

Abstract
Perturbation of Disrupted-In-Schizophrenia-1 (DISC1) and D-serine/NMDA receptor hypofunction have both been implicated in the pathophysiology of schizophrenia and other psychiatric disorders. In the present study, we demonstrate that these two pathways intersect with behavioral consequences. DISC1 binds to and stabilizes serine racemase (SR), the enzyme that generates D-serine, an endogenous co-agonist of the NMDA receptor. Mutant DISC1 fails to bind to SR, facilitating ubiquitination and degradation of SR and a decrease in D-serine production. To elucidate DISC1-SR interactions in vivo, we generated a mouse model of selective and inducible expression of mutant DISC1 in astrocytes, the main source of D-serine in the brain. Expression of mutant DISC1 down-regulates endogenous DISC1 and decreases protein but not mRNA levels of SR, resulting in diminished production of D-serine. In contrast, mutant DISC1 does not alter levels of ALDH1L1, connexins, GLT-1 or binding partners of DISC1 and SR, LIS1 or PICK1. Adult male and female mice with life-long expression of mutant DISC1 exhibit behavioral abnormalities consistent with hypofunction of NMDA neurotransmission. Specifically, mutant mice display greater responses to an NMDA antagonist, MK-801, in open field and pre-pulse inhibition of the acoustic startle tests and are significantly more sensitive to the ameliorative effects of D-serine. These findings support a model wherein mutant DISC1 leads to SR degradation via dominant-negative effects, resulting in D-serine deficiency that diminishes NMDA neurotransmission thus linking DISC1 and NMDA pathophysiologic mechanisms in mental illness.
Keywords: DISC1, serine racemase, astrocytes, D-serine, schizophrenia, MK-801
Introduction
Substantial evidence supports a model of schizophrenic pathophysiology involving decreased neurotransmission at NMDA-glutamate receptors. For instance, the psychotomimetic effects of drugs that block NMDA receptors, such as phencyclidine and MK-801, resemble schizophrenic behaviors observed in patients1, 2. The NMDA receptor is unique in that its activation requires stimulation by two agonists, one of which is glutamate while the other was first thought to be glycine. Accumulating evidence indicates that the D-isomer of serine is a physiologic agonist for the ‘glycine’ site of the receptor at many synapses and may well be the predominant endogenous ligand for this site in certain brain regions, including the cerebral cortex and hippocampus3–6. Thus D-amino acid oxidase (DAAO), which, at physiologic pH, selectively degrades D-serine but not glycine, virtually abolishes NMDA neurotransmission3. Also, NMDA transmission and associated behaviors are altered in mice with deletion of serine racemase (SR)7–10, the enzyme that generates D-serine from L-serine11, 12. One of the most striking features of D-serine is its major localization in astrocytes ensheathing synapses, especially in areas of the brain enriched in NMDA receptors13–15. Thus, D-serine appears to be a novel neurotransmitter-like molecule, often referred to as a “glio-transmitter.”
Clinical evidence for an association of D-serine with schizophrenia includes reports of diminished serum and cerebrospinal fluid levels of D-serine in schizophrenic patients16–18, polymorphisms of the SR gene in schizophrenia19, as well as polymorphisms of DAAO and its regulator G7220 in the disease. In several controlled clinical trials, administration of D-serine or glycine alleviates schizophrenic symptoms, especially in conjunction with classic neuroleptics1, 21–24.
Many candidate genes have been linked to schizophrenia and other psychoses25, 26, but for most of these, definitive evidence for a specific alteration in the gene as pathogenic has been lacking. Porteus and associates27, 28 discovered that disruption of two genes by a balanced chromosomal translocation (1;11) in a Scottish family, designated Disrupted-In-Schizophrenia-1 and -2 (DISC1 and DISC2) co-segregates with schizophrenia, major depression and bipolar disorder, providing compelling evidence for a specific association of DISC1 abnormalities and psychosis. Recently, DISC1 variants and polymorphisms have been consistently associated with major psychiatric disorders29.
The Scottish chromosomal translocation was predicted to lead to truncated DISC1 protein. Although only the existence of transcripts was confirmed and expression of the truncated protein has not been conclusively demonstrated or refuted, multiple forms of truncated DISC1 proteins can perturb cellular functions29, 30 and animal models expressing truncated DISC1 display behavioral abnormalities reminiscent of aspects of schizophrenia and/or other neuropsychiatric disorders31–34. Both dominant-negative and haploinsufficiency mechanisms can similarly perturb DISC1-interacting protein complexes, resulting in loss of function of DISC129. Thus, studying effects of mutant DISC1 on brain and behavior may provide valuable mechanistic insights into the pathogenesis of psychiatric disorders.
Studies of DISC1 reveal a role in neurodevelopment, with most studies focused on neuronal functions29, 35, 36. Recent reports showing expression of DISC1 in glial cells, including astrocytes, support a role for DISC1 in astrocytic functions37–39, consistent with recent appreciation of glial dysfunction in the pathophysiology of psychiatric disease40–44. We have employed a mouse model of inducible expression of mutant DISC1 to down-regulate the level of endogenous DISC1 selectively in astrocytes. We demonstrate physiologic binding of DISC1 to SR. Disruption of this binding by mutant DISC1 leads to increased ubiquitination and degradation of SR in astrocytes. The decreased protein level of SR in astrocytes of DISC1 mutant mice results in diminished production of D-serine and behavioral abnormalities consistent with hypofunction of NMDA neurotransmission. These findings support a model in which disruption of DISC1-SR interaction elicits NMDA receptor hypofunction by depleting SR and D-serine.
Materials and Methods
Animals
Our mouse model of inducible expression of human mutant DISC1 (ΔC-hDISC1) is based on the Tet-off system (Supplementary Figure 1) as previously described45. In this study, expression of ΔC-hDISC1 was under the control of the GFAP promoter46. Mice were generated by mating GFAP-tTA mice (B6.Cg-Tg(GFAP-tTA)110Pop/J; Jackson Lab, Bar Harbor, ME) with single transgenic mutant DISC1 mice (lines 1302B and 70) as described previously45. All mice have been backcrossed to the C57BL/6j background for at least 12 generations. Both male and female mice were used in all experiments. Mouse pups were weaned on postnatal day (PND) 21, genotyped and housed in sex-matched groups of five in standard mouse cages on a 12-h light/dark cycle at a room temperature of 23°C with free access to food and water in accordance with Johns Hopkins University Animal Care and Use Committee guidelines.
Behavioral tests
Behavioral tests were performed on mice of 3–6 months of age. The interval between different behavioral tests was at least one week. The tests were performed in the following order: open field test, elevated plus maze, Y maze, pre-pulse inhibition (PPI) of the acoustic startle response, and drug challenge tests. The behavioral protocols used were described in our previous publications45, 47. Drug-induced activity in the open field was assessed over a 90-min period using activity chambers with infrared beams (Instruments Inc., San Diego, CA) as previously described45. Animals were initially habituated to the chambers for 30 minutes followed by a single injection of saline that was followed by administration of MK-801 or D-amphetamine intraperitoneally (i.p.) at a dose of 0.3 mg/kg and 2.5 mg/kg, respectively. Total locomotor activity was automatically recorded and analyzed. In the case of D-serine treatment, the same protocol was used except that MK-801 administration was followed 5 minutes later by injection with D-serine at the dose of 2.7 g/kg (i.p.). Immediately afterwards mice were placed back in the activity chambers. Drug-induced impairment of PPI was evaluated using the same doses and routes of administration for MK801 and D-serine.
Cells and Transfections
HEK-293, HT-22 cells and N2A cells were grown in a humid atmosphere of 5% CO2 at 37°C in DMEM supplemented with 10% FBS, L-glutamine (2mM), penicillin (100units/ml), and streptomycin (100 µg/ml). Primary astrocytes were grown in DMEM supplemented with 10% FBS, L-glutamine (2 mM), penicillin (100 units/ml), streptomycin (100 µg/ml) and sodium pyruvate (1 mM). Primary astrocytes were prepared from mouse cortex as described45. The purity of the culture was assessed by immunofluorescence using GFAP as an astrocyte marker and 85–95% of the cells are GFAP positive. Dissociated cortical neurons were prepared from E18 mice as described48. HEK-293 cells were transfected with polyfect (Qiagen, Hilden, Germany) and HT-22 and N2A cells were transfected with lipofectamine (Invitrogen, Grand Island, NY).
Western Blotting
For western blot assays, mice were euthanized at embryonic day (E) 17, postnatal days (P) 0, 7 and 21 or as adults upon completion of behavioral tests to evaluate expression of mutant ΔC-hDISC1 protein. Brains were quickly removed and frontal cortex was isolated on ice-cold phosphate buffered saline (PBS) and frozen on dry ice and kept at −80°C until used. These samples were assayed for expression of ΔC-hDISC1, endogenous mouse DISC1 and mouse serine racemase (SR)45. Membranes were probed with anti-myc antibody (1:1000) to assess expression of mutant hDISC1 tagged with myc45, two types of anti-mouse DISC1 antibodies for endogenous DISC1 (1:500)45, anti-LIS1 antibody (1:1000), or anti-PICK1 (1:1000) overnight at 4°C. Antibodies used were monoclonal to myc (Santa Cruz Biotechnology, Santa Cruz, CA), monoclonal to LIS1 (Sigma, St Louis, MO), rabbit polyclonal to PICK1 (Abcam, Cambridge, MA) followed by corresponding peroxidase-conjugated goat anti-mouse (1:1000, Kierkegaard Perry Labs, Gaithersburg, MD) or sheep anti-rabbit (1:2500, GE Healthcare, Waukesha, WI) secondary antibodies. The optical density (O.D.) of protein bands on each digitized image was normalized to the O.D. of the loading control (β-tubulin, 1:10000, Cell signaling, Danvers, MA,). Densitometry was done using ImageJ software. Normalized values were used for analyses.
Reagents
L-serine, D-serine, MG-132, cycloheximide, MK-801 and D-amphetamine were purchased from Sigma (St. Louis, MO). Monoclonal mouse serine racemase (SR) antibody was from BD Biosciences (San Jose, CA), which is highly specific for SR tested by immunofluorescence (Supplementary Figure 2). In some western blotting analyses, SR antiserum immunized in rabbit raised against mouse SR (mSR) was used, a generous gift from Dr. Herman Wolosker from the Technion, Israel Institute of Technology. Both antibodies detected a single band of ~38 kDa in the homogenates of brains from WT but not from SR knockout mice. Mouse monoclonal anti-GFAP antibody (GA5 Mouse mAb #3670) was from Cell Signaling (Danvers, MA); rabbit polyclonal anti-GFAP antibody (ab7779) was from Abcam (Cambridge, MA); rabbit anti-mouse DISC1 antibodies were previously described30, 45.
Protein Binding Assays
Immunoprecipitation was carried out 48 h after transfecting HEK-293, HT-22 or N2A cells with different constructs. The cells were harvested in lysis buffer (50 mM Tris·HCl, pH 7.8, 150 mM NaCl, 1% Triton-X100, 1 mM EDTA, 1 mM PMSF, 10% glycerol and protease inhibitor tablet (Roche, Basel, Switzerland)). Alternatively, primary astrocytes were harvested in the lysis buffer and sonicated briefly three times to solubilize the proteins. After spinning down, HA or SR antibodies were added to the supernatant containing 500–700 µg of protein and incubated at 4°C overnight. The next day, protein G beads (Sigma, St. Louis, MO) were added to the mixture for 3 h at 4°C and washed with washing buffer (lysis buffer with 300 mM NaCl) three times. Bound proteins were analyzed by western blotting with myc or SR antibodies.
In vitro binding assay was carried out by transfecting HEK-293 cells with HA-tagged DISC1 or ΔC-hDISC1. 48 h after transfection, the cells were lysed and spun down. The supernatant was incubated with HA-affinity beads (Sigma, St. Louis, MO) overnight. The beads were washed three times in the washing buffer the next day. Mouse cortices were homogenized in lysis buffer and the homogenate was centrifuged. The supernatant was mixed with HA-affinity beads pre-incubated with cell lysates overnight at 4°C. The beads were washed with washing buffer three times before analyzed by western blotting using SR antibodies.
Immunohistochemistry of brain sections
The procedure was carried out as previously described49 with minor modifications. In short, 6-week old mice were deeply anesthetized with pentobarbital sodium (100 mg/kg body weight, i.p.), transcardially perfused with 0.1 M phosphate buffer (PB; pH 7.4) with heparin (10000 U/L), and then perfused with 4.0% paraformaldehyde in 0.1 M PB. The brains were dissected out and postfixed in 4.0% paraformaldehyde in 0.1 M PB for an additional 2–3 h at room temperature. After cryoprotection with 30% sucrose in 0.1M PB for 48 h, the brains were mounted on a freezing microtome and cut into 30-µm-thick sections. Some sections were stained with cresyl violet to evaluate histopathology of the brain in mutant mice. Adjacent sections were used for immunostaining. For immunofluorescence, after blocking for 1 h at room temperature, the sections were incubated with primary antibodies (mouse monoclonal anti-SR, 1:100; rabbit anti-GFAP 1:1000; mouse anti-GFAP, 1:500; and rabbit anti-DISC1 (mExon3 Ab), 1:400) overnight at 4°C. The sections were then incubated with Alexa 488-, 568-labeled species-specific secondary antibodies (Invitrogen, Carlsbad, CA) diluted at 1:600 for 1 h at room temperature. Images were taken with a Zeiss LSM 510 confocal laser scanning microscope at the Johns Hopkins University Neuroscience Multiphoton /Electrophysiology Core Facility.
Immunocytochemistry of primary astrocytes
Primary astrocytes were prepared as previously described45. Primary astrocytes were passed once and allowed to grow until confluency. Astrocytes were then collected for western blotting experiments or fixed in cold methanol and stained with rabbit anti-GFAP antibody (1:500), anti-SR antibody, anti- endogenous DISC1 antibody or anti-myc MAB (1:400) followed by either FITC-conjugated or Cy3-conjugated secondary antibodies (1:200, Chemicon, Temecula, CA). Images were taken using a confocal microscope.
D-serine production from cells
To measure the production of D-serine from HEK-293 cells, 24 h after transfection, the cell culture media was replaced with fresh media containing 10 mM L-serine. After 24 h, the media was harvested, spun down at 16,000 × g for 10 min and the supernatant stored at −80°C. To measure the production of D-serine from primary astrocytes, L-serine was added to the cell culture media to a final concentration of 4 mM and the cells allowed to grow for another 48 h. The level of D-serine was measured with a spectrophotometric assay previously described50. To determine the specific activity of SR, D-serine level in the media was normalized by SR O.D., which was obtained from western blotting analysis of the cell lysates. HPLC measurement for D-serine, as previously described51, was also employed in addition to the spectrophotometric assay to confirm the results.
SR activity assay
Primary astrocytes lysates were spun down at 14,000 × g for 20 min and the supernatant concentrated with endogenous amino acids removed by passing through Amicon Ultra-4 3 kDa Centrifugal Filter Units (Millipore, Billerica, MA) two times. The lysates were then incubated with reaction buffer containing 10 mM L-serine as previously described52. The resulting D-serine production was measured using the spectrophotometric assay described above. D-serine levels were normalized by SR O.D. in the lysates to yield specific activity of SR in the lysates.
D-serine measurement in mouse brain
High-performance liquid chromatography was used to detect endogenous levels of L-serine and D-serine in the mouse brain as described51.
Statistical analyses
Results are expressed as mean ± standard error of the mean (±SEM) throughout. The effects of mutant DISC1 on expression of various protein markers in the western blotting experiments were analyzed with Student’s t-test or Wilcoxon non-parametric test, if normal distribution test failed, and the Bonferroni correction made by adjusting the α level depending on the number of markers measured. The effects of mutant DISC1 on mouse behaviors were evaluated in male and female mice separately with a mixed model of analysis of variance (ANOVA), with the genetic background, treatment, sex and time of testing (if applicable) as independent variables. Significant effects were explored further with lower levels ANOVAs and/or post hoc comparisons. p<0.05 was used for the significance level.
Results
DISC1 binds to SR
We first explored binding of DISC1 to SR utilizing overexpressed proteins in HEK-293 cells (Figure 1a). Wild-type mouse and human DISC1 bind robustly to mouse and human SR, respectively. A pathogenic mutant form of human DISC1 (a C-terminus truncated protein, ΔC-hDISC1) displays substantially reduced binding to human SR, with binding to mouse SR virtually abolished. We also examined binding of these proteins in HT-22, a mouse hippocampal neuronal cell line, and observe similar results (Figure 1b). We observe co-precipitation of endogenous DISC1 and SR from mouse primary astrocyte cultures, indicating that these two proteins interact physiologically (Figure 1c). The observed binding seems to be direct, as mouse SR in brain lysates binds to both human and mouse DISC1 purified from HEK-293 cells (Figure 1d).
Figure 1. SR interacts with DISC1 and co-localizes in primary astrocytes.
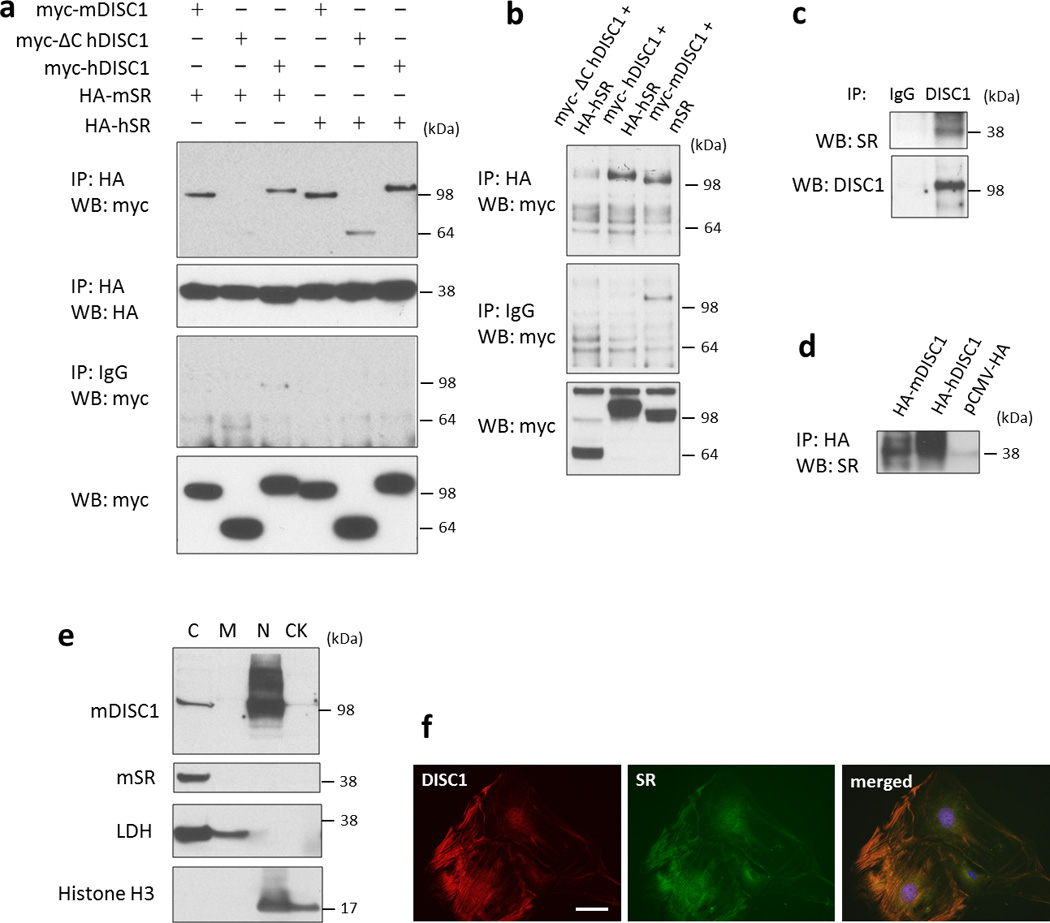
[a] Co-immunoprecipitation of overexpressed DISC1 and SR in HEK-293 cells. Note that full-length DISC1 (100 kDa) binds to SR, but not the truncated mutant DISC1 (64 kDa). mSR, mouse SR; hSR, human SR. Cell lysates containing 500 ug of protein were used for co-immunoprecipitation.
[b] Co-immunoprecipitation of DISC1 and SR in HT22 cells.
[c] Endogenous DISC1 binds to SR in mouse primary astrocytes. Cell lysates containing 700 ug of protein were used for co-immunoprecipitation.
[d] DISC1 immobilized to HA affinity beads binds to SR in mouse brain lysates. pCMV-HA is used as a control.
[e] Subcellular fractionation of primary astrocytes indicates that DISC1 is present in both the nuclear (N) and cytosolic (C) fraction while SR is exclusively in the cytosolic (C) fraction. C, cytosol; M, membrane; N, nucleus; CK, cytoskeleton.
[f] Immunofluorescent staining of DISC1 and SR in the mouse primary astrocytes, scale bar - 10 µm.
Substantial evidence indicates that mutant DISC1 exerts its pathogenic actions in a dominant-negative manner, binding to wild-type DISC1 and disrupting its function30–32, 34, 45, 53. Accordingly, we examined influences of mutant DISC1 on the binding of wild-type DISC1 to SR. Overexpression of mutant DISC1 in HEK-293 cells reduces the binding of wild-type DISC1 to SR (Supplementary Figure 3).
Utilizing subcellular fractionation, we evaluated localizations of DISC1 and SR in different subcellular compartments in primary astrocytes. Consistent with prior reports on localizations of DISC1 and SR in astrocytes11, 37, 54, we observe both nuclear and cytosol localizations for DISC1 and exclusive cytosolic localizations for SR. (Figure 1e). Neither DISC1 nor SR are detected in the membrane or cytoskeletal fractions. Immunofluorescent staining in primary astrocytes also reveals co-localization for the cytosolic pool of DISC1 and SR (Figure 1f).
Mutant DISC1 depletes SR and D-serine
DISC1 is thought to function primarily as a scaffold, regulating the disposition of other proteins29, 55. If DISC1 serves as a scaffold for SR, then expression of mutant DISC1, which binds to SR with much lower affinity, might affect SR. In three cell lines, i.e., HEK-293 cells, HT-22 cells and N2A cells, a mouse neuroblastoma cell line, we monitored the effects of mutant DISC1 on levels of SR (Figure 2a). In comparison to wild-type DISC1, mutant DISC1 substantially lowers levels of SR protein in all three cell lines (Figure 2a, b and c). Mutant DISC1, even when co-expressed with wild-type DISC1, also depletes SR in a dose-dependent manner, consistent with a dominant-negative action (Supplementary Figure 4).
Figure 2. Mutant DISC1 depletes SR.
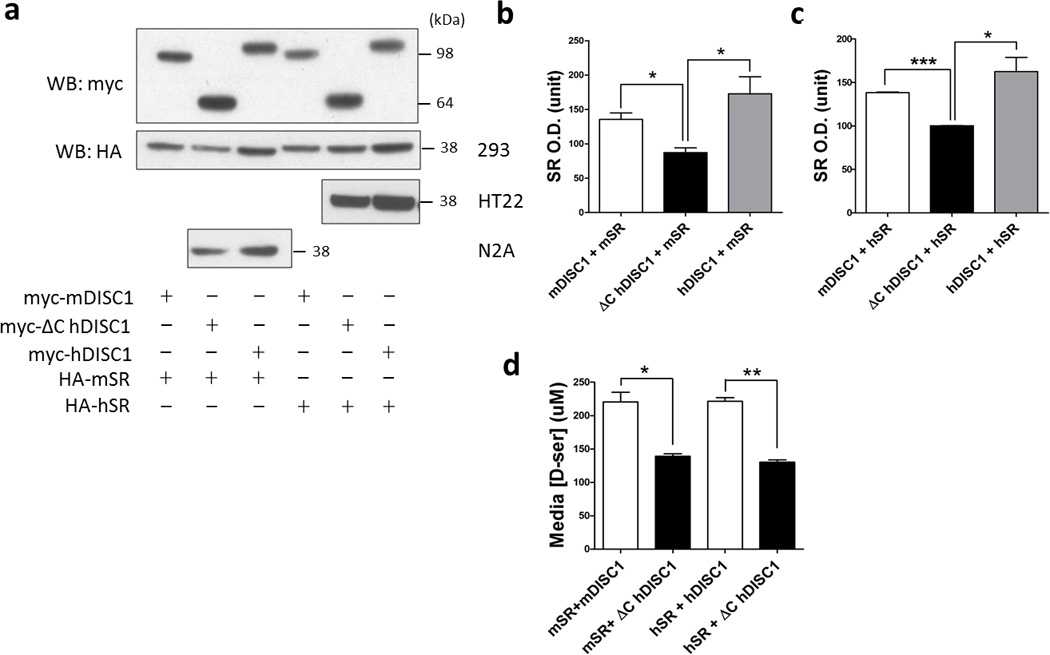
[a] SR protein is depleted when co-expressed with mutant DISC1 in HEK-293, HT-22 and N2A cell lines compared to full-length DISC1.
Quantification of mouse [b] and human SR protein [c] when co-expressed with different forms of DISC1 in HEK-293 cells. mSR, mouse SR; hSR, human SR. Data are representative of 4 independent experiments, *p<0.05, ***p<0.0005.
[d] Mutant DISC1 decreases D-serine production. L-serine (10 mM) was added to the cell culture media of HEK-293 cells transfected with different constructs and 24 h later, D-serine in the media was measured by the spectrophotometric assay. *p<0.05, **p<0.005.
To ascertain whether depletion of SR by mutant DISC1 affects the generation of D-serine, we incubated the same cell lines with L-serine and monitored formation of D-serine (Figure 2d). Both for mouse and human SR, overexpression of mutant DISC1 is associated with a 40% decline in D-serine levels. The extent of diminished production of D-serine is comparable to that of decreased protein levels of SR in these samples, implying that the intrinsic catalytic activity of SR enzyme protein is not altered by mutant DISC1.
Transgenic expression of mutant DISC1 in astrocytes depletes SR and D-serine
D-serine has been predominantly found in astrocytes13–15, 54, and recent studies have demonstrated astrocytic expression of DISC137, 38, 56. To study DISC1-SR interactions in vivo, we first confirmed co-expression of DISC1 and SR in astrocytes in the mouse brain (Figure 3a). We generated transgenic mice that express mutant DISC1 under the control of the GFAP promoter using a tet-off inducible system as previously described (Supplementary Figure 1). Consistent with the reported activity of the GFAP promoter and the main features of the tet-off system46, we detect expression of mutant DISC1 in the brains of double- but not single-transgenic mice (Supplementary Figure 5a), in astrocytes but not in neurons (Figure 3b) with expression of mutant DISC1 regulated by administration of doxycycline (Supplementary Figure 5b). We also confirm expression of mutant DISC1 in astrocytes in the brain (Figure 3c) and co-localization of mutant DISC1 and endogenous mouse DISC1 in primary astrocytes (Supplementary Figure 5c). Mutant DISC1 expression in the mouse brain peaks at around E18 followed by a decline during the postnatal period (Figure 3d), in agreement with the developmental time course of endogenous DISC157. This developmental pattern supports the use of new born mice for the culture of primary astrocytes. Furthermore, using a reporter mouse line (B6; SJL-Tg (tetop-lacZ) 2Mam/J, Jackson Lab, catalog # 002621), we find that mutant DISC1 is widely expressed in the brain, with higher levels of expression detected in the cortex, hippocampus and hindbrain (Supplementary Figure 5d), similar to the spatial expression pattern of endogenous DISC157. In addition, endogenous DISC1 and SR co-precipitate in primary astrocytes, which confirms the physiological binding of the two. This co-precipitation is diminished in primary astrocytes derived from mutant DISC1 mice (Supplementary Figure 6).
Figure 3. SR level is decreased in primary astrocytes and brain tissue from the GFAP-ΔC hDISC1 mice.
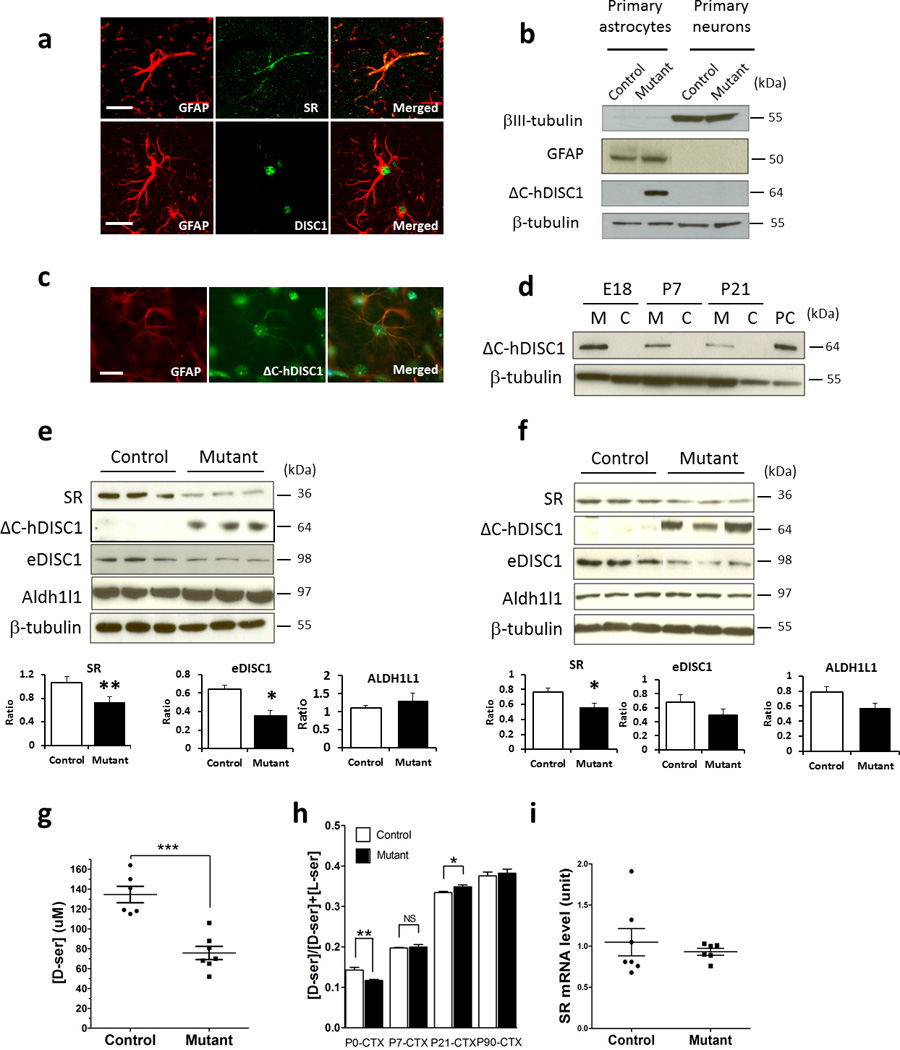
[a] Both DISC1 and SR were expressed in astrocytes in 6-week-old mouse brain. DISC1 and SR immunofluorescence was carried out with glial marker GFAP in the mouse forebrain sections, scale bar - 20 µm;
[b] ΔC-hDISC1 is expressed in the astrocytes of the mutant mouse but not in the neurons or in the control mouse;
[c] Immunofluorescence showing that mutant DISC1 is expressed in astrocytes of the mouse hippocampus, scale bar - 10 µm;
[d] Expression time course of ΔC-hDISC1 in the mutant mouse forebrain, with the highest expression level around E18. PC, positive control, brain tissue from a mouse that expresses mutant DISC1 under the CAMKII promoter;
[e] Expression of SR, ΔC-hDISC1, endogenous mouse DISC1 and an astrocytic marker, ALDH1L1, in samples from primary astrocytes. Below, quantitative evaluation of expression of these markers, n=7–10 per group, *p<0.05, ** p<0.005 vs. controls, Student’s t-test. The exposure time of the ΔC-hDISC1 and endogenous mouse DISC1 blots is the same.
[f] Expression of SR, ΔC-hDISC1, endogenous mouse DISC1 and an astrocytic marker, ALDH1L1, in samples from forebrain area of newborn mice. Below, quantitative evaluation of expression of these markers, n=7–9 per group, *p<0.05 vs. controls, Student’s t-test.
[g] D-serine level in the culture media of primary astrocytes. L-serine was added to the culture media to a final concentration of 4 mM and D-serine level was measured 48 h later. Control, n=6; mutant, n=7, *** p<0.0005;
[h] D-serine level (expressed as [D-serine]/[total serine]) in the brain of P0, P7, P21 and P90 mice determined by HPLC. Each bar represents ~4–6 mice. In the P0 mouse cortex, mutant has ~20% decrease in D-serine level compared to the control, while in the P21 mouse cortex and hippocampus, there is a small but significant increase, *p<0.05, ** p<0.005, NS, not significant. CTX - cortex.
[i] SR mRNA level in primary astrocytes is unaltered as measured by quantitative Real Time-PCR. Control, n=7; mutant, n=6.
The destabilization of DISC1-SR binding by mutant DISC1 might be anticipated to elicit SR depletion. We measured levels of SR, endogenous DISC1, aldehyde dehydrogenase-1L1 (ALDH1L1), a reliable astrocytic marker58, LIS1, a DISC1 interacting partner29, PICK1, a SR interacting partner59 and connexins-43 and GLT-1 as additional astrocytic markers in primary astrocytes as well as in the mouse forebrain (Figure 3e and f). In both preparations, we observe a significant decrease in protein levels of SR and endogenous DISC1 in mutant samples. By contrast, there are no significant alterations in expression of ALDH1L1, LIS1, PICK1, connexins-43 or GLT-1 in primary astrocytes or forebrain samples from mutant mice (Figure 3e and f and Supplementary Figure 7 and data not shown). SR levels are also diminished in another transgenic mutant DISC1 mouse line (i.e., line 70 as described previously45), arguing against putative non-specific insertion effects of the transgene. Moreover, no significant changes in SR level occur in primary neurons derived from mouse embryos with selective neuronal expression of mutant DISC1 (Supplementary Figure 8).
The decreased protein level of SR in the primary astrocytes of DISC1 mutant mice is associated with a substantial (~45%) decrease in generation of D-serine from L-serine (Figure 3g). We detect a modest (~20%) but significant decrease in forebrain levels of D-serine in mutant newborn mice, with no significant alteration in forebrain samples collected at P7 and P90 and a small but significant increase at P21 (Figure 3h). In contrast, no significant difference in the level of L-serine and glycine, another co-agonist of the NMDA receptor, was detected (Supplementary Figure 9). To ascertain whether attenuated catalytic activity of SR protein contributes to the decreased production of D-serine in mutant astrocytes, we assayed SR activity in vitro by quantifying D-serine formation per unit of SR protein (Supplementary Figure 10). We detect no alteration in SR activity per unit of SR protein.
Mutant DISC1 increases the turnover of SR by enhancing its ubiquitination
We explored mechanisms whereby mutant DISC1 affects levels of SR. Mutant DISC1 does not appear to affect transcription of SR, as mRNA levels of SR in primary mutant astrocytes are unaltered (Figure 3i). To assess a potential influence of DISC1 upon the stability of SR, we monitored the rate of decline of SR in HEK-293 cells treated with cycloheximide (Figure 4a). SR levels decline much more rapidly in the presence of mutant than wild-type DISC1. Thus, 24 h after treatment, SR protein levels decline by 50% in cells transfected with mutant DISC1 compared to a 15% decrease in SR levels in cells that express wild-type DISC1.
Figure 4. Mutant DISC1 depletes SR protein by increasing its ubiquitination level.
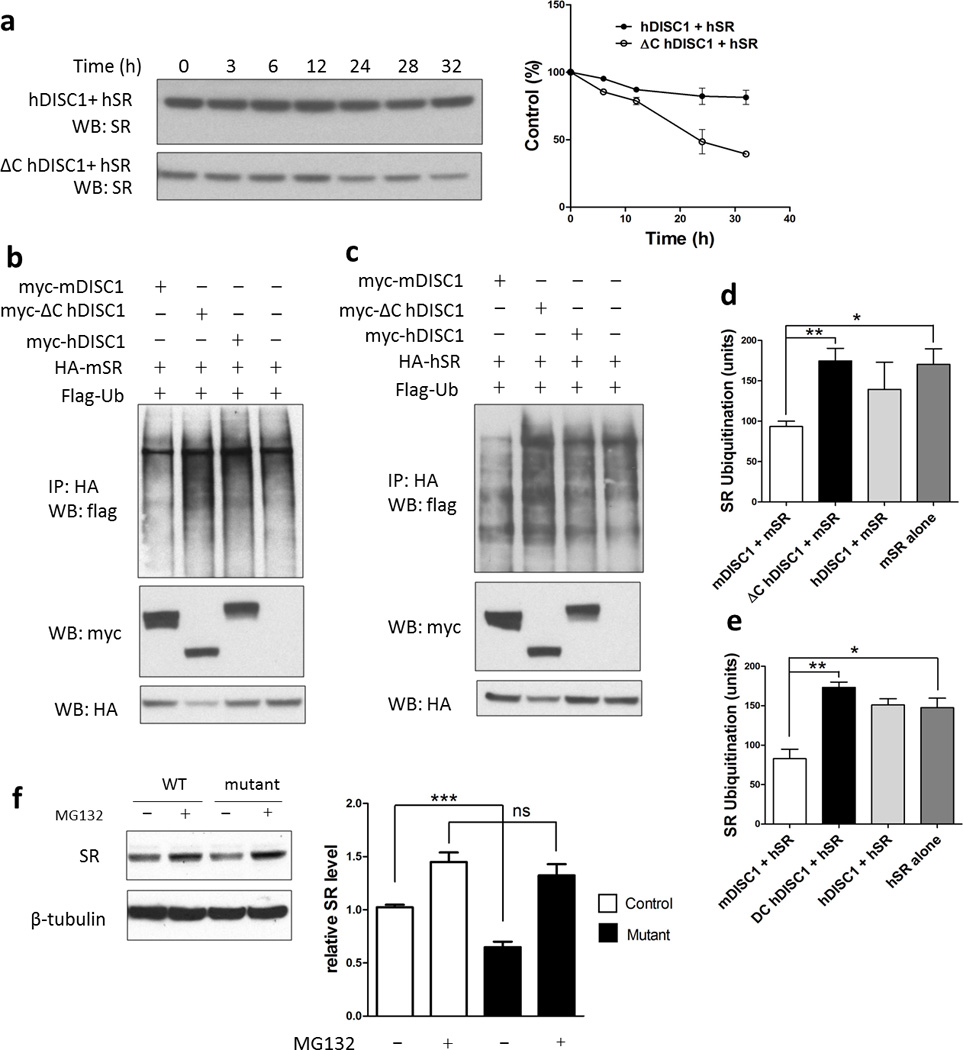
[a] Cycloheximide chase assay. HEK-293 cells were treated with 70 mg/ml cycloheximide 24–36 h after transfection and SR level was quantified. Protein levels were expressed as percentage of the level at time 0. The half-life of SR is significantly shorter when co-expressed with mutant DISC1.
[b,c] In vivo ubiquitination assay of mouse [b] and human SR [c] in HEK-293 cells.
The levels of mouse and human SR ubiquitination are higher when co-expressed with mutant DISC1 as quantified in [d] and [e], respectively.
Primary astrocytes [f] were treated with the proteasome inhibitor MG-132 (30 µM for 10 h), and relative SR protein levels are quantified below the blots. The value of SR level in primary astrocytes from control mice before treatment has been normalized to 1.0.
Wolosker and associates60 have reported that SR is physiologically ubiquitinated. We evaluated the ubiquitination of human and mouse SR in HEK-293 cells (Figure 4b and c). Overexpression of wild-type DISC1 diminishes SR ubiquitination, consistent with the notion that DISC1 acts as a scaffold to stabilize SR. By contrast, overexpression of mutant DISC1 elicits almost a doubling of SR ubiquitination compared to wild-type DISC1 (Figure 4d and e). This increase in ubiquitination of SR is associated with a 40% decline in levels of SR protein, indicating that the ubiquitination leads to SR degradation. Similar results are observed for mouse and human preparations of SR (Figure 4d and e).
If mutant-DISC1-induced degradation of SR is mediated by the proteasomal system, proteasomal inhibition ought to increase SR levels. Treatment with the proteasomal inhibitor MG-132 completely restores the low level of SR in primary astrocytes from mutant mice to a level comparable to the wild-type mice (Figure 4f). We also examined several validated DISC1-interacting partners, including NUDEL, CREB2, GSK3β, Kal7, LIS1 and PDE4b. Among those, NUDEL and CREB2 are degraded through the ubiquitin/ proteasome pathway, but their degradation is not differentially affected by wild-type or mutant DISC1 expression (Supplementary Figure 11).
Enhanced behavioral response to NMDA antagonism in mutant DISC1 mice is rescued by D-serine
Our biochemical studies established that mutant DISC1 destabilizes and depletes SR, leading to decreased levels of D-serine. As D-serine activates NMDA receptors, we wondered whether mutant mice would display behavioral alterations consistent with diminished NMDA receptor transmission. We treated mice with an NMDA antagonist, MK-801 (0.3 mg/kg, i.p.), which is commonly used to probe behavioral consequences of NMDA receptor dysfunction61. MK-801 elicits substantially greater locomotor stimulation in male and female mutant mice compared to control mice (Figure 5a and b). Repeated measures ANOVA reveals a significant group effect for male mice, F(1,479)=5.9, p<0.05; and female mice, F(1,429)=5.3, p<0.05. We also evaluated the influence of D-serine on MK-801-induced hyperactivity in mice. Our pilot experiments had shown that a high dose of D-serine (i.e., 2.7g/kg, i.p.24, 62–64) is needed to decrease MK-801-induced hyperactivity in mice. D-serine reduces the hyperactivity of mutants much more than wild-type animals (Figure 5a and b). Specifically, in male mice, D-serine does not alter hyperactivity in control mice but diminishes locomotor activity of mutant mice by 70–80%. ANOVA for the treatment data in male mice shows a significant time × group × treatment interaction, F(23,911)=1.6, p=0.041. A lower level ANOVA for the treatment data demonstrates a significant effect of treatment with D-serine in the mutant group, F(1,311)=53.5, p<0.001 but not the control one, F=1.3, p=0.3. In female mice, D-serine treatment reduces locomotor activity in both control and mutant mice but to a greater extent in mutant mice. ANOVA for the treatment data reveals a significant effect of treatment for control female mice, F(1,477)=14.8, p<0.001, and mutant female mice, F(1,527)=60.3, p<0.01. Comparing the data for locomotor activity after D-serine treatment between control and mutant female mice indicates a significant group by time interaction, F(23,575)=1.7, p=0.023. Post-hoc comparisons reveal a significantly greater locomotor activity in control mice vs. mutant mice at 20–23 time intervals (Figure 5a and b).
Figure 5. DISC1 mutant mice exhibit greater responses to an NMDA antagonist, MK-801, and D-serine treatment.
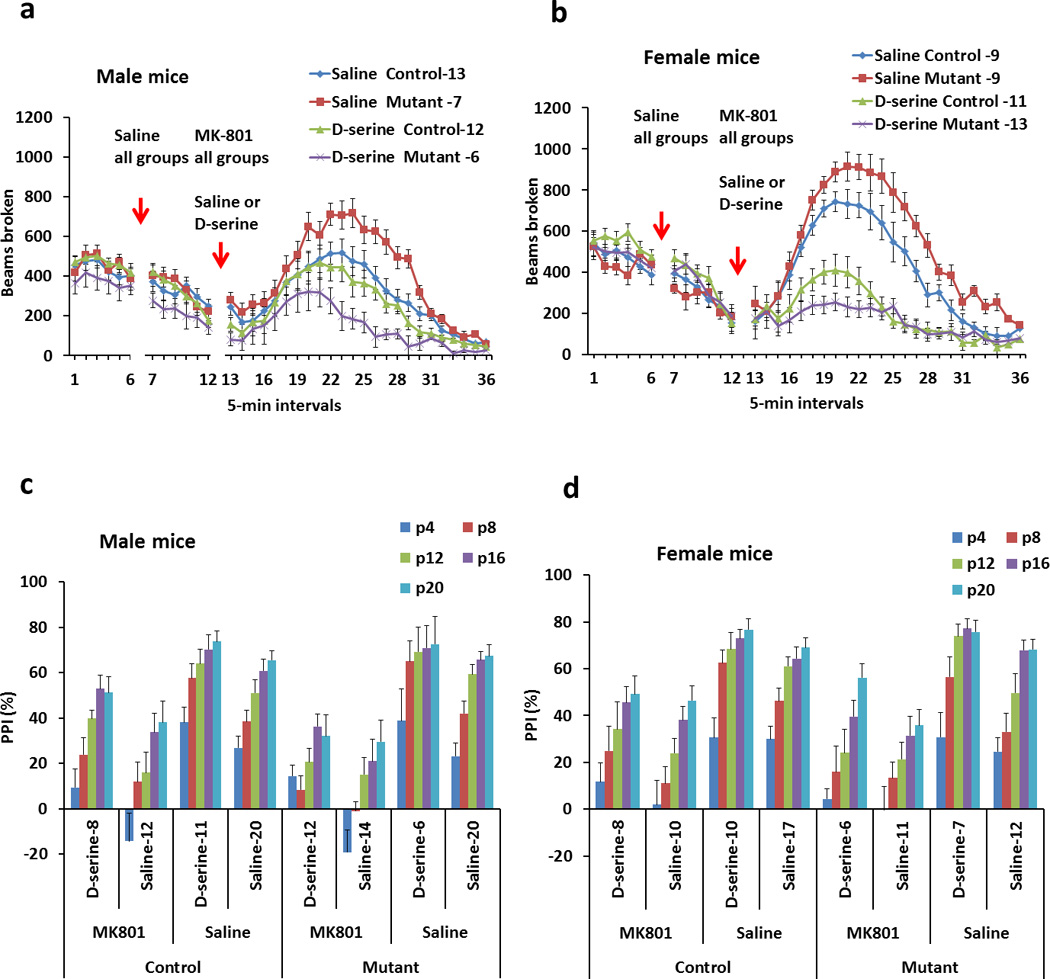
[a] Locomotor activity in open field of male mice before and after treatment with MK-801 (0.3 mg/kg, i.p.) followed by saline or D-serine injections (2.7 g/kg, i.p.). Compared to control male mice, mutant male mice display significantly greater activity in response to MK-801. Compared to control male mice, mutant mice also exhibit significantly greater sensitivity to the ameliorative effects of D-serine. Numbers of mice in each group are indicated on the graph.
[b] Locomotor activity in open field of female mice before and after treatments with MK-801 (0.3 mg/kg, i.p.) followed by saline or D-serine injections (2.7 g/kg, i.p.). Compared to control female mice, mutant female mice exhibit significantly higher locomotor activity in response to MK-801. Compared to control female mice, mutant female mice show significantly greater sensitivity to the ameliorative effects of D-serine. Numbers of mice in each group are indicated on the graph.
[c] PPI of the acoustic startle of male mice before and after treatment with MK-801 (0.3 mg/kg, i.p.) followed by saline or D-serine injections (2.7 g/kg, i.p.). p4–20 are the intensities of pre-pulses above the background noise level (70dB). Compared to control male mice, mutant male mice display greater MK-801 induced impairment in PPI at P4 and P8. Compared to control male mice, mutant mice also exhibit significantly greater sensitivity to the ameliorative effects of D-serine at P4. Numbers of mice in each group are indicated on the graph.
[d] PPI of the acoustic startle of female mice before and after treatment with MK-801 (0.3 mg/kg, i.p.) followed by saline or D-serine injections (2.7 g/kg, i.p.). Numbers of mice in each group are indicated on the graph.
We also assessed the effects of MK-801 in a rodent test for sensorimotor gating, pre-pulse inhibition (PPI) of the acoustic startle response. We find no baseline differences between two groups in PPI for either male or female mice (Figure 5c and d). As expected, administration of MK-801 (0.3 mg/kg, i.p.) disrupts PPI in both groups of animals. Three-way ANOVA of the PPI data for male mice shows a significant effect of MK-801 treatment, F(1,313)=160.2, p<0.01 and the group by treatment interaction, F(1,313)=4.3, p=0.04. A greater decrease in PPI is observed in mutant male mice at the intensities of pre-pulses of 4 dB (a strong trend to group effect- F(1,61)=2.92, p=0.09) and 8 dB (a significant group by pre-pulse interaction – F(1,61)=4.23, p=0.044)(Figure 5c). Three-way ANOVA for female mice detects a significant and comparable effect of MK-801 treatment on PPI between two groups, F(1,249)=100.9, p<0.01. We also evaluated the effects of D-serine on MK-801-induced impairment of PPI (Figure 5d). D-serine administration (2.7 g/kg, i.p.) increases MK-801-reduced PPI in both groups of male mice, with only mutant mice showing a significant treatment by PPI type interaction, F(4,124)=2.46, p=0.05 and a stronger response to D-serine at the 4-dB pre-pulse 4 (p<0.05). D-serine treatment of female mice leads to a moderate increase in MK-801-diminished PPI (Figure 5d). The observed behavioral alterations are not attributed to alterations of NMDA receptor protein, as levels of NR1, NR2A and NR2B are not altered in cortex of the mutant mice (data not shown).
In contrast to MK-801 treatment, amphetamine, used to probe dopaminergic synaptic neurotransmission65, produces a comparable increase in locomotor activity in control and mutant mice of both sexes (Supplementary Figure 12a). ANOVAs of the treatment data show a significant effect of time for male mice, F(23,640)=29.7, p<0.001, and female mice, F(23,476)=9.4, p<0.001. We find no significant differences between mutant and control groups in anxiety in elevated plus maze, spontaneous alternations or spatial recognition memory assessed in Y maze (Supplementary Figure 12b–d). In addition, we observe no gross effects of mutant DISC1 on the cytoarchitectonics and layering of the hippocampus or cerebellum in mice (Supplementary Figure 13).
Discussion
The present study reveals a role for DISC1 in astrocytes involving interactions between DISC1 and SR. DISC1 binds physiologically to SR and appears to stabilize it. In vitro and in vivo expression of mutant human DISC1 in astrocytes prevents the binding of wild-type DISC1 to SR in a dominant-negative manner, leading to decreased levels of SR and diminished production of D-serine. These molecular alterations are associated with behavioral consequences reflecting decreased NMDA neurotransmission.
In the past, DISC1 was thought to be primarily localized to neurons. Recent studies have revealed that DISC1 is also expressed in glial cells, including astrocytes37, 38, 56. To evaluate a link of DISC1 to D-serine, which is localized to astrocytes, we generated transgenic mice that express mutant DISC1 selectively in astrocytes. Functional relationships of DISC1 and SR in astrocytes are indicated by the depletion of astrocytic SR associated with expression of mutant DISC1.
Our finding that concurrent depletion of D-serine in the brain of newborn pups and SR in astrocytic preparations from mutant DISC1 mice establishes that astrocytic SR gives rise to a large proportion of D-serine in the brain. The relative roles of neurons or astrocytes in generating D-serine has been unclear, as D-serine is largely astrocytic13–15, 54 while recent studies reveal SR is largely neuronal49, 54, 66. The rate-limiting enzyme in the genesis of L-serine is 3-phosphoglycerate dehydrogenase (PHGDH), which is highly concentrated in astrocytes with negligible levels in neurons67. Wolosker and associates68 suggested that L-serine generated in astrocytes by PHGDH enters neurons where it is converted to D-serine and is then accumulated into astrocytes by a D-serine transporter Alanine-Serine-Cysteine Transporter (ASCT). Alternatively, even if glial levels of SR are lower than those of neurons, the higher levels of the substrate L-serine in astrocytes would enable astrocytic SR to be the primary source of astrocytic D-serine. In support of this latter notion, D-serine levels in the brain are depleted in mice with targeted deletion of PHGDH69. Oliet and collaborators5 demonstrated that in the hypothalamic supraoptic nucleus (SON), the degree of astrocytic coverage of neurons determines the level of glycine site occupancy on the NMDA receptor by D-serine. Another study established that Long Term Potentiation (LTP) is determined by the release of D-serine from astrocytes70. Thus in studies of D-serine function, astrocytes have appeared to be dominant. Several protein binding partners of SR—protein interacting with C-kinase 1 (PICK1), glutamate receptor interacting protein (GRIP) and Golga359, 60, 71, 72, all bind to SR in glia and exert their regulatory functions through glia. In the present study, we demonstrated that 50% depletion of astrocytic SR could lead to a 20% decrease in the forebrain D-serine level, implying that at least about 40% of the D-serine in the forebrain is produced by astrocytes.
Our findings indicate that full-length DISC1 binds to SR and stabilizes it, probably by decreasing the ubiquitination of SR (Figure 4b–e). Expression of mutant DISC1 led to increased ubiquitination and degradation of SR. Exact mechanisms whereby mutant DISC1 facilitates SR ubiquitination are unclear. Conceivably, mutant DISC1 binds to full-length DISC1 and leads to decreased expression of full-length protein in a dominant-negative manner. Indeed, previous studies have shown that mutant DISC1 binds to full-length DISC1 with high affinity and depletes it30, 45, 53. Our data demonstrate reduced expression of endogenous DISC1 in primary astrocytes from mutant mice (Figure 3e and f). As a result, degradation of full-length DISC1 results in its diminished binding to SR and attenuated stabilization of the enzyme.
Interestingly, the effect of mutant DISC1 on SR appears to be astrocyte-specific. We examined SR protein level in the primary cortical neurons prepared from mouse embryos with selective neuronal expression of mutant DISC145 and failed to detect any changes in SR levels (Supplementary Figure 8). Thus, while DISC1 and SR occur both in glia and neurons, evidence for their functional interaction is greater in glia though a neuronal role might also be important. Another intriguing observation is that D-serine levels in the mutant mouse brain are only altered at a very early stage (P0), but not later in development (P7 and P21, Figure 4h). This could imply that astrocytic SR is more important in generating D-serine in early development, while neuronal SR may play a greater role in adulthood. Studies that identified neuronal localizations of SR employed older animals49, 54, whereas the present data were generated using primary astrocytes derived from newborn mice. This implies that astrocytic SR impacts D-serine metabolism most prominently at early developmental stages. Such a notion is supported by studies of PICK1 knockout mice. PICK1 binds and activates SR in glia59, 71. A 30%-decrease of D-serine level was observed only at P7 in PICK1 KO mice while unaltered levels were found in 8-week-old mice71.
A developmental effect of mutant DISC1 on SR and D-serine production would also be consistent with the established role of DISC1 in neurodevelopment29, 55. DISC1 expression peaks at around E13.5, decreases postnatally and is maintained at a low level until P35, gradually increasing thereafter57. This is consistent with the time course of mutant DISC1 expression in our mouse model (Figure 3d). Accordingly, interactions of DISC1 and SR in astrocytes might predominate in early development. Interestingly, there is an apparent compensation of D-serine levels in the mutant mouse with time, especially from P7 to early adulthood. At P0, forebrain D-serine levels of mutant mice were decreased, at P7 they were the same as controls, while there was a modest increase at P21 in both the cortex and hippocampus (Figure 3h and data not shown). This suggests that astrocytic depletion of SR leads to D-serine depletion in newborns. Neuronal SR could then compensate so that in adult mice, total brain D-serine levels are not reduced in mutant mice. However, the D-serine compensation by neuronal SR was apparently insufficient to alleviate the neurobehavioral abnormalities observed in adult mice. Presumably, because of its different disposition, this neuronal D-serine is less effective than glial D-serine in activating NMDA receptors. The depletion of SR and D-serine was associated with behavioral alterations in DISC1 mutant mice. Mutant mice displayed much stronger locomotor activity and greater disruption of PPI in response to an NMDA antagonist, MK-801. The enhanced pharmacological effects in transgenic mice may indicate supersensitivity of NMDA receptors, reminiscent of NMDA transmission alterations in patients73–75. The supersensitivity to NMDA-related agents appeared to be selective, as mutant mice did not display altered responses to amphetamine, which has a more direct effect on dopaminergic neurotransmission.
Many models for the pathophysiology of mental illnesses, including schizophrenia, involve aberrant disposition of neurotransmitters. A wide array of drugs that are beneficial in psychiatric illness act via one or another neurotransmitter. DISC1 has been implicated in a number of processes that are involved in neurodevelopment29, 55. These studies have largely addressed neuronal DISC1 influencing intracellular organelles and signaling pathways. As the involved organelles and pathways are also present in astrocytes as in neurons, DISC1 may exert physiological influences in astrocytes. Presumably, in normal organisms, DISC1 also functions to stabilize SR, maintaining a normal level of D-serine in intact brains. To the best of our knowledge, the present findings are the first to identify interactions of DISC1 with a neurotransmitter-generating enzyme. Moreover, this study is the first to explore the function of DISC1 in glia relevant to the pathophysiology of schizophrenia, adding to a growing appreciation of roles for astrocytes in psychiatric diseases41–44. Our results may have therapeutic consequences. Several studies have demonstrated therapeutic effects of D-serine administration to schizophrenics1, 21–24. Our data imply that DISC1 stabilizes SR and facilitates formation of D-serine. Enhancing stability of the DISC1-SR complex should be beneficial in patients with DISC1 variants. Given the diverse abnormalities in D-serine metabolism reported in schizophrenics, stabilization of the DISC1-SR complex may be relevant to a much broader population of psychotic patients than just those with DISC1 mutations.
In conclusion, the present study demonstrates physiologic binding of DISC1 to SR and disruption of this binding by mutant DISC1, resulting in increased ubiquitination and degradation of SR in astrocytes. Mutant DISC1-induced decreased levels of SR in astrocytes give rise to attenuated production of D-serine and behavioral abnormalities consistent with hypofunction of NMDA neurotransmission. As abnormalities in DISC1 and NMDA transmission are prominently associated with psychosis, our findings linking these two themes may facilitate conceptualization of aberrations underlying schizophrenic behavior.
Supplementary Material
Acknowledgement
We thank members of the S.H.S. and M.V.P laboratories for help and critical comments, B. Paul, M. Pucak and K. Ishizuka for technical support, R. Xu and H. Wall for scientific discussions and H. Wolosker for providing the SR antiserum. This work is supported by ARRA MH083728 (to M.V.P.), The Brain and Behavior Research Foundation Independent Investigator Award (to M.V.P.), US Public Health Service Grant MH18501 (to S.H.S.) and Young Investigator Award (to B.A.), NIMH P50 Conti Center Grant (to M.V.P.), The Cell Science Research Foundation Japan (to J.N.), NIH grants of P50 MH094268 (to A.S.), P20 MH084018 (to A.S.), R01 MH069853 (to A.S.), R01 MH092443 (to A.S.), R21 MH085226 (to A.S.), RC1 MH088753 (to A.S.); NARSAD grants (to A.S.), SR/RUSK foundation (to A.S.), Stanley foundation (to A.S.), Maryland Stem Cell Research Fund (to A.S.) and in part by MH084020.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. The American journal of psychiatry. 2001;158(9):1367–1377. doi: 10.1176/appi.ajp.158.9.1367. [DOI] [PubMed] [Google Scholar]
- 2.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. The American journal of psychiatry. 1991;148(10):1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- 3.Mothet JP, Parent AT, Wolosker H, Brady RO, Jr, Linden DJ, Ferris CD, et al. D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(9):4926–4931. doi: 10.1073/pnas.97.9.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shleper M, Kartvelishvily E, Wolosker H. D-serine is the dominant endogenous coagonist for NMDA receptor neurotoxicity in organotypic hippocampal slices. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25(41):9413–9417. doi: 10.1523/JNEUROSCI.3190-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, et al. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125(4):775–784. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- 6.Gustafson EC, Stevens ER, Wolosker H, Miller RF. Endogenous D-serine contributes to NMDA-receptor-mediated light-evoked responses in the vertebrate retina. Journal of neurophysiology. 2007;98(1):122–130. doi: 10.1152/jn.00057.2006. [DOI] [PubMed] [Google Scholar]
- 7.Basu AC, Tsai GE, Ma CL, Ehmsen JT, Mustafa AK, Han L, et al. Targeted disruption of serine racemase affects glutamatergic neurotransmission and behavior. Molecular psychiatry. 2009;14(7):719–727. doi: 10.1038/mp.2008.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mori H, Inoue R. Serine racemase knockout mice. Chemistry & biodiversity. 2010;7(6):1573–1578. doi: 10.1002/cbdv.200900293. [DOI] [PubMed] [Google Scholar]
- 9.DeVito LM, Balu DT, Kanter BR, Lykken C, Basu AC, Coyle JT, et al. Serine racemase deletion disrupts memory for order and alters cortical dendritic morphology. Genes, brain, and behavior. 2011;10(2):210–222. doi: 10.1111/j.1601-183X.2010.00656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sullivan SJ, Esguerra M, Wickham RJ, Romero GE, Coyle JT, Miller RF. Serine racemase deletion abolishes light-evoked NMDA receptor currents in retinal ganglion cells. The Journal of physiology. 2011;589(Pt 24):5997–6006. doi: 10.1113/jphysiol.2011.217059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolosker H, Blackshaw S, Snyder SH. Serine racemase: a glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(23):13409–13414. doi: 10.1073/pnas.96.23.13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Miranda J, Santoro A, Engelender S, Wolosker H. Human serine racemase: moleular cloning, genomic organization and functional analysis. Gene. 2000;256(1–2):183–188. doi: 10.1016/s0378-1119(00)00356-5. [DOI] [PubMed] [Google Scholar]
- 13.Puyal J, Martineau M, Mothet JP, Nicolas MT, Raymond J. Changes in D-serine levels and localization during postnatal development of the rat vestibular nuclei. The Journal of comparative neurology. 2006;497(4):610–621. doi: 10.1002/cne.21016. [DOI] [PubMed] [Google Scholar]
- 14.Williams SM, Diaz CM, Macnab LT, Sullivan RK, Pow DV. Immunocytochemical analysis of D-serine distribution in the mammalian brain reveals novel anatomical compartmentalizations in glia and neurons. Glia. 2006;53(4):401–411. doi: 10.1002/glia.20300. [DOI] [PubMed] [Google Scholar]
- 15.Schell MJ, Brady RO, Jr, Molliver ME, Snyder SH. D-serine as a neuromodulator: regional and developmental localizations in rat brain glia resemble NMDA receptors. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1997;17(5):1604–1615. doi: 10.1523/JNEUROSCI.17-05-01604.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hashimoto K, Fukushima T, Shimizu E, Komatsu N, Watanabe H, Shinoda N, et al. Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Archives of general psychiatry. 2003;60(6):572–576. doi: 10.1001/archpsyc.60.6.572. [DOI] [PubMed] [Google Scholar]
- 17.Yamada K, Ohnishi T, Hashimoto K, Ohba H, Iwayama-Shigeno Y, Toyoshima M, et al. Identification of multiple serine racemase (SRR) mRNA isoforms and genetic analyses of SRR and DAO in schizophrenia and D-serine levels. Biological psychiatry. 2005;57(12):1493–1503. doi: 10.1016/j.biopsych.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 18.Bendikov I, Nadri C, Amar S, Panizzutti R, De Miranda J, Wolosker H, et al. A CSF and postmortem brain study of D-serine metabolic parameters in schizophrenia. Schizophrenia research. 2007;90(1–3):41–51. doi: 10.1016/j.schres.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 19.Morita Y, Ujike H, Tanaka Y, Otani K, Kishimoto M, Morio A, et al. A genetic variant of the serine racemase gene is associated with schizophrenia. Biological psychiatry. 2007;61(10):1200–1203. doi: 10.1016/j.biopsych.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 20.Boks MP, Rietkerk T, van de Beek MH, Sommer IE, de Koning TJ, Kahn RS. Reviewing the role of the genes G72 and DAAO in glutamate neurotransmission in schizophrenia. European neuropsychopharmacology : the journal of the European College of Neuropsychopharmacology. 2007;17(9):567–572. doi: 10.1016/j.euroneuro.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 21.Heresco-Levy U, Javitt DC, Ebstein R, Vass A, Lichtenberg P, Bar G, et al. D-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biological psychiatry. 2005;57(6):577–585. doi: 10.1016/j.biopsych.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 22.Tsai G, Yang P, Chung LC, Lange N, Coyle JT. D-serine added to antipsychotics for the treatment of schizophrenia. Biological psychiatry. 1998;44(11):1081–1089. doi: 10.1016/s0006-3223(98)00279-0. [DOI] [PubMed] [Google Scholar]
- 23.Kantrowitz JT, Malhotra AK, Cornblatt B, Silipo G, Balla A, Suckow RF, et al. High dose D-serine in the treatment of schizophrenia. Schizophrenia research. 2010;121(1–3):125–130. doi: 10.1016/j.schres.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Labrie V, Wong AH, Roder JC. Contributions of the d-serine pathway to schizophrenia. Neuropharmacology. 2012;62(3):1484–1503. doi: 10.1016/j.neuropharm.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 25.Sawa A, Snyder SH. Schizophrenia: neural mechanisms for novel therapies. Molecular medicine (Cambridge, Mass) 2003;9(1–2):3–9. [PMC free article] [PubMed] [Google Scholar]
- 26.Hamilton SP. Schizophrenia candidate genes: are we really coming up blank? The American journal of psychiatry. 2008;165(4):420–423. doi: 10.1176/appi.ajp.2008.08020218. [DOI] [PubMed] [Google Scholar]
- 27.St Clair D, Blackwood D, Muir W, Carothers A, Walker M, Spowart G, et al. Association within a family of a balanced autosomal translocation with major mental illness. Lancet. 1990;336(8706):13–16. doi: 10.1016/0140-6736(90)91520-k. [DOI] [PubMed] [Google Scholar]
- 28.Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Human molecular genetics. 2000;9(9):1415–1423. doi: 10.1093/hmg/9.9.1415. [DOI] [PubMed] [Google Scholar]
- 29.Brandon NJ, Sawa A. Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nature reviews Neuroscience. 2011;12(12):707–722. doi: 10.1038/nrn3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kamiya A, Kubo K, Tomoda T, Takaki M, Youn R, Ozeki Y, et al. A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nature cell biology. 2005;7(12):1167–1178. doi: 10.1038/ncb1328. [DOI] [PubMed] [Google Scholar]
- 31.Shen S, Lang B, Nakamoto C, Zhang F, Pu J, Kuan SL, et al. Schizophrenia-related neural and behavioral phenotypes in transgenic mice expressing truncated Disc1. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28(43):10893–10904. doi: 10.1523/JNEUROSCI.3299-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hikida T, Jaaro-Peled H, Seshadri S, Oishi K, Hookway C, Kong S, et al. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(36):14501–14506. doi: 10.1073/pnas.0704774104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clapcote SJ, Lipina TV, Millar JK, Mackie S, Christie S, Ogawa F, et al. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron. 2007;54(3):387–402. doi: 10.1016/j.neuron.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 34.Li W, Zhou Y, Jentsch JD, Brown RA, Tian X, Ehninger D, et al. Specific developmental disruption of disrupted-in-schizophrenia-1 function results in schizophrenia-related phenotypes in mice. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(46):18280–18285. doi: 10.1073/pnas.0706900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaaro-Peled H, Hayashi-Takagi A, Seshadri S, Kamiya A, Brandon NJ, Sawa A. Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends in neurosciences. 2009;32(9):485–495. doi: 10.1016/j.tins.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramsey AJ, Milenkovic M, Oliveira AF, Escobedo-Lozoya Y, Seshadri S, Salahpour A, et al. Impaired NMDA receptor transmission alters striatal synapses and DISC1 protein in an age-dependent manner. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(14):5795–5800. doi: 10.1073/pnas.1012621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seshadri S, Kamiya A, Yokota Y, Prikulis I, Kano S, Hayashi-Takagi A, et al. Disrupted-in-Schizophrenia-1 expression is regulated by beta-site amyloid precursor protein cleaving enzyme-1-neuregulin cascade. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(12):5622–5627. doi: 10.1073/pnas.0909284107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuroda K, Yamada S, Tanaka M, Iizuka M, Yano H, Mori D, et al. Behavioral alterations associated with targeted disruption of exons 2 and 3 of the Disc1 gene in the mouse. Human molecular genetics. 2011;20(23):4666–4683. doi: 10.1093/hmg/ddr400. [DOI] [PubMed] [Google Scholar]
- 39.Eastwood SL, Walker M, Hyde TM, Kleinman JE, Harrison PJ. The DISC1 Ser704Cys substitution affects centrosomal localization of its binding partner PCM1 in glia in human brain. Human molecular genetics. 2010;19(12):2487–2496. doi: 10.1093/hmg/ddq130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sawa A, Pletnikov MV, Kamiya A. Neuron-glia interactions clarify genetic-environmental links in mental illness. Trends in neurosciences. 2004;27(6):294–297. doi: 10.1016/j.tins.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 41.Henn FA, Henn SW. The psychopharmacology of astroglial cells. Progress in neurobiology. 1980;15(1):1–17. doi: 10.1016/0301-0082(80)90014-3. [DOI] [PubMed] [Google Scholar]
- 42.Stevens B. Neuron-astrocyte signaling in the development and plasticity of neural circuits. Neuro-Signals. 2008;16(4):278–288. doi: 10.1159/000123038. [DOI] [PubMed] [Google Scholar]
- 43.Hamidi M, Drevets WC, Price JL. Glial reduction in amygdala in major depressive disorder is due to oligodendrocytes. Biological psychiatry. 2004;55(6):563–569. doi: 10.1016/j.biopsych.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 44.Schwarcz R, Pellicciari R. Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. The Journal of pharmacology and experimental therapeutics. 2002;303(1):1–10. doi: 10.1124/jpet.102.034439. [DOI] [PubMed] [Google Scholar]
- 45.Pletnikov MV, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov MV, Huang H, et al. Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Molecular psychiatry. 2008;13(2):173–186. 115. doi: 10.1038/sj.mp.4002079. [DOI] [PubMed] [Google Scholar]
- 46.Lin TN, Wang Q, Simonyi A, Chen JJ, Cheung WM, He YY, et al. Induction of secretory phospholipase A2 in reactive astrocytes in response to transient focal cerebral ischemia in the rat brain. Journal of neurochemistry. 2004;90(3):637–645. doi: 10.1111/j.1471-4159.2004.02540.x. [DOI] [PubMed] [Google Scholar]
- 47.Ayhan Y, Abazyan B, Nomura J, Kim R, Ladenheim B, Krasnova IN, et al. Differential effects of prenatal and postnatal expressions of mutant human DISC1 on neurobehavioral phenotypes in transgenic mice: evidence for neurodevelopmental origin of major psychiatric disorders. Molecular psychiatry. 2011;16(3):293–306. doi: 10.1038/mp.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang BN, Ahmad AS, Saleem S, Patterson RL, Hester L, Dore S, et al. Death-associated protein kinase-mediated cell death modulated by interaction with DANGER. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30(1):93–98. doi: 10.1523/JNEUROSCI.3974-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miya K, Inoue R, Takata Y, Abe M, Natsume R, Sakimura K, et al. Serine racemase is predominantly localized in neurons in mouse brain. The Journal of comparative neurology. 2008;510(6):641–654. doi: 10.1002/cne.21822. [DOI] [PubMed] [Google Scholar]
- 50.Mustafa AK, Kumar M, Selvakumar B, Ho GP, Ehmsen JT, Barrow RK, et al. Nitric oxide S-nitrosylates serine racemase, mediating feedback inhibition of D-serine formation. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(8):2950–2955. doi: 10.1073/pnas.0611620104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hashimoto A, Nishikawa T, Oka T, Takahashi K, Hayashi T. Determination of free amino acid enantiomers in rat brain and serum by high-performance liquid chromatography after derivatization with N-tert.-butyloxycarbonyl-L-cysteine and o-phthaldialdehyde. Journal of chromatography. 1992;582(1–2):41–48. doi: 10.1016/0378-4347(92)80300-f. [DOI] [PubMed] [Google Scholar]
- 52.Balan L, Foltyn VN, Zehl M, Dumin E, Dikopoltsev E, Knoh D, et al. Feedback inactivation of D-serine synthesis by NMDA receptor-elicited translocation of serine racemase to the membrane. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(18):7589–7594. doi: 10.1073/pnas.0809442106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pletnikov MV, Xu Y, Ovanesov MV, Kamiya A, Sawa A, Ross CA. PC12 cell model of inducible expression of mutant DISC1: new evidence for a dominant-negative mechanism of abnormal neuronal differentiation. Neuroscience research. 2007;58(3):234–244. doi: 10.1016/j.neures.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 54.Kartvelishvily E, Shleper M, Balan L, Dumin E, Wolosker H. Neuron-derived D-serine release provides a novel means to activate N-methyl-D-aspartate receptors. The Journal of biological chemistry. 2006;281(20):14151–14162. doi: 10.1074/jbc.M512927200. [DOI] [PubMed] [Google Scholar]
- 55.Porteous DJ, Millar JK, Brandon NJ, Sawa A. DISC1 at 10: connecting psychiatric genetics and neuroscience. Trends in molecular medicine. 2011;17(12):699–706. doi: 10.1016/j.molmed.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Austin CP, Ma L, Ky B, Morris JA, Shughrue PJ. DISC1 (Disrupted in Schizophrenia-1) is expressed in limbic regions of the primate brain. Neuroreport. 2003;14(7):951–954. doi: 10.1097/01.wnr.0000074342.81633.63. [DOI] [PubMed] [Google Scholar]
- 57.Schurov IL, Handford EJ, Brandon NJ, Whiting PJ. Expression of disrupted in schizophrenia 1 (DISC1) protein in the adult and developing mouse brain indicates its role in neurodevelopment. Molecular psychiatry. 2004;9(12):1100–1110. doi: 10.1038/sj.mp.4001574. [DOI] [PubMed] [Google Scholar]
- 58.Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28(1):264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fujii K, Maeda K, Hikida T, Mustafa AK, Balkissoon R, Xia J, et al. Serine racemase binds to PICK1: potential relevance to schizophrenia. Molecular psychiatry. 2006;11(2):150–157. doi: 10.1038/sj.mp.4001776. [DOI] [PubMed] [Google Scholar]
- 60.Dumin E, Bendikov I, Foltyn VN, Misumi Y, Ikehara Y, Kartvelishvily E, et al. Modulation of D-serine levels via ubiquitin-dependent proteasomal degradation of serine racemase. The Journal of biological chemistry. 2006;281(29):20291–20302. doi: 10.1074/jbc.M601971200. [DOI] [PubMed] [Google Scholar]
- 61.Deutsch SI, Rosse RB, Mastropaolo J. Behavioral approaches to the functional assessment of NMDA-mediated neural transmission in intact mice. Clinical neuropharmacology. 1997;20(5):375–384. doi: 10.1097/00002826-199710000-00001. [DOI] [PubMed] [Google Scholar]
- 62.Nilsson M, Carlsson A, Carlsson ML. Glycine and D-serine decrease MK-801-induced hyperactivity in mice. Journal of neural transmission (Vienna, Austria : 1996) 1997;104(11–12):1195–1205. doi: 10.1007/BF01294720. [DOI] [PubMed] [Google Scholar]
- 63.Kanahara N, Shimizu E, Ohgake S, Fujita Y, Kohno M, Hashimoto T, et al. Glycine and D: -serine, but not D: -cycloserine, attenuate prepulse inhibition deficits induced by NMDA receptor antagonist MK-801. Psychopharmacology. 2008;198(3):363–374. doi: 10.1007/s00213-008-1151-6. [DOI] [PubMed] [Google Scholar]
- 64.Hashimoto A, Chiba S. Effect of systemic administration of D-serine on the levels of D- and L-serine in several brain areas and periphery of rat. European journal of pharmacology. 2004;495(2–3):153–158. doi: 10.1016/j.ejphar.2004.05.036. [DOI] [PubMed] [Google Scholar]
- 65.Meyer U, Feldon J. Epidemiology-driven neurodevelopmental animal models of schizophrenia. Progress in neurobiology. 2010;90(3):285–326. doi: 10.1016/j.pneurobio.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 66.Rosenberg D, Kartvelishvily E, Shleper M, Klinker CM, Bowser MT, Wolosker H. Neuronal release of D-serine: a physiological pathway controlling extracellular D-serine concentration. The FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010;24(8):2951–2961. doi: 10.1096/fj.09-147967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jeon GS, Choi DH, Lee HN, Kim DW, Chung CK, Cho SS. Expression of L-serine biosynthetic enzyme 3-phosphoglycerate dehydrogenase (Phgdh) and neutral amino acid transporter ASCT1 following an excitotoxic lesion in the mouse hippocampus. Neurochemical research. 2009;34(5):827–834. doi: 10.1007/s11064-008-9831-5. [DOI] [PubMed] [Google Scholar]
- 68.Wolosker H. Serine racemase and the serine shuttle between neurons and astrocytes. Biochimica et biophysica acta. 2011;1814(11):1558–1566. doi: 10.1016/j.bbapap.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 69.Yang JH, Wada A, Yoshida K, Miyoshi Y, Sayano T, Esaki K, et al. Brain-specific Phgdh deletion reveals a pivotal role for L-serine biosynthesis in controlling the level of D-serine, an N-methyl-D-aspartate receptor co-agonist, in adult brain. The Journal of biological chemistry. 2010;285(53):41380–41390. doi: 10.1074/jbc.M110.187443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Henneberger C, Papouin T, Oliet SH, Rusakov DA. Long-term potentiation depends on release of D-serine from astrocytes. Nature. 2010;463(7278):232–236. doi: 10.1038/nature08673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hikida T, Mustafa AK, Maeda K, Fujii K, Barrow RK, Saleh M, et al. Modulation of D-serine levels in brains of mice lacking PICK1. Biological psychiatry. 2008;63(10):997–1000. doi: 10.1016/j.biopsych.2007.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim PM, Aizawa H, Kim PS, Huang AS, Wickramasinghe SR, Kashani AH, et al. Serine racemase: activation by glutamate neurotransmission via glutamate receptor interacting protein and mediation of neuronal migration. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(6):2105–2110. doi: 10.1073/pnas.0409723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Breier A, Su TP, Saunders R, Carson RE, Kolachana BS, de Bartolomeis A, et al. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc Natl Acad Sci U S A. 1997;94(6):2569–2574. doi: 10.1073/pnas.94.6.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moncrieff J. A critique of the dopamine hypothesis of schizophrenia and psychosis. Harv Rev Psychiatry. 2009;17(3):214–225. doi: 10.1080/10673220902979896. [DOI] [PubMed] [Google Scholar]
- 75.Krystal JH, D'Souza DC, Mathalon D, Perry E, Belger A, Hoffman R. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology (Berl) 2003;169(3–4):215–233. doi: 10.1007/s00213-003-1582-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.