

Abstract
While the thermodynamic effects of trimethylamine oxide (TMAO), urea and guanidine hydrochloride (GdnHCl) on protein stability are well understood, the underlying mechanisms of action are less well characterized and, in some cases, even under debate. Herein, we employ the stretching vibration of two infrared (IR) reporters, i.e., nitrile (C≡N) and carbonyl (C=O), to directly probe how these cosolvents mediate the ability of water to form hydrogen bonds with the solute of interest, e.g., a peptide. Our results show that these three agents, despite having different effects on protein stability, all act to decrease the strength of the hydrogen bonds formed between water and the infrared probe. While the behavior of TMAO appears to be consistent with its protein-protecting ability, those of urea and GdnHCl are inconsistent with their role as protein denaturants. The latter is of particular interest as it provides strong evidence indicating that although urea and GdnHCl can perturb the hydrogen-bonding property of water, their protein-denaturing ability does not arise from a simple indirect mechanism.
Keywords: TMAO, Urea, Guanidine hydrochloride, Nitrile stretching vibration, Hydrogen bond, denaturant
1. INTRODUCTION
It is well known that both the stability and solubility of a protein in aqueous solution can be altered by the addition of certain cosolvents, such as TMAO and urea. While there is a large body of research1–37 on how such stabilizing/destabilizing effects might take place, a coherent view of the underlying mechanism of action at the molecular level has not been reached. For example, it is still under debate whether urea and GdnHCl, both of which are commonly used as protein denaturants, affect the structure and dynamics of the solvent water molecules to such an extent that would significantly reduce the strength of stabilizing interactions in proteins.38,39
One of the distinctive physical properties of water is its strong ability to form hydrogen bonds (H-bonds), these interactions are essential for protein folding. As such, many previous studies40–45 have focused on investigating whether commonly encountered protectants (e.g., TMAO) and denaturants (e.g., urea and GdnHCl) affect the H-bond network of water molecules and, if so, to what extent. For example, Vanderkooi and co-workers have demonstrated, using IR spectroscopy and molecular dynamics (MD) simulations,46 that the addition of TMAO results in a significant change in the vibrational spectrum of water, whereas the addition of urea has little effect. While such studies have provided significant insights into our understanding of how a given cosolvent interacts with water, and how such interactions might change the structure of water, they offered little, if any, direct information on how the cosolvent of interest mediates the strength of the H-bonds formed between water and a third molecular component (e.g., a protein) in solution. Because water-protein H-bonding interactions are known to play a critical role in determining not only protein stability, but also protein solubility, it would therefore be very useful if one could devise a method that allows a direct assessment of how a given cosolvent mediates the ability of water to form H-bonds with proteins. It is well known that the vibrational frequency of nitriles is sensitive to local solvation environment51–66 and, especially, H-bonding interactions67–69 and thus is ideally suited to serve as a site-specific probe in this regard. Herein, we use this H-bonding probe to directly evaluate how the addition of TMAO, urea or GdnHCl affects the water-solute H-bonds of interest.
2. EXPERIMENTAL SECTION
Reagent-grade urea, GdnHCl, TMAO, and NaCl were purchased from Aldrich (Saint Louis, MO) and used as received. Their concentrations were determined by the refractive index of the corresponding solution. The PheCN-Peptide was synthesized on a PS3 peptide synthesizer (Protein Technologies), purified by reverse-phase liquid chromatography, and verified by mass spectrometry. The peptide concentration was determined optically using the absorbance of the peptide solution at 280 nm and a molar extinction coefficient of 850 cm−1 M−1. All FTIR spectra at a resolution of 1 cm−1 were collected on a Magna-IR 860 spectrometer (Nicolet, WI). The details of the setup have been described elsewhere.70 The pathlength was 53 μm for all measurements, except when 13C-urea was used as a cosolvent, where a 14 μm sample cell was used.
3. RESULTS AND DISCUSSION
Acetonitrile (ACN) is the simplest organic nitrile compound, whose stretching mode has been extensively studied and characterized in a large number of solvents.71–74 It is found that the nitrile group readily forms H-bonds with protic solvents and, more importantly for the current study, its stretching frequency shows a correlation with the H-bonding ability of the solvent, with the relation that a higher frequency corresponds to a stronger C≡N-solvent H-bond.74 For example, in water the C≡N stretching band of ACN is centered at 2259.8 cm−1, whereas in methanol it shifts to 2259.0 cm−1.73 Therefore, ACN constitutes a convenient model system to probe whether the addition of a third molecular component will affect the strength of the C≡N–H2O H-bond(s).
As shown (Figure 1), the nitrile stretching vibrational band of ACN in water collected at room temperature clearly shows a red-shift upon addition of TMAO (4.8 M), from 2260.0 to 2258.4 cm−1. This result provides the first direct indication that TMAO, which by itself cannot form H-bonds with the nitrile group, can effectively reduce the strength of the C≡N–H2O H-bond(s). To further confirm this finding, we also repeated the measurements at other temperatures. As indicated (Figure 2), the abovementioned red-shift is maintained over the entire temperature range of the experiment. Furthermore, in both pure water and the TMAO solution, the center frequency of the nitrile stretching band decreases with increasing temperature, due presumably to thermally induced weakening of the corresponding C≡N–H2O H-bond(s).76
Figure 1.

The C≡N stretching vibrational bands of ACN (~10 mM) in water and 4.8 M TMAO aqueous solution, as indicated. These data were collected at 25.9 and 25.1 °C, respectively. For easy comparison, the spectrum obtained in TMAO solution has been scaled.
Figure 2.

Temperature dependence of the peak frequency of the C≡N stretching vibrational band of ACN (~50 mM) obtained under different solvent conditions, as indicated. The concentrations of the cosolvent or cosolute in each case were: [TMAO] = 4.8 M, [Urea] = 5.2 M, [GdnHCl] = 5.1 M, and [NaCl] = 5.0 M.
What is more interesting to note is that urea and GdnHCl also induce a similar red-shift in the nitrile stretching band of ACN (Figure 2). This result is rather unexpected as both cosolvents are well known to have an opposite effect on protein solubility and stability compared to TMAO. On the other hand, addition of NaCl leads to a slight but measurable increase in the nitrile stretching frequency (Figure 2). Thus, taken together, these results corroborate our expectation that the nitrile stretching vibration can be used to probe cosolvent induced changes in local H-bond environment.
It is well recognized that the solute-solvent interaction energy is influenced by many factors and thus is difficult to determine, especially based on spectroscopic data alone.77 On the other hand, it is possible to quantify the change in local electric field and thus the change in solvent-solute electrostatic interaction energy using experimentally determined vibrational frequency shift of an appropriate vibrator and its Stark tuning rate. However, application of the C≡N stretching vibration in this regard is not straightforward as Cho and coworkers have shown that the nitrile stretching frequency shift caused by C≡N–H2O H-bonding interactions cannot be quantitatively described by a simple linear Stark effect relationship.78 Therefore, below we only seek to obtain a qualitative estimate of the extent to which the ACN-water electrostatic interaction energy changes in response to addition of a given cosolvent, by assuming that the vibrational frequency shift of the nitrile moiety can be described by its Stark tuning rate. For ACN, this assumption may not be totally unreasonable as the computational results of Cho and coworkers51 have suggested that the electric field vector components parallel to the C≡N molecular axis are the dominant factors of the nitrile stretching frequency shift induced by C≡N–H2O H-bonding interactions. The Stark tuning rate of the nitrile stretching vibration of ACN has been determined to be 0.43 cm−1/(MV/cm).79,80 By assuming the permanent dipole moment of ACN (3.92 D) to be independent of solvent conditions, a simple calculation indicates that the addition of TMAO (4.8 M) results in a decrease in the ACN-water electrostatic interaction energy due to decreased H-bonding interactions by approximately 0.69 kcal/mol. While this value cannot be simply interpreted as the TMAO-induced change in the solvation energy of ACN, its trend is nevertheless consistent with the role of TMAO as a protein protectant.
The ability of urea and GdnHCl to decrease the nitrile stretching frequency of ACN seems to be inconsistent with their protein-denaturing abilities. To verify the findings obtained with ACN, we further measured the effect of the abovementioned cosolvents on the nitrile stretching frequency of a pentapeptide that contains an unnatural amino acid, p-cyanophenylalanine (PheCN). Similar to that of ACN, the nitrile stretching mode of PheCN has been shown to be sensitive to the local hydration status of peptides and proteins and, hence, can be used as a local H-bond reporter.67,76,81 The primary reason that we employ a short peptide (sequence: GK-PheCN-TV, hereafter referred to as PheCN-Peptide), instead of a model protein, is to avoid the denaturant-induced conformational transitions of the latter, which could complicate data interpretation.
As shown (Figure 3), similar to that observed for ACN, addition of TMAO induces a red-shift in the nitrile stretching band of the PheCN-Peptide, in comparison with that obtained in pure water. This result further corroborates the above notion that TMAO, with its inability to directly form a H-bond with the nitrile group, must mediate the nitrile-water interaction by reducing the corresponding H-bond strength. Perhaps more important is that, consistent with the results obtained with ACN, both urea and GdnHCl induce a similar, albeit smaller, red-shift in the nitrile stretching frequency of the PheCN-Peptide (Figure 4). In comparison, addition of NaCl results in a blue-shift in the nitrile stretching frequency and hence a strengthening of the corresponding C≡N–H2O H-bond(s). While the current spectroscopic results do not allow a direct assessment of the effect of the added cosolvents on water-water H-bonding interactions, those obtained with NaCl appear to be consistent with several previous studies,82 which demonstrated that with increasing NaCl concentration, the strength of water-water H-bonds decreases.83,84
Figure 3.

The C≡N stretching vibrational bands of the PheCN peptide (~15 mM) in water and 4.8 M TMAO aqueous solution, as indicated. These data were collected at 27.7 and 25.2 °C, respectively. For easy comparison, the spectrum obtained in TMAO solution has been normalized to that obtained in water.
Figure 4.
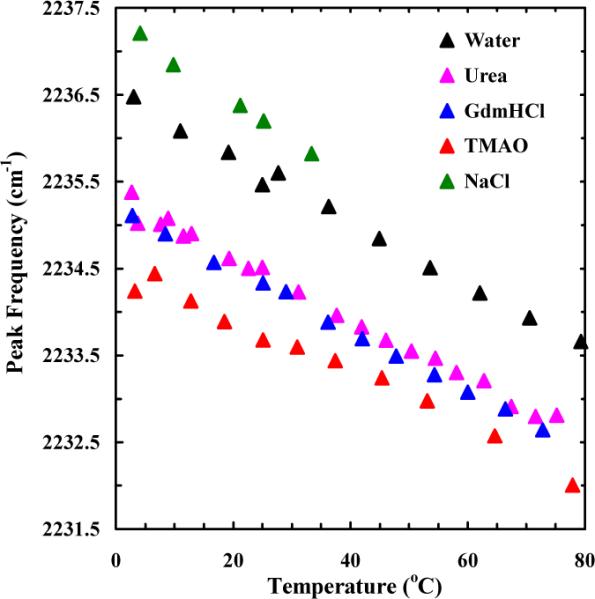
Temperature dependence of the peak frequency of the C≡N stretching vibrational band of the PheCN-Peptide obtained under different solvent conditions, as indicated. The concentrations of the cosolvent or cosolute in each case were: [TMAO] = 4.8 M, [Urea] = 5.2 M, [GdnHCl] = 5.1 M, and [NaCl] = 5.0 M.
Despite having distinctively different effects on protein stability, our results suggest that TMAO and urea both act to decrease the C≡N–H2O H-bond strength. To verify that this effect is not specific to the nitrile probe, we further studied the effect of TMAO and urea on the amide I' band of N-methylacetamide (NMA), a compound that contains a single amide unit and has been widely used as the simplest model of protein backbone.85,86 The amide I band of polypeptides arises mainly from the backbone C=O stretching vibrations and is an established IR probe of protein conformations,87 due to its sensitivity to various structural determinants, such as H-bonding interactions. Unlike nitriles, forming a stronger H-bond with the solvent will lead to a decrease in the amide I' vibrational frequency of NMA.
As shown (Figure 5), the amide I' band of NMA obtained in TMAO solution is shifted to higher wavenumber compared to that obtained in D2O, akin to the dehydration-induced blue-shift of the amide I' band of helical peptides.88 Thus, this result indicates that addition of TMAO decreases the strength of the H-bond(s) formed between the carbonyl group and water molecules, in agreement with the simulation study of Gao and coworker.89 As indicated (Figure 5), 13C-urea shows a similar effect (but to a lesser extent). It should be noted that because 12C-urea absorbs strongly in the amide I' region, 13C-urea was used herein to allow the measurement of part of the amide I' band of interest. This result is consistent with the results of Cremer and coworkers,90 which demonstrated that urea and its methyl derivatives can cause the amide stretching vibration of Poly(N-isopropylacrylamide) to shift to higher frequencies. Taken together, the C≡N and C=O results therefore argue strongly that the observed decrease/increase in the strength of solute–H2O H-bonds is not specific to the spectroscopic reporter used; rather, it intimately reports the perturbation of the cosolvent of interest on water's hydrogen bonding ability.
Figure 5.

FTIR spectra of NMA in the amide I' region at 25.0 °C under different solvent conditions, as indicated. The concentrations of TMAO and 13C-urea were 4.7 and 5.1 M, respectively. Since the C=O stretching vibrational band of 12C-urea overlaps strongly with that of NMA, 13C-urea was used in the current case. The NMA concentrations were ~50 mM for the 13C-urea experiment and ~10 mM for other measurements. For easy comparison, the spectra obtained in TMAO and 13C-urea solutions have been normalized to that obtained in D2O.
Our observation that TMAO decreases the solute-water hydrogen bonding interactions is consistent with its ability to protect proteins from unfolding. Water always seeks to form additional H-bonds, e.g., with any exposed protein amide units; hence, decreasing the protein-water H-bonding interaction strength is expected to strengthen the intra-protein H-bonds, as observed in a recent MD simulation,91 and consequently increasing the stability of the folded state. In addition, our results are in agreement with several previous studies.39,92,93 For example, Vanderkooi and co-workers have shown that addition of TMAO to a mixture of 5% H2O and 95% D2O leads to a significant broadening, primarily at the low-frequency side of the OH stretching band, indicative of an enhancement of the population of a more strongly H-bonded water species.46 It is reasonable to assume that such water species would form weaker H-bonds with other solute molecules, as observed in the present case. Similarly, our TMAO results are corroborated by the molecular dynamics (MD) simulation study of Gao and co-workers,88 which indicated that TMAO enhances the tetrahedral water structure and weakens the interactions between the amide carbonyl group and water molecules.94 In addition, our results suggest that when considering the fact that proteins normally contain a large number of sites capable of forming hydrogen bonds with water, the TMAO-induced weakening of protein-water H-bonds could play an important role in the overall stabilizing effect of TMAO, which is entirely consistent with the notion that TMAO acts to make water a poor solvent for the protein backbone.42,95,96
Many studies have shown that urea and GdnHCl are only weakly hydrated and, consequently, have little effect on the water-water H-bonding network.81,97,98 Thus, it is surprising that our results indicate that these cosolvents not only have a significant effect on the C≡N–H2O H-bond(s), but they act to reduce the strength of such H-bonds. In addition, the data obtained with NMA (Figure 5) suggests that urea could also decrease the interaction between water and protein backbone carbonyls, though probably to a lesser extent compared to TMAO. Considering the fact that both urea and GdnHCl are strong protein denaturants, these results are rather intriguing. In other words, based on these results alone one might, for example, predict that urea is a protecting osmolyte, instead of a denaturing one. Thus, these results provide strong evidence in support of the notion that urea and GdnHCl destabilize proteins via specific binding interactions98,99 and, in particular, the conclusion reached by Zhou and coworkers that the electrostatic (hydrogen-bonded) interactions only plays a relatively minor (even negative) role in urea-induced protein denaturation.37 Furthermore, our results are consistent with the viewpoint that for a given denaturing agent a direct correlation may not exist between its denaturation efficiency and its ability to modify water structure.82,100
Since both urea and guanidinium are hydrogen bond donors, it is possible that the observed vibrational frequency shifts do not report a change in the corresponding solute-water H-bonding environment. Instead, they correspond to the formation of a new type of H-bond, i.e., that formed between the nitrile moiety and denaturant. While we cannot entirely rule out this possibility, we believe this is an unlikely scenario, at least in the case of the PheCN-Peptide. This is because ample evidences in the literature suggest that urea and guanidinium show preferential binding to hydrophobic sidechains (PheCN in the current case), in a parallel or side-to-side fashion,97 which is incompatible with a hydrogen-bonded configuration in which the denaturant and PheCN are aligned in a head-to-tail manner. In addition, if the denaturants participate directly to form either new or additional H-bonds, the nitrile stretching frequency is expected to increase, which is not observed.
In light of the current findings, it would be quite useful to design new experiments that would allow a more direct visualization of the specific interactions between denaturants and proteins. In principle, such interactions could be studied in great detail by multidimensional infrared spectroscopy, for example, by examining the vibrational couplings between two well chosen vibrators, one on the peptide (such as a nitrile group on the sidechain or an amide carbonyl on the backbone), and another on the cosolvent (such as urea's carbonyl group).
4. CONCLUSIONS
In summary, we have investigated how TMAO, urea and GdnHCl affect the hydrogen bonding interactions between a solute molecule of interest and water via the vibrational frequency shift of a nitrile IR probe, aiming to shed new insight into the protecting or denaturing mechanism of these cosolvents. Our results demonstrated that all three cosolvents act to decrease the strength of the hydrogen bonds formed between water and the nitrile moiety. We believe that this is an important finding as it suggests that (1) both urea and GdnHCl are capable of perturbing the structure of water, at least the water-water interactions in the hydration shell of the solute, (2) simply knowing how a given cosolvent affects the structure and dynamics of water is probably insufficient to allow an accurate prediction of its protecting or denaturing efficiency, and (3) more specifically, urea and GdnHCl denature proteins through specific interactions with the targets.
ACKNOWLEDGMENTS
We gratefully acknowledge financial support from the National Institutes of Health (GM-065978) and a National Science Foundation Graduate Research Fellowship to IMP (DGE-0822).
REFERENCES
- (1).Tanford C. J. Am. Chem. Soc. 1964;86:2050–2059. [Google Scholar]
- (2).Tanford C. Adv. Protein Chem. 1970;24:1–95. [PubMed] [Google Scholar]
- (3).Anfinsen CB. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- (4).Greene RF, Pace CN. J. Biol. Chem. 1974;249:5388–5393. [PubMed] [Google Scholar]
- (5).Yancey PH, Clark ME, Hand SC, Bowlus RD, Somero GN. Science. 1982;217:1214–1222. doi: 10.1126/science.7112124. [DOI] [PubMed] [Google Scholar]
- (6).Pace CN. Methods Enzymol. 1986;131:266–80. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- (7).Alonso DOV, Dill KA. Biochemistry. 1991;30:5974–5985. doi: 10.1021/bi00238a023. [DOI] [PubMed] [Google Scholar]
- (8).Duffy EM, Kowalczyk PJ, Jorgensen WL. J. Am. Chem. Soc. 1993;115:9271–9275. [Google Scholar]
- (9).From NB, Bowler BE. Biochemistry. 1998;37:1623–1631. doi: 10.1021/bi970620l. [DOI] [PubMed] [Google Scholar]
- (10).Bolen DW, Baskakov IV. J. Mol. Biol. 2001;310:955–963. doi: 10.1006/jmbi.2001.4819. [DOI] [PubMed] [Google Scholar]
- (11).Uversky VN, Li J, Fink AL. FEBS Lett. 2001;509:31–35. doi: 10.1016/s0014-5793(01)03121-0. [DOI] [PubMed] [Google Scholar]
- (12).Timasheff SN. Biophys. Chem. 2002;101:99–111. doi: 10.1016/s0301-4622(02)00188-6. [DOI] [PubMed] [Google Scholar]
- (13).Schellman JA. Biophys. Chem. 2002;96:91–101. doi: 10.1016/s0301-4622(02)00009-1. [DOI] [PubMed] [Google Scholar]
- (14).Bennion BJ, Daggett V. Proc. Natl. Acad. Sci. U.S.A. 2004;101:6433–6438. doi: 10.1073/pnas.0308633101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Moglich A, Krieger F, Kiefhaber T. J. Mol. Biol. 2005;345:153–162. doi: 10.1016/j.jmb.2004.10.036. [DOI] [PubMed] [Google Scholar]
- (16).Auton M, Bolen DW. Proc. Natl. Acad. Sci. U.S.A. 2005;102:15065–15068. doi: 10.1073/pnas.0507053102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Athawale MV, Dordick JS, Garde S. Biophys. Chem. 2005;89:858–866. doi: 10.1529/biophysj.104.056671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Caballero-Herrera A, Nordstrand K, Berndt KD, Nilsson L. Biophys. J. 2005;89:842–857. doi: 10.1529/biophysj.105.061978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Street TO, Bolen DW, Rose GD. Proc. Natl. Acad. Sci. U.S.A. 2006;103:13997–14002. doi: 10.1073/pnas.0606236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Auton M, Holthauzen LMF, Bolen DW. Proc. Natl. Acad. Sci. U.S.A. 2007;104:15317–15322. doi: 10.1073/pnas.0706251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Di Michele A, Freda M, Onori G, Paolantoni M, Santucci A, Sassi P. J. Phys. Chem. B. 2006;110:21077–21085. doi: 10.1021/jp068055w. [DOI] [PubMed] [Google Scholar]
- (22).Paul S, Patey GN. J. Phys. Chem. B. 2007;111:7932–7933. doi: 10.1021/jp0733668. [DOI] [PubMed] [Google Scholar]
- (23).Hua L, Zhou R, Thirumalai D, Berne BJ. Proc. Natl. Acad. Sci. U.S.A. 2008;105:16928–16933. doi: 10.1073/pnas.0808427105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Paul S, Patey GN. J. Phys. Chem. B. 2008;112:11106–11111. doi: 10.1021/jp803956s. [DOI] [PubMed] [Google Scholar]
- (25).Athawale MV, Sarupria S, Garde S. J. Phys. Chem. B. 2008;112:5661–5670. doi: 10.1021/jp073485n. [DOI] [PubMed] [Google Scholar]
- (26).Zangi R, Zhou RH, Berne BJ. J. Am. Chem. Soc. 2009;131:1535–1541. doi: 10.1021/ja807887g. [DOI] [PubMed] [Google Scholar]
- (27).Meersman F, Bowron D, Soper AK, Koch MHJ. Biophys. J. 2009;97:2559–2566. doi: 10.1016/j.bpj.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kuffel A, Zielkiewicz J. J. Chem. Phys. 2010;133 doi: 10.1063/1.3464768. [DOI] [PubMed] [Google Scholar]
- (29).Canchi DR, Paschek D, Garcia AE. J. Am. Chem. Soc. 2010;132:2338–2344. doi: 10.1021/ja909348c. [DOI] [PubMed] [Google Scholar]
- (30).Zhang Y, Cremer PS. Annu. Rev. Phys. Chem. 2010;61:63–83. doi: 10.1146/annurev.physchem.59.032607.093635. [DOI] [PubMed] [Google Scholar]
- (31).Holthauzen LMF, Roesgen J, Bolen DW. Biochemistry. 2010;49:1310–1318. doi: 10.1021/bi9015499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hu CY, Lynch GC, Kokubo H, Pettitt BM. Proteins: Struct., Funct., Bioinf. 2010;78:695–704. doi: 10.1002/prot.22598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Auton M, Roesgen J, Sinev M, Holthauzen LMF, Bolen DW. Biophys. Chem. 2011;159:90–99. doi: 10.1016/j.bpc.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Guinn EJ, Pegram LM, Capp MW, Pollock MN, Record MT., Jr. Proc. Natl. Acad. Sci. U.S.A. 2011;108:16932–16937. doi: 10.1073/pnas.1109372108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Canchi DR, Garcia AE. Biophys. Chem. 2011;100:1526–1533. doi: 10.1016/j.bpj.2011.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Rossky PJ. Proc. Natl. Acad. Sci. U.S.A. 2008;105:16825–16826. doi: 10.1073/pnas.0809224105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Yang Z, Xiu P, Shi B, Hua L, Zhou R. J. Phys. Chem. B. 2012;116:8856–886. doi: 10.1021/jp304114h. [DOI] [PubMed] [Google Scholar]
- (38).Rezus YLA, Bakker HJ. Proc. Natl. Acad. Sci. U.S.A. 2006;103:18417–18420. doi: 10.1073/pnas.0606538103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bakulin AA, Pshenichnikov MS, Bakker HJ, Petersen C. J. Phys. Chem. A. 2011;115:1821–1829. doi: 10.1021/jp107881j. [DOI] [PubMed] [Google Scholar]
- (40).Bennion BJ, Daggett V. Proc. Natl. Acad. Sci. U.S.A. 2003;100:5142–5147. doi: 10.1073/pnas.0930122100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Rezus YLA, Bakker HJ. Phys. Rev. Lett. 2007;99 doi: 10.1103/PhysRevLett.99.148301. [DOI] [PubMed] [Google Scholar]
- (42).Bolen DW, Rose GD. Annu. Rev. Biochem. 2008;77:339–362. doi: 10.1146/annurev.biochem.77.061306.131357. [DOI] [PubMed] [Google Scholar]
- (43).Guo F, Friedman JM. J. Phys. Chem. B. 2009;113:16632–16642. doi: 10.1021/jp9072284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Panuszko A, Bruzdziak P, Zielkiewicz J, Wyrzykowski D, Stangret J. J. Phys. Chem. B. 2009;113:14797–14809. doi: 10.1021/jp904001m. [DOI] [PubMed] [Google Scholar]
- (45).Rezus YLA, Bakker HJ. J. Phys. Chem. B. 2009;113:4038–4044. doi: 10.1021/jp805458p. [DOI] [PubMed] [Google Scholar]
- (46).Sharp KA, Madan B, Manas E, Vanderkooi JM. J. Chem. Phys. 2001;114:1791–1796. [Google Scholar]
- (47).Lindquist BA, Corcelli SA. J. Phys. Chem. B. 2008;112:6301–6303. doi: 10.1021/jp802039e. [DOI] [PubMed] [Google Scholar]
- (48).Weeks CL, Polishchuk A, Getahun Z, DeGrado WF, Spiro TG. J. Raman Spectrosc. 2008;39:1606–1613. doi: 10.1002/jrs.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Boxer SG. J. Phys. Chem. B. 2009;113:2972–2983. doi: 10.1021/jp8067393. [DOI] [PubMed] [Google Scholar]
- (50).Taskent-Sezgin H, Chung J, Patsalo V, Miyake-Stoner SJ, Miller AM, Brewer SH, Mehl RA, Green DF, Raleigh DP, Carrico I. Biochemistry. 2009;48:9040–9046. doi: 10.1021/bi900938z. [DOI] [PubMed] [Google Scholar]
- (51).Choi J-H, Oh K-I, Lee H, Lee C, Cho M. J. Chem. Phys. 2008;128:134506–134513. doi: 10.1063/1.2844787. [DOI] [PubMed] [Google Scholar]
- (52).Aschaffenburg DJ, Moog RS. J. Phys. Chem. B. 2009;113:12736–12743. doi: 10.1021/jp905802a. [DOI] [PubMed] [Google Scholar]
- (53).Waegele MM, Tucker MJ, Gai F. Chem. Phys. Lett. 2009;478:249–253. doi: 10.1016/j.cplett.2009.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Liu J, Strzalka J, Tronin A, Johansson JS, Blasie JK. Biophys. J. 2009;96:4176–4187. doi: 10.1016/j.bpj.2009.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Lindquist BA, Furse KE, Corcelli SA. Phys. Chem. Chem. Phys. 2009;11:8119–8132. doi: 10.1039/b908588b. [DOI] [PubMed] [Google Scholar]
- (56).McMahon HA, Alfieri KN, Clark CAA, Londergan CH. J. Phys. Chem. Lett. 2010;1:850–855. doi: 10.1021/jz1000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Waegele MM, Gai F. J. Phys. Chem. Lett. 2010;1:781–786. doi: 10.1021/jz900429z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Marek P, Mukherjee S, Zanni MT, Raleigh DP. J. Mol. Biol. 2010;400:878–888. doi: 10.1016/j.jmb.2010.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Lee H, Choi J-H, Cho M. Phys. Chem. Chem. Phys. 2010;12:12658–12669. doi: 10.1039/c0cp00214c. [DOI] [PubMed] [Google Scholar]
- (60).Fafarman AT, Sigala PA, Herschlag D, Boxer SG. J. Am. Chem. Soc. 2010;132:12811–12813. doi: 10.1021/ja104573b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Hu W, Webb LJ. J. Phys. Chem. Lett. 2011;2:1925–1930. [Google Scholar]
- (62).Gai XS, Coutifaris BA, Brewer SH, Fenlon EE. Phys. Chem. Chem. Phys. 2011;13:5926–5930. doi: 10.1039/c0cp02774j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Zhang Z, Guo Y, Lu Z, Velarde L, Wang H-F. J. Phys. Chem. C. 2012;116:2976–2987. [Google Scholar]
- (64).Stafford AJ, Walker DM, Webb LJ. Biochemistry. 2012;51:2757–2767. doi: 10.1021/bi201225p. [DOI] [PubMed] [Google Scholar]
- (65).Bagchi S, Fried SD, Boxer SG. J. Am. Chem. Soc. 2012;134:10373–10376. doi: 10.1021/ja303895k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Zimmermann J, Thielges MC, Seo YJ, Dawson PE, Romesberg FE. Angew. Chem. Int. Ed. 2011;50:8333–8337. doi: 10.1002/anie.201101016. [DOI] [PubMed] [Google Scholar]
- (67).Getahun Z, Huang CY, Wang T, De Leon B, DeGrado WF, Gai F. J. Am. Chem. Soc. 2003;125:405–411. doi: 10.1021/ja0285262. [DOI] [PubMed] [Google Scholar]
- (68).Ghosh A, Remorino A, Tucker MJ, Hochstrasser RM. Chem. Phys. Lett. 2009;469:325–330. doi: 10.1016/j.cplett.2008.12.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Waegele MM, Culik RM, Gai F. J. Phys. Chem. Lett. 2011;2:2598–2609. doi: 10.1021/jz201161b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Huang CY, Getahun Z, Zhu YJ, Klemke JW, DeGrado WF, Gai F. Proc. Natl. Acad. Sci. U.S.A. 2002;99:2788–2793. doi: 10.1073/pnas.052700099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Purcell KF, Drago RS. J. Am. Chem. Soc. 1966;88:919–924. [Google Scholar]
- (72).Fawcett WR, Liu GJ, Kessler TE. J. Phys. Chem. 1993;97:9293–9298. [Google Scholar]
- (73).Reimers JR, Hall LE. J. Am. Chem. Soc. 1999;121:3730–3744. [Google Scholar]
- (74).Mountain RD. J. Phys. Chem. B. 2010;114:16460–16464. doi: 10.1021/jp105272q. [DOI] [PubMed] [Google Scholar]
- (75).Eaton G, Penanunez AS, Symons MCR. J. Chem. Soc., Faraday Trans. 1 F. 1988;84:2181–2193. [Google Scholar]
- (76).Huang CY, Wang T, Gai F. Chem. Phys. Lett. 2003;371:731–738. [Google Scholar]
- (77).Lee H, Lee G, Jeon J, Cho M. J. Phys. Chem. A. 2012;116:347–357. doi: 10.1021/jp209709e. [DOI] [PubMed] [Google Scholar]
- (78).Choi J-H, Cho M. J. Chem. Phys. 2011;134:154513–1545254. doi: 10.1063/1.3580776. [DOI] [PubMed] [Google Scholar]
- (79).Andrews SS, Boxer SG. J. Phys. Chem. A. 2000;104:11853–11863. [Google Scholar]
- (80).Andrews SS, Boxer SG. J. Phys. Chem. A. 2002;106:469–477. [Google Scholar]
- (81).Tucker MJ, Getahun Z, Nanda V, DeGrado WF, Gai F. J. Am. Chem. Soc. 2004;126:5078–5079. doi: 10.1021/ja032015d. [DOI] [PubMed] [Google Scholar]
- (82).Mountain RD, Thirumalai D. J. Phys. Chem. B. 2004;108:19711–19716. [Google Scholar]
- (83).Collins KD. Methods. 2004;34:300–311. doi: 10.1016/j.ymeth.2004.03.021. [DOI] [PubMed] [Google Scholar]
- (84).Sharp KA, Vanderkooi JM. Acc. Chem. Res. 2010;43:231–239. doi: 10.1021/ar900154j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Ham S, Kim JH, Lee H, Cho MH. J. Chem. Phys. 2003;118:3491–3498. [Google Scholar]
- (86).DeCamp MF, DeFlores L, McCracken JM, Tokmakoff A, Kwac K, Cho M. J. Phys. Chem. B. 2005;109:11016–11026. doi: 10.1021/jp050257p. [DOI] [PubMed] [Google Scholar]
- (87).Serrano AL, Waegele MM, Gai F. Protein Sci. 2012;21:157–170. doi: 10.1002/pro.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Mukherjee S, Chowdhury P, Gai F. J. Phys. Chem. B. 2007;111:4596–4602. doi: 10.1021/jp0689060. [DOI] [PubMed] [Google Scholar]
- (89).Wei H, Fan Y, Gao YQ. J. Phys. Chem. B. 2010;114:557–568. doi: 10.1021/jp9084926. [DOI] [PubMed] [Google Scholar]
- (90).Sagle LB, Zhang Y, Litosh VA, Chen X, Cho Y, Cremer PS. J. Am. Chem. Soc. 2009;131:9304–9310. doi: 10.1021/ja9016057. [DOI] [PubMed] [Google Scholar]
- (91).Hwang S, Shao Q, Williams H, Hilty C, Gao YQ. J. Phys. Chem. B. 2011;115:6653–6660. doi: 10.1021/jp111448a. [DOI] [PubMed] [Google Scholar]
- (92).Di Michele A, Freda M, Onori G, Santucci A. J. Phys. Chem. A. 2004;108:6145–6150. [Google Scholar]
- (93).Reddy PM, Taha M, Venkatesu P, Kumar A, Lee M-J. J. Chem. Phys. 2012;136:234904. doi: 10.1063/1.4729156. [DOI] [PubMed] [Google Scholar]
- (94).Zou Q, Bennion BJ, Daggett V, Murphy KP. J. Am. Chem. Soc. 2002;124:1192–1202. doi: 10.1021/ja004206b. [DOI] [PubMed] [Google Scholar]
- (95).Tran HT, Mao A, Pappu RV. J. Am. Chem. Soc. 2008;130:7380–7392. doi: 10.1021/ja710446s. [DOI] [PubMed] [Google Scholar]
- (96).Cho SS, Reddy G, Straub JE, Thirumalai D. Phys. Chem. B. 2011;115:13401–13407. doi: 10.1021/jp207289b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Mason PE, Neilson GW, Dempsey CE, Barnes AC, Cruickshank JM. Proc. Natl. Acad. Sci. U.S.A. 2003;100:4557–4561. doi: 10.1073/pnas.0735920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Mason PE, Neilson GW, Enderby JE, Saboungi ML, Dempsey CE, MacKerell AD, Brady JW. J. Am. Chem. Soc. 2004;126:11462–11470. doi: 10.1021/ja040034x. [DOI] [PubMed] [Google Scholar]
- (99).Canchi DR, Garcia AE. Biophys. J. 2011;100:1526–1533. doi: 10.1016/j.bpj.2011.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Batchelor JD, Olteanu A, Tripathy A, Pielak GJ. J. Am. Chem. Soc. 2004;126:1958–1961. doi: 10.1021/ja039335h. [DOI] [PubMed] [Google Scholar]