

Abstract
TGF-β plays a dual role in epithelial carcinogenesis with the potential to either suppress or promote tumor progression. We found that levels of Smad3 mRNA, a critical mediator of TGF-β signaling, are reduced by ~60% in human breast cancer. We therefore used conditionally immortalized mammary epithelial cells (IMECs) of differing Smad3 genotypes to quantitatively address the Smad3 requirement for different biological responses to TGF-β. We found that a two-fold reduction in Smad3 gene dosage led to complex effects on TGF-β responses; the growth inhibitory response was retained, the pro-apoptotic response was lost, the migratory response was reduced and the invasion response was enhanced. Loss of the pro-apoptotic response in the Smad3+/- IMECs correlated with loss of Smad3 binding to the Bcl-2 locus, while retention of the growth inhibitory response in Smad3+/- IMECs correlated with retention of Smad3 binding to the c-Myc locus. Addressing the integrated outcome of these changes in vivo, we showed that reduced Smad3 levels enhanced metastasis in two independent models of metastatic breast cancer. Our results suggest that different biological responses to TGF-β in the mammary epithelium are differentially affected by Smad3 dosage, and that a mere two-fold reduction in Smad3 is sufficient to promote metastasis.
Introduction
Transforming growth factor βs (TGF-βs) are widely expressed, pleiotropic growth factors that modulate a variety of key biological responses. In the adult animal, the TGF-βs are important in maintaining tissue homeostasis and in orchestrating response to stress or injury, and perturbations in the TGF-β signaling pathway have been implicated in a wide variety of pathological processes. In carcinogenesis, TGF-βs play a complex dual role in many epithelial tissues including the mammary gland, as evidenced by numerous pre-clinical studies and correlative clinical data (reviewed in (1-4)). Early in tumorigenesis, tumor suppressive effects of TGF-βs generally dominate, involving biological responses such as inhibition of proliferation, induction of apoptosis or replicative senescence, maintenance of genomic stability and regulation of differentiation. However in advanced disease, resistance to the tumor suppressive effects of TGF-β frequently develops, due to altered expression and/or activity of components of the TGF-β signaling pathway, or to progression-related changes that specifically affect the effector arm of the tumor suppression program. At the same time, tumor cell-autonomous responses to TGF-β that promote progression, such as invasion and migration, are enhanced or unmasked by the accumulating genetic and epigenetic changes in the tumor. Furthermore, increased TGF-β in the tumor bed leads to tumor-promoting effects of TGF-β on the microenvironment, such as suppression of immunesurveillance and enhanced angiogenesis.
The canonical TGF-β signaling pathway involves TGF-β induced phosphorylation of two central mediators, Smad2 and Smad3, which translocate to the nucleus and regulate gene expression (5;6). Smad3 appears to be the more important mediator in the adult organism, since Smad2 is either dispensible, or actually opposes Smad3, in many biological responses (7-9). However, unlike Smad2, Smad3 is rarely mutated or deleted in human cancer (10). Here we have shown that human breast tumors have Smad3 mRNA levels that are reduced approximately two-fold compared to the adjacent normal breast tissue. We have used a genetic approach to ask what effect this reduction in Smad3 has on TGF-β responses and tumorigenesis in the breast epithelium. Unexpectedly, we found that a mere two-fold reduction in Smad3 significantly affected multiple TGF-β responses in vitro in a variety of different ways. In vivo, the integrated outcome of these effects was an enhancement of metastasis in two different breast cancer models.
Materials and Methods
Cell lines and genetic modification
Conditionally immortalized mouse mammary epithelial cells (IMECs) with zero, one or two alleles of Smad3 were generated by intercrossing the Smad3DelEx8 knockout mouse (11) and the H-2Kb-tsA58 transgenic “Immortomouse”, which broadly expresses a temperature sensitive SV-40 large T antigen (12). The IMEC cells were generated and propagated as previously described (7). Briefly, IMECS were expanded and propagated under permissive conditions: growth at 33°C (5% CO2) in Ham’s F12 medium supplemented with 10% fetal bovine serum (FBS), 10 ng/ml EGF, 5 μg/ml insulin, 1 μg/ml hydrocortisone, 5 ng/mL cholera toxin, 50 μg/ml gentamycin (“complete” medium) and 30 units/ml interferon gamma (IFNγ). For assays, cells were switched to non-permissive conditions by growing at 37°C (5% CO2) in the absence of IFNγ for 2-4 days prior to the assay, to provide sufficient time for T antigen expression to fully decay. For the Smad3+/- genotype that was the focus of this study, three independent IMEC isolates were generated: EAK4, IMEC49 and IMEC 52. Mvt-1, a metastatic mouse mammary cell line derived from a mammary tumor that arose in an MMTV-c-Myc/VEGF bitransgenic mouse (13) was a gift of the late Dr. Robert Dickson (Georgetown University), and was cultured at 37°C (5% CO2) in DMEM with 10% FBS. For genetic modification of the Mvt-1 line to knockdown Smad3, short hairpin RNAs (shRNA) against Smad3 (5’-GGCCATCACCACGCAGAAC -3’) or GFP (5’-AAGACCCGCGCCGAGGTGAAG-3’) were cloned into the pLKO.1 lentiviral vector. To add back Smad3 to the Smad3+/- IMECs, human Smad3 was cloned into a modified pFUGW lentiviral backbone by Gateway recombinational cloning, with expression driven by the CMV13 promoter. A stuffer sequence was used in place of Smad3 to generate the control construct. In both cases, transduced cells were selected with puromycin for 5 days, and in vivo experiments were performed within 3-5 passages. For knockdown and add-back experiments, Smad3 protein levels were quantitated by SimpleWestern™ (see below).
Conventional Western blot, Simple Western™ analyses and zymography
Conventional Western blot analyses, and gelatin zymography to assess matrix metalloproteinase (MMP) activity, were performed essentially as previously described (7). For Western blots of phospho-Smads, serum-starved cells were treated with 2 ng/ml TGF-β for 1h. For Western blots of other TGF-β target proteins, serum-starved cells were treated for 24h. Antibodies used were as follows: Smad2, Zymed # 51-1300 or Invitrogen #511300 (1:1000); Phospho-Smad2, Cell Signaling #3108S (1:1000); Smad3, Abcam #ab28379 (1:1000); Phospho-Smad3, Epitomics #1880-1 (1:1000); β-actin, Sigma #A1978 (1:5000); c-myc, Active Motif #39012 (1:500); Bcl-2, Cell Signaling (1:1000); cleaved Caspase 3, Cell Signaling #9661 (1:1000); p15INK4B, Santa Cruz Biotechnology #sc-613 (1:1000); p21Cip1, Santa Cruz Biotechnology #sc-6246 (1:1000). Membranes were blocked with 5% BSA (Smad2) or 5% nonfat dry milk (all others).
In select cases Smad3 protein levels were quantitated using the automated capillary-based Simon SimpleWestern™ System (ProteinSimple; Santa Clara, CA). With this technique, automation and the elimination of the blotting step allows more accurate and reproducible assessment of protein levels. 60 ng of cell lysate protein in reducing buffer with fluorescent molecular weight standards was loaded into each capillary, and proteins were separated by molecular weight through stacking and separation matrices for 45 minutes at 250 V. Proteins were immobilized to capillary walls using proprietary, photoactivated capture chemistry. Capillaries were then incubated with a blocking reagent and target proteins were immunoprobed with rabbit anti-Smad3 (Epitomics #1735-1) and anti-α-tubulin (Cell Signaling Technology #2125) primary antibodies and horseradish peroxidase-conjugated anti-rabbit secondary antibodies (Jackson ImmunoResearch). A mixture of luminol and peroxide was added, the resulting chemiluminescent signal was captured by a CCD camera, and the signal intensities were quantified and analyzed using Compass Software (ProteinSimple). During analysis, the Smad3 signal was normalized to the α-tubulin loading control in the same samples.
Quantitative Real-time PCR
Real time PCR reactions were run according to the manufacturer’s instructions (Brilliant SYBR Green master Mix, Stratagene) using a BioRad CFX96 Real Time System. Expression was normalized to either PPIA or PPIB. For QRT-PCR of TGF-β targets related to the epithelial-to-mesenchymal (EMT) transition, IMEC cells were treated with TGF-β for 48h. NMuMG cells, which are very sensitive to TGF-β-induced EMT, were treated with TGF-β for 24h and served as a positive control. Primer pairs for the various targets are given in Supplementary Table S1.
Smad3 expression in human breast tissues
Total RNA from tumor and normal adjacent breast tissue from ten female breast cancer patients was purchased from Oncomatrix Inc., and Smad3 mRNA was assessed by RT-QPCR and normalized to PPIA. Patient characteristics are given in Supplementary Table S2. A meta-analysis to investigate the relationship of Smad3 mRNA expression to clinical parameters across 8 independent breast cancer cohorts was performed using the online GOBO tool (http://co.bmc.lu.se/gobo) (14). For immunohistochemistry to assess which cell types in the tumor express Smad3, unstained sections of an ER-negative, lymph node positive, grade 2 breast cancer, and matched adjacent normal tissue from the same patient were purchased from Capital Biosciences (Rockville, MD). Immunolocalization of Smad3 using rabbit anti-Smad3 from Abcam (cat # ab28379) at 0.04 μg/ml was performed as previously described (15). Prior to immunostaining, antigen retrieval in 1 mM EDTA (pH 8) was carried out at 95° C for 10 min following deparaffinization of sections. A mouse embryo section was stained as a positive control, and omission of primary antibody served as the negative control.
Smad3 Chromatin Immunoprecipitation (ChIP)
Serum-starved cells were treated with 2ng/ml TGF-β or vehicle for 1h, prior to dual cross-linking with 2mM di-(N-succinimidyl)glutarate (Thermo Scientific) and 1% formaldehyde, followed by sonication and immunoprecipitation of sonicated DNA with anti-Smad3 antibody (#28379 Chip Grade, AbCam). After immunoprecipitation, ChIPed DNA was quantitated by Q-PCR. Primers (5’-ACAGGACTTCTGCAAATGCT-3’ and 5’-AACCAGAGATCTCAAGAGCA-3’) were used to amplify a 98bp fragment encompassing the GC-rich repeat of the mouse Bcl-2 P2 promoter (16). Primers (5’-CGACTCGCCTCACTCCAGCTC-3’ and 5’-GTCCGCTCACTCCCTCTGTC-3’) were designed to amplify a 175bp fragment corresponding to the region in the mouse c-Myc gene that is homologous to the repressive Smad binding element (RSBE) in the human c-MYC promoter (17). Results are expressed as the fold enrichment of ChIPed DNA compared to input DNA for 3 biological replicates, with the Smad3 null cells serving as an internal negative control for non-specific pull-down.
Growth inhibition, apoptosis, cell migration and invasion assays
Relative rates of proliferation of the IMEC lines were assessed by real-time imaging of cell culture confluency using an IncuCyte™ Imaging system (Essen Instruments, Ann Arbor, MI). Phase contrast images were taken every 3 hours over a period of 30 hours and the culture confluency at each timepoint was calculated and plotted. Inhibition of cell proliferation by TGF-β was assessed by [3H]-thymidine incorporation, and apoptosis was quantitated using the Cell Death Detection ELISA kit (Roche). Cell migration and invasion assays were carried out using the Transwell ® System without or with Matrigel respectively (8μm, BD Biosciences). Conditions for all assays were as previously described (7).
Tumorigenesis in transgenic and syngeneic transplant models
All animal studies were performed under institutionally approved animal study protocols. Smad3+/- mice (11) were intercrossed with MMTV-PyMT transgenic mice (18) to generate oncogenically-initiated cohorts that were wild-type or heterozygous for Smad3. The Smad3+/- mice were on an inbred, mixed C57Bl/6/BlackSwiss/129Sv background while the MMTV-PyMT mice were pure FVB/NJ. Tumorigenesis and metastasis in this transgenic model were assessed in the F1 offspring of the intercross to control for background strain effects. It should be noted that the overall incidence of lung metastases was lower on this mixed genetic background (~40%) than is seen with the pure FVB/N strain (~90%) (18). This feature of the mixed background made it possible to see a stimulatory effect of the Smad3+/- genotype on metastasis incidence. Mice were euthanized 40 days after initial detection of a palpable mammary tumor and tissues were harvested for molecular and pathological analysis. Lung metastases in histological sections were enumerated by a pathologist. For the syngeneic transplant studies, 100,000 Mvt-1 cells (derived from a metastatic tumor in an MMTV-c-Myc/VEGF bitransgenic mouse and syngeneic to the FVB/N strain (13)) were surgically implanted in the #4 mammary gland of 6-8 week old Smad3 +/+ or Smad3 +/- mice that had been backcrossed for 10 generations onto the FVB/NJ background. Tumor-bearing mice were euthanized after 7 weeks and lungs were harvested for assessment of metastatic burden as above.
Results and Discussion
Although Smad3 is rarely, if ever, deleted in human breast cancer (10), we wished to assess whether more subtle alterations in Smad3 expression levels might occur. We found that Smad3 mRNA expression was reduced in 9 out of 10 cases of human breast cancer when compared with matched adjacent normal samples, for an average reduction in Smad3 of ~60% in the tumor (Fig. 1A,B). In a meta-analysis of 1412 human breast cancer cases with Affymetrix U133A gene expression data in the GOBO database (14), Smad3 mRNA levels were significantly lower in the highest grade (Grade 3) tumors compared with lower grade tumors (Fig. 1C). Furthermore, Smad3 mRNA was also significantly lower in the poorer prognosis ER-negative breast cancers than in ER-positive tumors in the same dataset (Fig. 1D). We have previously shown that the majority of Smad3 mRNA in the mouse mammary gland is associated with the epithelial compartment (19). To confirm the presence of Smad protein in the epithelial compartment in human breast cancer, we immunostained a matched breast cancer and adjacent normal breast tissue from the same patient. The results show that Smad3 is highly expressed in the normal ductal epithelium, and that lower expression is observed in the parenchymal cells of a breast cancer from the same patient (Fig. 1E). Together, these observations all suggest that a reduction in Smad3 levels in the tumor parenchyma might contribute to breast cancer progression. Our mRNA data are also in agreement with a previous immunohistochemical study which showed significant correlations between a decrease in Smad3 nuclear abundance and high tumor grade and hormone receptor negativity in breast cancer (20).
Figure 1. Smad3 expression in human breast cancer.

A. Smad3 mRNA levels in paired human breast cancers and adjacent normal tissue, normalized to Cyclophilin A (PPIA). Clinical Stage is indicated. Statistics: Paired t-test. B. Box plot showing Smad3 mRNA levels for grouped samples in A. Statistics: Fisher Exact test. C. Box plots showing meta-analysis of Smad3 gene expression as a function of tumor grade in 1412 human breast cancers using GOBO database and analysis tools. D. Meta-analysis of Smad3 gene expression as a function of estrogen receptor (ER) status in 1620 human breast cancers, as in C. E. Smad3 immunostaining of an ER-negative, stage II human breast cancer and adjacent normal tissue. Mouse embryonic lung was the positive control. Negative control; no primary antibody. Scale bar represents 50μm.
To ask what effect a two-fold reduction in Smad3 levels might have on TGF-β responses in the mammary epithelium, we generated conditionally immortalized mouse mammary epithelial cells (IMECs) of the three Smad3 genotypes, Smad3 +/+, Smad3+/- and Smad3-/- (7). This genetic approach allowed us to precisely achieve a reduction in Smad3 levels (in the Smad3+/- IMECs) that was comparable to that seen in the human tumors. At the protein level, Smad3 expression and TGF-β-induced Smad3 phosphorylation were reduced in the Smad3+/- cells and completely absent in the Smad3-/- cells, while Smad2 expression and activation were essentially unchanged (Fig. 2A). Quantitation of Smad3 protein in the Smad3+/- IMECs using the SimpleWestern™ technology showed that Smad3 protein was reduced to 47 +/- 7% (n= 3 independent determinations) of wildtype levels (Fig. 2B). Morphologically, Smad3+/+ and Smad3+/- cells showed similar, loosely organized, colonies in culture (Fig. 2C), while Smad3-/- cells formed colonies with clearly defined boundaries, as seen previously (7). Proliferation rates were broadly similar between the three cell lines (Fig. 2D).
Figure 2. Molecular and biological characterization of IMECs with different Smad3 levels.

A. Western blot showing relative levels of total and C-terminally phosphorylated Smad2 and Smad3 in IMECs of different Smad3 (S3) genotypes. B. Electrophoretogram (left) and corresponding “virtual blot” image (right) of SimpleWestern analyses of IMEC lysates simultaneously probed for Smad3 and alpha-tubulin as an internal loading control. Smad3/tubulin ratios normalized to the Smad3 wildtype condition are shown below the virtual blot image. C. Phase contrast images of IMEC morphology in monolayer culture. D. Growth characteristics of the IMEC cultures, assessed by quantitative imaging of culture confluence in real time using an IncuCyte Imaging system. Results are mean +/- SEM (n=3).
We next used these cells to examine the effect of Smad3 dosage on two tumor suppressive effects of TGF-β (cell proliferation and apoptosis), and two pro-progression responses (migration and invasion). While all responses to TGF-β were essentially lost in Smad3 null cells, the intermediate Smad3+/- genotype showed a complex pattern of responses. Smad3 +/- IMECs retained their growth inhibitory response to TGF-β (Fig. 3A), but completely lost their pro-apoptotic response (Fig. 3B). The loss of the pro-apoptotic response in the Smad3+/- cells was seen for 3 independent cell isolates (Fig. 3C). To confirm that this loss was Smad3-dependent and not a result of compensatory changes in the Smad3+/- cells, we used lentiviral transduction to add back Smad3 to wildtype levels (Fig. 3D). Addback of Smad3 restored the apoptotic response to TGF-β to the same level as in the Smad3+/+ cells (Fig. 3E).
Figure 3. Biological responses to TGF-β in IMECs with different Smad3 levels.
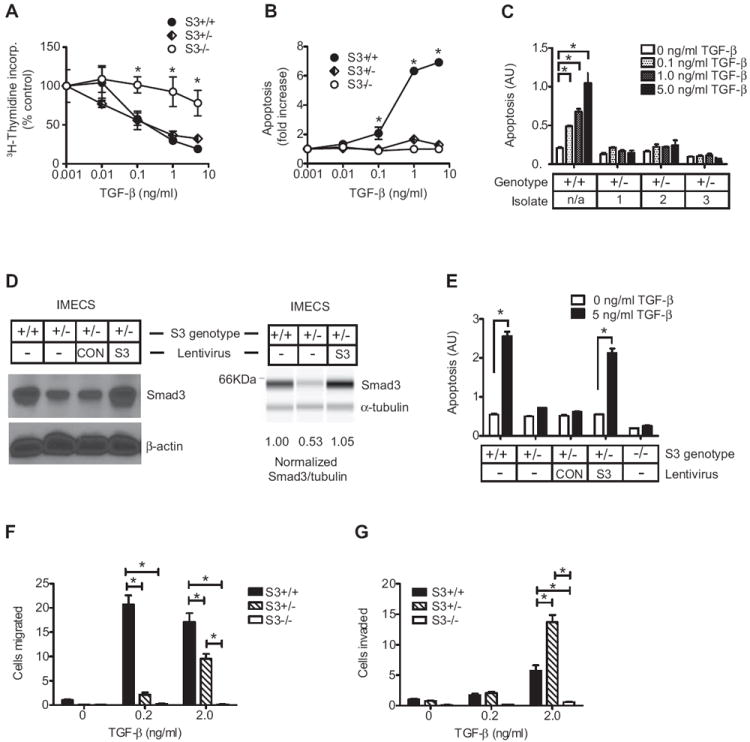
A,B. Effect of Smad3 genotype on growth inhibitory (A) or apoptotic (B) responses of IMECs to TGF-β. Results are mean +/- SEM for 3 determinations and are normalized to the untreated control for each genotype. * p<0.05, two-way ANOVA. C. The lack of an apoptotic response in Smad3+/- cells was confirmed in three independent isolates. Results are mean +/- SEM for 3 determinations and are presented without normalization to show similarity in basal apoptosis levels between genotype groups. D. Conventional Western (left) and SimpleWestern “virtual blot” (right) showing quantitative restoration of Smad3 to wildtype levels in Smad3+/- cells following lentiviral-mediated addback of Smad3. CON is a control lentivirus with stuffer sequence in place of Smad3. E. Restoration of apoptotic response to TGF-β on add-back of Smad3 to Smad3+/- IMECs. Results are expressed as in C. F,G. Effect of Smad3 genotype on migration (F) and invasion (G) responses to TGF-β. Results are mean +/- SEM for a representative experiment (n=10-20 fields /condition) normalized to the untreated Smad3+/+ condition. * p<0.05, two-way ANOVA with Bonferroni correction.
In contrast to this pattern of either full retention (growth inhibition) or complete loss (apoptosis) of response to TGF-β in the Smad3+/- cells, a different pattern was observed for the pro-progression responses. Smad3+/- cells showed a reduced migratory response to TGF-β (Fig. 3F), while the invasion response in Smad3+/- cells was at least as strong, and often stronger, than in Smad3+/+ cells (Fig. 3G). The unexpected enhancement of invasion in Smad3+/- cells was seen in 3 independent experiments at one or more of the two TGF-β concentrations tested (data not shown). Thus a two-fold reduction in Smad3 dosage can alter the outcome of TGF-β signaling in ways that differ dramatically depending on the response considered. The pattern of change in biological responses to TGF-β at the different Smad3 levels is summarized in Table 1.
Table 1.
Summary of effects of Smad3 gene dosage on biological responses to TGF-β
| Biological activity | Class of response | Response to TGF-β in IMECS of different genotypes | ||
|---|---|---|---|---|
| S3+/+ | S3+/- | S3-/- | ||
| Growth inhibition | Tumor suppression | + | + | - |
| Apoptosis | Tumor suppression | + | - | - |
| Migration | Pro-progression | + | +/- | - |
| Invasion | Pro-progression | + | + to ++ | - |
To address why the anti-proliferative and pro-apoptotic effects of TGF-β were differentially sensitive to loss of one Smad3 allele, we examined the known Smad3 targets c-Myc (17) and Bcl-2 (16;21). In agreement with the biological response outcome data, TGF-β effectively suppressed c-Myc protein levels in both Smad3+/+ and Smad3+/- cells (Fig. 4A). Expression of two other major cell cycle-related targets of TGF-β, the cyclin-dependent kinase inhibitors p21(Waf1/Cip1) and p15INK4B, was unaffected (data not shown). In contrast to the expression pattern seen with c-Myc, Bcl-2 protein was down-regulated by TGF-β only in Smad3+/+ and not Smad3+/- cells (Fig. 4B). As expected, down-regulation of Bcl-2 in Smad3+/+ cells was associated with an increase in cleaved caspase 3, a marker of apoptosis (Fig. 4B). Thus TGF-β-induced repression of c-Myc and Bcl-2 shows patterns of Smad3 dependency that correlate well with the observed biological outcomes.
Figure 4. TGF-β-induced Smad3 occupancy of c-Myc and Bcl-2 enhancer elements at different Smad3 levels.

A,B. Western blots showing TGF-β regulation of c-Myc protein (A) and Bcl-2 protein (B) in IMECs of different Smad3 genotypes. Cleaved caspase-3 reflects apoptosis. β-actin is the loading control. C,D. ChIP for Smad3 binding at the indicated sites in the c-Myc locus (C) and Bcl-2 locus (D) in response to TGF-β treatment of IMECs of the three Smad3 genotypes. TGF-β induced Smad3 binding was assessed by ChIP-qPCR, and results are expressed as fold enrichment of ChIPed DNA compared with input DNA. The Smad3-/- cells serve as a negative control. In the locus diagrams, white indicates untranslated regions and grey indicates coding regions within the specified exons. Arrows indicate approximate positions of primer pairs used. Results are mean +/- SEM (n=3). * p<0.05, t-test. E. Schematic summary of Smad3 occupancy of the enhancer elements and the transcriptional and biological response outcomes at different levels of Smad3.
We then asked whether TGF-β induced binding of Smad3 to these target gene loci was affected by Smad3 gene dosage. TGF-β represses transcription of the human c-MYC locus via binding of Smad3 to a repressive Smad binding element (RSBE) in the first exon (17). By ChIP analysis we showed strong TGF-β-induced Smad3 occupancy of the homologous region in the mouse c-Myc enhancer in both Smad3+/+ and Smad3+/- IMEC cells, consistent with the protein and biological response data (Fig. 4C). Transcriptional repression of the Bcl-2 locus involves Smad3 binding to a repressive GC-rich region in the first exon (16). In contrast to results at the c-Myc locus, we found that TGF-β treatment induced Smad3 binding to the GC-rich region of the Bcl-2 enhancer only in Smad3+/+ but not in Smad3+/- IMECs (Fig. 4D). In Smad3+/+ cells, binding of Smad3 to the GC-rich region of the Bcl-2 locus was ~ 4x weaker than binding of Smad3 to the RSBE in the c-Myc locus (compare fold enrichments) suggesting that Smad3 binds with lower affinity to the Bcl-2 enhancer than the c-Myc enhancer. Thus the reduction in Smad3 protein in the Smad3+/- state leads to selective loss of Smad3 from the low affinity repressive site in the Bcl-2 enhancer, but retention at the higher affinity repressive site in the c-Myc enhancer. The net effect is loss of the pro-apoptotic response, but retention of the growth inhibitory response in Smad3+/- cells (schematized in Fig. 4E).
The migration and invasion responses are molecularly more complex than the growth inhibitory and pro-apoptotic responses, involving many different molecular steps and processes. To address possible mechanisms underlying the unexpectedly enhanced invasion response to TGF-β in Smad3+/- cells, we first assessed MMP activity by gelatin zymography. While most MMPs were unaffected or undetectable, TGF-β induced MMP9 activity in Smad3 wildtype but not Smad3+/- cells, a pattern that cannot explain the enhanced invasion response (Suppl. Fig. 1). In some cell types, TGF-β can induce a strong epithelial-to-mesenchymal transition (EMT), which is thought to be important for invasion responses (22). Like the majority of breast cancer cell lines (23), the IMECs do not show a strong morphological EMT in response to TGF-β (7). However, we tested whether TGF-β might induce any of the molecular changes characteristic of a partial EMT, and whether the patterns of regulated gene expression in cells of the three different Smad3 genotypes correlated with the observed pattern of the invasion response. In a preliminary screen for effects of TGF-β on mRNA levels for Cdh1, Cdh2, Cdh3, Snai1, Snai2, Twist1, Zeb1 and Vimentin, only Cdh2 and Snai2 emerged as possible candidates with an appropriate pattern of response (Suppl. Fig. 2A). Snai2 did not validate in subsequent independent experiments, but Cdh2 showed an expression pattern like that of Bcl2, in that it was suppressed in Smad3+/+ but not Smad3+/- cells (Suppl. Fig. 2B). Cdh2 has been previously shown to promote invasion (24), so we hypothesize that the enhanced TGF-β-induced invasion seen in Smad3+/- cells likely involves a combination of selective loss of TGF-β-mediated repression of invasion-promoting components such as Cdh2, and/or selective loss of TGF-β mediated induction of inhibitors of invasion. Identification of additional contributing components will best be addressed through unbiased genome-wide approaches in future studies.
Our in vitro data suggested that reduced Smad3 levels might tip the balance of TGF-β responses in favor of tumor progression, with the apoptotic response lost and the invasion response enhanced. To address the integrated effect of the reduction in Smad3 levels on the tumorigenic process in vivo, we crossed the MMTV-PyMT transgenic mouse model of metastatic breast cancer (18) with Smad3 +/- mice (11). As expected, Smad3 was decreased by 50-70% at the mRNA (Fig. 5A) and protein (Fig. 5B) levels in primary tumors from MMTV-PyMT × Smad3+/- mice. There was no difference in primary tumorigenesis between oncogenically initiated Smad3 +/+ and Smad3 +/- mice (Fig. 5C). However Smad3+/- mice had a significantly higher incidence of metastases compared to wild-type mice (45% vs 20%, p= 0.02; Fisher Exact test) (Fig. 5D). Thus a mere two-fold reduction in Smad3 levels can significantly enhance metastatic efficiency in vivo.
Figure 5. Effect of reduced Smad3 levels on tumorigenesis and metastasis.

A,B. Smad3 mRNA (A) and protein (B) in tumors from MMTV-PyMT mice of different Smad3 genotypes. For mRNA, results are mean +/- SEM for 4 tumors/group. Protein levels were assessed by conventional Western, with β-actin as the loading control. C,D. Primary tumorigenesis (C) and cumulative lung metastasis incidence (D) in MMTV-PyMT mice of Smad3 +/+ (n=47) and Smad3+/- (n=39) genotypes. Metastasis incidence was statistically different between the genotype groups at the endpoint (p=0.02, Fisher Exact test). E. Metastasis of Smad3+/+ Mvt-1 tumor cells following orthotopic implantation into Smad3+/+ (n=19) or Smad3+/- (n=22) hosts. Results are median +/- interquartile range, Mann-Whitney U-test. F. Conventional Western blot (left) and quantitative SimpleWestern virtual blot (right) of Smad3 levels following shRNA knockdown in Mvt-1 cells. G. Metastasis of Mvt-1 cells transduced with shSmad3 (test) or shGFP (control) lentiviruses and then orthotopically implanted into Smad3+/+ hosts (15 mice/group). Statistics are as in E.
The Smad3+/- mouse used in the above studies was from a germline knockout model, and thus had reduced Smad3 levels in all tissues. To address whether the metastasis-promoting effect of reduced Smad3 was due to the reduction of Smad3 in the tumor parenchyma or microenvironment, we used the Mvt-1 transplantable model of metastatic murine breast cancer (13). To assess the microenvironmental contribution, Mvt-1 were orthotopically transplanted into syngeneic, immunocompetent Smad3+/+ and Smad3+/- host mice. Mvt-1 cells gave significantly increased numbers of metastases in the lungs of the Smad3+/- compared with Smad3+/+ mice (Fig. 5E). To assess the tumor parenchymal contribution, we knocked down Smad3 levels in Mvt-1 tumor cells with shRNA (Fig. 5F). Quantitation by Simon SimpleWestern showed that Mvt-1-shSmad3 cells had 50% less Smad3 protein than Mvt-1-shGFP control cells, so knockdown was comparable to that seen in the genetic model. We saw significantly increased metastasis in Mvt-1-shSmad3 cells compared with control Mvt-1 cells following orthotopic transplantation into Smad3+/+ hosts (Fig. 5G). Together these results confirm that a reduction in Smad3 can increase metastasis in an independent breast cancer model, and suggest that reduced Smad3 in both the tumor parenchyma and the tumor microenvironment can contribute independently to enhanced metastatic efficiency.
Overall, our data suggest that a mere 2–fold reduction in Smad3 levels can significantly impact on tumor progression. In this study, we looked at just four of the many biological responses to TGF-β that are potentially relevant to tumor development. However, our results illustrate the important general principle that relatively small reductions in Smad3 levels can cause a spectrum of different effects on the pattern of TGF-β responses, with individual outcomes ranging from total loss of response (apoptosis), or reduction in response (migration), through no effect (growth inhibition), to enhancement of response (invasion). For the growth inhibitory and apoptotic responses, we were able to show that the impact of Smad3 reduction on the biological readout correlated with differences in TGF-β-induced Smad3 occcupancy at enhancer elements of key target genes for the responses. The data are consistent with a mechanism in which different target enhancers have different affinities for Smad3, such that a small reduction in Smad3 levels causes a selective loss of Smad3 from some enhancers (eg. Bcl-2) but not from others (eg. c-Myc). In a similar manner, activin/nodal signaling through Smad2 was recently shown to cause transcriptional activation of different sets of target genes depending on the quantitative level of Smad2 activation, thereby eliciting different biological fates in the embryonic stem cell (21).
The integrated result of the various, sometimes opposing, effects of reduced Smad3 dosage on TGF-β responses in vivo is likely to be highly dependent on the specific constellation of TGF-β responses that are engaged and dominant in a given context. Here we have shown that the net outcome of Smad3 reduction in the mammary epithelium is an enhancement of metastatic progression, at least in the context of oncogenic activation of the Src (PyMT model) or Myc (Mvt1 model) pathways. However, it remains to be seen how general this phenomenon is. In the DMBA/TPA initiated skin carcinogenesis model, global deletion of one allele of Smad3 resulted in a significant decrease in primary tumor incidence, though effects on metastasis were not reported (25;26). Further study will be necessary to determine whether Smad3 agonists might be useful for the prevention or treatment of metastasis in specific cancer subpopulations.
Supplementary Material
Acknowledgments
We acknowledge the expert assistance of Dr. Mario Anzano, Anthony Vieira and Hao Du in the Animal Core. We thank the members of the Cancer Biology of TGF-beta Section and Dr. Kent Hunter for helpful discussions and critical reading of the manuscript, Dr. Akira Ooshima and Dr. Miriam Anver for pathology expertise, and Abigail Collett for technical assistance.
Financial support: This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research Z01 BC 005785.
Footnotes
Conflict of Interest: None
Reference List
- 1.Massague J. TGFβ in Cancer. Cell. 2008;134(2):215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barcellos-Hoff MH, Akhurst RJ. Transforming growth factor-β in breast cancer: too much, too late. Breast Cancer Res. 2009;11(1):202. doi: 10.1186/bcr2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan AR, Alexe G, Reiss M. Transforming growth factor-β signaling: emerging stem cell target in metastatic breast cancer? Breast Cancer Res Treat. 2009;115(3):453–495. doi: 10.1007/s10549-008-0184-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pardali K, Moustakas A. Actions of TGF-β as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta. 2007;1775(1):21–62. doi: 10.1016/j.bbcan.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Schmierer B, Hill CS. TGFβ-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8(12):970–82. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 6.Brown KA, Pietenpol JA, Moses HL. A tale of two proteins: differential roles and regulation of Smad2 and Smad3 in TGF-β signaling. J Cell Biochem. 2007;101(1):9–33. doi: 10.1002/jcb.21255. [DOI] [PubMed] [Google Scholar]
- 7.Kohn EA, Du Z, Sato M, Van Schyndle CM, Welsh MA, Yang YA, et al. A novel approach for the generation of genetically modified mammary epithelial cell cultures yields new insights into TGFβ signaling in the mammary gland. Breast Cancer Res. 2010;12(5):R83. doi: 10.1186/bcr2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kretschmer A, Moepert K, Dames S, Sternberger M, Kaufmann J, Klippel A. Differential regulation of TGF-β signaling through Smad2, Smad3 and Smad4. Oncogene. 2003;22(43):6748–63. doi: 10.1038/sj.onc.1206791. [DOI] [PubMed] [Google Scholar]
- 9.Dunn NR, Koonce CH, Anderson DC, Islam A, Bikoff EK, Robertson EJ. Mice exclusively expressing the short isoform of Smad2 develop normally and are viable and fertile. Genes Dev. 2005;19(1):152–63. doi: 10.1101/gad.1243205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 11.Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 1999;18(5):1280–91. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jat PS, Noble MD, Ataliotis P, Tanaka Y, Yannoutsos N, Larsen L, et al. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci U S A. 1991;88(12):5096–100. doi: 10.1073/pnas.88.12.5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pei XF, Noble MS, Davoli MA, Rosfjord E, Tilli MT, Furth PA, et al. Explant-cell culture of primary mammary tumors from MMTV-c-Myc transgenic mice. In Vitro Cell Dev Biol Anim. 2004;40(1-2):14–21. doi: 10.1290/1543-706X(2004)40<14:ECOPMT>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 14.Ringner M, Fredlund E, Hakkinen J, Borg A, Staaf J. GOBO: gene expression-based outcome for breast cancer online. PLoS One. 2011;6(3):e17911. doi: 10.1371/journal.pone.0017911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Figueroa JD, Flanders KC, Garcia-Closas M, Anderson WF, Yang XR, Matsuno RK, et al. Expression of TGF-β signaling factors in invasive breast cancers: relationships with age at diagnosis and tumor characteristics. Breast Cancer Res Treat. 2010;121(3):727–35. doi: 10.1007/s10549-009-0590-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang YA, Zhang GM, Feigenbaum L, Zhang YE. Smad3 reduces susceptibility to hepatocarcinoma by sensitizing hepatocytes to apoptosis through downregulation of Bcl-2. Cancer Cell. 2006;9(6):445–57. doi: 10.1016/j.ccr.2006.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frederick JP, Liberati NT, Waddell DS, Shi Y, Wang XF. Transforming growth factor β-mediated transcriptional repression of c-myc is dependent on direct binding of Smad3 to a novel repressive Smad binding element. Mol Cell Biol. 2004;24(6):2546–59. doi: 10.1128/MCB.24.6.2546-2559.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12(3):954–61. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang YA, Tang B, Robinson G, Hennighausen L, Brodie SG, Deng CX, et al. Smad3 in the mammary epithelium has a nonredundant role in the induction of apoptosis, but not in the regulation of proliferation or differentiation by transforming growth factor-β. Cell Growth Differ. 2002;13(3):123–30. [PubMed] [Google Scholar]
- 20.Jeruss JS, Sturgis CD, Rademaker AW, Woodruff TK. Down-regulation of activin, activin receptors, and Smads in high-grade breast cancer. Cancer Res. 2003;63(13):3783–90. [PubMed] [Google Scholar]
- 21.Lee KL, Lim SK, Orlov YL, Yit LY, Yang H, Ang LT, et al. Graded Nodal/Activin signaling titrates conversion of quantitative phospho-Smad2 levels into qualitative embryonic stem cell fate decisions. PLoS Genet. 2011;7(6):e1002130. doi: 10.1371/journal.pgen.1002130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu J, Lamouille S, Derynck R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009;19(2):156–72. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown KA, Aakre ME, Gorska AE, Price JO, Eltom SE, Pietenpol JA, et al. Induction by transforming growth factor-β1 of epithelial to mesenchymal transition is a rare event in vitro. Breast Cancer Res. 2004;6(3):R215–R231. doi: 10.1186/bcr778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cavallaro U. N-cadherin as an invasion promoter: a novel target for antitumor therapy? Curr Opin Investig Drugs. 2004;5(12):1274–8. [PubMed] [Google Scholar]
- 25.Tannehill-Gregg SH, Kusewitt DF, Rosol TJ, Weinstein M. The roles of Smad2 and Smad3 in the development of chemically induced skin tumors in mice. Vet Pathol. 2004;41(3):278–82. doi: 10.1354/vp.41-3-278. [DOI] [PubMed] [Google Scholar]
- 26.Li AG, Lu SL, Zhang MX, Deng C, Wang XJ. Smad3 knockout mice exhibit a resistance to skin chemical carcinogenesis. Cancer Res. 2004;64(21):7836–45. doi: 10.1158/0008-5472.CAN-04-1331. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.