Figure 2. Bid is expressed in neurons and astrocytes, and cleaved Bid is located to the mitochondria in Parkinson disease (PD) brains.

(A) Bid expression was visualized in neurons labeled with antibody against NeuN. (B) Bid expression was found in activated astrocytes identified with antibody against glial fibrillary acid protein. (C) Little Bid expression was found in activated microglia labeled with antibody CD45. Bars: 20 μm. (D) A full-length Bid around 22 kD and the cleaved Bid (tBid) around 14 kD were detected in cytoplasmic fractions of samples from the temporal cortex. (E) Spot density analysis showed increased levels of the full-length Bid and tBid in PD brain samples (ap < 0.05, bp < 0.01). (F) Western blot demonstrated tBid fragments in the mitochondrial fractions from the temporal cortex of PD brains, Cox IV as a mitochondrial loading control. (G) Spot density analysis of increased levels of the full-length Bid and tBid in the mitochondria of PD brain samples (ap < 0.05, bp < 0.01). NPC = nonpathologic control.
Cytochrome c is released from the mitochondria in PD brains.
The mitochondria from the temporal cortex were isolated. Western blot was performed and results showed that levels of cytoplasmic cytochrome c were increased in PD brain samples after normalization to β-actin level (figure 3, A and B, p < 0.01). However, the levels of cytochrome c in mitochondria were decreased after normalization to Cox IV level (figure 3, C and D, p < 0.01), suggesting that the translocation of tBid to mitochondria causes a significant cytochrome c release from the mitochondria.
Figure 3. Cytochrome c is released from mitochondria in Parkinson disease (PD) brains.

(A) Western blot showed cytochrome c release in cytoplasmic fractions of samples from the temporal cortex. (B) Spot density analysis showed increased levels of cytoplasmic cytochrome c release in PD brain samples (ap < 0.01). (C) Cytochrome c was detected in the mitochondrial fraction from temporal cortex using Cox IV as the mitochondrial loading control. (D) Spot density analysis showed decreased levels of cytochrome c in the mitochondria from PD brain samples (bp < 0.01). NPC = nonpathologic control.
Activity of caspase-3 is increased in PD brains.
To address the particular role of caspases in TNFRI-mediated destructive signal transduction pathway, we used both Western blot and procaspase-3 activity assay. Three bands were detected using Western blot, 1 fragment of procaspase-3 (32 kD), and 2 cleaved forms of proteins (21 kD and 12 kD). There was an increase of procaspase-3 and its cleaved fragments in the temporal cortex of PD brains compared to NPC brains (figure 4A). Procaspase-3 activity was detected and we found an increased caspase activity in PD brains compared to NPC brains, which is in agreement with the Western blot results (figure 4B).
Figure 4. Cleaved caspase-3 fragments and caspase-3 activity are increased in Parkinson disease (PD) brains.

(A) Full-length procaspase-3 (32 kD) and cleaved fragments (21 kD and 12 kD) were detected in the samples from the temporal cortex. These cleaved forms were little found in the nonpathologic control (NPC) brains. (B) Caspase-3 activity in mitochondria was elevated in mitochondria of PD brains compared to NPC samples (ap < 0.05).
No changes are found in Bid signal components between controls without dementia and preclinical AD among the patients with PD.
According to the current criteria for a pathologic diagnosis of preclinical AD, in the 15 cases of patients with PD listed in table e-1, 6 cases of patients with PD accompany the cortical changes of preclinical AD (moderate/frequent neuritic plaques and Braak III/IV of neurofibrillary tangles). To clarify the possible influences of preclinical AD-like changes in patients with PD, the values of each protein band density of Western blotting were measured. Results showed no significant differences in the expression levels of TNFRI, TRADD, Bid, tBid, cytochrome c, and procaspase 3 and cleaved caspase 3 between no dementia and brain preclinical AD changes among all patients with PD.
DISCUSSION
Available evidence suggests that patients with PD exhibit neurodegeneration not only in the dopaminergic neurons in SN, but also other brain regions including the cortex. The mechanisms underlying the neurodegenerative process remain elusive. In the present study, we found that the cleavage of Bid and its truncated tBid translocation to the mitochondria are implicated in the TNFRI-mediated signaling pathway in the temporal cortex of PD brains. These findings might provide a mechanistic link to how the death receptor leads to the activation of Bid, the dysfunction of mitochondria, and the consequent activation of caspases in the cortex regions of PD brains (figure e-1).
We previously reported that TNFRI levels are elevated in the cortex from AD brains.25 Here, we found an elevated expression of TNFRI and its adaptor protein, TRADD, in the temporal cortex of parkinsonian brains. All of these suggest a possible association of death receptors with cell death in neurodegenerative disorders. A likely interaction partner for TRADD may be FADD, which can then interact with procaspase-8. Activation of procaspase-8 through self-cleavage leads to a series of downstream events, including activation of procaspase-3 and induction of mitochondrial damage.26,27
Bid is a unique BH3-only molecule of Bcl-2 family,15 which is largely present in cytosol and is involved in the cellular destructive process mediated by TNFRI death receptor. A truncated fragment tBid is generated from a proteolytic digestion at Asp-59, a site with a conserved caspase-8-cleavage sequence.28 Upon the stimulation of a particular death signal, tBid translocates to the mitochondria and induces cytochrome c release.14,29–31 This is similar to a previous report showing that cells depleted of Bid by immunoprecipitation are no longer able to induce cytochrome c release.32 Bid-induced cytochrome c release is mediated by 2 different mechanisms. One is responsible for the initial release of free cytochrome c in the intermembrane space through a Bak-dependent mechanism. The other causes a dramatic remodeling of mitochondrial cristae, which mobilize cytochrome c storage. Effects of tBid on the mitochondria may not be limited to the induction of cytochrome c release, but may include other changes in mitochondrial physiology such as mitochondrial redistribution and loss of mitochondrial ψΔm in the cells.33
This portion of cytochrome c constitutes the majority (85%) of total release.34 The released cytochrome c then binds with Apaf-1 to form the apoptosome, a multimeric Apaf-1 and cytochrome c complex. Only the caspase-9 binding to the apoptosome is able to efficiently cleave and activate downstream executioner caspases such as caspase-3.16,35 These executioner caspases will then cleave intracellular substrates, leading to chromatin condensation and DNA fragmentation. As shown in figure e-1, TNFRI can directly activate caspase-3 through caspase-8. This might be due to the amplification of caspase activation and the involvement of additional mitochondrial dysfunction.
Our finding demonstrated that Bid is largely activated in the neurons of the temporal cortex from patients with PD as well as the activation of the entire TNFRI-Bid death signaling pathway in the cortex relevant to nonmotor PD. The results are similar to the observation of TNFRI-engaged hepatocyte apoptosis in which Bid is required for TNFα- and Fas-induced apoptosis through a mitochondrial pathway.34 Our data might in part explain the possibly emerging cognitive decline associated with cortical pathogenesis in PD.
Supplementary Material
ACKNOWLEDGMENT
The samples are from the Brain and Tissue Bank, Sun Health Research Institute. The BBDP is supported by the National Institute of Neurological Disorders and Stroke (U24NS072026 National Brain and Tissue Resource for PD and Related Disorders), the National Institute on Aging (P30AG19610 Arizona AD Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer's Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901, and 1001 to the Arizona PD Consortium), and the Michael J. Fox Foundation for Parkinson's Research.
GLOSSARY
- AD
Alzheimer disease
- BBDP
brain and body donation program
- DD
death domain
- FADD
Fas-associated death domain
- GFAP
glial fibrillary acid protein
- NPC
nonpathologic control
- PD
Parkinson disease
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SN
substantia nigra
- TNF
tumor necrosis factor
- TNFRI
tumor necrosis factor death receptor I
- TRADD
tumor necrosis factor death receptor I–associated death domain protein
Footnotes
Editorial, page 1750
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Dr. Jiang and Dr. He performed biochemical and immunochemical experiments, participated in data analyses, and wrote the initial manuscript draft. Dr. Adler and Dr. Shill made clinical diagnosis. Dr. Beach made pathologic evaluation. Dr. Shen and Dr. Li initiated the project and designed the experiments, and participated in data analyses and overviewed manuscript drafting.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1. Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron 2003;39:889–909 [DOI] [PubMed] [Google Scholar]
- 2. Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 2004;3:205–214 [DOI] [PubMed] [Google Scholar]
- 3. Hunot S, Hirsch EC. Neuroinflammatory processes in Parkinson's disease. Ann Neurol 2003;53(suppl 3):S49–S58; discussion S58–S60 [DOI] [PubMed] [Google Scholar]
- 4. Jenner P, Olanow CW. The pathogenesis of cell death in Parkinson's disease. Neurology 2006;66:S24–S36 [DOI] [PubMed] [Google Scholar]
- 5. Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature 2006;443:796–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hartmann A, Mouatt-Prigent A, Faucheux BA, Agid Y, Hirsch EC. FADD: a link between TNF family receptors and caspases in Parkinson's disease. Neurology 2002;58:308–310 [DOI] [PubMed] [Google Scholar]
- 7. Jiang H, Luan Z, Wang J, Xie J. Neuroprotective effects of iron chelator Desferal on dopaminergic neurons in the substantia nigra of rats with iron-overload. Neurochem Int 2006;49:605–609 [DOI] [PubMed] [Google Scholar]
- 8. Nagatsu T, Sawada M. Inflammatory process in Parkinson's disease: role for cytokines. Curr Pharm Des 2005;11:999–1016 [DOI] [PubMed] [Google Scholar]
- 9. McCoy MK, Martinez TN, Ruhn KA, et al. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson's disease. J Neurosci 2006;26:9365–9375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology 1988;38:1285–1291 [DOI] [PubMed] [Google Scholar]
- 11. Hald A, Lotharius J. Oxidative stress and inflammation in Parkinson's disease: is there a causal link? Exp Neurol 2005;193:279–290 [DOI] [PubMed] [Google Scholar]
- 12. Hirsch EC, Hunot S, Hartmann A. Neuroinflammatory processes in Parkinson's disease. Parkinsonism Relat Disord 2005;11(suppl 1):S9–S15 [DOI] [PubMed] [Google Scholar]
- 13. Mogi M, Togari A, Kondo T, et al. Caspase activities and tumor necrosis factor receptor R1 (p55) level are elevated in the substantia nigra from parkinsonian brain. J Neural Transm 2000;107:335–341 [DOI] [PubMed] [Google Scholar]
- 14. Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998;94:481–490 [DOI] [PubMed] [Google Scholar]
- 15. Kaufmann T, Tai L, Ekert PG, et al. The BH3-only protein bid is dispensable for DNA damage- and replicative stress-induced apoptosis or cell-cycle arrest. Cell 2007;129:423–433 [DOI] [PubMed] [Google Scholar]
- 16. Wang X. The expanding role of mitochondria in apoptosis. Genes Dev 2001;15:2922–2933 [PubMed] [Google Scholar]
- 17. Beach TG, Sue LI, Walker DG, et al. The Sun Health Research Institute Brain Donation Program: description and experience, 1987–2007. Cell Tissue Bank 2008;9:229–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. Neurobiol Aging 1997;18:S1–S2 [PubMed] [Google Scholar]
- 19. Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD): part II: standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–486 [DOI] [PubMed] [Google Scholar]
- 20. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259 [DOI] [PubMed] [Google Scholar]
- 21. McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005;65:1863–1872 [DOI] [PubMed] [Google Scholar]
- 22. Beach TG, Adler CH, Lue L, et al. Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 2009;117:613–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. He P, Zhong Z, Lindholm K, et al. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer's mice. J Cell Biol 2007;178:829–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He P, Liu Q, Wu J, Shen Y. Genetic deletion of TNF receptor suppresses excitatory synaptic transmission via reducing AMPA receptor synaptic localization in cortical neurons. FASEB J 2012;26:334–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheng X, Yang L, He P, Li R, Shen Y. Differential activation of tumor necrosis factor receptors distinguishes between brains from Alzheimer's disease and non-demented patients. J Alzheimers Dis 2010;19:621–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yin XM. Bid, a critical mediator for apoptosis induced by the activation of Fas/TNF-R1 death receptors in hepatocytes. J Mol Med 2000;78:203–211 [DOI] [PubMed] [Google Scholar]
- 27. Jin Z, El-Deiry WS. Distinct signaling pathways in TRAIL- versus tumor necrosis factor-induced apoptosis. Mol Cell Biol 2006;26:8136–8148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thornberry NA, Rano TA, Peterson EP, et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem 1997;272:17907–17911 [DOI] [PubMed] [Google Scholar]
- 29. Eskes R, Desagher S, Antonsson B, Martinou JC. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol 2000;20:929–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Desagher S, Osen-Sand A, Nichols A, et al. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol 1999;144:891–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yuan J, Lipinski M, Degterev A. Diversity in the mechanisms of neuronal cell death. Neuron 2003;40:401–413 [DOI] [PubMed] [Google Scholar]
- 32. Gross A, Yin XM, Wang K, et al. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem 1999;274:1156–1163 [DOI] [PubMed] [Google Scholar]
- 33. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998;94:491–501 [DOI] [PubMed] [Google Scholar]
- 34. Zhao Y, Ding WX, Qian T, Watkins S, Lemasters JJ, Yin XM. Bid activates multiple mitochondrial apoptotic mechanisms in primary hepatocytes after death receptor engagement. Gastroenterology 2003;125:854–867 [DOI] [PubMed] [Google Scholar]
- 35. Yuan J, Yankner BA. Apoptosis in the nervous system. Nature 2000;407:802–809 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.