

Abstract
Glucocorticoids (GC) are implicated in the development of metabolic syndrome, and patients with GC excess share many clinical features, such as central obesity and glucose intolerance. In patients with obesity or type 2 diabetes, systemic GC concentrations seem to be invariably normal. Tissue GC concentrations determined by the hypothalamic-pituitary-adrenal (HPA) axis and local cortisol (corticosterone in mice) regeneration from cortisone (11-dehydrocorticosterone in mice) by the 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) enzyme, principally expressed in the liver. Transgenic mice have demonstrated the importance of 11β-HSD1 in mediating aspects of the metabolic syndrome, as well as HPA axis control. In order to address the primacy of hepatic 11β-HSD1 in regulating metabolism and the HPA axis, we have generated liver-specific 11β-HSD1 knockout (LKO) mice, assessed biomarkers of GC metabolism, and examined responses to high-fat feeding. LKO mice were able to regenerate cortisol from cortisone to 40% of control and had no discernible difference in a urinary metabolite marker of 11β-HSD1 activity. Although circulating corticosterone was unaltered, adrenal size was increased, indicative of chronic HPA stimulation. There was a mild improvement in glucose tolerance but with insulin sensitivity largely unaffected. Adiposity and body weight were unaffected as were aspects of hepatic lipid homeostasis, triglyceride accumulation, and serum lipids. Additionally, no changes in the expression of genes involved in glucose or lipid homeostasis were observed. Liver-specific deletion of 11β-HSD1 reduces corticosterone regeneration and may be important for setting aspects of HPA axis tone, without impacting upon urinary steroid metabolite profile. These discordant data have significant implications for the use of these biomarkers of 11β-HSD1 activity in clinical studies. The paucity of metabolic abnormalities in LKO points to important compensatory effects by HPA activation and to a crucial role of extrahepatic 11β-HSD1 expression, highlighting the contribution of cross talk between GC target tissues in determining metabolic phenotype.
The regulation of metabolism and energy homeostasis is partly controlled through blood glucocorticoid (GC) levels, such that in times of starvation or stress, GC mobilize adipose fat stores and amino acids from muscle to support hepatic gluconeogenesis to maintain energy and glucose supplies (1, 2). GC have been implicated in the development of metabolic syndrome (including obesity, type 2 diabetes, and dyslipidemia) (3), and GC excess (Cushing's syndrome), whether exogenous or endogenous in cause, offers a paradigm of metabolic syndrome, because patients share many clinical features such as central obesity, glucose intolerance, and premature cardiovascular mortality (4). However, in patients with obesity or type 2 diabetes, systemic GC concentrations are invariably normal (5).
GC concentrations are determined by the activity of the hypothalamic-pituitary-adrenal (HPA) axis and through peripheral cortisol production by the 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) enzyme (6). 11β-HSD1 is a reduced nicotinamide adenine dinucleotide phosphate (NADPH)-dependent endoplasmic reticulum luminal enzyme that regenerates cortisol (corticosterone in mice) from inactive cortisone [11-dehydrocorticosterone (11-DHC) in mice], thereby increasing tissue concentrations independently of circulating levels (6). Within the endoplasmic reticulum lumen, 11β-HSD1 is dependent upon the activity of the enzyme hexose-6-phosphate dehydrogenase (H6PDH) to generate a high NADPH concentration, such that in H6PDH knockout (KO) mice, the loss of NADPH generation results in 11β-HSD1 activity switching from GC regeneration to GC inactivation (cortisol or corticosterone to cortisone or 11-DHC) (7). 11β-HSD1 is expressed in many mouse tissues, most notably the liver, adipose, muscle, adrenal, and pancreas. However, the principal site of expression is considered to be the liver, with 10-fold greater expression and enzyme activity per unit mass than any other (6).
Recently, in vitro, in vivo, and clinical data have collectively demonstrated the importance of local cortisol generation via 11β-HSD1 in hepatic and adipose tissue in mediating aspects of the metabolic syndrome (3, 6). A number of rodent transgenic models have highlighted the potential benefits to 11β-HSD1 inhibition: global 11β-HSD1KO mice resist obesity on high-fat diet, have improved hepatic insulin sensitivity, an improved lipid profile, and healthier fat distribution and function (8–11). H6PDHKO mice, in which GC sensitivity is greatly diminished, also display improved glucose homeostasis and resist fat accumulation on a high-fat diet (12, 13). Conversely, 11β-HSD1 overexpression in adipose tissue completely recapitulates the metabolic syndrome with obesity, insulin resistance, hypertension, and dyslipidemia. In this model, delivery of active GC into the liver from the portal vein is markedly increased (14, 15). Finally, 11β-HSD1 liver overexpression produces mild hepatic insulin resistance without obesity (16). Together these models provide compelling evidence for targeted therapeutic inhibition of the 11β-HSD1 enzyme as a means to ameliorate aspects of the metabolic syndrome in humans.
In this regard, a number of pharmaceutical companies have demonstrated proof of concept in preclinical trials using 11β-HSD1 inhibitors, with weight loss, improved glucose tolerance, and beneficial lipid profiles in diet-induced obese and diabetic mouse models (17–20). Human clinical data using the nonselective 11β-HSD1 inhibitor carbenoxolone, and more selective inhibitors in recent phase IIb trials, demonstrate weight loss and reduced blood glucose in patients with type 2 diabetes poorly controlled by metformin (21, 22).
The 11β-HSD1KO, endorsed by mutations in 11β-HSD1 and H6PDH enzymes in man, illustrates the close interplay between cortisol metabolism and normal HPA axis function. 11β-HSD1KO mice have abnormal HPA axis control and enhanced circadian HPA drive (23). However, crossing the 11β-HSD1 liver-specific-overexpressing mouse with the 11β-HSD1KO appears to recover normal HPA function, suggesting that hepatic GC regeneration is important in setting HPA tone (24). Furthermore, H6PDHKO mice also have enhanced GC clearance and display activation of the HPA axis (25). Humans with deficiency of 11β-HSD1 and H6PDH, termed cortisone reductase deficiency (CRD) and “apparent” CRD respectively, present with hyperandrogenic disorders, with a diagnosis of premature pubarche in childhood and PCOS in adult females (26, 27). The increased cortisol clearance rate in both CRD and apparent CRD patients is offset by an ACTH-mediated HPA axis activation and adrenal cortisol secretion, concurrent with excess adrenal androgen production.
To address the primacy of hepatic 11β-HSD1 in regulating metabolic and HPA consequences of altered GC metabolism, we have generated liver-specific 11β-HSD1KO (LKO) mice, assessed biomarkers of GC metabolism, and subjected them to a high-fat diet challenge.
Materials and Methods
Animal generation, maintenance, and dietary manipulation
Previously, a conditional HSD11B1 allele was generated by flanking exon 5 with LoxP sites and from this global 11β-HSD1KO derived (28). To generate LKO, floxed homozygous HSD11B1 mice on a mixed C57BL/6J/129SvJ background were crossed with Albumin-Cre transgenic mice (on a C57BL/6J background, targeting Cre expression to hepatocytes) (29). This generated mice devoid of 11β-HSD1 activity in the liver. Genotyping PCR was carried out on ear clip DNA using gene specific primers (5′-3′) P1-GGGAGCTTGCTTACAGCATC, P2-CATTCTCAAGGTAGATTGAACTCTG, and P3-TCCATGCAATCAACTTCTCG. P1+P2 give a 138-bp product for a wild-type allele and a 172-bp product for an allele containing a 3′ LoxP site. P1 and P3 give a 279-bp product for an allele after Cre recombination and exon 5 removal.
All studies were conducted on male LKO, HSD1KO, and control mice group-housed under controlled temperature (21–23 C) and light (12-h light, 12-h dark cycle; lights on at 0700 h). Mice had ad libitum access to water and for 18 wk from 6 wk of age were fed either control diet (11% kcal fat-D12328; Research Diets, New Brunswick, NJ) or high-fat diet (58% kcal fat-D12331). Animal procedures were approved under the British Home Office Animals (Scientific Procedures) Act 1986 and through the Local Animal Ethics Committee.
Metabolic parameters
Unstressed corticosterone and insulin were measured toward the end of the study by tail bleeds into lithium-heparin tubes (Sarstedt, Leicester, UK) between 0800 and 0900 h. Blood was centrifuged at 6000 × g for 10 min and plasma separated and frozen in liquid N2. Corticosterone was measured using a corticosterone immunoassay kit (Cambridge Biosciences, Cambridge, UK). Insulin was measured using the Ultra Sensitive Mouse Insulin ELISA kit (Crystal Chem, Inc., Downers Grove, IL). Circulating glucose was measured by tail-nick using a glucometer (Accu-Chek Aviva; Roche, Burgess Hill, UK). Intraperitoneal glucose tolerance tests (GTT) and insulin tolerance tests (ITT) were performed to assess changes in glucose and insulin sensitivity. For GTT, groups were fasted for 16 h, and fasted for 4 h for ITT, a basal blood sample taken, followed by IP injection of either glucose (1 g/kg; Abbott, Abbott Park, IL) or insulin (0.75 U/kg; Sigma, St. Louis, MO). Blood samples were taken by tail-nick at 15, 30, and 60 for ITT and additional 90 and 120 min for GTT after the injection and blood glucose measured by glucometer. Triglyceride, high-density lipoprotein (HDL) and low-density lipoprotein (LDL) cholesterol, and nonesterified free fatty acids (FFA) were measured according to the manufacturer's instructions in plasma from terminal bleeds (BioVision, Inc., Mountain View, CA).
Liver morphology and lipid content levels
Livers were fixed in 10% neutral buffered formalin, paraffin embedded, and cut into 10 μm sections and stained with hematoxylin and eosin or with Oil Red O (Sigma) to identify neutral lipids, cholesterol, and fatty acids. Images shown are at ×40 magnification. To measure hepatic triglyceride content, 100 mg of liver tissue was homogenized in 1 ml of 5% Nonidet P-40 in water, samples were heated to 80–100 C in a water bath for 2–5 min, and then allowed to cool to room temperature. The heating step was repeated to solubilize all triglyceride, upon which samples were centrifuged for 2 min to remove any insoluble material. Samples were diluted 10-fold with dH2O before being subjected to a triglyceride assay, according to the manufacturer's instructions (BioVision, Inc.).
Cortisone acetate challenge and cortisol generation after gavage
At 8–12 wk of age, LKO, HSD1KO, and control mice were administered a cortisone acetate suspension in water (0.25 mg in 200 μl) by gavage. After 15 min, a 150 μl blood sample was taken by tail venesection, and cortisone metabolism was assessed by gas chromatography-mass spectrometry (GC/MS) analysis. Briefly, 20 ng of d4-cortisol (internal standard) and 2 ml distilled water were added to 40 μl of mouse serum. The solution was heated to 55 C for 20 min to denature proteins then cooled, steroids were extracted using C18 sep-pak columns (Waters Chromatography, Milford, MA), and eluted in 4 ml of methanol. Solvent was evaporated under nitrogen at 55 C. Three drops of a 2% methyoxine-pyridine solution were added to each sample and heated to 55 C for 1 h to derivitise ketone groups. The solution was evaporated under nitrogen at 55 C and 50 μl of N-Trimethylsilylimidazole added. Samples were then heated at 120 C overnight to derivatize hydroxyl groups. Two aliquots of a standard solution containing cortisol, cortisone, d4-cortisol, tetra-hydrocortisone, and α-cortolone were also derivatized. Steroids were extracted into cyclohexane, vortexed, 2 ml distilled water added, centrifuged for 5 min at 1500 rpm, and the bottom layer removed. A further 2 ml of distilled water were added, centrifuged, the top layer removed, and cyclohexane evaporated. Standards and samples were dissolved in 50 μl cyclohexane, injected into the GC/MS, and analyzed as previously described (26). Cortisol, cortisone, d4-cortisol, tetra-hydrocortisone, and α-cortolone were quantified against the standard solution using d4F as the internal standard.
Western blot analysis
Liver microsomes were prepared from 8- to 10-wk-old mice by differential centrifugation techniques as described previously (7); 30 μg of microsomal protein were then run on 11% SDS-PAGE. After electrophoresis, proteins were transferred onto Immobilon polyvinyl difluoride membranes (Millipore, Bedford, MA) at 100 V for 1 h, in a buffer containing 25 mm Tris, 200 mm glycine, and 20% (vol/vol) methanol. The membrane was blocked in PBS containing 0.1% (vol/vol) Tween 20 and 20% (wt/vol) skimmed milk powder, washed, and then incubated with anti-11β-HSD1 (generated in house) sheep polyclonal antibody, diluted 1/1000 overnight at 4 C in PBS containing 0.05% (vol/vol) Tween 20. The membrane was then washed and incubated with goat antisheep secondary antibody diluted 1/25,000 in PBS containing 0.05% (vol/vol) Tween 20. Detection was by enhanced chemiluminescence (Amersham Biosciences, Bucks, UK). Anticalreticulin antibody was used as a loading control (27).
Analysis of urinary steroid metabolites
Urine was collected from 8- to 12-wk-old male mice on filter paper from five members of each group (LKO, HSD1KO, and control). Steroid extraction and analysis was carried out using GC/MS as previously described (7). Data are represented as a proportion of metabolites containing an 11-OXO group, (i.e. metabolites of 11-DHC), the remaining GC metabolites containing an 11-OH group are therefore metabolites of corticosterone.
11β-HSD1 activity assay
11β-HSD1 reductase activity was measured in liver explants in 8- to 10-wk-old mice. Mice were killed by cervical dislocation and liver tissue immediately removed. Tissue (2–4 mg wet weight) was incubated at 37 C in serum-free DMEM containing 100 nm 11-DHC and 60,000 cpm 3H-11-DHC for 1 h, medium was then removed, steroids extracted, and enzyme activity analyzed as previously described (28). All assays were performed in duplicate with n = 4 per group and data expressed as pmol/mg·h conversion. Corticosterone and 11-DHC were purchased from Sigma-Aldrich (Poole, UK). 3H-corticosterone (specific activity 1 mCi/ml) was purchased from Amersham Biosciences (Amersham, UK). 3H-11-DHC was synthesized “in house” from 3H-corticosterone.
RNA analysis and real-time PCR
Liver was homogenized with a PowerGen 125 homogenizer (Fisher Scientific, Loughborough, UK). RNA was extracted using TriReagent (Sigma, Poole, UK) according to the manufacturer's protocol. RNA quality was assessed by 1% agarose gel electrophoresis and quantified spectrophotometrically. Two-step RT-PCR was performed using 1 μg of RNA, random hexamers, and Multiscribe Reverse Transcriptase kit (Applied Biosystems, Cheshire, UK). Real-time PCR was carried out as previously described (28). Gene expression was assessed using prevalidated specific TaqMan gene expression assays and Universal PCR Master Mix (Applied Biosystems). Expression levels were normalized to the housekeeping genes glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and 18S in all tissues except adipose, in which only GAPDH was used. Data are expressed as arbitrary units using the following transformation: expression = 1000 × [2−Δct]. Fold change in expression was calculated using 2−ΔΔCt, where ΔCt = (Ct value of gene of interest) − (Ct value of GAPDH/18S). Data are expressed as fold change with respect to wild-type animals.
Statistical analysis
Statistical comparisons were performed using Prism 4 (GraphPad, CA). Data are presented as mean ± sem with statistical significance defined as P < 0.05. Two-way ANOVA followed by Bonferroni's multiple comparison post hoc test was used to compare between groups for changes in expression of genes. One-way ANOVA followed by Bonferroni's multiple comparison post hoc test was used to compare between groups, and the GTT data were analyzed using repeated measures ANOVA.
Results
Generation and validation of LKO mice
Expression of Cre recombinase under control of the albumin promoter excised exon 5 (coding the catalytic domain) specifically in hepatocytes, rendering the allele null (Fig. 1A), confirmed by assessing recombination between the LoxP sites in a range of tissues using genomic DNA PCR. Liver was the only tissue to show recombination, with other tissues (adipose, muscle, lung, kidney, and heart remaining normal for conditional HSD11B1 alleles) (Fig. 1B). Liver 11β-HSD1 mRNA was reduced 60-fold in LKO compared with controls; all other tissues studied had 11β-HSD1 mRNA similar to controls (Fig. 1C). Western blotting for 11β-HSD1 in liver microsomes showed a complete lack of protein, with a calreticulin used as a loading control (Fig. 1D). We assessed 11β-HSD1 activity by measuring 11-DHC to corticosterone conversion in liver microsomes and muscle tissue explants and used HSD1KO mice as an additional control. In LKO and HSD1KO, liver microsome activity was almost undetectable (<98% control activity) (Fig. 1E). In LKO mice, 11β-HSD1 activity in muscle explants was similar to control levels, whereas in the HSD1KO, it was undetectable (Fig. 1E).
Fig. 1.

Targeting strategy and LKO validation. A, Targeting strategy; exon 5 was flanked with LoxP sites and bred with the liver-specific Albumin-Cre transgenic. B, Genomic DNA PCR showing recombination is specific to the liver, giving a null allele for 11β-HSD1. C, mRNA levels in liver, adipose, lung, and muscle in control vs. LKO mice. D, Western blotting in two pairs of control and LKO mice livers showing no detectable 11β-HSD1 protein in the LKO. E, 11β-HSD1 reductase activity in control, LKO and global 11β-HSD1KO hepatic microsomes, and muscle explants. LKO and 11β-HSD1KO hepatic microsomes were devoid, whereas LKO muscle had normal levels of activity. Alb-Cre, Albumin-Cre transgenic; A.U., arbitrary units; Con., control.
Biomarkers of in vivo 11β-HSD1 activity
To assess the impact of LKO on systemic GC metabolism, we conducted a cortisone challenge test. Cortisone acetate was administered by oral gavage, and at 15 min, a tail blood sample was processed to assess cortisol generation. LKO mice were able to regenerate approximately 35–40% of the cortisol that control mice generate, despite hepatic loss of 11β-HSD1 (control, 1800 ± 125 nm vs. LKO, 650 ± 87 nm), whereas HSD1KO were unable to regenerate cortisol (Fig. 2A); 10- and 30 min bleeds after cortisone acetate gavage gave similar relative results (data not shown). This suggests that extrahepatic 11β-HSD1 activity is important for the rapid regeneration of cortisol in this experiment.
Fig. 2.
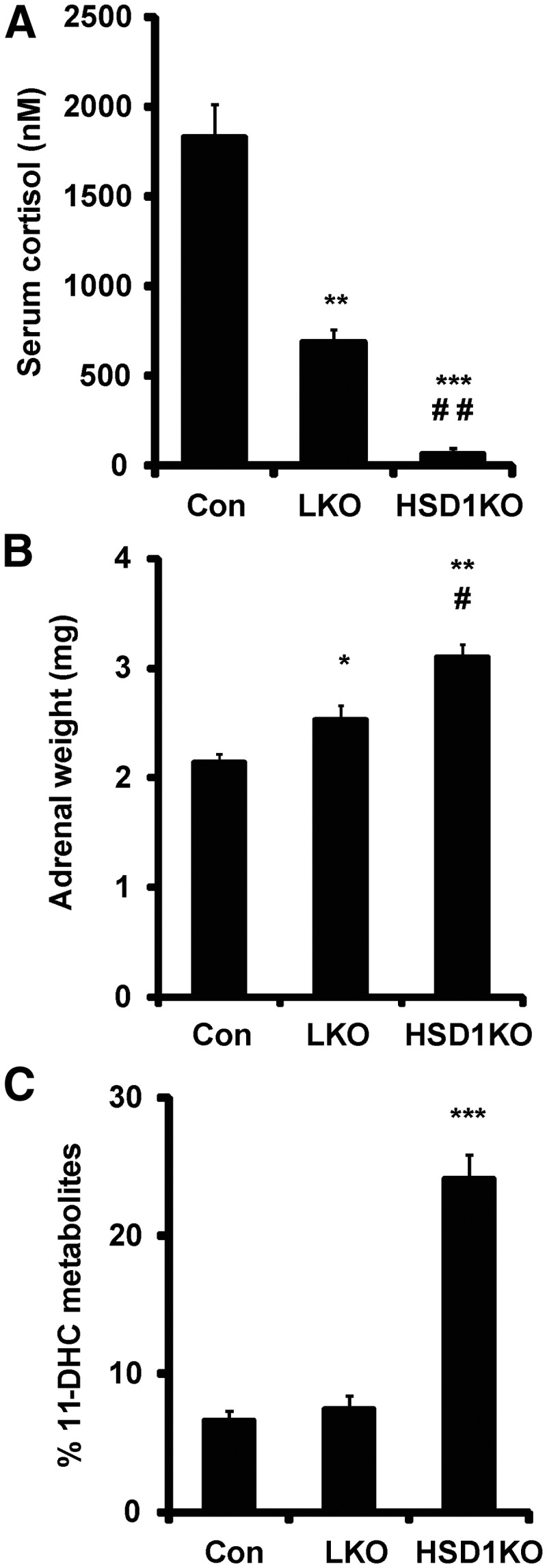
Assessment of in vivo 11β-HSD1 activity and adrenal function. A, Cortisone challenge. Control, LKO, and 11β-HSD1KO mice were gavaged with 200 μl suspension of cortisone acetate (0.5 mg) and serum collected by tail venesection after 20 min and cortisol generation quantified by GC/MS analysis. B, Adrenal weights in control, LKO, and 11β-HSD1KO mice (n = 9–10, 25–28 wk, *, P < 0.05; **, P < 0.01 vs. control; #, P < 0.05; ##, P < 0.01 vs. LKO). C, Analysis of urine by GC/MS in control, LKO, and 11β-HSD1KO mice showing the % of 11-DHC metabolites present (n = 6, 12–14 wk; ***, P < 0.001 vs. control). Con, Control.
We measured unstressed morning corticosterone levels and found no difference between control and LKO (Table 1). However, we did measure adrenal size and found that compared with control mice, LKO mice had moderately but significantly enlarged (∼18%) glands, with HSD1KO adrenals even more so (∼45%) (Fig. 2B), indicative of chronic stimulation of the adrenal gland. To further assess systemic GC metabolism, we examined urinary GC metabolites derived from inactive 11-DHC or active corticosterone. Previously, we have demonstrated that control mice excrete the majority of their GC as metabolites of corticosterone with less than 10% being excreted as metabolites of 11-DHC, whereas in HSD1KO mice, 20–25% of GC metabolites are 11-DHC derived (28). Surprisingly, in urine from LKO mice, the proportion of 11-DHC metabolites was similar to the controls (Fig. 2C), suggesting that a lack of hepatic 11β-HSD1 activity does not impinge on the proportion of 11-DHC or corticosterone metabolites produced in the liver, or that this urinary ratio might be more driven by extrahepatic 11β-HSD1.
Table 1.
Serum corticosterone, glucose, and lipid parameters
| Control |
LKO |
|||
|---|---|---|---|---|
| LF | HF | LF | HF | |
| Corticosterone (ng/ml) | 35 ± 10 | 53 ± 17 | ||
| Glucose (mm) | ||||
| Fed | 7.5 ± 0.2 | 7.5 ± 0.4 | 8 ± 0.2 | 7.3 ± 0.3 |
| Fasted | 5.2 ± 0.3 | 6.1 ± 0.5a | 4.8 ± 0.3 | 5.9 ± 0.5a |
| Triglyceride (μm) | 44 ± 14 | 280 ± 28a | 54 ± 12 | 330 ± 12a |
| FFA (μm) | 26 ± 6 | 34 ± 6a | 24 ± 4 | 33 ± 3a |
| LDL cholesterol (μm) | 336 ± 29 | 621 ± 50a | 310 ± 40 | 491 ± 44a |
| HDL cholesterol (μm) | 39 ± 7 | 160 ± 35 | 68 ± 17b | 122 ± 28 |
LF, Low fat; HF, high fat.
P < 0.05 compared with LF.
P < 0.05 compared with control.
Metabolic parameters on low-fat and high-fat diet
LKO exhibited no difference in the rate of weight gain compared with control on either low-fat or high-fat feeding (Fig. 3A). Similarly, liver, kidney, epididymal, and renal fat pad weights between LKO and control were unchanged (Table 2). We also assessed the expression of peroxisome proliferator-activated receptor gamma, uncoupling protein 2, fatty acid synthase (FAS), Acetyl-CoA carboxylase 1, hormone sensitive lipase, adiponectin, leptin, and resistin, lipid homeostasis genes fat known to be either GC regulated or altered in expression in the epididymal fat of 11β-HSD1KO mice (10). On high-fat diet, we detected no difference in the expression levels of these genes between LKO and control mice. Food intake on normal chow diet over a 4-wk period was marginally reduced in LKO compared with control mice with an average daily consumption of 3.6 vs. 4.1 g/d per mouse (Fig. 3B). LKO had normal fed blood glucose levels on both low-fat and high-fat diets (Table 2). They also had normal fasting glucose, and on a high-fat diet, fasting glucose was similarly elevated in both LKO and control mice compared with low fat-fed groups, indicating a degree of insulin resistance in both (Table 1). LKO mice also displayed normal glucose tolerance on a low-fat diet (Fig. 3C). However, on a high-fat diet, LKO resisted glucose intolerance having enhanced glucose clearance 30 min after glucose injection (Fig. 3D). An additional group of LKO and control mice fed a high-fat diet was subjected to an ITT. However, the LKO were not more tolerant of insulin as anticipated, showing no difference in response compared with control (Fig. 3E). In the fed state, LKO mice had normal insulin levels on a low-fat diet. However, on high-fat diet, insulin levels in control mice increased 5-fold, whereas in LKO mice, they only increased 3-fold. This constitutes a significant 40% reduction in circulating insulin in LKO mice and may be a result of the mildly decreased food intake (Fig. 3F). Fasting insulin levels were normal in LKO mice on a low-fat diet and significantly increased in both LKO and control mice on a high-fat diet, indicative of similar levels of insulin resistance (Fig. 3G).
Fig. 3.

Metabolic parameters on low- and high-fat diet. In all cases n = 7–9 with data collected after 18 wk on diet. A, Growth curve on low-fat (LF) and high-fat (HF) diet in control and LKO mice. B, Average daily food intake (n = 5 from 6 wk). Glucose tolerance in control and LKO mice on LF (C) and HF (D) diet (**, P < 0.01 vs. control). E, Insulin tolerance in control and LKO mice on HF diet. Fed (F) and fasted (G) insulin on LF and HF diets (**, P < 0.01 vs. HF control; #, P < 0.05 vs. fed; ††, P < 0.01 vs. LF). Control, Con.
Table 2.
Tissues weights
| Control |
LKO |
|||
|---|---|---|---|---|
| LF | HF | LF | HF | |
| Liver (g) | 1.80 ± 0.20 | 2.21 ± 0.31 | 1.83 ± 0.12 | 2.16 ± 0.34 |
| Kidney (g) | 0.24 ± 0.02 | 0.22 ± 0.05 | 0.25 ± 0.03 | 0.26 ± 0.03 |
| Epididymal fat (g) | 0.92 ± 0.18 | 1.73 ± 0.32a | 0.64 ± 0.14 | 1.76 ± 0.22a |
| Renal fat (g) | 0.33 ± 0.11 | 1.43 ± 0.25a | 0.37 ± 0.17 | 1.28 ± 0.33a |
LF, Low fat; HF, high fat.
P < 0.05 compared with LF.
Glucose homeostasis gene expression in response to high-fat diet and fasting
We examined the expression of hepatic phosphoenolpyruvate carboxykinase (PEPCK), glucose-6-phosphatase catalytic subunit (G6Pase), glucokinase, and peroxisome proliferator-activated receptor gamma coactivator 1α mRNA levels and determined them to be unaltered in LKO compared with control mice on a high-fat diet, suggesting that there is no major change to the gluconeogenic pathway or a major regulator of hepatic glucose homeostasis (Fig. 4A). Fasting is a stress status that invokes GC stimulation of hepatic glucose output. We assessed the expression of PEPCK and G6Pase in fed and overnight-fasted LKO and control mice on a regular chow diet. In either nutritional state, there was no difference in expression levels between LKO and controls, with mRNA induction to fasting being similar in both (Fig. 4B), corroborating the notion that loss of hepatic 11β-HSD1 activity has minimal impact upon liver-mediated glucose homeostasis.
Fig. 4.

Hepatic gene expression: glucose homeostasis. A, Relative glucose homeostasis gene mRNA levels in control and LKO mice on high-fat (HF) diet. B, Fold induction from fed to fasting state of G6Pase and PEPCK (*, P < 0.05; **, P < 0.01 vs. fed state). Con, Control; PGC1a, peroxisome proliferator-activated receptor gamma coactivator 1-α; GK, glucokinase.
Hepatic triglyceride content and plasma lipid profile
Staining of liver with hematoxylin and eosin (H&E) and Oil Red O shows no difference in hepatic fat content in LKO mice on either diet. A high-fat diet did induced pathology associated with steatosis equally in both LKO and control livers (Fig. 5A). Biochemical analyses confirmed that although the high-fat diet greatly increased hepatic triglyceride content, it was to a similar degree in both LKO and control mice (Fig. 5A), indicating that LKO mice have no protection from steatosis.
Fig. 5.

Hepatic triglyceride accumulation and gene expression. A, Representative H&E staining depicts pathology associated with hepatic steatosis, which was similar between control and LKO sections (by light microscope at ×40 magnification). Graph shows liver triglyceride content on low-fat (LF) and high-fat (HF) diet in control and LKO mice (*, P < 0.05 vs. LF diet). B, Relative lipogenic gene mRNA levels in control and LKO mice on HF diet (*, P < 0.05 vs. control). C, Relative expression of mRNA for genes associated sensitive to GC, cholesterol metabolism, and β-oxidation. D, Representative H&E and Oil Red O staining of 16-h-fasted control and LKO livers showing similar levels of steatosis (by light microscope at ×40 magnification). SREBP1c, Sterol regulatory element-binding protein 1c; FAS, fatty acid synthase; ACC1, acetyl-CoA carboxylase 1; ACC2, acetyl-CoA carboxylase 2; SCD1, stearoyl-coenzyme A desaturase 1; ELOVL6, elongation of long-chain fatty acids family member 6; H6PDH, hexose-6-phospahte dehydrogenase; AGT, angiotensinogen; FGA, fibrinogen alpha; LDLr, low density lipoprotein receptor; HMG, 3-hydroxy-3-methyl-glutaryl-CoA reductase; LIPC, hepatic lipase; PPARa, peroxisome proliferator-activated receptor alpha; CPT1a, carnitine palmitoyltransferase 1a; Con, control; GC, glucocorticoid related genes; Chol, cholesterol metabolism related genes; Beta-Ox, beta oxidation related genes.
We assessed serum triglyceride, FFA, and LDL and HDL cholesterol levels in LKO and controls (Table 1). LKO mice on a low-fat diet had a significant increase in HDL cholesterol compared with control mice, and although it was also elevated compared with controls on a high-fat diet, it did not retain statistical significance. For all other lipid measurements, no significant differences were observed.
To investigate molecular consequences of altered hepatic GC levels, we examined the expression of genes involved in lipid metabolism. On a low-fat diet, no changes were detected (data not shown). On a high-fat diet, a series of genes associated with hepatic lipogenesis were assessed. We detected a significant reduction in the expression of the sterol regulatory element-binding protein 1c transcript in LKO, a major transcriptional regulator of lipogenesis. However, of the enzymes involved in lipogenesis (including FAS and ACC1), only ElovL6 was down-regulated but not to a significant level (Fig. 5B), supporting the similar levels of triglyceride accumulation in LKO and control liver. To further examine altered hepatic GC signaling, we determined the expression of other genes associated with hepatic lipid homeostasis, including GC-regulated genes previously shown to be altered in expression in 11β-HSD1 transgenic models (Fig. 5C) (9, 16). We did not observe any significant changes in gene expression associated with lipid homeostasis or lipid oxidation, and although LDL receptor mRNA was increased, it did not reach significance. Finally, H6PDH, the enzyme controlling the redox set-point of 11β-HSD1 activity, mRNA was unchanged, and mRNA for angiotensinogen (AGT) and fibrinogen 1α, previously shown to be GC sensitive, were also unchanged in LKO mice on high-fat diet (Fig. 5C) (9, 16). It has been reported that HSD1KO mice develop transient steatosis on regular chow diet after an overnight fast (9), so we examined this in LKO mice by histology and Oil Red O staining (Fig. 5D). Both LKO and control livers accumulated significant lipid content after an overnight fast. However, there was no particular increase in transient lipid accumulation in LKO over controls.
Discussion
Comprehensive studies, ranging from in vitro, in vivo, preclinical, to human, all suggest that attenuation of GC signaling, through 11β-HSD1 inhibition in metabolically important tissue such as the liver, is a therapeutically achievable strategy to ameliorate the glucose intolerance, obesity, fatty liver, and hypertension associated with metabolic syndrome.
LKO mice have almost a complete loss of hepatic 11β-HSD1 activity and therefore are unable to regenerate GC. Surprisingly, mice gavaged with a bolus of cortisone are able to regenerate a significant amount of cortisol (35–40% that of control mice) in a short period of time. Because 11β-HSD1 has previously been shown to be the sole cortisone reductase in mice, one might have anticipated, due to the loss of 11β-HSD1 via first pass hepatic metabolism, that this value would be much lower (8). It may be that the intestinal mucosa or other intestinal sites express 11β-HSD1. Similarly, vascular wall and endothelium, and blood cell types, have been shown to express 11β-HSD1, and this may account for some of the GC regeneration, although some cells of these tissues also express 11β-HSD2 and therefore subject to cortisol inactivation also (30–32). The kidney is the site in which the majority of inactive GC substrate for 11β-HSD1 (i.e. cortisone or 11-DHC) is produced through the activity of the enzyme 11β-HSD2 (33). 11β-HSD1 is expressed in distinct renal areas, such as the proximal tubule and interstitial cells of the medulla, such that reductase activity here may contribute toward setting the proportions of 11-DHC/corticosterone metabolites in LKO mice (34, 35). However, there is evidence that kidney 11β-HSD1 activity is mainly dehydrogenase in nature and, therefore, may contribute little to the reactivation of GC (35–37). This has the implication that, when assessing 11β-HSD1 activity using cortisol generation tests in humans, extrahepatic tissue can greatly affect result interpretation, which will be important for understanding the mode of action and clinical benefit of 11β-HSD1 therapeutics.
If the liver is the major site of 11β-HSD1-mediated GC regeneration, then we would anticipate that urinary metabolites would reflect a transition toward 11β-HSD1KO values with a greater proportion of 11-DHC metabolites compared with control. However, assessment of urinary GC metabolites in LKO mice demonstrated normal values. The reasons for this are unclear; it may suggest that the proportion of circulating corticosterone and 11-DHC clearance via hepatic routes are established upon entry into the liver, such that 11β-HSD1 metabolism of 11-DHC is a more terminal event as hepatic flux transitions across the lobule to the central vein. This idea is strengthened by the apparent concentration of 11β-HSD1 immunostaining in hepatocytes adjacent to the central vein, which decrease moving toward the portal vein (34, 38) .
LKO mice have normal morning corticosterone levels in the systemic circulation; importantly, they appear to have adrenal glands approximately 20% larger than controls, although not as large as HSD1KO adrenals. This suggests that there is subtle HPA axis stimulation to increase GC production to correct for the lack of 11β-HSD1-mediated regenerated GC exiting the liver. HPA stimulation would redress corticosterone delivery to the liver via splanchnic and portal drainage reflecting the lack of urinary metabolite phenotype. The HPA axis defect associated with 11β-HSD1KO mice can be rescued when crossed with liver-specific HSD1-overexpressing mice (24), thus it seems that hepatic GC output can modulate HPA axis sensitivity. LKO mice are on a mixed C57BL/6J/129SvJ background and Carter et al. (39) demonstrated that background is important in governing responses to 11β-HSD1 deletion. C57BL/6J mice have adrenal hypertrophy but normal HPA activity, in contrast to 129SvJ mice, which have a more exaggerated adrenal hypertrophy with reduced GC negative feedback. LKO mice display the level of adrenal hypertrophy associated with the C57BL/6J background, but the 129SvJ contribution may account for the observed trend toward increased corticosterone levels and could impact upon metabolic outcome. Together, these findings have implications for human studies using biomarkers of 11β-HSD1 inhibition and the potential long term effects upon HPA regulation and response to stress.
The effects of liver selective 11β-HSD1KO on metabolic parameters appear to be relatively minor and only then on high-fat feeding. Food intake is altered in 11β-HSD1KO mice (40), and we measured a slight decrease in average daily food intake, however, this did not reach significance. Although chronic changes in food intake may be important, we believe that it would not explain the changes observed in the high fat-fed LKO mice. There was no effect upon body weight, adiposity, or the expression levels of lipid homeostasis genes measured in the epididymal depot. In terms of glucose homeostasis, although there was a modest improvement in glucose tolerance and lower postprandial insulin levels, fasting glucose and insulin, and insulin tolerance in LKO and control mice on a high-fat diet were indistinguishable, indicating a similar degree of diet-induced insulin resistance. Although it would be easy to dismiss the lack of significant difference in absolute fasting blood glucose concentrations, within the current protocol, we have not been able to measure glucose production rate (as distinct from circulating concentrations). In the fasting state, endogenous glucose production is largely from the liver, and we would have hypothesized that this would be decreased in LKO mice. However, any changes in endogenous glucose production rate might be offset by alterations in insulin and glucagon secretion in the absence of any changes in glucose sensing or glucose disposal. Considering that we have only altered 11β-HSD1 expression within the liver, other tissues where this might be important (including pancreas and skeletal muscle) should be identical in both LKO and controls. This contrasts with the 11β-HSD1KO, where there may be additional dysregulation of insulin secretion and increased glucose disposal thus lowering absolute glucose concentrations (41, 42).
Hepatic lipid accumulation was present in LKO mice to a similar level to that of controls. The serum lipid profile in LKO was also equivalent to controls, with only HDL cholesterol being marginally elevated in low fat-fed LKO mice, a difference that was lost on high-fat feeding. There was no difference in gene expression levels between LKO and control mice fed a low- or high-fat diet. The only consistent finding was for lower SREBP1c expression in the liver of high fat-fed LKO mice. However, the downstream target genes associated with this transcription factor, such as those controlling de novo lipogenesis, were unaltered in expression, thus the significance of this finding is uncertain. Indeed, the response to fasting in the regulation of gluconeogenic gene expression, which GC are well known to augment, were also unchanged and support the lack of difference in fasting blood glucose in both LKO and control mice. AGT gene expression has been found to be increased in the liver of adipose and liver 11β-HSD1-overexpressing mice, where it contributes to hypertension (15, 16). In LKO mice, AGT mRNA was unchanged, suggesting that expression is not affected by a lack 11β-HSD1 and GC regeneration.
11β-HSD1 is expressed in a number of other tissues important for metabolic regulation, such as the central nervous system and adipose tissue (6, 15, 43, 44). In humans, there is evidence of a pathological role for elevated 11β-HSD1 activity in adipose tissue, and adipose overexpression of 11β-HSD1 in mice leads to elevated intraadipose GC levels that can spill into the liver, although there is debate as to the actual potential for GC spillover to the liver from adipose depots in human (reviewed comprehensively in Refs. 3, 43). Thus, it may be the case that local effects in adipose tissue are sufficient to propagate full metabolic syndrome and not spillover of adipose GC in to the liver per se, supported in liver 11β-HSD1-overexpressing mice, in which increased intrahepatic GC regeneration does not drive glucose intolerance or changes in fasting glucose. Further to this, patients with the metabolic syndrome and obese rodent strains demonstrate unchanged or even reduced liver 11β-HSD1 levels. Therefore, in this model and those in which hepatic 11β-HSD1 is not elevated, the liver is “innocent” in determining aspects of metabolic dysreguation and responsive to endocrine and portal GC delivery, a notion in keeping with the idea that extrahepatic 11β-HSD1 activity is as important for determining metabolic phenotype (3, 45, 46).
A recent study used antisense oligos against 11β-HSD1 and suggested preferential hepatic inhibition resulting in improved plasma and hepatic lipid levels, reduced lipogenesis rates, and reduced hepatic triglyceride accumulation on high-fat diet (47). Although these data are compelling, it was also noted that the antisense sequence down-regulated 11β-HSD1 significantly in most adipose depots and lungs, in effect making this a partial-global KO with little data describing glucose homeostasis or HPA axis defects. Another study targeted an 11β-HSD1-selective inhibitor (BVT.2733) (48) coupled to a white adipose tissue vasculature homing peptide and compared it with an equimolar, nontargeted dose (49). They showed that adipose targeted inhibitor improved body weight, glucose tolerance, and decreased fat cell size. Significantly, adipose targeted inhibitor also had the capacity to improve hepatic metabolism decreasing PEPCK and increasing carnitine palmitoyltransferase I and PPARα expression. In inhibitor-alone-treated mice, the effects were less pronounced and most likely reflect adipose-mediated effects, thus reduced adipose 11β-HSD1 can improve hepatic function and does not necessarily rely upon direct GC regulation in the liver. The pathogenesis of hepatic steatosis and progression to steatohepatitis and cirrhosis remains an area of intense research (50). Various processes contribute to lipid accumulation within the liver, including FFA delivery from adipose tissue with subsequent reesterification as well as de novo lipogenesis. Alterations in lipid accumulation are balanced by use through β-oxidation and V packaging. Data suggest that genetic and pharmacological inhibition of 11β-HSD1 can decrease hepatic steatosis (47, 51–53). Importantly however, both small interfering RNA technology and selective 11β-HSD1 inhibitors do not offer exclusive tissue selectivity and decrease activity and expression in both liver and fat. Within our LKO, we failed to observe any decrease in liver fat accumulation. This may well highlight the important cross talk between adipose tissue and liver, suggesting that decreased 11β-HSD1 in adipose tissue limits lipolysis, and this in turn will decrease FFA delivery to the liver, and this might be the most important factor in limiting hepatic steatosis (54). Specifically and exclusively inhibiting 11β-HSD1 within the liver plays a relatively minor role.
Preclinical and human studies using various selective and nonselective 11β-HSD1 inhibitors (21) have been encouraging, but the need to understand the specific sites and modes of action is paramount if the concept of reducing tissue-specific GC generation as a means to improve metabolic health is to fulfill its promise. The discrepant changes in biomarkers of GC metabolism and action have significant implications for their use in clinical studies, where they have been regarded as providing an accurate reflection of hepatic enzyme activity. Finally, selective loss of liver 11β-HSD1 highlights that extrahepatic sites of activity, and cross talk between GC target tissues may be of greater importance in determining global metabolic phenotype.
Acknowledgments
We thank Cedric Shackleton for interpretation of the urinary GC/MS data and Lianne Abrahams for technical support.
This work was supported by the Biotechnology and Biological Sciences Research Council David Philips Fellowship BB/G023468/1 (to G.G.L.), a Welcome Trust Program Grant 082809 (to P.M.S. and E.A.W.), and an ERC Advanced Grant 20090506 (to P.M.S.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AGT
- Angiotensinogen
- CRD
- cortisone reductase deficiency
- 11-DHC
- 11-dehydrocorticosterone
- FAS
- fatty acid synthase
- FFA
- free fatty acid
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GC
- glucocorticoid
- GC/MS
- gas chromatography-mass spectrometry
- G6Pase
- glucose-6-phosphatase catalytic subunit
- GTT
- glucose tolerance test
- HDL
- high-density lipoprotein
- H&E
- hematoxylin and eosin
- HPA
- hypothalamic-pituitary-adrenal
- H6PDH
- hexose-6-phosphate dehydrogenase
- 11β-HSD1
- 11β-hydroxysteroid dehydrogenase type 1
- ITT
- insulin tolerance test
- KO
- knockout
- LDL
- low-density lipoprotein
- LKO
- liver-specific 11β-HSD1KO
- NADPH
- reduced nicotinamide adenine dinucleotide phosphate
- PEPCK
- phosphoenolpyruvate carboxykinase.
References
- 1. Nordlie RC, Foster JD, Lange AJ. 1999. Regulation of glucose production by the liver. Annu Rev Nutr 19:379–406 [DOI] [PubMed] [Google Scholar]
- 2. Pilkis SJ, Granner DK. 1992. Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Physiol 54:885–909 [DOI] [PubMed] [Google Scholar]
- 3. Gathercole LL, Stewart PM. 2010. Targeting the pre-receptor metabolism of cortisol as a novel therapy in obesity and diabetes. J Steroid Biochem Mol Biol 122:21–27 [DOI] [PubMed] [Google Scholar]
- 4. Friedman TC, Mastorakos G, Newman TD, Mullen NM, Horton EG, Costello R, Papadopoulos NM, Chrousos GP. 1996. Carbohydrate and lipid metabolism in endogenous hypercortisolism: shared features with metabolic syndrome X and NIDDM. Endocr J 43:645–655 [DOI] [PubMed] [Google Scholar]
- 5. Fraser R, Ingram MC, Anderson NH, Morrison C, Davies E, Connell JM. 1999. Cortisol effects on body mass, blood pressure, and cholesterol in the general population. Hypertension 33:1364–1368 [DOI] [PubMed] [Google Scholar]
- 6. Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper MS, Hewison M, Stewart PM. 2004. 11β-Hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev 25:831–866 [DOI] [PubMed] [Google Scholar]
- 7. Lavery GG, Walker EA, Draper N, Jeyasuria P, Marcos J, Shackleton CH, Parker KL, White PC, Stewart PM. 2006. Hexose-6-phosphate dehydrogenase knock-out mice lack 11β-hydroxysteroid dehydrogenase type 1-mediated glucocorticoid generation. J Biol Chem 281:6546–6551 [DOI] [PubMed] [Google Scholar]
- 8. Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, Jamieson P, Best R, Brown R, Edwards CR, Seckl JR, Mullins JJ. 1997. 11β-Hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci USA 94:14924–14929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morton NM, Holmes MC, Fiévet C, Staels B, Tailleux A, Mullins JJ, Seckl JR. 2001. Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11β-hydroxysteroid dehydrogenase type 1 null mice. J Biol Chem 276:41293–41300 [DOI] [PubMed] [Google Scholar]
- 10. Morton NM, Paterson JM, Masuzaki H, Holmes MC, Staels B, Fievet C, Walker BR, Flier JS, Mullins JJ, Seckl JR. 2004. Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11β-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes 53:931–938 [DOI] [PubMed] [Google Scholar]
- 11. Wamil M, Battle JH, Turban S, Kipari T, Seguret D, de Sousa Peixoto R, Nelson YB, Nowakowska D, Ferenbach D, Ramage L, Chapman KE, Hughes J, Dunbar DR, Seckl JR, Morton NM. 2011. Novel fat depot-specific mechanisms underlie resistance to visceral obesity and inflammation in 11β-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes 60:1158–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bujalska IJ, Hewitt KN, Hauton D, Lavery GG, Tomlinson JW, Walker EA, Stewart PM. 2008. Lack of hexose-6-phosphate dehydrogenase impairs lipid mobilization from mouse adipose tissue. Endocrinology 149:2584–2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lavery GG, Hauton D, Hewitt KN, Brice SM, Sherlock M, Walker EA, Stewart PM. 2007. Hypoglycemia with enhanced hepatic glycogen synthesis in recombinant mice lacking hexose-6-phosphate dehydrogenase. Endocrinology 148:6100–6106 [DOI] [PubMed] [Google Scholar]
- 14. Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, Flier JS. 2001. A transgenic model of visceral obesity and the metabolic syndrome. Science 294:2166–2170 [DOI] [PubMed] [Google Scholar]
- 15. Masuzaki H, Yamamoto H, Kenyon CJ, Elmquist JK, Morton NM, Paterson JM, Shinyama H, Sharp MG, Fleming S, Mullins JJ, Seckl JR, Flier JS. 2003. Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J Clin Invest 112:83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paterson JM, Morton NM, Fievet C, Kenyon CJ, Holmes MC, Staels B, Seckl JR, Mullins JJ. 2004. Metabolic syndrome without obesity: hepatic overexpression of 11β-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc Natl Acad Sci USA 101:7088–7093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alberts P, Nilsson C, Selen G, Engblom LO, Edling NH, Norling S, Klingström G, Larsson C, Forsgren M, Ashkzari M, Nilsson CE, Fiedler M, Bergqvist E, Ohman B, Björkstrand E, Abrahmsen LB. 2003. Selective inhibition of 11β-hydroxysteroid dehydrogenase type 1 improves hepatic insulin sensitivity in hyperglycemic mice strains. Endocrinology 144:4755–4762 [DOI] [PubMed] [Google Scholar]
- 18. Hermanowski-Vosatka A, Balkovec JM, Cheng K, Chen HY, Hernandez M, Koo GC, Le Grand CB, Li Z, Metzger JM, Mundt SS, Noonan H, Nunes CN, Olson SH, Pikounis B, Ren N, Robertson N, Schaeffer JM, Shah K, Springer MS, Strack AM, Strowski M, Wu K, Wu T, Xiao J, Zhang BB, Wright SD, Thieringer R. 2005. 11β-HSD1 inhibition ameliorates metabolic syndrome and prevents progression of atherosclerosis in mice. J Exp Med 202:517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lloyd DJ, Helmering J, Cordover D, Bowsman M, Chen M, Hale C, Fordstrom P, Zhou M, Wang M, Kaufman SA, Véniant MM. 2009. Antidiabetic effects of 11β-HSD1 inhibition in a mouse model of combined diabetes, dyslipidaemia and atherosclerosis. Diabetes Obes Metab 11:688–699 [DOI] [PubMed] [Google Scholar]
- 20. Park JS, Rhee SD, Kang NS, Jung WH, Kim HY, Kim JH, Kang SK, Cheon HG, Ahn JH, Kim KY. 2011. Anti-diabetic and anti-adipogenic effects of a novel selective 11β-hydroxysteroid dehydrogenase type 1 inhibitor, 2-(3-benzoyl)-4-hydroxy-1,1-dioxo-2H-1,2-benzothiazine-2-yl-1-phenylethano ne (KR-66344). Biochem Pharmacol 81:1028–1035 [DOI] [PubMed] [Google Scholar]
- 21. Rosenstock J, Banarer S, Fonseca VA, Inzucchi SE, Sun W, Yao W, Hollis G, Flores R, Levy R, Williams WV, Seckl JR, Huber R. 2010. The 11-β-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care 33:1516–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Walker BR, Connacher AA, Lindsay RM, Webb DJ, Edwards CR. 1995. Carbenoxolone increases hepatic insulin sensitivity in man: a novel role for 11-oxosteroid reductase in enhancing glucocorticoid receptor activation. J Clin Endocrinol Metab 80:3155–3159 [DOI] [PubMed] [Google Scholar]
- 23. Harris HJ, Kotelevtsev Y, Mullins JJ, Seckl JR, Holmes MC. 2001. Intracellular regeneration of glucocorticoids by 11β-hydroxysteroid dehydrogenase (11β-HSD)-1 plays a key role in regulation of the hypothalamic-pituitary-adrenal axis: analysis of 11β-HSD-1-deficient mice. Endocrinology 142:114–120 [DOI] [PubMed] [Google Scholar]
- 24. Paterson JM, Holmes MC, Kenyon CJ, Carter R, Mullins JJ, Seckl JR. 2007. Liver-selective transgene rescue of hypothalamic-pituitary-adrenal axis dysfunction in 11β-hydroxysteroid dehydrogenase type 1-deficient mice. Endocrinology 148:961–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rogoff D, Ryder JW, Black K, Yan Z, Burgess SC, McMillan DR, White PC. 2007. Abnormalities of glucose homeostasis and the hypothalamic-pituitary-adrenal axis in mice lacking hexose-6-phosphate dehydrogenase. Endocrinology 148:5072–5080 [DOI] [PubMed] [Google Scholar]
- 26. Lavery GG, Walker EA, Tiganescu A, Ride JP, Shackleton CH, Tomlinson JW, Connell JM, Ray DW, Biason-Lauber A, Malunowicz EM, Arlt W, Stewart PM. 2008. Steroid biomarkers and genetic studies reveal inactivating mutations in hexose-6-phosphate dehydrogenase in patients with cortisone reductase deficiency. J Clin Endocrinol Metab 93:3827–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lawson AJ, Walker EA, Lavery GG, Bujalska IJ, Hughes B, Arlt W, Stewart PM, Ride JP. 2011. Cortisone-reductase deficiency associated with heterozygous mutations in 11β-hydroxysteroid dehydrogenase type 1. Proc Natl Acad Sci USA 108:4111–4116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Semjonous NM, Sherlock M, Jeyasuria P, Parker KL, Walker EA, Stewart PM, Lavery GG. 2011. Hexose-6-phosphate dehydrogenase contributes to skeletal muscle homeostasis independent of 11β-hydroxysteroid dehydrogenase type 1. Endocrinology 152:93–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. 1999. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic β cell-specific gene knock-outs using Cre recombinase. J Biol Chem 274:305–315 [DOI] [PubMed] [Google Scholar]
- 30. Chapman KE, Coutinho AE, Gray M, Gilmour JS, Savill JS, Seckl JR. 2009. The role and regulation of 11β-hydroxysteroid dehydrogenase type 1 in the inflammatory response. Mol Cell Endocrinol 301:123–131 [DOI] [PubMed] [Google Scholar]
- 31. Gilmour JS, Coutinho AE, Cailhier JF, Man TY, Clay M, Thomas G, Harris HJ, Mullins JJ, Seckl JR, Savill JS, Chapman KE. 2006. Local amplification of glucocorticoids by 11β-hydroxysteroid dehydrogenase type 1 promotes macrophage phagocytosis of apoptotic leukocytes. J Immunol 176:7605–7611 [DOI] [PubMed] [Google Scholar]
- 32. Hadoke PW, Lindsay RS, Seckl JR, Walker BR, Kenyon CJ. 2006. Altered vascular contractility in adult female rats with hypertension programmed by prenatal glucocorticoid exposure. J Endocrinol 188:435–442 [DOI] [PubMed] [Google Scholar]
- 33. Draper N, Stewart PM. 2005. 11β-Hydroxysteroid dehydrogenase and the pre-receptor regulation of corticosteroid hormone action. J Endocrinol 186:251–271 [DOI] [PubMed] [Google Scholar]
- 34. Brereton PS, van Driel RR, Suhaimi Fb, Koyama K, Dilley R, Krozowski Z. 2001. Light and electron microscopy localization of the 11β-hydroxysteroid dehydrogenase type I enzyme in the rat. Endocrinology 142:1644–1651 [DOI] [PubMed] [Google Scholar]
- 35. Gomez-Sanchez EP, Romero DG, de Rodriguez AF, Warden MP, Krozowski Z, Gomez-Sanchez CE. 2008. Hexose-6-phosphate dehydrogenase and 11β-hydroxysteroid dehydrogenase-1 tissue distribution in the rat. Endocrinology 149:525–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brem AS, Bina RB, King T, Chobanian MC, Morris DJ. 1997. Influence of dietary sodium on the renal isoforms of 11 β-hydroxysteroid dehydrogenase. Proc Soc Exp Biol Med 214:340–345 [DOI] [PubMed] [Google Scholar]
- 37. Gong R, Morris DJ, Brem AS. 2008. Human renal 11β-hydroxysteroid dehydrogenase 1 functions and co-localizes with COX-2. Life Sci 82:631–637 [DOI] [PubMed] [Google Scholar]
- 38. Ricketts ML, Verhaeg JM, Bujalska I, Howie AJ, Rainey WE, Stewart PM. 1998. Immunohistochemical localization of type 1 11β-hydroxysteroid dehydrogenase in human tissues. J Clin Endocrinol Metab 83:1325–1335 [DOI] [PubMed] [Google Scholar]
- 39. Carter RN, Paterson JM, Tworowska U, Stenvers DJ, Mullins JJ, Seckl JR, Holmes MC. 2009. Hypothalamic-pituitary-adrenal axis abnormalities in response to deletion of 11β-HSD1 is strain-dependent. J Neuroendocrinol 21:879–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Densmore VS, Morton NM, Mullins JJ, Seckl JR. 2006. 11β-Hydroxysteroid dehydrogenase type 1 induction in the arcuate nucleus by high-fat feeding: a novel constraint to hyperphagia? Endocrinology 147:4486–4495 [DOI] [PubMed] [Google Scholar]
- 41. Morgan SA, Sherlock M, Gathercole LL, Lavery GG, Lenaghan C, Bujalska IJ, Laber D, Yu A, Convey G, Mayers R, Hegyi K, Sethi JK, Stewart PM, Smith DM, Tomlinson JW. 2009. 11β-Hydroxysteroid dehydrogenase type 1 regulates glucocorticoid-induced insulin resistance in skeletal muscle. Diabetes 58:2506–2515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Swali A, Walker EA, Lavery GG, Tomlinson JW, Stewart PM. 2008. 11β-Hydroxysteroid dehydrogenase type 1 regulates insulin and glucagon secretion in pancreatic islets. Diabetologia 51:2003–2011 [DOI] [PubMed] [Google Scholar]
- 43. Morton NM. 2010. Obesity and corticosteroids: 11β-hydroxysteroid type 1 as a cause and therapeutic target in metabolic disease. Mol Cell Endocrinol 316:154–164 [DOI] [PubMed] [Google Scholar]
- 44. Wyrwoll CS, Holmes MC, Seckl JR. 2011. 11β-Hydroxysteroid dehydrogenases and the brain: from zero to hero, a decade of progress. Front Neuroendocrinol 32:265–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Morton NM, Seckl JR. 2008. 11β-Hydroxysteroid dehydrogenase type 1 and obesity. Front Horm Res 36:146–164 [DOI] [PubMed] [Google Scholar]
- 46. Stewart PM, Boulton A, Kumar S, Clark PM, Shackleton CH. 1999. Cortisol metabolism in human obesity: impaired cortisone–>cortisol conversion in subjects with central adiposity. J Clin Endocrinol Metab 84:1022–1027 [DOI] [PubMed] [Google Scholar]
- 47. Li G, Hernandez-Ono A, Crooke RM, Graham MJ, Ginsberg HN. 2011. Effects of antisense-mediated inhibition of 11β-hydroxysteroid dehydrogenase type 1 on hepatic lipid metabolism. J Lipid Res 52:971–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Alberts P, Engblom L, Edling N, Forsgren M, Klingström G, Larsson C, Rönquist-Nii Y, Ohman B, Abrahmsén L. 2002. Selective inhibition of 11β-hydroxysteroid dehydrogenase type 1 decreases blood glucose concentrations in hyperglycaemic mice. Diabetologia 45:1528–1532 [DOI] [PubMed] [Google Scholar]
- 49. Liu J, Wang L, Zhang A, Di W, Zhang X, Wu L, Yu J, Zha J, Lv S, Cheng P, Hu M, Li Y, Qi H, Ding G, Zhong Y. 2011. Adipose tissue-targeted 11β-hydroxysteroid dehydrogenase type 1 inhibitor protects against diet-induced obesity. Endocr J 58:199–209 [DOI] [PubMed] [Google Scholar]
- 50. Postic C, Girard J. 2008. The role of the lipogenic pathway in the development of hepatic steatosis. Diabetes Metab 34:643–648 [DOI] [PubMed] [Google Scholar]
- 51. Berthiaume M, Laplante M, Festuccia WT, Cianflone K, Turcotte LP, Joanisse DR, Olivecrona G, Thieringer R, Deshaies Y. 2007. 11β-HSD1 inhibition improves triglyceridemia through reduced liver VLDL secretion and partitions lipids toward oxidative tissues. Am J Physiol Endocrinol Metab 293:E1045–E1052 [DOI] [PubMed] [Google Scholar]
- 52. Berthiaume M, Laplante M, Festuccia W, Gélinas Y, Poulin S, Lalonde J, Joanisse DR, Thieringer R, Deshaies Y. 2007. Depot-specific modulation of rat intraabdominal adipose tissue lipid metabolism by pharmacological inhibition of 11β-hydroxysteroid dehydrogenase type 1. Endocrinology 148:2391–2397 [DOI] [PubMed] [Google Scholar]
- 53. Berthiaume M, Laplante M, Festuccia WT, Berger JP, Thieringer R, Deshaies Y. 2010. Preliminary report: pharmacologic 11β-hydroxysteroid dehydrogenase type 1 inhibition increases hepatic fat oxidation in vivo and expression of related genes in rats fed an obesogenic diet. Metabolism 59:114–117 [DOI] [PubMed] [Google Scholar]
- 54. Tomlinson JW, Sherlock M, Hughes B, Hughes SV, Kilvington F, Bartlett W, Courtney R, Rejto P, Carley W, Stewart PM. 2007. Inhibition of 11β-hydroxysteroid dehydrogenase type 1 activity in vivo limits glucocorticoid exposure to human adipose tissue and decreases lipolysis. J Clin Endocrinol Metab 92:857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]