

Abstract
With the breakthrough crystallization of the bacterial leucine transporter protein LeuT, the first available X-ray structure for the neurotransmitter/sodium symporter family, development of 3-D computational models is suddenly essential for structure–function studies on the plasmalemmal monoamine transporters (MATs). LeuT-based MAT models have been used to guide elucidation of substrate and inhibitor binding pockets, and molecular dynamics simulations using these models are providing insight into conformations involved in the substrate translocation cycle. With credible MAT models finally in hand, structure-based virtual screening for novel ligands is yielding lead compounds toward the development of new medications for psychostimulant dependence, attention deficit hyperactivity, depression, anxiety, schizophrenia, and other disorders associated with dopamine, norepinephrine, or serotonin dysregulation.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-012-9391-0) contains supplementary material, which is available to authorized users.
KEY WORDS: homology model, molecular dynamics, monoamine transporter, virtual screening
INTRODUCTION
Monoamine transporters (MATs), integral to the maintenance of neurotransmitter homeostasis, are responsible for the synaptic reuptake of these chemical messengers that terminates a neurotransmission event. MATs are members of the SLC6 protein family that also includes the GABA transporters and glycine transporters. An extensive review on the SLC6 family has recently been published (1). Monoamine neurotransmitter imbalances have been linked to drug dependence, mood abnormalities, and other neurological disorders (2–7). Given the involvement of dopamine, norepinephrine, and serotonin in numerous disease states, advances in the understanding of MAT structure, function, and regulation remain a priority. The field has waited two decades, since the publication of cDNAs encoding the MAT proteins, for reliable in silico (“virtual”) 3-D MAT models that predict ligand binding sites and other structural aspects that could inform bench experimentation. This review focuses on MAT protein computational advances that have followed the elucidation of an X-ray structure for the MAT homolog LeuT (8). A leucine transporter from Aquifex aeolicus, a thermophilic bacterium common to undersea volcanic vents, now universally serves as the template for in silico MAT model design.
Prior to the availability of a relevant crystal structure template, most MAT computational efforts were ligand-based studies focusing on quantitative structure–activity relationships and 3-D pharmacophore generation. These ligand-based models relied on established MAT ligands and their associated SAR, and provided some predictive power for the study of ligand–transporter interactions (9–17). Caveats exist with ligand-based studies, including the assumption that all training set compounds (experimentally established ligands for a given MAT) bind at the same site of the same protein in a similar conformation. Additionally, models relying on biological data are at the mercy of any experimental variations (e.g., lab-to-lab, cell type, or radioligand displaced.) Following the crystallization of LeuT (pdb:2a65), reliable 3-D homology models for MATs rapidly surfaced and are proving to be invaluable in the study of transporter structure and function. These MAT models have guided elucidation of ligand binding sites, inter- and intramolecular interactions, substrate translocation pathways, and potential MAT conformations. Recent structure-based virtual screening (VS) efforts have identified putative ligands of novel scaffold that can serve, or be further developed, as MAT probes or lead compounds for drug discovery.
As a brief overview of the evolution, current state, and utility of MAT computational models, the development of LeuT crystal structure-based MAT models are first discussed. This is followed by prediction of MAT substrate, inhibitor and ion cofactor binding pockets, MD simulations regarding substrate translocation and MAT conformations, and structure-based VS toward discovery of novel MAT ligands and lead compound therapeutics. Attempts at developing MAT protein models prior to the availability of the LeuT crystal structure will not be discussed due to the uncertainty of these models, as the X-ray structures of NhaA and LacY were later found to not adequately resemble the MATs (18–23).
Structure of the MAT Homolog LeuT
The LeuT protein crystal structure provided the first reliable MAT template for computational studies. LeuT is a distant but bona fide MAT homolog, with approximately 20% primary amino acid sequence identity (Supplementary Figure 1). The LeuT crystal structure revealed an inverted symmetry between TMs 1–5 and TMs 6–10, with TMs 11 and 12 not proximal to the substrate/ion permeation pore and ligand binding sites. The TM 1 and TM 6 α-helices are unwound at their approximate midpoints, near the center of the lipid bilayer, to create the primary substrate binding pocket and Na+ binding site. Access to this substrate binding site is controlled by TM 1—TM 10 ionic and TM 3—TM 8 hydrophobic bridges comprising an extracellular-facing gate, and an ionic pre-TM 1–intracellular loop (IL) 4 interaction that forms the intracellular gate (Fig. 1). The original LeuT-leucine cocrystal (8) launched a wave of new MAT models that continue to be corroborated by biophysical studies.
Fig. 1.
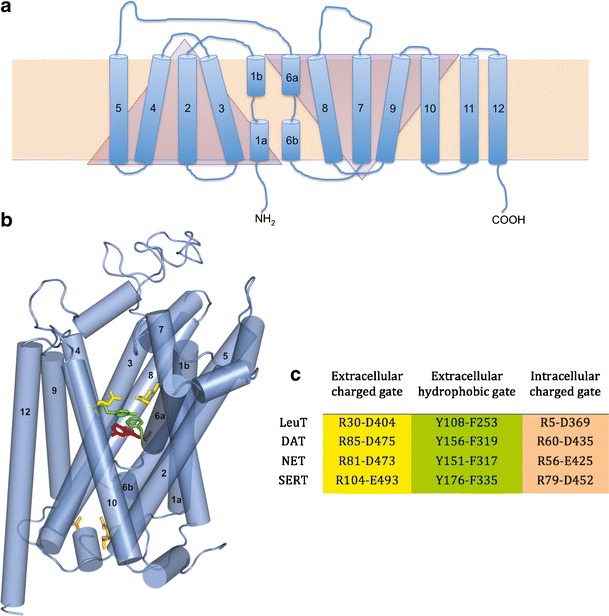
LeuT and MAT topology. a 2D representation of LeuT (blue) in the cell membrane bilayer (beige rectangle). TM domains (cylinders) 1–5 and 6–10 are symmetrical but inverted motifs (triangles). b 3D representation of hSERT based on the LeuT (pdb:2A65) crystal structure. The primary substrate binding site (represented by serotonin; magenta) is flanked by gating residues to the extracellular (green, yellow) and intracellular (peach) sides. c LeuT and MAT gating residues, color-coded to match b
Construction of LeuT-Based MAT Homology Models
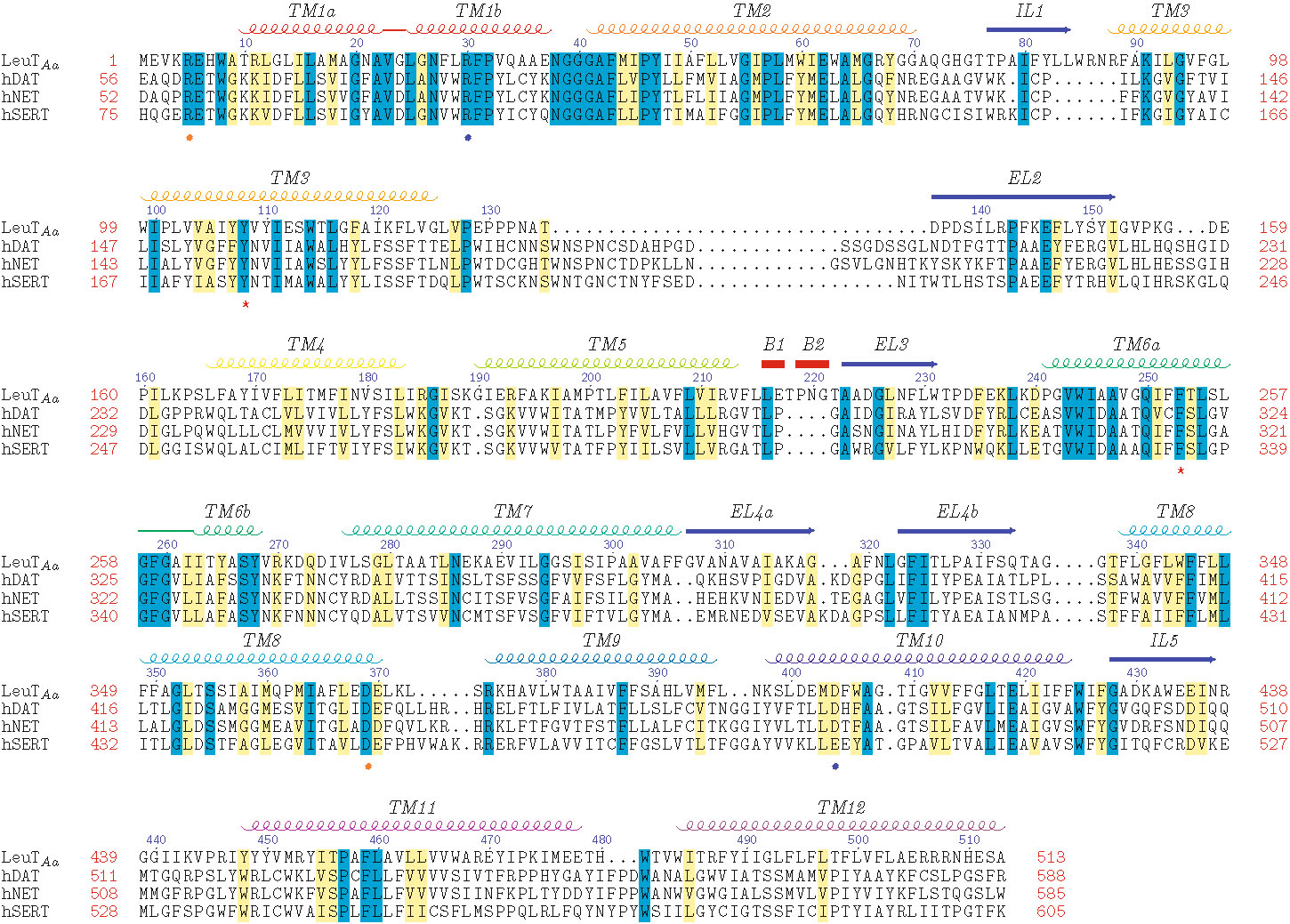
With the advent of the LeuT crystal structure, numerous MAT homology models have been built. Because the quality of such models is dependent on the MSA employed, development of an MSA using LeuT as the template protein is one of the most important steps of the model building process. MSAs are generated by first aligning regions conserved among the proteins, making use of available biological data, followed by the alignment of nonconserved regions. Several MAT models are based on a comprehensive alignment of 344 NSS proteins (Supplementary Figure 1) (24). The evolutionarily conserved regions of NSS proteins are primarily located in the TM domains; non-transmembranous extracellular and intracellular connecting loops are less conserved, and the N- and C-terminal tails are highly sequence-variable. The NSS substrate binding pocket is predicted to be within a substrate/ion translocation pore formed by the highly conserved regions of TM domains 1, 3, 5, 6, 8, and 10 (Supplementary Figure 2) (25).
Once 3-D MAT homology models have been constructed using the MSA in combination with the template crystal structure, model refinement is an iterative endeavor that employs initial validations including Ramachandran and 3-D fold analyses. Details regarding the construction of DAT, NET, and SERT models published to date are included in Tables I, II, and III, respectively.
Table I.
DAT Homology Models
| Reference | Year | Template (PDB ID) | Sequence alignment | Sequence identity (%) | Software |
|---|---|---|---|---|---|
| (18) | 1994 | None | Developed | – | MIDAS |
| (22) | 2003 | NhaA | Developed | 7 | MIDAS plus |
| (20) | 2006 | LacY (1pv6) | Developed | 5 | ICM |
| (51) | 2007 | LeuT | Developed | 20.4 | Insight II |
| (52) | 2008 | LeuT | Developed | – | MOE, Robetta |
| 3D-JIGSAW | |||||
| (38) | 2008 | LeuT | Developed | – | Malign, MODELLER |
| (62) | 2008 | LeuT | Beuming | 20 | MODELLER |
| (70) | 2009 | LeuT | Beuming | 20 | MODELLER |
| (65) | 2009 | LeuT | Beuming | 20 | ICM |
| (64) | 2010 | LeuT (2qju) | Beuming | 20 | MODELLER, MOE |
– information not provided
Table II.
NET Homology Models
| Reference | Year | Template (PDB ID) | Sequence alignment | Sequence identity (%) | Software |
|---|---|---|---|---|---|
| (21) | 2006 | NhaA | Developed | 7 | ICM |
| (20) | 2006 | LacY (1pv6) | Developed | 5 | ICM |
| (30) | 2007 | LeuT | Beuming | 20 | MODELLER |
| (55) | 2011 | DAT | Beuming | 66 | Insight II |
| (34) | 2011 | LeuT | Beuming | 20 | – |
| Present work | 2011 | LeuT (3f3a) | Beuming | 20 | DS 2.5.1, APBS |
| (50) | 2011 | LeuT | Beuming | 27 | MODELLER |
– information not provided
Table III.
SERT Homology Models
| Reference | Year | Template (PDB ID) | Sequence alignment | Sequence identity (%) | Software |
|---|---|---|---|---|---|
| (19) | 2001 | – | Developed | – | PHD, WHATIF |
| (21) | 2003 | DAT | Developed | 80 | ICM |
| (22) | 2003 | NhaA | Developed | 7 | ICM |
| (20) | 2006 | LacY (1pv6) | Developed | 5 | ICM |
| (57) | 2006 | LeuT | Developed | 20 | ICM |
| (58) | 2006 | LeuT | Yamashita | 20.5 | SCWRL |
| (31) | 2007 | LeuT | Developed | – | MODELER |
| (45) | 2007 | LeuT | Beuming | 20 | NEST |
| (38) | 2008 | LeuT | Developed | – | Malign |
| (72) | 2008 | LacY (1pv6) | Developed | – | ICM, Bioedit |
| LeuT (2a65) | |||||
| (59) | 2008 | LeuT | Developed | 22.9 | MODELER |
| (35) | 2009 | LeuT | Beuming | 17 | ROSETTA suite |
| (65) | 2009 | LeuT | Beuming | 20 | ICM |
| (66) | 2010 | LeuT (3f3a) | Beuming | 20 | MODELLER |
| (32) | 2010 | LeuT (3f3a) | Beuming | 20 | MODELLER |
| (73) | 2010 | LeuT | Beuming | 20 | MODELLER |
| (47) | 2010 | LeuT | Beuming | 20 | MOE 2007.08 |
| (42) | 2011 | LeuT | Beuming | 20 | ICM |
| (48) | 2011 | LeuT | Developed | 23.9 | DS 2.5.1 |
– information not provided
MAT Substrate Pockets
Dopamine Transporter
The first LeuT-based DAT model focused on substrate interaction with the transporter and the substrate permeation pathway. Dopamine was postulated to favor a DAT location analogous to the leucine binding site of LeuT. Free energy calculations for the proposed binding mode of dopamine in DAT were in close agreement with experimental values (51). The next reported DAT model was used to dock dopamine and d-amphetamine; two non-overlapping substrate pockets were identified. In addition to the previously identified dopamine binding site corresponding to the leucine site of LeuT, a putative second site was located in the “extracellular vestibule,” a MAT region to the extracellular side of the original dopamine binding site and above the extracellular gate. In keeping with the original LeuT–leucine crystal structure, the gate was closed in the DAT model. It was proposed that this vestibular site could serve as a temporary staging area for dopamine. Upon opening of the gate, dopamine would progress along the permeation pathway to the more interior binding site corresponding to that of leucine in LeuT (52). In separate work, LeuT crystallization combined with [3H]-leucine binding pharmacology appeared to confirm the presence of interior and vestibular substrate pockets. For LeuT, a second substrate molecule must occupy the vestibular site to trigger release of the first substrate molecule from the primary (more interior) site and into the cytoplasm. The primary and secondary substrate sites have been labeled S1 and S2, respectively (Fig. 2) (26,53). For LeuT (54) and SERT (27), the necessity for a second substrate binding site has been challenged: crystal structures reveal an S2 (vestibular) site may be occupied by inhibitor ligands or certain detergents molecules, but an X-ray snapshot of two substrate molecules simultaneously occupying both S1 and S2 has not been observed (1).
Fig. 2.
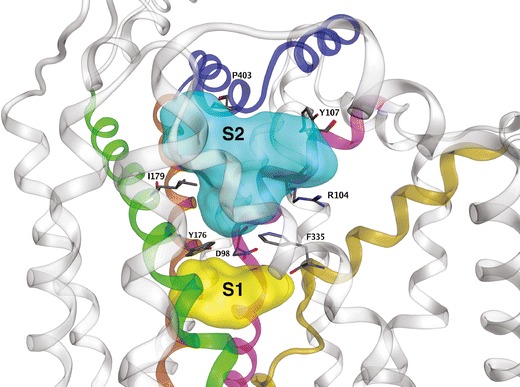
Ligand-accessible regions of SERT. Regions available to ligands in the S1 (yellow) and S2 (cyan) pockets are displayed as surfaces. Selected residues that define each pocket are displayed as sticks (atomtype color). For clarity, only the main TM contributors to SERT ligand binding pockets are highlighted: TM 1 (pink), TM 3 (green), TM 6 (gold), TM 8 (brown) and EL 4 (blue)
Norepinephrine Transporter
As has been the case for essentially all aspects of MAT structure–function research, computational modeling of the NET has lagged behind that of the DAT and SERT. Given the similarity (67%) between DAT and NET polypeptide sequences, a LeuT-based DAT model (28,51) was used as a template to construct a NET model. Norepinephrine was found to dock into the S1 site (55). Two distinct binding pockets were identified (Nolan, T.L., unpublished) using a NET model based on the open-to-out LeuT crystal structure 3f3a (56). Adaptive Poisson–Boltzmann Solver (APBS) calculations of selected docking poses for established NET ligands were compared with experimental data. A secondary substrate binding site was suggested, again in the extracellular vestibule (Fig. 3).
Fig. 3.
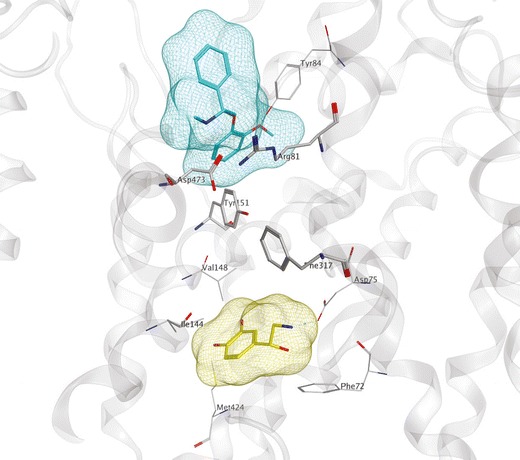
LeuT-based NET computational model. S1 (yellow surface) and S2 (cyan surface) binding sites of NET (gray) are shown occupied by norepinephrine (yellow) and nisoxetine (cyan), respectively. Binding site residues (atomtype, line) and extracellular gate-forming residues (atomtype, stick) are shown. Interactions between gating residues separate the S1 and S2 pockets
Serotonin Transporter
Of the three MATs, the SERT has received most of the computational attention. A SERT model ligand binding pocket consisting of residues from TMs 1, 3, 6, and 8 and corresponding to the leucine binding site of LeuT was found, using the ICM pocket finder, to be important for substrate recognition (57). A similar conclusion was reached from a study in which LeuT and SERT substrate binding sites were compared. It was suggested that the larger serotonin substrate may be accommodated by smaller SERT side chains present in S1 relative to those in LeuT and that the presence of the SERT TM 1 aspartate residue D98 may be important as a substrate interaction (58). The paired mutant–ligand analog complementation method has also been used to study the binding of serotonin in the SERT S1 site. Using an induced-fit method, serotonin analogs were docked into each of three independently generated hSERT models. Poses were experimentally analyzed by characterizing 13 SERT point mutants with respect to binding of serotonin analogs; interactions between serotonin and residues D98 (TM1), A173 (TM3), and T439 (TM8) were suggested (59). An alternative orientation of serotonin in S1 has been suggested from mutagenesis and modeling data. Human and Drosophila SERT models were built, and support vector machine (SVM) models were derived from experimental binding inhibition data for serotonin analogs at hSERT and dSERT to identify differences conferring species selectivity. The more favorable poses, supported by SVM models and mutagenesis data, oriented the serotonin indole nitrogen toward the intracellular side of the bilayer, near the hSERT Y176 (dSERT Y171) residue of the extracellular-facing gate (35). A comparable serotonin orientation has been suggested from separate computational work (60).
Finally, DAT, NET, and SERT model S1 docking of the cognate monoamine substrate yielded poses that suggest very different alternate substrate binding orientations. In “ionic mode,” the Na1 atom pairs with the TM 1 aspartate (D98 of hSERT). A 180° reorientation of the substrate in S1 aligns Na1 with the catechol hydroxyls of dopamine or norepinephrine or the indole hydroxyl of serotonin, in what is termed “chelation mode” (38). While the ionic mode of monoamine–MAT interaction is more widely accepted, there is experimental evidence for the chelation mode (39).
MAT Inhibitor Pockets
There are now several reported LeuT crystal structures, with most containing an inhibitor bound in the extracellular vestibule or in a region between the S1 and S2 sites (29,40,43,44,56). Unless otherwise specified, MAT models discussed herein were based on the original LeuT–leucine 2a65 crystal structure (8).
Dopamine Transporter
Although the outward- (extracellular-)facing LeuT X-ray structures have an accessible extracellular vestibule, the presence of a substrate molecule in the S1 pocket renders an occluded conformation. Docking of cocaine to a LeuT-based DAT model resulted in cocaine binding in the S2 pocket, perhaps because the extracellular gate to S1 was closed (61). Manually opening this gate by rotating its side chains away from the substrate permeation pore allowed cocaine to sink deeper in the bilayer but with only modest encroachment in the S1 pocket (unpublished data). Using molecular dynamics (MD), a similar cocaine binding site within the DAT was obtained, between S1 and S2. The authors concluded that dopamine and cocaine favor non-overlapping binding sites and proposed that cocaine binding in this quasi-S2 pocket impairs substrate transporter by blocking its pathway to S1 (28).
Using steered MD with a DAT model based on their own MSA, amphetamine as well as analogs of the tropane-based inhibitors cocaine and benztropine were observed to reach the S1 pocket. Access of the inhibitors to S1 was verified pharmacologically, as the presence of a Zn2+ ion in its engineered DAT binding site to the extracellular side of S1 was able to trap the cocaine and benztropine analogs within S1. The authors concluded that a single, dopamine-overlapping binding site exists for cocaine (62). Subsequent work resulted in a similar conclusion for benztropine and its analogs (63). A DAT model based on the LeuT crystal containing the TCA desipramine (pdb:2qju) was used to examine the binding of bivalent phenethylamines. Docking studies suggested that these large, flexible compounds span the core of the protein, simultaneously occupying the S1 and S2 pockets. W84L (TM1) and D313N (TM6) DAT mutants indicated that the bivalent compounds preferred the inward-facing conformation. Multiple low-affinity substrate interaction sites were proposed to exist throughout the translocation pore (64). In a study focused on defining drug interactions at the MATs, electrostatic potential surfaces for DAT were calculated followed by docking of cocaine and clomipramine into the S1 and S2 sites, respectively (65).
Norepinephrine Transporter
With the recent focus on norepinephrine reuptake inhibitors (NRIs) and norepinephrine–dopamine reuptake inhibitors as therapeutics, attention to the NET is expected to increase. A LeuT-based NET homology model was created to visualize the effects of site-directed mutations on the binding of both desipramine and the snail venom conopeptide χ-MrIA, both noncompetitive NET-selective inhibitors. Several residues important for χ-MrIA binding were located in the extracellular vestibule. The combination of NET model docking and mutagenesis led to the conclusion that the binding sites for desipramine and the conotoxin overlap, while the conotoxin and substrate sites are fully separate (30). The size and bulk of conopeptide χ-MrIA, a 13 residue peptide with two intramolecular disulfide bridges, probably restricts its access to any region except the vestibule. In separate work, the cocaine analogs RTI-33 and RTI-113 were found to dock just above the NET S1 pocket, with the C–3 phenyl group dangling below the extracellular gate salt bridge; this positioning essentially coincided with the TCA binding site (66). Based on docking studies, the Y151F mutation was generated for NET (corresponding to F155 in DAT) and shown to increase the sensitivity of NET to RTI-113 while the reciprocal mutation in DAT (F155Y) decreased sensitivity. Similar to their previous DAT study (28), the authors concluded that cocaine occupies a NET binding site distinct from its S1 substrate site (55). It should be noted, however, that the S1 site was occupied by norepinephrine during the docking experiment.
Serotonin Transporter
Binding of the TCAs to the SERT was studied using a SERT model based on the open-to-out LeuT crystal structure 3f3a (56). Ligand docking analysis was in agreement with site-directed mutagenesis data in suggesting the presence of a vestibular low-affinity TCA binding site. The docking poses position the tricyclic rings on the floor of the vestibular S2 pocket, with the amine tail potentially extending into the S1 site (66). Regarding SSRI ligands, both the open-to-out (3f3a) and the occluded (2a65) LeuT x-ray structures have been employed to develop SERT docking poses. SSRI ligands do not appear to bind with high affinity in the S2/vestibular regions, based on reports from several groups (and unpublished results from this laboratory) that SERT mutations in these regions have no appreciable effect on SSRI binding (33,59). The high affinity S-enantiomer of citalopram (escitalopram) was thus docked into the S1 site of SERT, and 64 point mutations were created to experimentally test poses suggesting escitalopram occupancy of S1. Mutation of N177 (TM3) and F341 (TM6), S1 SERT residues previously unassociated with ligand binding, produced significant affinity changes and were proposed to be critical for escitalopram binding (32). The structural determinants conferring inhibitor selectivity for SERT and NET have also been addressed using the SSRI citalopram and the NRI talopram, respectively. Mutational data suggested that the SSRI binding pocket overlaps the S1 substrate pocket, whereas talopram bound at a separate site (34).
MAT Ion Binding Sites
Structure-based computational efforts are more difficult for ion cofactors than for MAT substrates and inhibitors because of the many potential interaction sites for ions. Placement of the Na+ and Cl− ions in a MAT model is therefore especially reliant on the LeuT data. Residues interacting with the two bound Na+ ions in LeuT crystal structures are conserved among the NSS proteins. MAT investigators manually place Na+ ions in the LeuT-corresponding positions of the DAT and in the Na1 position of LeuT for the NET and SERT given that these two proteins appear to need only one Na+ ion for substrate translocation.
In contrast, less is known about the binding domain of the Cl− ion, as it is not essential for LeuT and other NSS members including Tyt1 and TnaT. Hypothesizing that a negatively charged LeuT residue could serve the role of Cl− in the MATs, an elegant study used the multi-conformer continuum electrostatics method to calculate the pKa of membrane-buried acidic residues (45). The negatively charged E290 side chain of LeuT, replaced by a charge-neutral residue in the Cl−-dependent transporters (serine in MAT proteins), was identified as a candidate Cl- surrogate. When the Cl− ion was incorporated into a SERT model at the position correlating with that of LeuT, the ion was coordinated by the hydroxyl and amine hydrogens of Y121 (TM 2), S336 (TM 6), N368, and S372 (TM 7). This located Cl− within 5 Å of Na1, suggesting that Cl− in this position stabilized binding of Na+ (Fig. 4). Experimentally, substitution of SERT N368 or S372 with other charge-neutral side chains decreased transport by increasing the Km for Cl−. Substitution of N368 or S372 with a negatively charged side chain, however, produced a Cl−-independent transporter, albeit of reduced activity (45). Similar studies with GABA transporters are in agreement with respect to location of the Cl− binding site and suggest that a Cl− ion or a precisely positioned TM 7 negatively charged residue surrogate is required for transport (46,67). Additionally, the SERT TM 1 N101 residue does not coordinate with the Cl− ion but appears to restrict ion flux above the observed 1:1:1 serotonin/Na+/Cl− stoichiometry (36).
Fig. 4.
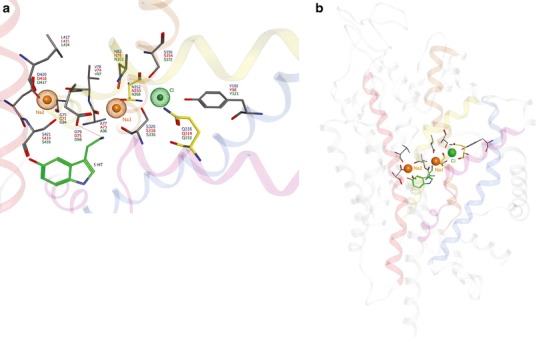
Proposed MAT ion binding sites. a Top and b side views of the sodium (orange) and chloride (green) binding sites are shown relative to S1 substrate position (serotonin; green). Participating side chains (atomtype) are annotated for hDAT (blue), hNET (red) and hSERT (green). Yellow side chains indicate a variance between the hSERT models of Zomot et al. ( 67). (Gln332) and Forrest et al. ( 45). (Asn368) with respect to the chloride ion binding site. The helices of TM1 (yellow), TM2 (blue), TM6 (pink), TM7 (brown) and TM8 (red) are involved in ion interactions
MAT Conformations and Substrate Translocation
The most likely mechanism of substrate translocation across the lipid bilayer is via an “alternating access” model (25,41). By opening extracellular- and intracellular-facing gates of the MAT in alternating fashion, the substrate and co-transporting ions are admitted into and briefly held in an inner chamber (essentially the S1 pocket), then released to travel to the other side of the plasma membrane. Because one of the two gates is always closed, uncontrolled ion flux across this pore in the lipid bilayer is avoided, and the ion gradients key to driving substrate transporter are retained (25). At least three MAT conformations are required: outward-facing (extracellular gate open), substrate-occluded (both gates closed, trapping the substrate), and inward-facing (intracellular gate open) (Fig. 5).
Fig. 5.
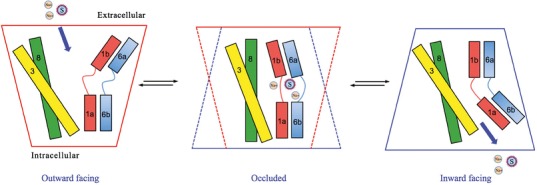
Alternating access model of substrate translocation. Transmembrane (TM) helices 1, 3, 6 and 8 that define the substrate translocation pore are displayed as cylinders. The minimum required conformations of MATs during substrate translocation are shown. An outward-facing conformation allows entry of substrate and ions (S and Na+, respectively) into the transporter from the extracellular space. The occluded conformation restricts access from both the extracellular and intracellular environments. Conversion to the inward-facing conformation allows the release of the substrate and ion into the cytoplasm
Dopamine Transporter
DAT MD simulations in the absence and presence of dopamine have been performed to study substrate passage through the protein. Dopamine binding affinity was evaluated using MM-PBSA calculations (−6.4 kcal/mol) and found to be similar to the experimental binding energy (−7.4 kcal/mol). Substrate permeation appears to involve the formation and breakage of the extracellular gate R85 (TM1)—D476 (TM10) salt bridge (51). Separate MD work corroborates the importance of the DAT extracellular salt bridge. While formation of the LeuT extracellular gate was actually promoted by the presence of substrate, the DAT TM1-TM10 salt bridge was less likely to be intact. Differences between LeuT and DAT in the movement and unwinding of EL4 were also identified (68). The DAT substrate translocation pathway has also been studied using a steered molecular dynamics approach. Dopamine was placed in S1 and pulled in both extracellular and intracellular directions, identifying a temporary binding event at a site corresponding to S2. Simultaneous S1 and S2 dopamine occupancy triggered passage of S1-held dopamine into the cytoplasm (69).
The role of the conserved DAT pre-TM1 residue T62 in the substrate translocation cycle was examined using free energy perturbation (FEP). T62 of the phosphoacceptor site of a “RETW” consensus sequence was replaced with aspartate to mimic the phosphothreonine side chain. In the FEP simulation, the T62D DAT mutant favored an inward-facing conformation, an experimentally supported finding indicating that posttranslational modifications of the N-terminal tail affect DAT conformation (70).
Serotonin Transporter
SERT recognition of the inhibitor and anxiolytic buspirone was addressed using two hSERT models based on templates representing different conformations. Specifically, the outward-facing LeuT was used along with the inward-facing LacY (pdb:1pv6) due to the lack of an inward-facing LeuT crystal structure. (LeuT has recently been crystallized in an inward-facing conformation (71).) Docking of buspirone analogs to the LeuT-based model suggested two discrete binding sites corresponding to what are now termed S1 and S2; these sites were postulated to represent high- and low-affinity binding modes, respectively. MD simulations were performed on the LacY-based model in an effort to identify potential SERT conformational states (72). In another study, MD trajectory analysis was used to detect movement of TM7 SERT residues during substrate and ion translocation. The upper (extracellular-proximal) half of TM7, containing key residues for Cl− and Na2 binding, experienced movement of ~3 A. MD findings were experimentally tested using scanning cysteine accessibility mutagenesis to identify solvent-exposed residues. Na2 vacating this region was postulated to allow rotation of TM7 so that V366 and M370 are exposed to the permeation pathway (73). MD simulations also indicated SERT conformational rearrangements in response to serotonin or citalopram binding. Specifically, the TM3 external gate residue Y176 was shown to adopt different conformations to accommodate changes in the S1 pocket as a result of ligand binding (31). MD simulations and mutagenesis from the same laboratory indicated that D98 (TM1) and I172 (TM3) were important residues for binding of serotonin and citalopram derivatives (60). MD comparison of unoccupied, serotonin-bound and escitalopram-bound SERT indicated the formation and breakage of ionic interactions between TM6, TM8, and IL1 during substrate translocation (42).
As discussed above for the DAT, the N-terminal tail was also investigated for influence on SERT conformation using MD simulations. Models of wild-type, T81A and T81D SERT differed with respect to access to the inward (cytoplasmic)-facing side of S1. T81A SERT, lacking phosphorylation potential at this position, favored the inward-facing conformation, which reduced substrate turnover and amphetamine-induced serotonin efflux. Unexpectedly, introducing a phosphate mimic at this position via T81D mutation failed to preserve amphetamine-induced efflux (47). Finally, an intriguing study employing extended MD simulations tracked SERT conformation as a function of substrate/ion occupancy. Binding of one serotonin molecule triggered the SERT conformational shift from outward-facing to inward-facing, followed by intracellular solvation of Na2 in the S1 pocket and its release into the cytoplasm. Only then was the substrate seen to be solvated from the intracellular side and translocated into the cell. S2 substrate occupancy was found to be unnecessary for substrate transport (27).
MAT Structure-Based Virtual Screening for Novel Ligands
The utility of MAT homology models is not limited to mechanistic and binding studies. Recent reports demonstrate the value of using these models for the identification of novel chemotypes (Fig. 6). There have been very few reports describing the discovery of novel MAT ligands through structure-based VS. Combining a DAT S2 pocket structure-based pharmacophore with high-throughput VS of a chemical database containing over 140,000 compounds, ten compounds were selected for pharmacological evaluation based on score (Affinity dG in MOE), predicted pKi, visual inspection, and commercial availability. MI-4, 1, an ifenprodil analog (Table IV), was discovered to have affinity at all three MATs (74). A similar approach with the SERT utilized a four-feature pharmacophore spanning the S2 pocket and halogen binding pocket (29) to screen a subset of the ZINC database (49). From this, two structurally novel, selective SERT ligands, 2 and 3, were obtained (48). Additionally, this SERT model was used to suggest potential modifications for a VS hit compound. Specifically, docking and flexible alignment studies guided the hit-to-lead optimization of a non-selective MAT inhibitor, 4, discovered through DAT virtual screening efforts into a novel SSRI, 5 (75). Most recently, a database containing 6,436 available drugs (KEGG DRUG) (76) was screened using the S1 pocket of a LeuT-based NET model. Ten of the top-scoring 18 drugs were shown to inhibit NET function at micromolar levels (50), thereby providing an example of the potential for drug repurposing and the ability to uncover new drug-activity profiles via virtual screening. Furthermore, the above virtual screening studies emphasize the reliability of LeuT-based 3-D MAT models, in that these efforts led to the discovery of micromolar affinity hits. Identifying VS hit compounds using homology models or crystal structures is not always guaranteed; moreover, compounds discovered through VS using homology models are typically characterized by low micromolar affinity (37,77–83). The recent success of virtual screening using MAT models suggests that this method may also be useful when applied to other proteins of the SLC6 family given the familial similarities (84).
Fig. 6.
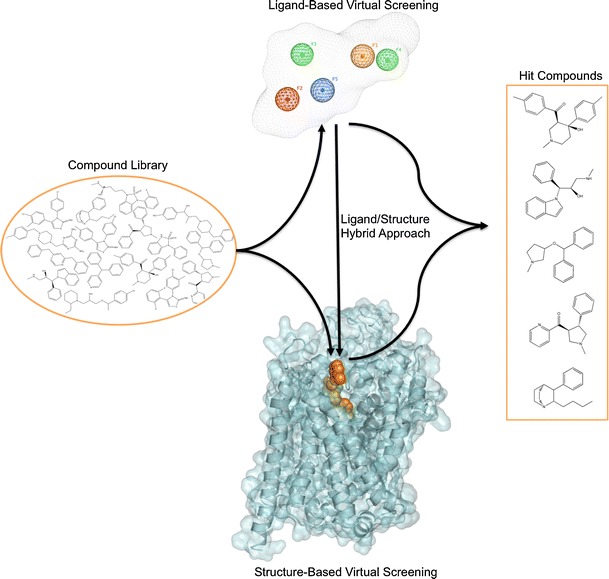
Virtual screening for novel MAT ligands. Structurally diverse “hit” compounds can be identified from large structural databases using ligand-based or structure-based VS approaches, or both (hybrid approach). The ligand-based method employs a 3D pharmacophore model; an example containing five features labeled F1–F5 with a volume constraint is shown. The structure-based method involves docking VS compounds into the binding site (orange VDW spheres) of a 3-D MAT homology model. For the hybrid approach, structure-based VS usually follows ligand-based VS
Table IV.
Novel MAT Ligands Discovered Through Virtual Screening and Molecular Modeling
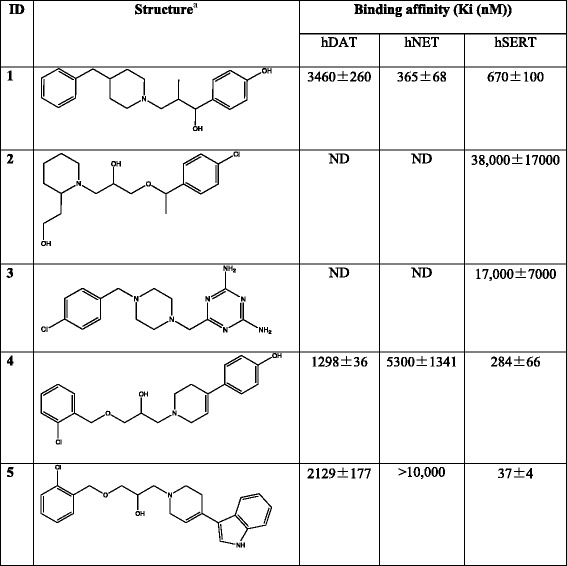
aStereochemistry for stereocenters is undefined
CONCLUSION
The elucidation of the LeuT crystal structure was a breakthrough for monoamine transporter research, serving as a template for the generation of MAT 3-D protein models. Through the use of these models and structure-based computational tools, novel MAT ligands have been discovered, insight into protein structure and aspects of ligand recognition has been gained, and details on the dynamic nature (including conformational states and possible mechanisms of transport) of these proteins have been uncovered. The longstanding question of where the various structural classes of inhibitors bind in the MATs is a central focus for many computational studies, as is defining the conformations involved in the substrate translocation cycle and the actual mechanism by which translocation occurs. It is also a matter of debate whether two substrate molecules are required for this translocation cycle. Structure-based VS efforts with MAT models are revealing ligands of such novel chemotypes that their identification could likely only have been achieved via computational approaches. Hit-to-lead optimization of these chemotypes will lead to superior CNS medications that display fewer adverse effects.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
{kind=link}
Beuming et al ( 24). primary amino acid sequence alignment of LeuT Aa with the human MATs. N- and C-terminal tails were not used for model building and are not shown. Amino acid residue position (red numbers) is indicated at the beginning and end of each row; LeuT residue increments of 10 (blue numbers) are indicated above the row; gaps in alignment are shown as dots. Identical (blue) and similar (yellow) aligned residues are highlighted. Secondary structural features for transmembrane domains (TM), intracellular loops (IL), extracellular loops (EL) and beta-strands (B) are displayed above the sequence rows. Charged residues of the extracellular (blue spheres) and intracellular (orange spheres) gates and residues of the hydrophobic extracellular gate (red stars) are noted below the sequence rows. (JPEG 2383 kb)
{kind=link}
SERT (green) embedded within a POPE lipid bilayer (van der Waals spheres). Hydrocarbon tails (gray) of the phospholipids are oriented toward the center of the cell membrane, arranged tail-to-tail; the polar head groups containing phosphorous (pink), oxygen (red) and nitrogen (blue) atoms are oriented toward the hydrophilic borders of the membrane. The translocation pore is defined by the primary (S1, yellow) and secondary (S2, cyan) substrate binding pockets and are displayed as surfaces. Ions and water molecules are hidden for the sake of clarity, as is the front portion of the lipid bilayer. (JPEG 2390 kb)
ACKNOWLEDGMENTS
This work was supported by the National Institute on Drug Abuse under award DA026530 (C.K.S.) and the National Institutes of Health, National Science Foundation, Department of Defense, and the U.S. Department of Education under awards DA27806 (J.D.M.), CHE-1005145(REU/ASSURE), CHE-0723109(MRI), and P116Z080180 (J.D.M.).
Abbreviations
- DAT
dopamine transporter
- GABA
gamma-aminobutyric acid
- MAT
monoamine transporter
- MD
molecular dynamics
- MSA
multiple sequence alignment
- NET
norepinephrine transporter
- NRI
norepinephrine reuptake inhibitor
- NSS
neurotransmitter/sodium symporter
- SERT
serotonin transporter
- SSRI
selective serotonin reuptake inhibitor
- TCA
tricyclic antidepressant
- TM
transmembrane
- VS
virtual screening
REFERENCES
- 1.Kristensen AS, Andersen J, Jorgensen TN, Sorensen L, Eriksen J, Loland CJ, et al. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol Rev. 2011;63(3):585–640. doi: 10.1124/pr.108.000869. [DOI] [PubMed] [Google Scholar]
- 2.Hahn MK, Blakely RD. Monoamine transporter gene structure and polymorphisms in relation to psychiatric and other complex disorders. Pharmacogenomics J. 2002;2:217–235. doi: 10.1038/sj.tpj.6500106. [DOI] [PubMed] [Google Scholar]
- 3.Heinz A, Mann K, Weinberger DR, Goldman D. Serotonergic dysfunction, negative mood states, and response to alcohol. Alcohol Clin Exp Res. 2001;25(4):487–495. doi: 10.1111/j.1530-0277.2001.tb02240.x. [DOI] [PubMed] [Google Scholar]
- 4.Klimek V, Stockmeier C, Overholser J, Meltzer HY, Kalka S, Dilley G, et al. Reduced levels of norepinephrine transporters in the locus coeruleus in major depression. J Neurosci. 1997;17(21):8451–8458. doi: 10.1523/JNEUROSCI.17-21-08451.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller GM, De La Garza R, II, Novak MA, Madras BK. Single nucleotide polymorphisms distinguish multiple dopamine transporter alleles in primates: Implications for association with attention deficit hyperactivity disorder and other neuropsychiatric disorders. Mol Psychiatry. 2001;6(1):50–58. doi: 10.1038/sj.mp.4000809. [DOI] [PubMed] [Google Scholar]
- 6.Ozaki N, Goldman D, Kaye WH, Greenberg BD, Lappalainen J, Rudnick G, et al. Serotonin transporter missense mutation associated with a complex neuropsychiatric phenotype. Mol Psychiatry. 2003;8:933–936. doi: 10.1038/sj.mp.4001365. [DOI] [PubMed] [Google Scholar]
- 7.Robertson D, Flattem N, Tellioglu T, Carson R, Garland E, Shannon JR, et al. Familial orthostatic tachycardia due to norepinephrine transporter deficiency. Ann N Y Acad Sci. 2001;940:527–543. doi: 10.1111/j.1749-6632.2001.tb03703.x. [DOI] [PubMed] [Google Scholar]
- 8.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl–dependent neurotransmitter transporters. Nature. 2005;437(7056):215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 9.Brust A, Palant E, Croker DE, Colless B, Drinkwater R, Patterson B, et al. χ-Conopeptide pharmacophore development: toward a novel class of norepinephrine transporter inhibitor (Xen2174) for pain. J Med Chem. 2009;52(22):6991–7002. doi: 10.1021/jm9003413. [DOI] [PubMed] [Google Scholar]
- 10.Chen C, Dyck B, Fleck BA, Foster AC, Grey J, Jovic F, et al. Studies on the SAR and pharmacophore of milnacipran derivatives as monoamine transporter inhibitors. Bioorg Med Chem Lett. 2008;18(4):1346–1349. doi: 10.1016/j.bmcl.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 11.Enyedy IJ, Sakamuri S, Zaman WA, Johnson KM, Wang S. Pharmacophore-based discovery of substituted pyridines as novel dopamine transporter inhibitors. Bioorg Med Chem Lett. 2003;13(3):513–517. doi: 10.1016/S0960-894X(02)00943-5. [DOI] [PubMed] [Google Scholar]
- 12.Enyedy IJ, Wang J, Zaman WA, Johnson KM, Wang S. Discovery of substituted 3,4-diphenyl-thiazoles as a novel class of monoamine transporter inhibitors through 3-D pharmacophore search using a new pharmacophore model derived from mazindol. Bioorg Med Chem Lett. 2002;12:1775–1778. doi: 10.1016/S0960-894X(02)00243-3. [DOI] [PubMed] [Google Scholar]
- 13.Enyedy IJ, Zaman WA, Sakamuri S, Kozikowski AP, Johnson KM, Wang S. Pharmacophore-based discovery of 3,4-disubstituted pyrrolidines as a novel class of monoamine transporter inhibitors. Bioorg Med Chem Lett. 2001;11(9):1113–1118. doi: 10.1016/S0960-894X(01)00132-9. [DOI] [PubMed] [Google Scholar]
- 14.Macdougall IJ, Griffith R. Pharmacophore design and database searching for selective monoamine neurotransmitter transporter ligands. J Mol Graph Model. 2008;26(7):1113–1124. doi: 10.1016/j.jmgm.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 15.Pratuangdejkul J, Schneider B, Jaudon P, Rosilio V, Baudoin E, Loric S, et al. Definition of an uptake pharmacophore of the serotonin transporter through 3D-QSAR analysis. Curr Med Chem. 2005;12(20):2393–2410. doi: 10.2174/0929867054864813. [DOI] [PubMed] [Google Scholar]
- 16.Pratuangdejkul J, Schneider B, Launay JM, Kellermann O, Manivet P. Computational approaches for the study of serotonin and its membrane transporter SERT: implications for drug design in neurological sciences. Curr Med Chem. 2008;15(30):3214–3227. doi: 10.2174/092986708786848523. [DOI] [PubMed] [Google Scholar]
- 17.Wang S, Sakamuri S, Enyedy IJ, Kozikowski AP, Deschaux O, Bandyopadhyay BC, et al. Discovery of a novel dopamine transporter inhibitor, 4-hydroxy-1-methyl-4-(4-methylphenyl)-3-piperidyl 4-methylphenyl ketone, as a potential cocaine antagonist through 3D-database pharmacophore searching. Molecular modeling, structure-activity relationships, and behavioral pharmacological studies. J Med Chem. 2000;43(3):351–360. doi: 10.1021/jm990516x. [DOI] [PubMed] [Google Scholar]
- 18.Edvardsen O, Dahl SG. A putative model of the dopamine transporter. Brain Res Mol Brain Res. 1994;27:265–274. doi: 10.1016/0169-328X(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 19.Ravna A, Edvardsen O. A putative three-dimensional arrangement of the human serotonin transporter transmembrane helices: a tool to aid experimental studies. J Mol Graph Model. 2001;20:133–144. doi: 10.1016/S1093-3263(01)00112-7. [DOI] [PubMed] [Google Scholar]
- 20.Ravna A, Sylte I, Kristiansen K, Dahl S. Putative drug binding conformations of monoamine transporters. Bioorg Med Chem. 2006;14(3):666–675. doi: 10.1016/j.bmc.2005.08.054. [DOI] [PubMed] [Google Scholar]
- 21.Ravna AW. Three-dimensional models of neurotransmitter transporters and their interactions with cocaine and S-citalopram. World J Biol Psychiatry. 2006;7:99–109. doi: 10.1080/15622970500402144. [DOI] [PubMed] [Google Scholar]
- 22.Ravna AW, Sylte I, Dahl SG. Molecular model of the neural dopamine transporter. J Comput Aided Mol Des. 2003;17:367–382. doi: 10.1023/A:1026116017725. [DOI] [PubMed] [Google Scholar]
- 23.Guan L, Kaback HR. Lessons from lactose permease. Annu Rev Biophys Biomol Struct. 2006;35:67–91. doi: 10.1146/annurev.biophys.35.040405.102005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beuming T, Shi L, Javitch JA, Weinstein H. A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol Pharm. 2006;70(5):1630–1642. doi: 10.1124/mol.106.026120. [DOI] [PubMed] [Google Scholar]
- 25.Rudnick G. Cytoplasmic permeation pathway of neurotransmitter transporters. Biochemistry. 2011;50(35):7462–7475. doi: 10.1021/bi200926b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. The mechanism of a neurotransmitter:sodium symporter-inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol Cell. 2008;30(6):667–677. doi: 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koldso H, Noer P, Grouleff J, Autzen HE, Sinning S, Schiott B. Unbiased simulations reveal the inward-facing conformation of the human serotonin transporter and Na ion release. PLoS Comput Biol. 2011;7(10). doi:10.1371/journal.pcbi.1002246. [DOI] [PMC free article] [PubMed]
- 28.Huang X, Gu HH, Zhan CG. Mechanism for cocaine blocking the transport of dopamine: insights from molecular modeling and dynamics simulations. J Phys Chem. 2009;113:15057–15066. doi: 10.1021/jp900963n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Z, Zhen J, Karpowich NK, Law CJ, Reith ME, Wang DN. Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nat Struct Mol Biol. 2009;16(6):652–657. doi: 10.1038/nsmb.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paczkowski FA, Sharpe IA, Dutertre S, Lewis RJ. X-conotoxin and tricyclic antidepressant interactions at the norepinephrine transporter define a new transporter model. J Biol Chem. 2007;282(24):17837–17844. doi: 10.1074/jbc.M610813200. [DOI] [PubMed] [Google Scholar]
- 31.Jorgensen AM, Tagmose L, Bogeso KP, Peters GH. Molecular dynamics simulations of Na+/Cl(−)-dependent neurotransmitter transporters in a membrane-aqueous system. ChemMedChem. 2007;2(6):827–840. doi: 10.1002/cmdc.200600243. [DOI] [PubMed] [Google Scholar]
- 32.Andersen J, Olsen L, Hansen KB, Taboureau O, Jorgensen FS, Jorgensen AM, et al. Mutational mapping and modeling of the binding site for (S)-citalopram in the human serotonin transporter. J Biol Chem. 2010;285(3):2051–2063. doi: 10.1074/jbc.M109.072587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andersen J, Taboureau O, Hansen KB, Olsen L, Egebjerg J, Stromgaard K, et al. Location of the antidepressant binding site in the serotonin transporter: importance of Ser-438 in recognition of citalopram and tricyclic antidepressants. J Biol Chem. 2009;284(15):10276–10284. doi: 10.1074/jbc.M806907200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andersen J, Stuhr-Hansen N, Zachariassen L, Toubro S, Hansen SM, Eildal JN, et al. Molecular determinants for selective recognition of antidepressants in the human serotonin and norepinephrine transporters. Proc Natl Acad Sci U S A. 2011;108(29):12137–12142. doi: 10.1073/pnas.1103060108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaufmann KW, Dawson ES, Henry LK, Field JR, Blakely RD, Meiler J. Structural determinants of species-selective substrate recognition in human and Drosophila serotonin transporters revealed through computational docking studies. Proteins. 2009;74(3):630–642. doi: 10.1002/prot.22178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Henry LK, Iwamoto H, Field JR, Kaufmann K, Dawson ES, Jacobs MT, et al. A conserved asparagine residue in transmembrane segment 1 (TM1) of serotonin transporter dictates chloride-coupled neurotransmitter transport. J Biol Chem. 2011;286(35):30823–30836. doi: 10.1074/jbc.M111.250308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Enyedy IJ, Ling Y, Nacro K, Tomita Y, Wu X, Cao Y, et al. Discovery of small-molecule inhibitors of Bcl-2 through structure-based computer screening. J Med Chem. 2001;44(25):4313–4324. doi: 10.1021/jm010016f. [DOI] [PubMed] [Google Scholar]
- 38.Xhaard H, Backstrom V, Denessiouk K, Johnson MS. Coordination of Na(+) by monoamine ligands in dopamine, norepinephrine, and serotonin transporters. J Chem Inf Model. 2008;48(7):1423–1437. doi: 10.1021/ci700255d. [DOI] [PubMed] [Google Scholar]
- 39.Wang W, Sonders MS, Ukairo OT, Scott H, Kloetzel MK, Surratt CK. Dissociation of high-affinity cocaine analog binding and dopamine uptake inhibition at the dopamine transporter. Mol Pharmacol. 2003;64(2):430–439. doi: 10.1124/mol.64.2.430. [DOI] [PubMed] [Google Scholar]
- 40.Zhou Z, Zhen J, Karpowich NK, Goetz RM, Law CJ, Reith MEA, et al. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science. 2007;317(5843):1390–1393. doi: 10.1126/science.1147614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitchell P. Osmochemistry of solute translocation. Res Microbiol. 1990;141:286–289. doi: 10.1016/0923-2508(90)90002-8. [DOI] [PubMed] [Google Scholar]
- 42.Gabrielsen M, Ravna AW, Kristiansen K, Sylte I. Substrate binding and translocation of the serotonin transporter studied by docking and molecular dynamics simulations. J Mol Model. 2011. doi:10.1007/s00894-011-1133-1. [DOI] [PMC free article] [PubMed]
- 43.Quick M, Lund Winther A-M, Shi L, Nissen P, Weinstein H, Javitch JA. Binding of an octylglucoside detergent molecule in the second substrate (S2) site of LeuT establishes an inhibitor-bound conformation. Proc Natl Acad Sci U S A. 2009;106(14):5563–5568. doi: 10.1073/pnas.0811322106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh SK, Yamashita A, Gouaux E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature. 2007;448(7156):952–956. doi: 10.1038/nature06038. [DOI] [PubMed] [Google Scholar]
- 45.Forrest LR, Tavoulari S, Zhang YW, Rudnick G, Honig B. Identification of a chloride ion binding site in Na+/Cl -dependent transporters. Proc Natl Acad Sci U S A. 2007;104(31):12761–12766. doi: 10.1073/pnas.0705600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tavoulari S, Rizwan AN, Forrest LR, Rudnick G. Reconstructing a chloride-binding site in a bacterial neurotransmitter transporter homologue. J Biol Chem. 2011;286(4):2834–2842. doi: 10.1074/jbc.M110.186064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sucic S, Dallinger S, Zdrazil B, Weissensteiner R, Jorgensen TN, Holy M, et al. The N terminus of monoamine transporters is a lever required for the action of amphetamines. J Biol Chem. 2010;285(14):10924–10938. doi: 10.1074/jbc.M109.083154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manepalli S, Geffert LM, Surratt CK, Madura JD. Discovery of novel selective serotonin reuptake inhibitors through development of a protein-based pharmacophore. J Chem Inf Model. 2011;51(9):2417–2426. doi: 10.1021/ci200280m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Irwin JJ, Shoichet BK. ZINC–a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45(1):177–182. doi: 10.1021/ci049714+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schlessinger A, Geier E, Fan H, Irwin JJ, Shoichet BK, Giacomini KM, et al. Struture-based discovery of prescription drugs that interact with the norepinephrine transporter, NET. Proc Natl Acad Sci U S A. 2011;108(38):15810–15815. doi: 10.1073/pnas.1106030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang X, Zhan C. How dopamine transporter interacts with dopamine: insights from molecular modeling and simulation. Biophys J. 2007;93(10):3627–3639. doi: 10.1529/biophysj.107.110924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Indarte M, Madura JD, Surratt CK. Dopamine transporter comparative molecular modeling and binding site prediction using the LeuTAa leucine transporter as a template. Proteins Struct Funct Bioinforma. 2008;70(3):1033–1046. doi: 10.1002/prot.21598. [DOI] [PubMed] [Google Scholar]
- 53.Zhao Y, Terry DS, Shi L, Quick M, Weinstein H, Blanchard SC, et al. Substrate-modulated gating dynamics in a Na(+)-coupled neurotransmitter transporter homologue. Nature. 2011;474(7349):109–113. doi: 10.1038/nature09971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Piscitelli CL, Krishnamurthy H, Gouaux E. Neurotransmitter/sodium symporter orthologue LeuT has a single high-affinity substrate site. Nature. 2010;468(7327):1129–1132. doi: 10.1038/nature09581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hill ER, Huang X, Zhan CG, Carroll FI, Gu HH. Interaction of tyrosine 151 in norepinephrine transporter with the 2-β group of cocaine analog RTI-113. Neuropharmacology. 2011;61(1–2):112–120. doi: 10.1016/j.neuropharm.2011.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singh SK, Piscitelli CL, Yamashita A, Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science. 2008;322(5908):1655–1661. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ravna AW, Jaronczyk M, Sylte I. A homology model of SERT based on the LeuT(Aa) template. Bioorg Med Chem Lett. 2006;16(21):5594–5597. doi: 10.1016/j.bmcl.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 58.Henry LK, Defelice LJ, Blakely RD. Getting the message across: a recent transporter structure shows the way. Neuron. 2006;49(6):791–796. doi: 10.1016/j.neuron.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 59.Celik L, Sinning S, Severinsen K, Hansen CG, Moller MS, Bols M, et al. Binding of serotonin to the human serotonin transporter. Molecular modeling and experimental validation. J Am Chem Soc. 2008;130(12):3853–3865. doi: 10.1021/ja076403h. [DOI] [PubMed] [Google Scholar]
- 60.Jorgensen AM, Tagmose L, Topiol S, Sabio M, Gundertofte K, Bogeso KP, et al. Homology modeling of the serotonin transporter: insights into the primary escitalopram-binding site. ChemMedChem. 2007;2(6):815–826. doi: 10.1002/cmdc.200600242. [DOI] [PubMed] [Google Scholar]
- 61.Indarte M, Madura JD, Surratt CK, editors. Comparative molecular modeling of the dopamine transporter using the leucine transporter LeuTAa as a template. Atlanta: Society for Neuroscience; 2006. [Google Scholar]
- 62.Beuming T, Kniazeff J, Bergmann ML, Shi L, Gracia L, Raniszewska K, et al. The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat Neurosci. 2008;11(7):780–789. doi: 10.1038/nn.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bisgaard H, Larsen MA, Mazier S, Beuming T, Newman AH, Weinstein H, et al. The binding sites for benztropines and dopamine in the dopamine transporter overlap. Neuropharmacology. 2011;60(1):182–190. doi: 10.1016/j.neuropharm.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schmitt KC, Mamidyala S, Biswas S, Dutta AK, Reith ME. Bivalent phenethylamines as novel dopamine transporter inhibitors: evidence for multiple substrate-binding sites in a single transporter. J Neurochem. 2010;112(6):1605–1618. doi: 10.1111/j.1471-4159.2010.06583.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ravna AW, Sylte I, Dahl SG. Structure and localisation of drug binding sites on neurotransmitter transporters. J Mol Model. 2009;15(10):1155–1164. doi: 10.1007/s00894-009-0478-1. [DOI] [PubMed] [Google Scholar]
- 66.Sarker S, Weissensteiner R, Steiner I, Sitte HH, Ecker GF, Freissmuth M, et al. The high-affinity binding site for tricyclic antidepressants resides in the outer vestibule of the serotonin transporter. Mol Pharmacol. 2010;78(6):1026–1035. doi: 10.1124/mol.110.067538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zomot E, Bendahan A, Quick M, Zhao Y, Javitch JA, Kanner BI. Mechanism of chloride interaction with neurotransmitter: sodium symporters. Nature. 2007;449(7163):726–730. doi: 10.1038/nature06133. [DOI] [PubMed] [Google Scholar]
- 68.Gedeon PC, Indarte M, Surratt CK, Madura JD. Molecular dynamics of leucine and dopamine transporter proteins in a model cell membrane lipid bilayer. Proteins Struct Funct Bioinforma. 2010;78(4):797–811. doi: 10.1002/prot.22601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shan J, Javitch JA, Shi L, Weinstein H. The substrate-driven transition to an inward-facing conformation in the functional mechanism of the dopamine transporter. PLoS One. 2011;6(1). doi:10.1371/journal.pone.0016350. [DOI] [PMC free article] [PubMed]
- 70.Guptaroy B, Zhang M, Bowton E, Binda F, Shi L, Weinstein H, et al. A juxtamembrane mutation in the N terminus of the dopamine transporter induces preference for an inward-facing conformation. Mol Pharmacol. 2009;75(3):514–524. doi: 10.1124/mol.108.048744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krishnamurthy H, Gouaux E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature. 2012;481(7382):469–474. doi: 10.1038/nature10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jaronczyk M, Chilmonczyk Z, Mazurek AP, Nowak G, Ravna AW, Kristiansen K, et al. The molecular interactions of buspirone analogues with the serotonin transporter. Bioorg Med Chem. 2008;16(20):9283–9294. doi: 10.1016/j.bmc.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 73.Wenthur CJ, Rodriguez GJ, Kuntz CP, Barker EL. Conformational flexibility of transmembrane helix VII of the human serotonin transporter impacts ion dependence and transport. Biochem Pharmacol. 2010;80(9):1418–1426. doi: 10.1016/j.bcp.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Indarte M, Liu Y, Madura JD, Surratt CK. Receptor-based discovery of a plasmalemmal monoamine transporter inhibitor via high throughput docking and pharmacophore modeling. ACS Chem Neurosci. 2010;1(3):223–233. doi: 10.1021/cn900032u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nolan TL, Lapinsky DJ, Talbot JN, Indarte M, Liu Y, Manepalli S, et al. Identification of a novel selective serotonin reuptake inhibitor by coupling monoamine transporter-based virtual screening and rational molecular hybridization. ACS Chem Neurosci. 2011;2(9):544–552. doi: 10.1021/cn200044x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Okuda S, Yamada T, Hamajima M, Itoh M, Katayama T, Bork P, et al. KEGG Atlas mapping for global analysis of metabolic pathways. Nucleic Acids Res. 2008;36:W423–W426. doi: 10.1093/nar/gkn282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300(5626):1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 78.Enyedy IJ, Lee S-L, Kuo AH, Dickson RB, Lin C-Y, Wang S. Structure-based approach for the discovery of Bis-benzamidines as novel inhibitors of matriptase. J Med Chem. 2001;44(9):1349–1355. doi: 10.1021/jm000395x. [DOI] [PubMed] [Google Scholar]
- 79.Li R, Chen X, Gong B, Selzer PM, Li Z, Davidson E, et al. Structure-based design of parasitic protease inhibitors. Bioorg Med Chem. 1996;4(9):1421–1427. doi: 10.1016/0968-0896(96)00136-8. [DOI] [PubMed] [Google Scholar]
- 80.Que X, Brinen LS, Perkins P, Herdman S, Hirata K, Torian BE, et al. Cysteine proteinases from distinct cellular compartments are recruited to phagocytic vesicles by Entamoeba histolytica. Mol Biochem Parasitol. 2002;119(1):23–32. doi: 10.1016/S0166-6851(01)00387-5. [DOI] [PubMed] [Google Scholar]
- 81.Rajnarayanan RV, Dakshanamurthy S, Pattabiraman N. “Teaching old drugs to kill new bugs”: structure-based discovery of anti-SARS drugs. Biochem Biophys Res Commun. 2004;321(2):370–378. doi: 10.1016/j.bbrc.2004.06.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Selzer PM, Chen X, Chan VJ, Cheng M, Kenyon GL, Kuntz ID, et al. Leishmania major: molecular modeling of cysteine proteases and prediction of new nonpeptide inhibitors. Exp Parasitol. 1997;87(3):212–221. doi: 10.1006/expr.1997.4220. [DOI] [PubMed] [Google Scholar]
- 83.Zuccotto F, Zvelebil M, Brun R, Chowdhury SF, Di Lucrezia R, Leal I, et al. Novel inhibitors of Trypanosoma cruzi dihydrofolate reductase. Eur J Med Chem. 2001;36(5):395–405. doi: 10.1016/S0223-5234(01)01235-1. [DOI] [PubMed] [Google Scholar]
- 84.Schlessinger A, Matsson P, Shima JE, Pieper U, Yee SW, Kelly L, et al. Comparison of human solute carriers. Prot Sci. 2010;19(3):412–428. doi: 10.1002/pro.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Beuming et al ( 24). primary amino acid sequence alignment of LeuT Aa with the human MATs. N- and C-terminal tails were not used for model building and are not shown. Amino acid residue position (red numbers) is indicated at the beginning and end of each row; LeuT residue increments of 10 (blue numbers) are indicated above the row; gaps in alignment are shown as dots. Identical (blue) and similar (yellow) aligned residues are highlighted. Secondary structural features for transmembrane domains (TM), intracellular loops (IL), extracellular loops (EL) and beta-strands (B) are displayed above the sequence rows. Charged residues of the extracellular (blue spheres) and intracellular (orange spheres) gates and residues of the hydrophobic extracellular gate (red stars) are noted below the sequence rows. (JPEG 2383 kb)
SERT (green) embedded within a POPE lipid bilayer (van der Waals spheres). Hydrocarbon tails (gray) of the phospholipids are oriented toward the center of the cell membrane, arranged tail-to-tail; the polar head groups containing phosphorous (pink), oxygen (red) and nitrogen (blue) atoms are oriented toward the hydrophilic borders of the membrane. The translocation pore is defined by the primary (S1, yellow) and secondary (S2, cyan) substrate binding pockets and are displayed as surfaces. Ions and water molecules are hidden for the sake of clarity, as is the front portion of the lipid bilayer. (JPEG 2390 kb)