

Abstract
Nanosuspensions, formulations based on the reduction of the active pharmaceutical ingredient (API) particle size in the sub-micron range and most typically around 100–200 nm, represent a valuable option for formulators to facilitate oral absorption of Biopharmaceutics Classification System class II and IV compounds. Their ability to increase the API dissolution rate and subsequent absorption and thus oral bioavailability has been demonstrated in preclinical and clinical settings. This review summarizes the current experience in the biopharmaceutic field with the use of nanosuspensions as oral delivery formulations. The principles behind nanosuspensions as well as the in vitro and in silico evaluation are discussed, while examples are presented highlighting both successes as well as limitations in their application as either toxicology or clinical formulations.
Key words: bioavailability enhancement, nanocrystals, nanoparticles, nanosizing, nanosuspensions
INTRODUCTION
It is estimated that up to 90% of new drug development candidates potentially exhibit poor aqueous solubility and are classified as class II or class IV compounds based on the Biopharmaceutics Classification System (BCS) (1). Many of these compounds have solubilities as low as 1–10 μg/mL (2). This trend toward low solubility compounds poses a significant challenge for formulators and biopharmaceutic scientists to develop dosage forms that would allow for sufficient oral absorption and therefore oral bioavailability of these drug candidates.
Poor oral bioavailability can represent a significant hurdle for development of drug candidates both in the preclinical and clinical settings. Perhaps the biggest impact in the preclinical setting is limiting the ability to obtain sufficient exposure in toxicology studies. This can necessitate very high compound doses to obtain necessary safety margins resulting in large consumption of active pharmaceutical ingredient (API). In the clinic, poor bioavailability can be associated with increased variability that can manifest itself also in the form of food effects. Similar to preclinical studies, low bioavailability can also result in utilization of higher doses to either establish clinical safety margins or even to achieve the intended pharmacological response. To address these issues, formulation scientists have been employing a variety of formulation approaches in both preclinical studies and as clinical formulations. Salt formation, particle size reduction, use of lipid vehicles and cosolvents in the form of liquid-filled capsules, complexation (e.g., cyclodextrins), and more recently amorphous solid dispersions are routinely employed to improve the solubilization of compounds in the gastrointestinal tract and thus subsequently improve their oral bioavailability.
While particle size reduction traditionally largely referred to micronization, more recently methodologies have been developed that allow for the preparation of API particles in the submicron range. The submicron API formulations, with particle sizes typically in the 100–200-nm range, are referred to as nanosuspensions or drug nanocrystals and have been attracting increasing interest as they have been demonstrated to result in increases in bioavailability significantly beyond what can be achieved with traditional micronization (3–8). These nanoparticles are stabilized in the formulation with the help of surfactants or polymers and have been successfully employed as both liquid nanosuspensions for the use in preclinical toxicology studies and in standard dosage forms, such as capsules or tablets, suitable for oral administration in the clinic and eventually in commercial products.
The utility of nanocrystalline API to improve bioavailability has been demonstrated in vitro in dissolution testing providing significantly increased dissolution rates and in vivo in both preclinical species as well as clinical trials where researchers have demonstrated improved bioavailability and/or reduction of food effect for BCS II and IV compounds. These successes have led to so far five marketed drug products utilizing crystalline API. However, despite the available examples, the biopharmaceutical understanding of these systems is still evolving. The bioavailability gains obtained are often times more significant than what is expected based on dissolution rate improvement. However, at the same time, not all compounds appear to be benefiting from nanosizing. With the incorporation of in silico tools in the biopharmaceutical research, new approaches that link in vitro and in vivo data are being explored that shed more light into the performance of these formulations.
This review summarizes the current experience in the biopharmaceutic field with the use of crystalline API nanoparticles as bioavailability improving technology for BCS II and IV compounds, for both toxicology application in the form of liquid nanosuspensions and for clinical formulation application more typically in the form of nanoparticle-based solid dosage forms. The current experience with in vitro and in silico evaluation of crystalline nanosuspensions is discussed. Specific case studies and clinical data are discussed to demonstrate the current knowledge around biopharmaceutical evaluation of these systems during drug development. The examples provided also include case studies where nanosuspensions may not represent the highest bioavailability formulation. Finally, an overview of the currently marketed products is provided.
PRINCIPLES OF NANOSUSPENSIONS
The rate of dissolution of a solid drug compound is directly proportional to the surface area available for dissolution. This is described by the Nernst–Brunner/Noyes–Whitney equation (Eq. 1) (9, 10).
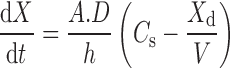 |
1 |
where dX/dt = dissolution rate, Xd = amount dissolved, A = particle surface area, D = diffusion coefficient, V = volume of fluid available for dissolution, Cs = saturation solubility, and h = effective boundary layer thickness.
Based on Eq. 1, it is clear that the two main parameters affecting the in vitro dissolution rate is the solubility of the compound (Cs) and the surface area (A). While the solubility of the drug compound will depend on its physical state (i.e., crystalline vs. amorphous, polymorphic form, salt vs. free form), the surface area term links the rate of dissolution to the bulk properties of the drug compound. Based on this principle, API particle size reduction to micron size range has been extensively used in the pharmaceutical industry as a tool to improve oral bioavailability of poor soluble (BCS II and IV) drug compounds. It has also been demonstrated that a decrease of the particle size down to the submicron range will further enhance dissolution rate due to a greater increase of the effective particle surface area (11). Numerous studies have demonstrated this advantage of nanoformulations over a conventional micronized API or un-milled API. Recently, Quinn et al. (12) have shown that for a gamma secretase inhibitor reduction of API particle size from 50 μm to 159 nm resulted in a surface area enhancement of 333-fold. This resulted in a significant enhancement in the dissolution rate of the nanosuspension as compared to the unmilled API. The nanosuspension was completely dissolved by 5 min; in contrast, the 50-μm API suspension showed only 6% dissolved at 60 min. Since this was a BCS-II compound, the enhance in dissolution rate for the nanosuspension led to a marked improvement in oral bioavailability in fasted beagle dogs as compared to the 50-μm API suspension (87% vs. 11%) (12). Similar results have also been shown by Wu et al. for aprepitant (13) and Miao et al. for cilostazol (14), among others.
In addition, as described by the Prandtl equation (Eq. 2), a decrease in the diffusion layer thickness (h) with increasing curvature of nanoparticles leads to a further increase in the dissolution rate (11).
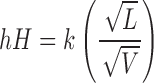 |
2 |
where L = length of the surface in the direction of flow, k = denotes a constant, V = relative velocity of the flowing liquid against a flat surface, and hH = the hydrodynamic boundary layer thickness.
In agreement with the Prandtl equation, Nystrom and Bisrat (15) have shown that for solid particles dispersed in a liquid medium under agitation, a decrease in particle size results in a thinner hydrodynamic layer around particles and hence lead to an increase in the dissolution rate. The enhancement of dissolution rate is especially pronounced for solid particles that have mean particle size of less than 2 μm. Thus, the combination of decrease in h and the increase in Cs would lead to an increase in the dissolution rate as per the Noyes–Whitney equation (Eq. 1).
In addition to the dissolution rate enhancement described above, an increase in the saturation solubility of the nanosized API is also expected (16), as described by the Freundlich–Ostwald equation (Eq. 3):
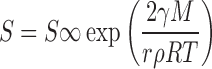 |
3 |
where S = saturation solubility of the nanosized API, S∞ = saturation solubility of an infinitely large API crystal, γ is the crystal-medium interfacial tension, M is the compound molecular weight, r is the particle radius, ρ is the density, R is a gas constant, and T is the temperature.
Based on this equation and assuming a molecular weight of 500, ρ = 1 g/mL and a γ value of 15–20 mN m−1 for the crystal-intestinal fluid interfacial tension, an approximately 10–15% increase in solubility is predicted for 100 nm API particles compared to the crystalline thermodynamic solubility of unmilled API. However, a more significant increase in solubility has been reported, e.g., Muller and Peters reported an increase of 50% in the solubility of an insoluble antimicrobial compound when the particle size was reduced from 2.4 μm to 800 or 300 nm (16). The higher than anticipated increase in solubility is unusual; the authors suggested that this may be related to the creation of high energy surfaces on the API crystals during the nanomilling process. This increased solubility will further increase the dissolution rate, and as a result, nanosuspensions often achieve significantly higher exposure levels compared to suspensions of micronized API, even when the same surfactants are used. Finally, the presence of surfactants in nanosuspension formulations would increase the surface wetting and would most likely result in further enhancement of the dissolution rates compared to micronized suspensions.
A recent hypothesis by Sugano (17) proposes that increase in bioavailability for nanoparticles as compared to formulations containing unmilled API might be due to a change in the effective intestinal permeability for nanosuspension formulations due to a decrease in the thickness of the unstirred water layer (UWL). UWL is a stagnant layer of water and mucus adjacent to the intestinal wall and is not completely separated from the well-stirred bulk fluid. The permeation through UWL thus in essence contributes to the overall effective intestinal permeability (Peff) of a compound (18). The theory of particle drifting hypothesizes that nanoparticles can diffuse into the UWL thus decreasing the effective thickness of the UWL (heff) and hence resulting in an increase of Peff (17). When these nanoparticles diffuse into the UWL, they can in effect form a reservoir of the drug in the UWL, and thus, the dissolving drug molecules from the surface of these particles could diffuse into the epithelial cell membrane. On increasing the dose and/or reducing the particle size, the number of drug particles in the UWL would be increased. This would increase the portion of the dissolved drug diffusing from the nanoparticles in the UWL. Since the diffusional distances from the nanoparticles in the UWL to the epithelial cell membrane is smaller than that from drug particles in the bulk fluid/UWL interface, when a drug reservoir exists in the UWL, the effective thickness of the UWL would decrease and so the effect of UWL as rate limiting to permeability would become smaller. This theory suggests that changes in effective permeability should be taken into account while building in silico models to predict bioperformance of nanoformulations.
IN VITRO EVALUATION OF NANOSUSPENSIONS
Since oral nanoformulations are designed with the goal to disperse in the stomach, redispersibility and dissolution testing in simulated gastric fluid (SGF) should be conducted to provide an initial estimate of the potential bioperformance. Redispersibility testing typically includes dilution of the nanoformulation in SGF or another suitable media and then measuring particle size to ensure that the nanoformulation would maintain its original particle size once ingested and/or reconstituted for dosing (Fig. 1) (19). Dissolution testing provides an estimate of the extent of dissolution rate enhancement for a nanoformulation as compared to other formulations. For insoluble compounds, where dissolution is expected to mainly occur in the small intestine, additional dissolution studies in fasted state simulated intestinal fluid (FaSSIF) will provide further insight on the expected bioperformance. During dissolution testing, care should be taken to ensure that undissolved drug particles are not assayed. Due to the small particle size for nanoformulations, filtering through smaller pore size filters (e.g., 0.1 μm) or ultracentrifugation to separate undissolved API should be implemented during sampling handling. For more details of dissolution testing of nanoformulations and methodologies, interested readers are referred to (12, 20).
Fig. 1.

Typical example of a nanosuspension formulation showing good redispersibility in simulated gastric fluid (SGF). The blue plot shows particle size distribution of the formulation after milling, and the red plot shows particle size distribution after dispersing in SGF
IN SILICO EVALUATION OF NANOSUSPENSIONS: PREDICTING PERFORMANCE OF NANOSUSPENSIONS
As discussed above, nanosizing can lead to an enhancement in the dissolution rate of the formulation as well as an increase in the saturation solubility of the API. Therefore, computational methods could be used to a priori predict potential increase in bioavailability of nanoformulations as compared to conventional micronized formulation. For example, Jinno et al. utilized a mixing tank model to predict the dissolution rates for cilostazol suspensions of different particle sizes prepared by using hammer-mill, jet-mill, and the NanoCrystal technology (21). The predicted dissolution rates showed good agreement with the experimental in vitro data and showed that the dissolution rate of cilostazol increased significantly with reduction in the particle size. The effect of such API or formulation properties on bioperformance has now become feasible through the advent of computational models that can simulate the oral absorption process. These absorption models are then linked to systemic pharmacokinetic models in order to predict of the pharmacokinetic profiles of different formulations. Such an approach was taken by Shono et al. to predict bioperformance of micronized and nanosized aprepitant formulations in human under fasted and fed conditions (22). In vitro dissolution tests of the nanoparticulate and microparticulate formulations of aprepitant were performed in fasted and fed state gastric and intestinal media. These data showed an enhancement in the rate and extent of aprepitant dissolution for the nanosuspension as compared to the micronized formulation. The dissolution data were then incorporated in a physiologically based pharmacokinetic model to predict the area under the plasma concentration curve (AUC) and Cmax for the aprepitant formulations under fasted and fed conditions in human. To better predict the bioperformance of formulations in fasted and fed states, a model was built that applied permeability restrictions to absorption. This model successfully predicted the plasma concentration vs. time profiles for aprepitant in both fasted and fed states as well as predicted the effect of dose and particle size on aprepitant pharmacokinetics (22). This work showed that the high dissolution rate achieved by nanosized aprepitant formulation was the primary reason for improvement in bioavailability under fasted state and also reduction in food effect as compared to the micronized aprepitant formulation. On the other hand, Sugano (17) was able to predict the effect of increase in dose and decrease in particle size on fraction absorbed of nanomilled griseofulvin, efavirenz, danazol, and cilostazol using the particle drifting theory, with reasonable accuracy. This model incorporates the concept of reduction in thickness of the UWL and hence reduction in the effective intestinal permeability as major driver for increase in bioavailability of nanosuspensions. These recent publications in the area of nanosuspension modeling show that bioavailability enhancement by nanosuspensions can be driven by combination of several processes (i.e., increase in dissolution rate, increase in saturation solubility, and reduction in effective permeability). As a result, developing accurate models to capture all these processes is complicated and is an evolving and exciting field of research.
EXPERIENCE WITH CRYSTALLINE NANOSUSPENSIONS IN VIVO: EVALUATION AS ORAL TOXICOLOGY FORMULATIONS FOR BCS II/IV COMPOUNDS
Obtaining sufficient exposures in preclinical species and understanding the exposure/safety profile of a drug development candidate compound is a necessity during drug development in order to be able to safely advance compounds in the clinic. It is generally desirable that the route of administration in toxicology studies matches that of the intended clinical practice. Thus, for orally administered compounds, toxicology studies have been traditionally a challenge for formulators given the high doses needed that often requires employment of formulation technologies to solubilize the test compound. Therefore, it comes as no surprise that the increase in the number of BCS class II/IV compounds in development has also impacted the formulation strategies employed in toxicology studies to both optimize exposures and allow the observation of toxicity signals as well as minimize unnecessary use of API to maximize doses (which was often the case if sufficient exposures or toxicity is not observed). While solubilization in toxicology studies have been traditionally obtained via the employment of high surfactant vehicles (e.g., 10% Tween 80), co-solvents (e.g., PEG 400), or via the use of lipid systems (e.g., Imwitor/Tween mixtures), nanosuspensions represent an attractive alternative provided they can obtain acceptable exposures. The aqueous nature of the nanosuspension facilitates dosing at higher volumes (thus allowing for higher doses), with less concerns around GI side effects compared to high surfactant systems or high lipid loads. Also given that nanosuspensions represent well-characterized suspensions of crystalline drug, concerns around API stability in the formulation that can arise with use of non-aqueous vehicles are minimized.
Reports Detailing the Application of Nanosuspensions for Toxicology Studies Are Not Common in the Literature. Perhaps a reason for this is the expectation that the primary role of nanosuspensions is the increase in dissolution rate while in most of the cases, the absorption at toxicology doses would be characterized as solubility-limited. However, interestingly enough and similar to the case with clinical formulations, nanosuspensions have proven to often result in exposures higher than what would be anticipated based on solubility limitations in these studies. In one of the earlier reported studies, Jia et al. demonstrated that nanosized (280 nm) carbendazim (aqueous solubility of 8 μg/mL) could allow for an approximately 2-fold reduction in dose (516 vs. 1,000 mg/kg) required to obtain comparable exposure to a micronized suspension (23). Hecq et al. demonstrated that nanonization, using high-pressure homogenization, resulted in 4-fold improvement in exposure compared to micronization (~90 μm) for a BCS II weak base (ucb-35440-3) when dosed to Wistar rats at 100 mg/kg (24). Most recently, Sigfridsson et al. reported successful increase in bioavailability for a weak acid (pKa 4.7) BCS II compound (25). Specifically the authors obtained up to 4.5-fold increase in AUC and approximately 3-fold increase in Cmax using a 190-nm nanosuspension compared to micronized API (12 μm) when dosed in rats at 225 mg/kg . While these reports focused on comparison of exposures to micronized API in a previous report, we demonstrated significant improvement in exposure for a BCS II compound (compound A, aqueous solubility <0.1 μg/mL, Caco-2 Papp = 19 × 10−6 cm/s) when nanosuspension was compared to a non-aqueous vehicle suspension (4). As seen in Fig. 2, the nanosuspension allowed for a higher dose and resulted in significant exposure increase; one could postulate that the nanosuspension formulation avoided uncontrolled precipitation in the GI tract and resulted in continuous absorption as evident by the late Tmax.
Fig. 2.

Pharmacokinetic comparison of a nanosuspension and a lipid-based vehicle of Merck compound A, a BCS II compound, in rats. The nanosuspension provided significant better oral bioavailability compared to the lipid vehicle. Prolong absorption was observed from the nanosuspension formulation. Modified from (4)
While the examples above focused on performance in a small animal model (rats), Euler et al. demonstrated the utility of nanosuspensions in increasing bioavailability in larger species, studying another weakly basic BCS class II compound (26). The authors observed an approximately 2-fold increase in exposure of the NanoMill® prepared suspension compared to jet-milled API in dogs at a dose of 50 mg/kg. Similarly as shown in Fig. 3, we previously reported an approximately 2-fold increase in exposure in Rhesus monkeys for a poorly soluble BCS II compound (compound B, aqueous solubility <1 μg/mL) across a wide dose range compared to micronized (5 μm) API (4).
Fig. 3.

Comparison of AUC for a nanosuspension and a micronized API suspension of a BCS II development candidate (Merck compound B) in rhesus monkeys. The nanosuspension resulted in improved exposures compared to the micronized suspension in a wide dose range, although at doses above 200 mg/kg no further gains in exposure were observed
Most of the reported successful outcomes for use of nanosuspension formulations in toxicology studies as well as in the clinic is with BCS II compounds. High permeability further facilitates very fast dissolution of nanosuspensions resulting in improved absorption. However, nanosuspensions can be also successfully employed for BCS IV compounds. Compound C is a Merck development candidate with aqueous solubility <0.1 μg/mL and FaSSIF solubility of approximately 2 μg/mL. The compound was intended for local action in the GI and that was reflected in the measured Caco-2 permeability being below 1 × 10−6 cm/s (lower than permeability of reference low permeability marker mannitol). However, for toxicology studies, some systemic exposure was desirable. Table I summarizes the outcome of an early screening study of toxicology formulations in Sprague-Dawley rats (n = 3/formulation). A nanosuspension formulation was tested against formulations ranging from a methylcellulose suspension to vehicles that provided significant solubilization of the API (160 mg/mL in PEG 400 and 24 mg/mL in Imwitor/Tween 50:50). Despite the apparent lack of solubilization capacity, the nanosuspension formulation resulted in exposures higher than the methylcellulose and the PEG 400 vehicles at the same dose (750 mg/kg) and comparable exposures to the Imwitor/Tween suspension which was tested at lower dose (375 mg/kg) due to syringeability limitations. Although relatively high variability was observed in the study, this was expected given both the solubility as well as the permeability limitations for this drug candidate. The nanosuspension formulation resulted in adequate exposures and thus was considered as a viable toxicology formulation candidate and could be considered preferable compared to the Imwitor/Tween suspension due to its aqueous nature and better syringeability that also allows for higher dose to be administered. The nanosuspension formulation showed a longer Tmax compared to the rest of the vehicles, perhaps indicating the possibility of prolonged absorption similarly to what we reported previously by for another drug development candidate (4]. In a case reported in the literature, Jia et al. demonstrated a significant increase in oral bioavailability for a poorly soluble antiviral compound PG301029 when utilizing nanoparticles (280 nm) (27). The permeability reported for this compound would have suggested a low permeability classification based on the BCS system (BCS class IV) although the final bioavailability obtained (~99%) may suggest a disconnect between cell-based permeability and in vivo permeability. Also contrary to the case of compound A, the nanosuspension provided significantly faster absorption with a shorter Tmax by approximately 3 h.
Table I.
Comparison of Pharmacokinetic Parameters for Four Test Toxicology Formulations for a BCS IV Compound, Tested in Sprague-Dawley rats (n = 3)
| Formulation | PEG 400 | 0.5% methocel w/ 0.24% SDS | Imwitor/Tween 50:50 | Nanosuspension |
|---|---|---|---|---|
| Dose (mg/kg) | 750 | 750 | 375 | 750 |
| Dosing volume (mL/kg) | 2 | 5 | 1 | 5 |
| AUC0–8h (nM h) | 47.3 ± 17.6 | 38.7 ± 18.6 | 222 ± 120 | 189 ± 83 |
| C max (nM) | 20.2 ± 7.5 | 13.0 ± 7.0 | 72.9 ± 43.3 | 317 ± 233 |
| T max (h) | 1.0 ± 0.5 | 2.3 ± 0.9 | 2.5 ± 1.8 | 8.3 ± 7.8 |
While significant variability was observed in the study, on average the nanosuspension formulation of compound C resulted in significant improvement in exposure over the PEG 400 and the methylcellulose vehicles and comparable exposure to the Imwitor/Tween suspension
PEG polyethylene glycol, SDS sodium dodecyl sulfate, AUC area under the plasma concentration curve
While the examples presented earlier and the available literature has demonstrated ability of nanosuspension formulations for different compounds to enhance bioavailability of BCS II/IV compounds in different species (although as discussed, majority of literature is in the rat), previous literature has not focused on the performance of the same formulation across species. The ability of the formulation to deliver consistent bioavailability gains in multiple species is a potentially important consideration during toxicology formulation selection, as availability of a single formulation can significantly expedite and simplify formulation preparation/manufacturing efforts, study timelines, and study data interpretation.
Although there is a quite long history of use of preclinical animal models as formulation screening tools, one cannot exclude the possibility of physiological differences of the gastrointestinal tract between different species affecting the behavior of formulations, including nanosuspensions. One such example is provided in Table II for Merck compound D. Compound D was identified as a human-specific active metabolite and would be characterized as a BCS IV compound with very poor aqueous solubility and low-moderate permeability (LLC-PK1 permeability of ~5 × 10−6 cm/s). To be able to assess safety of the compound, efforts were undertaken to obtain exposures in preclinical species by direct oral administration. Initial screening for compound D was undertaken in rats where the nanosuspension formulation that afforded a maximum feasible dose (MFD) of 640 mg/kg that was higher than the rest of the formulation candidates significantly (approximately 3-fold higher AUC) improved exposures over previously tested methocel or Imwitor/Tween formulations. While similar exposures were obtained when the compound was formulated in labrasol, the higher MFD and the aqueous nature favored the use of the nanosuspension formulation based on the rat screening study. However, when the same formulations were evaluated in beagle dogs, the increase in bioavailability previously seen with the nanosuspension did not materialize—the nanosuspension formulation resulted in identical exposures to the Imwitor/Tween vehicle despite the 2.8-fold (640 vs. 225 mg/kg) higher dose used. Same observation was seen in Rhesus monkeys (data not shown). A final test was conducted in Yucatan minipigs where on average the nanosuspension formulation at 640 mg/kg appeared to be resulting in a moderate increase (~60%) in exposure compared to the Imwitor/Tween suspension at 225 mg/kg.
Table II.
Comparison of Exposure of a Nanosuspension Formulation of Compound D Relative to Other Test Formulations in Sprague-Dawley Rats, Beagle Dogs, and Yukatan Minipigs
| Formulation | Dose (mg/kg) | AUC | |
|---|---|---|---|
| Rat (Sprague-Dawley) | 0.5% methocel | 150 | 3.7 |
| Imwitor/Tween | 400 | 5.3 | |
| Labrasol | 600 | 16.2 ± 4.3 | |
| Nanosuspension | 640 | 15.3 ± 3.8 | |
| Dog (Beagle) | Imwitor/Tween | 225 | 5.1 ± 0.8 |
| Labrasol 300 | 300 | 2.8 ± 1.2 | |
| Nanosuspension | 640 | 5.4 ± 1.1 | |
| Minipig (Yukatan) | Imwitor/Tween | 225 | 9.5 |
| Nanosuspension | 640 | 15.2 |
While the nanosuspension was generally one of the higher bioavailability formulations across all three species, the relative performance compared to other formulations significantly differed between species. Mean ± SE reported if n > 2. For minipig studies, average of two animals are reported
AUC area under the plasma concentration curve
While in the case of compound D, one could argue that the nanosuspension was consistently one of the best performing formulations, a more dramatic difference was observed for the previously discussed compound C. While for that compound the nanosuspension resulted in exposures comparable to the Imwitor/Tween in the rats (Table I) when the two formulations were tested in the dogs, the Imwitor/Tween suspension resulted in approximately 10-fold higher exposure compared to the nanosuspension (Table III). Of note is that the prolonged absorption that was seen in the rats as evident by the long Tmax was not seen in the dogs (Tmax < 1 h), suggesting potential differences in in vivo precipitation and/or gastrointestinal transit of the nanosuspension formulation in the two species.
Table III.
Comparison of Exposure of a Nanosuspension and an Imwitor/Tween Formulation for Compound C in Beagle Dogs
| Dose | Formulation | AUC | T max |
|---|---|---|---|
| 30 mg/kg | 0.5 mL/kg Imwitor/Tween | 194 ± 60 | 2.2 ± 1.8 |
| 100 mg/kg | 0.5 mL/kg Imwitor/Tween | 162 ± 60 | 2.0 ± 1.7 |
| 30 mg/kg | 5 mL/kg nanosuspension | 15.4 ± 3.2 | 0.75 ± 0.43 |
| 100 mg/kg | 5 mL/kg nanosuspension | 27.6 ± 35.7 | 0.83 ± 0.29 |
The nanosuspension resulted in significantly lower exposures compared to the Imwitor/Tween suspension despite providing comparable exposures in an earlier screening study in rats
AUC area under the plasma concentration curve
The data for compounds C and D clearly highlight the need for evaluation of formulations in multiple species. In both cases, the best performance of the nanosuspension was observed in rats, potentially indicating a more favorable absorption process for nanosuspensions in smaller species. However, it is worth noticing that the lack of translation of exposures between species was not specific to nanosuspensions as seen for example for the labrasol formulation for compound C which was one of the best performing formulation in the rats but resulted in the lowest exposure in the dogs. Therefore, more data across different compounds are required to fully understand whether consistent trends in nanosuspension performance across species can be established.
USE OF NANOSIZED API IN CLINICAL FORMULATIONS
The gradual shift toward less soluble drug candidates has led to a steady adoption of bioavailability enhancing formulation technologies including nanosized API also in clinical settings. Starting with the approval of RAPAMUNE® in 2001 which represented the first nanoparticulate-based commercial product (using Elan’s Nanocrystals® technology), five oral drug products currently on the market utilize a nanosizing technology to deliver the needed oral bioavailability for their respective active pharmaceutical ingredient. As seen in Table IV, these products cover four different BCS II or IV drug molecules across different therapeutic areas; four of the five products utilize the Nanocrystals® technology while one formulation (TriglideTM) utilizes the SkyePharma IDD®-P technology. Often times, the improved oral bioavailability is seen in the form of reduction of food effect (Table V). It is known that improved solubility in the presence of fat and increased bile salt concentrations, oftentimes accompanied with improved dissolution rate, is one of the most common mechanisms for the positive food effect seen for many BCS II/IV compounds. Hence, the significantly improved dissolution rate of the nanosized particles in the fasted state helps minimize and in some instances eliminate the fed and fasted state exposure differences.
Table IV.
Current Marketed Orally Administered Pharmaceutical Products for BCS II/IV Compounds Utilizing Nanosuspensions
| Product | Company | API | BCS class | Technology |
|---|---|---|---|---|
| RAPAMUNE® | Wyeth | Sirolimus | II | Nanocrystals® |
| EMEND® | Merck | Aprepitant | IV | Nanocrystals® |
| TRICOR® | Abbott | Fenofibrate | II | Nanocrystals® |
| MEGACE® ES | PAR Pharmaceutical | Megestrol acetate | II | Nanocrystals® |
| TRIGLIDETM | First Horizon Pharmaceutical | Fenofibrate | II | IDD®-P |
API active pharmaceutical ingredient, BCS Biopharmaceutics Classification System
Table V.
Mitigation of Positive Food Effect via the use of Nanosuspension Formulations in Commercial Products
| Product | Food effect (% increase AUC) for nanosuspension | Food effect (% increase AUC) for original/early formulations |
|---|---|---|
| EMEND® | 20% (125 mg), 9% (80 mg)a | 2.7-fold (100 mg) |
| TRICOR® | 5% (145 mg)a | 35% (160 mg) |
| MEGACE® ES | 36% (625 mg) | 2-fold (800 mg) |
| TRIGLIDETM | 14% (160 mg)a | 35% (160 mg) |
AUC area under the plasma concentration curve
aExposures within bioequivalence bounds
Similarly to the literature reports discussed in the toxicology formulation section, a good number of reports have detailed the performance of crystalline nanosuspensions at doses relevant to clinical administration. In the majority of the cases, evaluation is available in preclinical species where nanosuspensions demonstrate increase in bioavailability (4, 27–30) or reduction in food effect (12, 29). While these preclinical studies provide proof of concept for utilization of nanosized API, clinical data are not as commonly available in the literature. In one of the few available reports, Merisko-Liversidge et al. reported successful mitigation of the positive food effect associated with danazol via a nanocrystalline suspension (3). Specifically while the marketed product, Danocrine®, exhibited a six-fold positive food effect, the nanocrystalline suspension resulted in comparable exposures in the fasted and fed state (3). In the same study, the authors compared the bioavailability of the nanocrystalline API delivered as a dry-filled capsule or as a liquid nanosuspension. Interestingly, the former resulted in a loss of bioavailability relative to the pre-dispersed formulation, indicative of the potential challenges associated with final conversion of such formulations into solid dosage forms. As discussed in the in vitro evaluation section, redispersibility of the nanoparticle-based formulation in simulated intestinal fluids should be thoroughly studied to ensure maximal bioavailability from the solid nanocrystal formulation.
A comparison of the performance in preclinical species and in the clinic for a given formulation is of great interest to formulation and biopharmaceutic scientists. During drug development, early clinical formulation selection prior to first in human (FIH) studies often relies in pharmacokinetic studies in dogs. Especially for formulation technologies aimed to improve compound bioavailability such as nanosized API, for which in vitro dissolution methodologies are still evolving, such animal models may represent the major biopharmaceutical decision point. Once clinical data are available, the models can be assessed for their predictive ability and their utility for use to guide later stage formulation development.
Merck compound A, a BCS class II molecule with aqueous solubility <1 μg/mL, was discussed in the toxicology formulation section, and the toxicology formulation screening is shown in Fig. 2. Figure 4 demonstrates the comparison of the FIH formulation candidates in dogs at a dose of 10 mg/kg. It is evident that the results from the dog study mirror the outcome of the rat screening studies with the nanosuspension formulation resulting in the highest exposure while the liquid-filled capsules similar to the toxicology study resulted in low exposures potentially due to in vivo precipitation. The amorphous solid dispersion formulation also failed in this case to significantly improve compound bioavailability. Despite some loss of bioavailability once the nanosuspension was formulated to a solid dosage form (in this case a nanosolid capsule), the nanosized-based API formulation remained by a significant margin the highest bioavailability formulation and was advanced to the FIH formulation study. In the clinic, the nanocrystal formulation resulted in linear pharmacokinetics up to a dose of 1,600 mg, confirming the outcome of the preclinical studies.
Fig. 4.

Pharmacokinetic comparison of a nanosuspension to other FIH formulation candidates for Merck compound A. The nanosuspension provides the highest exposure compared to either a liquid-filled capsule or a solid dispersion formulation
Merck compound E represents an interesting case study comparing preclinical and clinical data. Compound E is also BCS class II compound with very poor aqueous solubility and low solubility in simulated intestinal fluids (FaSSIF solubility ~2 μg/mL). Nanosuspension was shown to provide high exposures during toxicology formulation screening (somewhat higher than a solid dispersion) and given the highest MFD was selected as the toxicology formulation. Given the positive experience with the toxicology formulation, a liquid nanosuspension formulation was also evaluated as a FIH formulation candidate. The FIH nanosuspension candidate was screened in a dog study at 15 mg/kg and resulted in approximately 3-fold higher exposures compared to formulated jet milled API. When dosed in the clinic as a liquid nanosuspension, approximately linear exposures were obtained up to 1,200-mg dose. However, despite the linear pharmacokinetic response, a significant food effect was observed at the 200-mg dose with approximately 4-fold increase in exposure. While the linear pharmacokinetic response could allow for a dose adjustment if needed to achieve pharmacokinetic targets, the observed food effect clearly indicated potential dissolution/solubility limitations for the nanosuspension which was not seen in preclinical species.
PERFORMANCE OF CRYSTALLINE NANOPARTICLES IN COMMERCIAL SOLID DOSAGE FORMS
Sirolimus is a macrocyclic lactone that is used as an immunosuppressive. The large molecular weight (914.2), high logP (clogP 4.26) and very low aqueous solubility (2.6 μg/mL) represent major challenges for oral absorption of the compound. It is a highly permeable (BCS II) compound. The original formulation for sirolimus was an oral solution (Phosal 50 PG® and polysorbate 80 being the major constituents) that requires storage under refrigeration. The RAPAMUNE® nanocrystalline formulation provided a modest increase in bioavailability relative to the solution formulation (27% increase); however, the two formulations have been shown to be clinically equivalent at the 2-mg dose (31). The nanoparticle-based formulation still demonstrates a moderate positive food effect (65% Cmax, 23% AUC) indicating that this formulation may still not represent a maximum oral bioavailability formulation although other factors past formulation may be also playing a role in this observed food effect. Thus, while in the case of RAPAMUNE, the gains in bioavailability with nanosizing are modest and could be considered non-critical given the demonstrated clinical equivalence, the nanosolid formulation, did enable the availability of a room temperature product and also a more convenient dosage form for administration.
Aprepitant is a weakly basic, lipophilic (logDpH = 7 = 4.8) and poorly soluble compound (solubility of 3–7 μg/mL across pH range 2–10). It exhibits moderate–high permeability in a standard Caco-2 assay of 7.8 × 10−6 cm/s and has been preliminarily classified as a BCS IV compound, although the moderate–high permeability would suggest an expected in vivo behavior closer to a BCS II compound. The compound exhibits a significant difference in solubility in fasted (FaSSIF) and fed (FeSSIF) simulated intestinal fluids (22) which resulted in a pronounced food effect for early formulations of aprepitant utilizing micronized API (approximately 3-fold increase at 100 mg and 4.5-fold increase in AUC at 300 mg). Since administration with food would not be an acceptable means to improve oral bioavailability of aprepitant, given the use for prevention of chemotherapy-induced nausea and vomiting, solubilization technologies were explored to improve aprepitant oral bioavailability, eventually leading to the development of the Nanocrystal® formulation that eliminated the clinical food effect (Table V).
The effect of nanosizing on the bioavailability of aprepitant has been previously reported. Wu et al. have detailed the preclinical biopharmaceutical evaluation that facilitated the development of the nanosuspension formulation (13). A Nanocrystal® suspension was prepared by ball-milling and formulated with 4% HPC-SL, 0.08% SDS, and 20% sucrose. The liquid suspension was dosed to beagle dogs and as seen in Fig. 5 resulted in 4.3-fold increase in exposure compared to a suspension of micronized (5.49 μm) API. The exposure of the nanosuspension matched that obtained with a liquid-filled capsule where API was pre-solubilized in a lipid/surfactant mixture. However, contrary to the nanosuspension that was amenable to conversion to a solid drug product, the liquid-filled capsule was not able to support the intended clinical dose due to API solubility limitations. Furthermore, as discussed by the authors, the nanosuspension eliminated the food effect observed with the micronized API suspension. The improved bioavailability of the nanosized API was confirmed in the clinic where 80 and 125 mg of aprepitant using dried nanosuspension in capsule showed no significant food effect (32) (Table V). It is worth noting that even the nanosized API formulation was not able to overcome the significant solubility limitations at higher doses where a significant positive food effect (fed/fasted AUC ratio of 2.7) was observed at a dose of 300 mg. However, this dose was not required for the final commercial product.
Fig. 5.

Aprepitant plasma concentration vs. time profiles in fasted male beagle dogs following administration of aprepitant formulations at a dose of 2 mg/kg. The nanosuspension formulation resulted in a significant (4.3-fold) increase in AUC over the conventional suspension of micronized API and matched the exposures of a liquid-filled capsule (lipid/surfactant) formulation
Megestrol acetate, a synthetic steroid, is another poorly soluble (aqueous solubility of 2 μg/mL), highly permeable (BCS class II) drug compound. The initial megestrol acetate formulation exhibited positive food effect with a high fat meal increasing AUC and Cmax of megestrol acetate by 2- and 7-fold, respectively, compared to fasted dosing. The nanosuspension formulation that was subsequently developed to address this observed food effect, improved the oral bioavailability and resulted in less fluctuation of exposure between fed and fasted state. Specifically for the nanosuspension formulation, the observed increases in AUC and Cmax were moderate, 36% and 48%, respectively (33). The 625- and 675-mg doses of the nanosuspension have been shown to be bioequivalent to 800 mg of the original suspension formulation under fed conditions (33).
Finally, Tricor® and TriglideTM are nanosuspension formulations developed to improve the oral bioavailability of fenofibrate. Tricor® utilizes the Elan Nanocrystal® technology while TriglideTM was developed using the SkyePharma Insoluble Drug Delivery Platform (IDD®-P) platform. Fenofibrate is a highly lipophilic (logP = 5.24), highly permeable compound that is practically insoluble in water (solubility <1 μg/mL). The micronized fenofibrate formulation exhibited a positive food effect with 35% higher exposure in the fed compared to the fasted state. Both Tricor and Triglide were able to eliminate this food effect resulting in dosage forms that are bioequivalent (AUC) in fed and fasted state (34–36). In the case of Tricor®, three 48-mg or one 145-mg tablets are equivalent to one 200-mg micronized fenofibrate capsule when administered under fed conditions. TRIGLIDE 160-mg tablet exhibits comparable total exposure (AUC) but 32% higher Cmax compared to the 200-mg micronized fenofibrate capsule following administration with a low-fat meal.
CONCLUSION AND FUTURE DIRECTIONS
The continuous trend toward more lipophilic, poorly water soluble candidates has necessitated the employment of bioavailability enhancing formulation technologies in both discovery and preclinical development, as well as in the clinic. Nanosuspensions, formulations of crystalline API with particle size in the submicron range, have been shown to increase oral bioavailability of compounds beyond what standard micronization milling techniques have achieved. The several examples in preclinical toxicology studies that were reviewed in this chapter clearly demonstrate the potential utility of this formulation approach. Five orally administered nanosuspension formulations are currently on the market with proven bioavailability benefits, such as reduction or elimination of food effect compared to previously used formulations. While not all aspects of nanosuspension behavior in vivo as related to the bioavailability increase are well understood, detailed dissolution studies in biorelevant media in conjunction with the use of absorption modeling can eventually lead to a quantitative prediction of nanosuspension clinical performance based on preclinical information, greatly facilitating the development of such formulations.
Acknowledgments
We would like to thank the following colleagues for their contributions in generating the data for Merck compounds described in this article—Kim Manser, Becky Nissley, Brian Marks, Iris Xie, Linda Rakes, Sunny Panmai, and Jennifer Hehman.
References
- 1.Benet LZ, Wu CY. Using a biopharmaceutics drug disposition classification system to predict bioavailability and elimination characteristics of new molecular entities. Somerset: NJDMDG; 2009. [Google Scholar]
- 2.Ku MS, Dulin W. A biopharmaceutical classification-based right-first-time formulation approach to reduce human pharmacokinetic variability and project cycle time from first-in-human to clinical proof-of-concept. Pharm Dev Technol. 2012; 17:285–302. [DOI] [PubMed]
- 3.Merisko-Liversidge E, Liversidge GG, Cooper ER. Nanosizing: a formulation approach for poorly-water-soluble compounds. Eur J Pharm Sci. 2003;18:113–120. doi: 10.1016/S0928-0987(02)00251-8. [DOI] [PubMed] [Google Scholar]
- 4.Kesisoglou F, Panmai S, Wu Y. Nanosizing—oral formulation development and biopharmaceutical evaluation. Adv Drug Deliv Rev. 2007;59:631–644. doi: 10.1016/j.addr.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Merisko-Liversidge E, Liversidge GG. Nanosizing for oral and parenteral drug delivery: a perspective on formulating poorly-water soluble compounds using wet media milling technology. Adv Drug Deliv Rev. 2011;63:427–440. doi: 10.1016/j.addr.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Merisko-Liversidge EM, Liversidge GG. Drug nanoparticles: formulating poorly water-soluble compounds. Toxicol Pathol. 2008;36:43–48. doi: 10.1177/0192623307310946. [DOI] [PubMed] [Google Scholar]
- 7.Shegokar R, Müller RH. Nanocrystals: industrially feasible multifunctional formulation technology for poorly soluble actives. Int J Pharm. 2010;399:129–39. doi: 10.1016/j.ijpharm.2010.07.044. [DOI] [PubMed] [Google Scholar]
- 8.Van Eerdenbrugh B, Van den Mooter G, Augustijns P. Top-down production of drug nanocrystals: nanosuspension stabilization, miniaturization and transformation into solid products. Int J Pharm. 2008;364:64–75. doi: 10.1016/j.ijpharm.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 9.Horter D, Dressman JB. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv Drug Deliv Rev. 2001;46:75–87. doi: 10.1016/S0169-409X(00)00130-7. [DOI] [PubMed] [Google Scholar]
- 10.Dressman JB, Amidon GL, Reppas C, Shah VP. Dissolution testing as a prognostic tool for oral drug adsorption: immediate release dosage forms. Pharm Res. 1998;15:11–22. doi: 10.1023/A:1011984216775. [DOI] [PubMed] [Google Scholar]
- 11.Mosharraf M, Nystrom C. The effect of particle size and shape on the surface specific dissolution rate of micronized practically insoluble drugs. Int J Pharm. 1995;122:35–47. doi: 10.1016/0378-5173(95)00033-F. [DOI] [Google Scholar]
- 12.Quinn K, Gullapalli RP, Merisko-Liversidge E, Goldbach E, Wong A, Liversidge GG, et al. A formulation strategy for gamma secretase inhibitor ELND006, a BCS class II compound: development of a nanosuspension formulation with improved oral bioavailability and reduced food effects in dogs. J Pharm Sci. 2012;101:1462–1474. doi: 10.1002/jps.23034. [DOI] [PubMed] [Google Scholar]
- 13.Wu Y, Loper A, Landis E, Hettrick L, Novak L, Lynn K, et al. The role of biopharmaceutics in the development of a clinical nanoparticle formulation of MK-0869: a beagle dog model predicts improved bioavailability and diminished food effect on absorption in human. Int J Pharm. 2004;285:135–46. doi: 10.1016/j.ijpharm.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Miao X, Sun C, Jiang T, Zheng L, Wang T, Wang S. Investigation of nanosized crystalline form to improve the oral bioavailability of poorly water soluble cilostazol. J Pharm Pharm Sci. 2011;14(2):196–214. doi: 10.18433/j3pw2w. [DOI] [PubMed] [Google Scholar]
- 15.Nystrom C, Bisrat M. Physiochemical aspects of drug release. VIII. The relation between particle size and surface specific dissolution rate in agitated suspensions. Int J Pharm. 1998;47:223–231. [Google Scholar]
- 16.Muller RH, Peters K. Nanosuspensions for the formulation of poorly soluble drugs I. Preparation by a size-reduction technique. Int J Pharm. 1998;160:229–237. doi: 10.1016/S0378-5173(97)00311-6. [DOI] [Google Scholar]
- 17.Sugano K. Possible reduction of effective thickness of intestinal unstirred water layer by particle drifting effect. Int J Pharm. 2010;387:103–109. doi: 10.1016/j.ijpharm.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 18.Sugano K. Theoretical investigation of passive intestinal membrane permeability using Monte Carlo method to generate drug-like molecule population. Int J Pharm. 2009;373:55–61. doi: 10.1016/j.ijpharm.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Chaubal MV, Popescu C. Conversion of nanosuspensions into dry powders by spray drying: a case study. Pharm Res. 2008;25(10):2302–2308. doi: 10.1007/s11095-008-9625-0. [DOI] [PubMed] [Google Scholar]
- 20.Kakran M, Shegokar R, Sahoo NG, Shaal LA, Li L, Müller RH. Fabrication of quercetin nanocrystals: comparison of different methods. Eur J Pharm Biopharm. 2012;80:113–21. doi: 10.1016/j.ejpb.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 21.Jinno J, Kamada N, Miyake M, Yamada K, Mukai T, Odomi M, et al. Effect of particle size reduction on dissolution and oral absorption of a poorly water-soluble drug, cilostazol, in beagle dogs. J Control Release. 2006;111:56–64. doi: 10.1016/j.jconrel.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 22.Shono Y, Jantratid E, Kesisoglou F, Reppas C, Dressmam JB. Forecasting in vivo oral absorption and food effect of micronized and nanosized aprepitant formulations in humans. Eur J Pharm Biopharm. 2010;76:95–104. doi: 10.1016/j.ejpb.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 23.Jia L, Wong H, Wang Y, Garza M, Weitman SD. Carbendazim: disposition, cellular permeability, metabolite identification, and pharmacokinetic comparison with its nanoparticle. J Pharm Sci. 2003;92:161–72. doi: 10.1002/jps.10272. [DOI] [PubMed] [Google Scholar]
- 24.Hecq J, Deleers M, Fanara D, Vranckx H, Boulanger P, Le Lamer S, et al. Preparation and in vitro/in vivo evaluation of nano-sized crystals for dissolution rate enhancement of ucb-35440-3, a highly dosed poorly water-soluble weak base. Eur J Pharm Biopharm. 2006;64:360–8. doi: 10.1016/j.ejpb.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 25.Sigfridsson K, Lundqvist AJ, Strimfors M. Particle size reduction for improvement of oral absorption of the poorly soluble drug UG558 in rats during early development. Drug Dev Ind Pharm. 2009;35:1479–86. doi: 10.3109/03639040903025855. [DOI] [PubMed] [Google Scholar]
- 26.Euler D, Frech P, Karki S, Cowden C, Pearce G, Mehta P, et al. Influence of physicochemical properties and intestinal region on the absorption of 3-fluoro-2-pyrimidylmethyl 3-(2,2-difluoro-2-(2-pyridyl)ethylamino)-6-chloropyrazin-2-one-1-acetamide , a water insoluble thrombin inhibitor, in dog. Int J Pharm. 2004;275:19–27. doi: 10.1016/j.ijpharm.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 27.Jia L, Wong H, Cerna C, Weitman SD. Effect of nanonization on absorption of 301029: ex vivo and in vivo pharmacokinetic correlations determined by liquid chromatography/mass spectrometry. Pharm Res. 2002;19:1091–6. doi: 10.1023/A:1019829622088. [DOI] [PubMed] [Google Scholar]
- 28.Kondo N, Iwao T, Masuda H, Yamanouchi K, Ishihara Y, Yamada N, et al. Improved oral absorption of a poorly water-soluble drug, HO-221, by wet-bead milling producing particles in submicron region. Chem Pharm Bull (Tokyo) 1993;41:737–40. doi: 10.1248/cpb.41.737. [DOI] [PubMed] [Google Scholar]
- 29.Liversidge GG, Cundy KC. Particle size reduction for improvement of oral bioavailability of hydrophobic drugs: I. Absolute oral bioavailability of nanocrystalline danazol in beagle dogs. Int J Pharm. 1995;125:91–97. doi: 10.1016/0378-5173(95)00122-Y. [DOI] [Google Scholar]
- 30.Fakes MG, Vakkalagadda BJ, Qian F, Desikan S, Gandhi RB, Lai C, et al. Enhancement of oral bioavailability of an HIV-attachment inhibitor by nanosizing and amorphous formulation approaches. Int J Pharm. 2009;370:167–74. doi: 10.1016/j.ijpharm.2008.11.018. [DOI] [PubMed] [Google Scholar]
- 31.Wyeth Pharmaceuticals, Inc. Rapamune(R) prescribing information. Philadelphia: Wyeth Pharmaceuticals; 2006.
- 32.Majumdar AK, Howard L, Goldberg MR, Hickey L, Constanzer M, Rothenberg PL, et al. Pharmacokinetics of aprepitant after single and multiple oral doses in healthy volunteers. J Clin Pharmacol. 2006;46:291–300. doi: 10.1177/0091270005283467. [DOI] [PubMed] [Google Scholar]
- 33.Par Pharmaceutical Companies, Inc. Megace(R) ES prescribing information. Woodcliff: Par Pharmaceutical Companies; 2005.
- 34.Abbott Laboratories. Nanoparticle technology now allows TriCor(R) to be taken with or without food. Abbott Park: Abbott Laboratories; 2004.
- 35.Abbott Laboratories. TriCor(R) prescribing information. Abbott Park: Abbott Laboratories; 2004.
- 36.First Horizon Pharmaceutical Corporation. TriglideTM prescribing information. Roswell: First Horizon Pharmaceutical Corporation; 2005.