

Summary
Protein ubiquitination is a highly conserved, central mechanism to regulate cellular events in all eukaryotes, such as proteasomal degradation, protein trafficking, DNA repair, synaptic plasticity and immune response. The consequence of protein ubiquitination is modulated by the structure of ubiquitin moiety attached on the substrates, including ubiquitin monomer and diverse polyubiquitin chains with different linkages (N-terminus, K6, K11, K27, K29, K33, K48 and K63). The development of ubiquitin-enrichment strategies coupled with sensitive mass spectrometry enables direct analysis of ubiquitinated proteins in cells, providing an invaluable tool for ubiquitin research. In this chapter we describe recent technology updates for analyzing tissue-specific ubiquitin conjugates in transgenic models, as well as targeted proteomics methods for quantifying different polyubiquitin chain linkages in any type of samples, including human tissues.
Keywords: ubiquitin, proteomics, mass spectrometry, tissue
1. Introduction
Ubiquitin (Ub) is an essential protein of 76 amino acids encoded by multiple genes in eukaryotes. Ubiquitin conjugation of proteins is carried out by the sequential action of ubiquitin activating (E1), conjugating (E2) and ligating (E3) enzymes, and the modification is reversed by deubiquitinating (DUB) proteases (1). This post-translational modification is involved in a wide range of cellular processes. Ubiquitination is linked to the turnover of an ever-growing number of proteins; it regulates protein trafficking, and is also widely used to transiently facilitate protein-protein interactions, but little is known yet regarding where and when are those proteins ubiquitinated in vivo, within the context of a whole organism.
Although ubiquitin is typically conjugated to the side chain of lysine residues on targeted proteins (Fig. 1), in some cases, non-lysine residues (e.g., N-terminal amine group and cysteine residues) function as alternative sites during ubiquitination (2, 3). Moreover, after the first ubiquitin molecule is attached to the substrates, additional Ub molecules can be mounted through any of the eight amine groups in the first molecule, including the N-terminus, K6, K11, K27, K29, K33, K48, and K63 (4–6), to form polyUb chains. These eight polyUb linkages may produce diverse chain structures (7–9), providing different structures for downstream recognition by Ub receptors and thus mediating specific signaling processes (10, 11). Therefore, analysis of polyUb chains on the substrates is instructive to understand the function of ubiquitination events.
Figure 1.

The chemistry of the ubiquitin family. During the posttranslational modification by Ub and Ub like proteins, the C-terminal carboxyl group forms an isopeptide bond with the amine group on the side chain of Lys residues. Alternatively, N-terminal amine, Cys residues and even some lipid and other small molecules also serve as acceptors in this reaction.
The ubiquitin pathway is essential for brain development and function, and its failure is associated with a number of neurodegenerative diseases (12, 13). For instance, some early-onset Parkinson’s disease cases are linked to mutations in the Parkin E3 ligase (14). Mutations in KLHL7, one subunit of a cullin E3 ligase, cause progressive degeneration of rod and cone photoreceptors in the retina; another E3 (gigaxonin) gene mutations are associated with giant axonal neuropathy (15); and UBE3A mutations are linked to the onset of Angelman syndrome (16). Different from ubiquitin conjugation enzymes, mutations in two DUB genes, UCHL1 and ATXN3, are also linked to Parkinson’s disease and spinal cerebral ataxia, respectively (16). In addition, ubiquitin-positive inclusion staining is viewed as a hallmark of pathology in a wide range of neurodegenerative disorders, suggesting its involvement in the disease development (12, 16). However, it is not known how changes in the Ubiquitin Proteasome System (UPS) during normal aging are related to pathological processes that occur during early stages of neurodegeneration. Studies that have attempted to measure global proteasome activity and address the issue of global age-related decrease in ubiquitin turnover remain inconclusive. The analysis of ubiquitination activities in a tissue or cell type-specific manner (e.g. in neuron) should be able to deepen the understanding of these topics.
Advances in mass spectrometry (MS) make it feasible to analyze proteins in the attomole range (17–20), providing unprecedented sensitivity for biochemical analysis of ubiquitin signaling. Prior to MS analysis, ubiquitinated proteins are often enriched by affinity approaches, including ubiquitin antibodies (21, 22), ubiquitin-binding proteins (23–26), or epitope-tagged ubiquitin (e.g., FLAG, HA, myc, His, and biotin tag) (27–29). Polyhistidine tagging allows the purification to be performed under denaturing conditions, in order to prevent protease activity, including that of the DUB proteases (4). An alternate method that works under denaturing conditions is biotin-tagging (30). The biotin-avidin interaction is far stronger and allows for much more stringent washes, resulting in minimal non-specific background. Furthermore, most organisms contain a limited number of endogenous biotinylated proteins that can be readily identified by mass spectrometry. A tagged ubiquitin construct containing both a His-tag and a biotin-accepting domain was shown to be efficiently biotinylated by endogenous enzymes in cell culture, but has not been reported yet to work in vivo, maybe due to the size of the used tag being far larger than ubiquitin itself (30).
Here we describe a detailed protocol for the efficient isolation of neuronal ubiquitin conjugates from specific tissues in flies (31). The system relies on the BirA (biotinylating) enzyme from E. coli expressed as a fusion protein with multiple copies of ubiquitin that bear a short biotinylatable motif at their N-terminus (Fig. 2). Endogenous DUBs process this polypeptide into individual ubiquitin molecules that are then modified by BirA in vivo. Due to the strength and the specificity of the avidin-biotin interaction, we have been able to isolate and enrich the neuronal ubiquitinated proteins from a multicellular organism up to levels not achieved previously by any other approaches. This allows to identify by mass spectrometry those neuronal proteins that are ubiquitinated in a specific tissue, and to resolve by western blotting whether they are mono or polyubiquitinated, even in the absence of proteasome inhibitors, therefore reporting on physiological ubiquitination levels. Furthermore, we present a targeted MS approach to quantify the polyUb linkages on isolated protein targets or in total tissue lysates, such as human postmortem brains (Fig. 3). This approach is highly specific and bypasses the requirement of antibody against polyUb chains.
Figure 2.
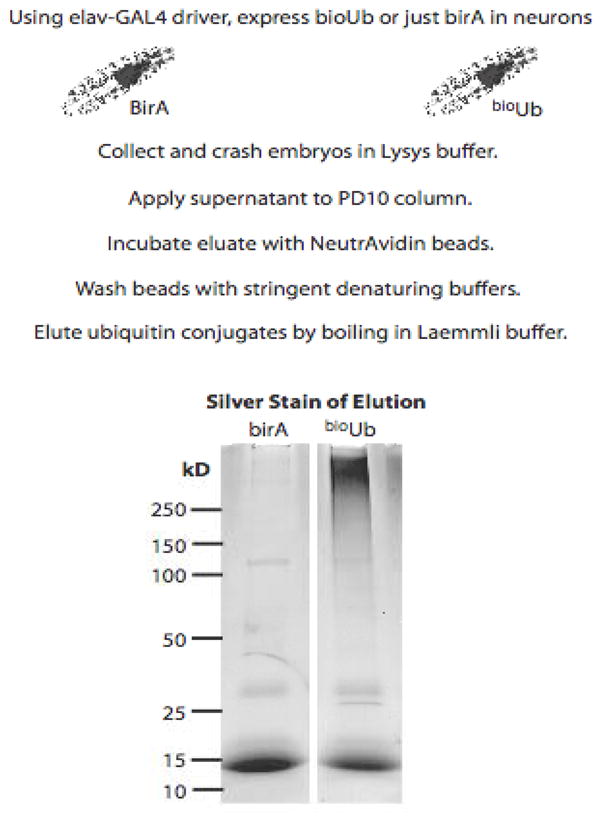
The strategy for analyzing neuron-specific protein ubiquitination in the fly. The neuronal elavGAL4 driver allows the expression of BirA (negative control) or Ub6-BirA in neurons using the GAL4/UAS system. Biotinylated Ub-conjugates are isolated and subjected to 1D SDS gel analysis followed by in-gel digestion and LC-MS/MS.
Figure 3.

Measurement of the abundance of Ub linkages by mass spectrometry. (A) The synthesis of stable isotope-labeled internal standard as exemplified by K48 linkages. (B) and (C) Representative AQUA analysis of K11 and K48 linkages, respectively.
2. Materials
2.1 Enrichment of neuron-specific ubiquitinated proteins in Drosophila
Fly lines expressing BirA (negative control) or biotin-Ub-BirA under the neuronal elavGAL4 driver (31) (see Note 1
Apple juice plates: Mix 72 g of agar with 1.5 L of cold water. Warm it up while stirring until it starts boiling. Add further 500 mL of water, 800 mL of apple juice, and 80 g of dextrose pre-dissolved in 40 mL of water. Stir for 1–2 min. Switch off the heater and keep stirring until the solution cools down to 60°C. Add 80 mL of 10% Nipagin solution (in 50% EtOH) and stir for 1 min. Pour into 90mm Petri dishes.
Yeast paste: from frozen yeast blocks, dilute if needed with a small amount of water.
PA 160/43 mesh: SAATIFIL polyamide, 160 microns mesh size (Fujifilm Sericol UK Ltd)
Fly collection cages: ~9 cm diameter, 16 cm high, blocked with PA 160/43 mesh at one end.
Embryo collection sieve made with the top half of a 50 mL tube, which is used to hold a disk of PA 160/43 mesh. The cap has been perforated to the whole diameter of the tube opening.
PBST: 0.1% Triton X-100 in PBS solution.
Bleach 50% in water.
Dounce tissue grinder 7 mL (Jencons).
N-ethylmaleimide (NEM, Sigma).
Embryo lysis buffer: 8 M Urea +1% SDS + 50 mM NEM in PBS.
25x P.I : 1 Protease inhibitors tablet (Roche Applied Science) in 2 mL Lysis/Binding buffer.
PD10 Desalting Columns (GE Healthcare); maximum sample volume 2.5 mL, recovery > 95%.
High Capacity NeutrAvidin-agarose beads (ThermoScientific); binding capacity ≥ 8 mg biotinylated BSA/ml of settled resin.
Binding buffer: 3 M Urea + 1 M NaCl + 0.25% SDS
Washing Buffer 1 (WB1): 8 M Urea + 0.25% SDS in PBS.
WB2: 6 M GdnHCl in PBS.
WB3: 6.4 M Urea + 1 M NaCl + 0.2% SDS in PBS.
WB4: 4 M Urea + 1 M NaCl + 10% isopropanol +10% EtOH + 0.2% SDS in PBS.
WB5: 8 M Urea + 1% SDS in PBS.
WB6: 2% SDS in PBS.
Elution buffer (4× Laemmli SDS loading buffer): 200 mM Tris, pH 6.8, 8% SDS, 40% glycerol, and 0.8 mg/mL of Bromophenol blue, with the addition of 100 mM DTT.
Minicolumn clarifying filters (Sartorious).
2.2 Analysis of the ubiquitinated proteins by mass spectrometry
Acetone (Sigma)
Iodoacetamide (IAA, Sigma)
SDS gel running apparatus
SDS poly acrylamide gels
Coomassie Blue G-250 staining buffer: 0.2% Brilliant Blue G250 (Sigma), 0.5% acetic acid (J.T. Baker) and 20% methanol (Sigma)
Razor blade, air incubator, and speedvac
Gel washing buffer: 50% acetonitrile (Sigma) and 50% 50 mM ammonium bicarbonate (Sigma)
Trypsin (Promega)
Extraction buffer: 5% formic acid (Fisher Scientific) and 50% acetonitrile (Sigma)
Trifluoroacetic acid (TFA) (B&J)
MS sample loading buffer: 6% acetic acid, 0.005% heptafluorobutyric acid, 5% acetonitrile, and 0.1% TFA
Methanol (Sigma)
100 μm i.d. × 12 cm fused-silica capillary C18 column (Magic C18AQ; particle size, 5 μm; pore size, 200 Å; Michrom Bioresources, Auburn, CA)
Buffer A: 0.4% acetic acid (J.T. Baker), 0.005% heptafluorobutyric acid (Sigma), and 5% acetonitrile (Sigma)
Buffer B: 0.4% acetic acid (J.T Baker), 0.005% heptafluorobutyric acid (Sigma), and 95% acetonitrile (Sigma)
LTQ-Orbitrap mass spectrometer (Thermo Finnigan, San Jose, CA) or other LC-MS/MS systems
2.3 Quantitative profiling of polyUb linkages in isolated ubiquitinated proteins, cells or tissues by mass spectrometry
The majority of materials are listed above with some additional items.
Cultured cells or tissues
Cell lysis buffer: 8 M Urea +1% SDS + 20 mM IAA in PBS
Glass beads (BioSpec Products, 0.5-mm diameter)
LTQ-Orbitrap mass spectrometer (Thermo Finnigan, San Jose, CA) or LC coupled with triple quadrupole (QqQ) mass spectrometers.
3. Methods
3.1 Enrichment of neuron-specific ubiquitinated proteins in Drosophila
Fly cages containing young flies from about 6 bottles are enclosed with Apple juice plates partially covered with yeast paste.
Allow flies to lay the embryos for 12 hr, change plates and let embryos age for further 9 hr.
Collect embryos gently using a brush, wash with PBST, water and dechorionate in 50% bleach solution for 3 min. Wash immediately with water and PBST, collect into tubes, flash-freeze and kept at −80°C until required.
Pre-wash 0.1 mL Neutravidin agarose beads with Binding buffer (see Note 2).
Crash 1–1.5 mL dechorionated embryos with 2.5 mL of Lysis buffer + 400 μL 25xPI (in Lysis buffer) using a 7 mL Dounce thissue homogeniser.
Centrifuge 1 min at 14 K at room temperature. Discard pellet.
Centrifuge the supernatant for 5 min at 14 K at 4°C. Repeat this step if needed.
Apply supernatant to a PD10 column preequilibrated with 25 mL Binding buffer (see Note 3).
Add 250 μL of 25xPI in Binding buffer to the eluate (3.5 mL, save 1 % for Western blotting analysis) and incubate with NeutrAvidin beads for 40 min at room temperature and 2 hr at 4°C.
Spin down the beads (2 min at 1000 rpm) and keep the supernatant as flow-through.
Wash beads in 15 mL tube with Washing buffers (12 mL each time): 3x WB1, 3x WB2, 1x WB3, 3x WB4, 1x WB1, 1x WB5 and 3x WB6 (see Note 4).
Boil beads for 5 min at 95°C with 40 μL Elution buffer.
Centrifuge-elute in minicolumn filter for 2 min at 14 K to recover Ub-conjugates (~65 μL) for mass spectrometry analysis.
Regular Western blotting analysis may be also performed to examine if one protein is ubiquitinated by comparing mass shift in the input with that in the eluted sample. The data may be able to differentiate monoubiquitination from polyubiquitination (polyUb chain on one ubiquitination site) and/or polymonoubiquitination (multiple monoubiquitin on different Lys sites).
3.2 Analysis of the ubiquitinated proteins by mass spectrometry
If the volume of eluted Ub-conjugates is too large (e.g. more than 100 μL), acetone precipitation may be performed to concentrate the sample. Add four times cold acetone (−20°C) of the sample volume, and then incubate at −20°C for 1 h. Centrifuge the precipitated proteins at 15,000 × g for 10 min. Dry and dissolve the pellet in Laemmli SDS loading buffer with 5 mM IAA and 8 M urea at room temperature. Do not heat the sample (see Note 2, 5 and 6).
Run the purified Ub-conjugate sample on 6–12% gradient gel and stain with Coomassie Blue.
Excise the gel lanes into multiple slices for in-gel digestion (32).
Cut the each gel slices into as small pieces as 1 mm3.
Wash the gel with gel washing buffer briefly and then dehydrate the gel with 100% acetonitrile.
Remove acetonitrile and completely dry the gel pieces in speedvac for 15 min.
Cover the gel pieces with trypsin solution on ice (12.5 ng/μl trypsin in 50 mM ammonium bicarbonate) to allow the gel rehydrate.
Incubate the sample at 37°C overnight and extract the digested peptides.
Dry the sample in speedvac and then resuspend into MS sample loading buffer.
Load the sample on LC-MS system (e.g. LTQ-Orbitrap) using an optimized protocol (33). Briefly, the peptides are loaded onto a C18 column and eluted during a 10–30% gradient. The eluted peptides are detected by Orbitrap (350–1500 m/z, 1,000,000 AGC target, 1,000 ms maximum ion time, resolution 60,000 fwhm) followed by ten data-dependent MS/MS scans in LTQ (2 m/z isolation width, 35% collision energy, 5,000 AGC target, 200 ms maximum ion time, see Note 7).
MS/MS spectra are searched against yeast database using the SEQUEST Sorcerer algorithm (version 2.0, SAGE-N) (34). Searching parameters included mass tolerance of precursor ions (±50 ppm) and product ion (±0.5 m/z), partially tryptic restriction, fixed modification of carboxyamidomethylated Cys (+57.0215 Da), dynamic modifications for oxidized Met (+15.9949), five maximal modification sites and three maximal missed cleavages (see Note 8).
The target-decoy strategy is used to evaluate false discovery rate (35, 36). In general, the protein false discovery rate is controlled less than 1% after filtering.
Validate the identified Ub-conjugates by western blotting (see Note 9, 10 and 11).
3.3 Quantitative profiling of polyUb linkages in isolated ubiquitinated proteins, cells or tissues by mass spectrometry
To quantify polyUb linkages on substrates, we employed the absolute quantification (AQUA) strategy (Fig. 3) (37), also termed LC-SRM/MRM (selected or multiple reaction monitoring). The first step is to generate stable-isotope labeled internal standards corresponding to ubiquitinated peptides by chemical synthesis or metabolic labeling. The stable-isotope labeled standards are added into any protein samples, such as purified Ub-conjugates, cell or tissue lysate. Trypsin digestion of ubiquitin moiety leads to the production of specific GG peptides from polyUb chains and modified substrates. The native peptides and internal standards are co-eluted during chromatography, but separated in a mass spectrometer. The MS instrument is set to SRM mode in which native peptides and internal standards are selected for fragmentation to generate product ions. Pairs of product ions are monitored and their ratios allowed for measurement of linkages in the original sample. The AQUA strategy circumvents antibody requirement to detect targeted proteins in complex mixtures. As it is not difficult to find unique peptides for any protein, this strategy can be extended to quantify nearly all proteins. The method is modified from a previously reported protocol (38, 39), listed in detail below.
Prepare a number of internal standards included labeled, synthetic GG-linked Ub peptides for absolute quantification (6), and metabolically labeled HEK293 cells (Lys +8.0142 Da and Arg +10.0083 Da), yeast or mouse tissues (Lys +6.0201 Da) for relative quantification (40) (see Note 12 and 13).
The synthetic peptides are used to optimize the LC-SRM conditions, such as elution gradient to determine retention time, fragmentation condition, and the selection of product ions (Table 1, see Note 14).
Labeled cell lysate (20 μg) is prepared in the Lysis buffer, spiked into unlabeled lysates mostly at a 1:1 ratio, and resolved on a 1D SDS gel. The gel region above 80 kDa containing the vast majority of polyUb species is used for analysis. If ultrahigh detection sensitivity is achieved in an optimized LC-SRM setting, the entire gel lane could be excised into a single gel band.
If labeled peptides are used as internal standards, the peptides are added in during trypsin digestion instead of the step of gel running.
Perform in-gel trypsin digestion to produce pairs of light and heavy peptides.
Analyze peptide pairs by reverse phase LC followed by MS, in which peptide ion pairs of interest are selected for fragmentation and quantified by intensity ratio of coeluting, related product ion pairs. The instrument is operated in the SRM or MRM mode.
The LC-SRM analysis can be performed on a number of instruments, such as a hybrid LTQ-Orbitrap mass spectrometer (Thermo Finnigan, San Jose, CA) or triple quadrupole (QqQ) mass spectrometers (see Note 15).
Table 1.
Ubiquitin quantification by the LC-SRM analysis
| Pepetide Names | Peptide Sequences | Synthetic peptides
|
SILAC peptides
|
SILAM peptides
|
Precursor Ions (m/z)
|
Product Ion (m/z)
|
RT (min) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Labeled AA | Mass Shift (Da) | Labeled AA | Mass Shift (Da) | Labeled AA | Mass Shift (Da) | z | Native | Synthetic | SILAC | SILAM | Ion Name | Native | Synthetic | SILAC | SILAM | |||
| Ub (unmodified, TLS) | TLSDYNIQK | L2 | 7.0606 | K9 | 8.0142 | K9 | 6.0201 | 2 | 541.2798 | 544.8101 | 545.2869 | 544.2899 | b1-2 | 215.1390 | 222.1996 | 215.1390 | 215.1390 | 13.1 |
| Ub (unmodified, TLS) | TLSDYNIQK | L2 | 7.0606 | K9 | 8.0142 | K9 | 6.0201 | 2 | 541.2798 | 544.8101 | 545.2869 | 544.2899 | y1-7 | 867.4207 | 867.4207 | 875.4349 | 873.4408 | 13.1 |
| Ub (linear GG tag) | (GG)MQIFVK | I3 | 7.0606 | K6 | 8.0142 | K6 | 6.0201 | 2 | 440.2415 | 443.7718 | 444.2486 | 443.2515 | b1-7 | 733.3702 | 740.4308 | 733.3702 | 733.3702 | 19.7 |
| Ub (linear, oxidative) | (GG)M*QIFVK | I3 | 7.0606 | K6 | 8.0142 | K6 | 6.0201 | 2 | 448.2389 | 451.7692 | 452.2460 | 451.2490 | b1-7 | 749.3651 | 756.4257 | 749.3651 | 749.3651 | 14.4 |
| Ub (K6) | MQIFVK(GG)TLTGK | L8 | 7.0606 | K6, K11 | 16.0284 | K6, K11 | 12.0403 | 2 | 690.3894 | 693.9197 | 698.4036 | 696.4095 | y1-8 | 1007.5884 | 1014.6490 | 1023.6168 | 1019.6287 | 21.7 |
| Ub (K6 oxidative) | M*QIFVK(GG)TLTGK | L8 | 7.0606 | K6, K11 | 16.0284 | K6, K11 | 12.0403 | 2 | 698.3869 | 701.9172 | 706.4011 | 704.4070 | y1-8 | 1007.5884 | 1014.6490 | 1023.6168 | 1019.6287 | 19.4 |
| Ub (myc tagged K6) | LISEEDLGMQIFVK(GG)TLTGK | L16 | 7.0606 | K14, K19 | 16.0284 | K14, K19 | 12.0403 | 3 | 746.0680 | 748.4215 | 751.4108 | 750.0814 | y2-17 | 1005.5142 | 1009.0446 | 1013.5284 | 1011.5344 | 28.9 |
| Ub (myc-K6 oxidative) | LISEEDLGM*QIFVK(GG)TLTGK | L16 | 7.0606 | K14, K19 | 16.0284 | K14, K19 | 12.0403 | 3 | 751.4011 | 753.7546 | 756.7439 | 755.4145 | y2-17 | 1013.5117 | 1017.0420 | 1021.5259 | 1019.5318 | 27.3 |
| Ub (K11, yeast) | TLTGK(GG)TITLEVESSDTIDNVK | V20 | 6.0520 | K5, K21 | 16.0284 | K5, K21 | 12.0403 | 3 | 793.4148 | 795.4321 | 798.7576 | 797.4282 | b2-20 | 1116.5657 | 1119.5917 | 1120.5728 | 1119.5758 | 22.1 |
| Ub (K11, human) | TLTGK(GG)TITLEVEPSDTIENVK | V20 | 6.0520 | K5, K21 | 16.0284 | K5, K21 | 12.0403 | 3 | 801.4269 | 803.4442 | 806.7697 | 805.4403 | y1-9 | 1002.5102 | 1008.5622 | 1010.5244 | 1008.5303 | 21.7 |
| Ub (K27, yeast) | TITLEVESSDTIDNVK(GG)SK | V15 | 6.0520 | K16, K18 | 16.0284 | K16, K18 | 12.0403 | 3 | 698.3585 | 700.3758 | 703.7013 | 702.3719 | y2-16 | 939.9682 | 942.9942 | 947.9824 | 945.9883 | 20.1 |
| Ub (K27, human) | TITLEVEPSDTIENVK(GG)AK | V15 | 6.0520 | K16, K18 | 16.0284 | K16, K18 | 12.0403 | 3 | 701.0390 | 703.0563 | 706.3818 | 705.0524 | y2-16 | 943.9889 | 947.0149 | 952.0031 | 950.0090 | 20.5 |
| Ub (K29, yeast) | SK(GG)IQDK | GG | 6.0520 | K2, K6 | 16.0284 | K2, K6 | 12.0403 | 2 | 416.7298 | 419.7558 | 424.7440 | 422.7499 | y1-4 | 503.2824 | 503.2824 | 511.2966 | 509.3025 | 7.4 |
| Ub (K29, human) | AK(GG)IQDK | GG | 6.0520 | K2, K6 | 16.0284 | K2, K6 | 12.0403 | 2 | 408.7323 | 411.7583 | 416.7465 | 414.7525 | y1-4 | 503.2824 | 503.2824 | 511.2966 | 509.3025 | 7.2 |
| Ub (K33) | IQDK(GG)EGIPPDQQR | P9 | 6.0520 | K4, R13 | 18.0225 | K4 | 6.0201 | 3 | 546.6129 | 548.6302 | 552.6204 | 548.6196 | y2-6 | 370.6879 | 373.7139 | 375.6921 | 370.6879 | 11.3 |
| Ub (K48) | LIFAGK(GG)QLEDGR | L8 | 7.0606 | K6, R12 | 18.0225 | K6 | 6.0201 | 2 | 730.8964 | 734.4267 | 739.9077 | 733.9065 | y2-10 | 617.8124 | 621.3427 | 626.8236 | 620.8224 | 19.5 |
| Ub (K63) | TLSDYNIQK(GG)ESTLHLVLR | L17 | 7.0606 | K9, R18 | 18.0225 | K9 | 6.0201 | 3 | 748.7376 | 751.0911 | 754.7451 | 750.7443 | y2-16 | 1015.5369 | 1019.0672 | 1024.5481 | 1018.5470 | 22.7 |
Labeled AA: the selected residue for stable isotopic labeling (e.g. L8, the eighth leucine residue); mass shift: the mass change due to isotopic labeling; synthetic peptides: chemically labeled during chemical reaction; SILAC peptides: metabolically labeled in SILAC experiments; SILAM peptides: metabolically labeled in SILAC mice; z: charge state; native: endogenous peptide ion without labeling; precursor ions: monoisotopic m/z values of native and labeled peptides; product ions, monoisotopic m/z values of monitored product ions in SRM; Ion name: the monitored ions in SRM (e.g. b1-2 is single charged b2 ion); RT: experimental retention time in one setting of reverse liquid LC-SRM (the retention time may change in another experimental setting and needs to be defined when changing the LC column); The total Ub level was calculated by adding the amount of TLS peptide to that of the GG-linked K63 peptide.
Acknowledgments
This work was partially supported by the National Institutes of Health grant (RR025822), and the American Cancer Society grant (RSG-09-181). UM is an Ikerbasque Research Professor.
Footnotes
The approach is based on the in vivo biotinylation of ubiquitin expressed uniquely in the nervous system using the GAL4/UAS system. We take advantage of the processing activity of endogenous deubiquitinating enzymes to digest a precursor containing both the tagged ubiquitin and the enzyme responsible for its biotinylation, the bacterial BirA enzyme. As a wide range of drivers for different tissue expression are available in the fly system, this strategy could be used to isolate ubiquitin conjugates from other tissues, as well as from different developmental stages. It is likely to be also applicable to other model organisms.
As solubilized urea is in equilibrium with ammonium cyanate that leads to carbamylation of amine groups in proteins, and the reaction is accelerated by heating, we generally use fresh urea solutions and do not heat urea solutions.
We are using the PD10 columns to eliminate free biotin, but also as a buffer exchange step. We equilibrate the column with Binding buffer, so the sample is exchanged into Binding buffer, ready to incubate with the beads.
These different wash buffers are used to almost eliminate background proteins under various denaturing conditions.
Iodoacetamide (IAA) or N-ethylmaleimide (NEM) can be used as a Cys-alkylation reagent to block most of DUB activities. At high temperatures (e.g. heating in SDS gel loading buffer), IAA modifies a fraction of Lys residues twice to form a tag of 114.0429 Da, the same mass of a GG tag generated by tryptic digestion of ubiquitin (41). Even chloroacetamide (41) might produce this artifact tag at high temperature (6), but NEM does not. At low temperature (i.e. room temperature or lower) or low dosage, this side reaction of IAA is essentially eliminated (6). As the most abundant ubiquitinated peptide in cells (K48-GG Ub peptide) could be distinguished from its iodoacetamide-modified artifact based on LC retention time and a specific neutral loss in MS/MS pattern, detecting if such an artifact peptide exists in samples would be used for quality control during analysis. During the experiment, although NEM/IAA was only added in the Lysis buffer, it could be added in all except the elution buffer to inhibit DUB activities.
Sample loss may occur during the concentration step by acetone precipitation, in particular for highly diluted samples. It is recommended to perform a testing experiment to examine the recovery of Ub-conjugates. Alternatively, one may minimize the volume of elution buffer and collect the eluent into multiple fractions, some of which will have higher concentration of Ub-conjugates and can be directly used for SDS gel electrophoresis without the concentration step.
The amount of the sample loaded on the column is dependent on the LC system used. In general, we load peptides digested from 1 μg protein onto a 12 cm × 70 μm ID column. Increasing gradient elution time may increase the number of identified proteins, but a plateau can be reached at a certain point due to broadening of elution peaks. More details may be found in a technical paper (33).
Although the Orbitrap allows the acquisition of high resolution data with mass accuracy within a few ppm or even subppm dependent on the setting and intensity of ion signal, wide window (50 ppm) is used during the search and a much narrower mass window is used later during data filtering to remove false positives. However, the narrow mass window may be applied during the search step, and then cannot be used for filtering (33).
Proteins that are not modified by ubiquitin are usually co-purified during the purification of Ub-conjugates, raising a main challenge on how to remove these false positives. The first effective approach is to reduce the contaminants during purification by introducing highly stringent buffers (e.g. 8 M urea, or 6 M GdnHCl); denaturing conditions are useful for not only minimizing contaminants but also inhibiting DUB activities. Only two types of tags (6xHis and biotin) on ubiquitin are suitable for denatured conditions (4, 42). Even under such stringent conditions, the contaminants may still contribute up to ~50% of the identified ubiquitinated proteome, since many contaminants exist in a minute amount but are still detectable by highly sensitive mass spectrometry (43). Moreover, the status of protein ubiquitination may be verified by traditional western blotting, virtual western blotting images reconstructed based on 1D gel and LC-MS/MS (43), and the ubiquitinated sites. The caveats of analyzing ubiquitinated lysine residues are discussed in another review paper (44).
Quantitative comparison of protein samples from negative control (e.g. cells expressing untagged ubiquitin) with cells expressing tagged ubiquitin is an alternative method to differentiate contaminants and Ub-conjugates. While the contaminants are proposed to be isolated from both sources at equal efficiency, the real Ub-conjugates are only enriched from cells carrying the tagged ubiquitin. This strategy has been successfully used for mapping SUMOylated proteins (45).
Development of specific antibodies to GG-tagged ubiquitinated peptides provides an independent method for enriching ubiquitinated species (46). This method allows the enrichment of ubiquitinated peptides instead of Ub-conjugates. The identification GG peptides validate the status of protein ubiquitination, although false discoveries may also exist in database search (47).
Although internal standard peptides work in the AQUA analysis, they do not represent true standards for proteins, because digestion efficiency is not accounted for. Labeled proteins are better standards in this type of study.
Metabolic labeling is performed using the SILAC (stable isotope labeling of amino acid in cell culture) protocol in cells (e.g. mammalian culture and yeast). To prevent significant heavy isotope-labeled Arg-Pro conversion, extra proline is often implemented. More recently, the SILAC strategy has also been applied to other model systems, including fly (48) and mouse (49). It will thus be possible to perform a similar analysis of ubiquitinated proteins in higher organisms.
Optimization of the LC-SRM conditions is crucial to improve the detection sensitivity of peptides of interest. As peptides ionization efficiency varies vastly and reliable detection is often influenced by the co-eluting peptides due to ion suppression, it is recommended to carry out pilot experiments to characterize the sensitivity.
The QqQ instrument has larger dynamic range (5–6 orders of magnitude) than the LTQ mass spectrometry (~3 orders of magnitude), but the detection sensitivity may vary upon the lab settings. A detailed comparison of the performance of these instruments has been recently reported (50).
References
- 1.Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005;6:79–87. doi: 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- 2.Ciechanover A, Ben-Saadon R. N-terminal ubiquitination: more protein substrates join in. Trends in cell biology. 2004;14:103–106. doi: 10.1016/j.tcb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Cadwell K, Coscoy L. Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science (New York, NY. 2005;309:127–130. doi: 10.1126/science.1110340. [DOI] [PubMed] [Google Scholar]
- 4.Peng J, Schwartz D, Elias JE, et al. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21:921–926. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- 5.Kirisako T, Kamei K, Murata S, et al. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006;25:4877–4887. doi: 10.1038/sj.emboj.7601360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu P, Duong DM, Seyfried NT, et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varadan R, Assfalg M, Haririnia A, et al. Solution conformation of Lys63-linked di-ubiqutin chain provides clues to functional diversity of polyubiquitin signaling. The Journal of biological chemistry. 2004;279:7055–7063. doi: 10.1074/jbc.M309184200. [DOI] [PubMed] [Google Scholar]
- 8.Virdee S, Ye Y, Nguyen DP, et al. Engineered diubiquitin synthesis reveals Lys29-isopeptide specificity of an OTU deubiquitinase. Nature chemical biology. 2010;6:750–757. doi: 10.1038/nchembio.426. [DOI] [PubMed] [Google Scholar]
- 9.Bremm A, Freund SM, Komander D. Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nature structural & molecular biology. 2010;17:939–947. doi: 10.1038/nsmb.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol. 2004;8:610–616. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains - from structures to functions. Nat Rev Mol Cell Biol. 2009;10:659–671. doi: 10.1038/nrm2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40:427–446. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- 13.Goldberg AL. Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem Soc Trans. 2007;35:12–17. doi: 10.1042/BST0350012. [DOI] [PubMed] [Google Scholar]
- 14.Bandopadhyay R, de Belleroche J. Pathogenesis of Parkinson’s disease: emerging role of molecular chaperones. Trends Mol Med. 2010;16:27–36. doi: 10.1016/j.molmed.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 15.Friedman JS, Ray JW, Waseem N, et al. Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa. Am J Hum Genet. 2009;84:792–800. doi: 10.1016/j.ajhg.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yi JJ, Ehlers MD. Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacological reviews. 2007;59:14–39. doi: 10.1124/pr.59.1.4. [DOI] [PubMed] [Google Scholar]
- 17.Peng J, Gygi SP. Proteomics: the move to mixtures. J Mass Spectrom. 2001;36:1083–1091. doi: 10.1002/jms.229. [DOI] [PubMed] [Google Scholar]
- 18.Cravatt BF, Simon GM, Yates JR., 3rd The biological impact of mass-spectrometry-based proteomics. Nature. 2007;450:991–1000. doi: 10.1038/nature06525. [DOI] [PubMed] [Google Scholar]
- 19.Gstaiger M, Aebersold R. Applying mass spectrometry-based proteomics to genetics, genomics and network biology. Nat Rev Genet. 2009;10:617–627. doi: 10.1038/nrg2633. [DOI] [PubMed] [Google Scholar]
- 20.Choudhary C, Mann M. Decoding signalling networks by mass spectrometry-based proteomics. Nat Rev Mol Cell Biol. 2010;11:427–439. doi: 10.1038/nrm2900. [DOI] [PubMed] [Google Scholar]
- 21.Matsumoto M, Hatakeyama S, Oyamada K, et al. Large-scale analysis of the human ubiquitin-related proteome. Proteomics. 2005;5:4145–4151. doi: 10.1002/pmic.200401280. [DOI] [PubMed] [Google Scholar]
- 22.Vasilescu J, Smith JC, Ethier M, Figeys D. Proteomic analysis of ubiquitinated proteins from human MCF-7 breast cancer cells by immunoaffinity purification and mass spectrometry. Journal of proteome research. 2005;4:2192–2200. doi: 10.1021/pr050265i. [DOI] [PubMed] [Google Scholar]
- 23.Layfield R, Tooth D, Landon M, et al. Purification of poly-ubiquitinated proteins by S5a-affinity chromatography. Proteomics. 2001;1:773–777. doi: 10.1002/1615-9861(200106)1:6<773::AID-PROT773>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 24.Weekes J, Morrison K, Mullen A, et al. Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics. 2003;3:208–216. doi: 10.1002/pmic.200390029. [DOI] [PubMed] [Google Scholar]
- 25.Maor R, Jones A, Nuhse TS, et al. Multidimensional protein identification technology (MudPIT) analysis of ubiquitinated proteins in plants. Mol Cell Proteomics. 2007;6:601–610. doi: 10.1074/mcp.M600408-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Bennett EJ, Shaler TA, Woodman B, et al. Global changes to the ubiquitin system in Huntington’s disease. Nature. 2007;448:704–708. doi: 10.1038/nature06022. [DOI] [PubMed] [Google Scholar]
- 27.Kirkpatrick DS, Denison C, Gygi SP. Weighing in on ubiquitin: the expanding role of mass-spectrometry-based proteomics. Nature cell biology. 2005;7:750–757. doi: 10.1038/ncb0805-750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu P, Peng J. Dissecting the ubiquitin pathway by mass spectrometry. Biochimica et biophysica acta. 2006;1764:1940–1947. doi: 10.1016/j.bbapap.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Guerrero C, Kaiser P, Huang L. Proteomics of proteasome complexes and ubiquitinated proteins. Expert review of proteomics. 2007;4:649–665. doi: 10.1586/14789450.4.5.649. [DOI] [PubMed] [Google Scholar]
- 30.Meierhofer D, Wang X, Huang L, Kaiser P. Quantitative analysis of global ubiquitination in HeLa cells by mass spectrometry. J Proteome Res. 2008;7:4566–4576. doi: 10.1021/pr800468j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Franco M, Seyfried NT, Brand AH, et al. A novel strategy to isolate ubiquitin conjugates reveals wide role of ubiquitination during neural development. Mol Cell Proteomics. 2010 doi: 10.1074/mcp.M110.002188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 33.Xu P, Duong DM, Peng J. Systematical optimization of reverse-phase chromatography for shotgun proteomics. Journal of proteome research. 2009;8:3944–3950. doi: 10.1021/pr900251d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eng J, McCormack AL, Yates JR., 3rd An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 35.Peng J, Elias JE, Thoreen CC, et al. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res. 2003;2:43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 36.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 37.Gerber SA, Rush J, Stemman O, et al. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:6940–6945. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirkpatrick DS, Hathaway NA, Hanna J, et al. Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat Cell Biol. 2006;8:700–710. doi: 10.1038/ncb1436. [DOI] [PubMed] [Google Scholar]
- 39.Xu P, Cheng D, Duong DM, et al. A proteomic strategy for quantifying polyubiquitin chain topologies. Israel J Chem. 2006;46:171–182. [Google Scholar]
- 40.Dammer EB, Na CH, Xu P, et al. Polyubiquitin linkage profiles in three models of proteolytic stress suggest etiology of Alzheimer disease. The Journal of biological chemistry. 2011 doi: 10.1074/jbc.M110.149633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nielsen ML, Vermeulen M, Bonaldi T, et al. Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat Methods. 2008;5:459–460. doi: 10.1038/nmeth0608-459. [DOI] [PubMed] [Google Scholar]
- 42.Tagwerker C, Flick K, Cui M, et al. A Tandem Affinity Tag for Two-step Purification under Fully Denaturing Conditions: Application in Ubiquitin Profiling and Protein Complex Identification Combined with in vivo Cross-Linking. Mol Cell Proteomics. 2006;5:737–748. doi: 10.1074/mcp.M500368-MCP200. [DOI] [PubMed] [Google Scholar]
- 43.Seyfried NT, Xu P, Duong DM, et al. Systematic approach for validating the ubiquitinated proteome. Anal Chem. 2008;80:4161–4169. doi: 10.1021/ac702516a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peng J. Evaluation of proteomic strategies for analyzing ubiquitinated proteins. BMB Rep. 2008;41:177–183. doi: 10.5483/bmbrep.2008.41.3.177. [DOI] [PubMed] [Google Scholar]
- 45.Golebiowski F, Matic I, Tatham MH, et al. System-wide changes to SUMO modifications in response to heat shock. Sci Signal. 2009;2:ra24. doi: 10.1126/scisignal.2000282. [DOI] [PubMed] [Google Scholar]
- 46.Xu G, Paige JS, Jaffrey SR. Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nature biotechnology. 2010;28:868–873. doi: 10.1038/nbt.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi Y, Xu P, Qin J. Ubiquitinated proteome: Ready for global? Mol Cell Proteomics. 2011 doi: 10.1074/mcp.R110.006882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sury MD, Chen JX, Selbach M. The SILAC fly allows for accurate protein quantification in vivo. Mol Cell Proteomics. 2010;9:2173–2183. doi: 10.1074/mcp.M110.000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kruger M, Moser M, Ussar S, et al. SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell. 2008;134:353–364. doi: 10.1016/j.cell.2008.05.033. [DOI] [PubMed] [Google Scholar]
- 50.Phu L, Izrael-Tomasevic A, Matsumoto ML, et al. Improved quantitative mass spectrometry methods for characterizing complex ubiquitin signals. Mol Cell Proteomics. 2010 doi: 10.1074/mcp.M110.003756. [DOI] [PMC free article] [PubMed] [Google Scholar]