

Abstract
With the advancement of biotechnology in the last two decades, optimized and novel modalities and platforms of biologic moieties have emerged rapidly in drug discovery pipelines. In addition, new technologies for delivering therapeutic biologics (e.g., needle-free devices, nanoparticle complexes), as well as novel approaches for disease treatments (e.g., stem cell therapy, individualized medicine), continue to be developed. While pharmacokinetic studies are routinely carried out for therapeutic biologics, experiments that elucidate underlying mechanisms for clearance and biodistribution or identify key factors that govern absorption, distribution, metabolism, and excretion (ADME) of biologics often are not thoroughly conducted. Realizing the importance of biologics as therapeutic agents, pharmaceutical industry has recently begun to move the research focus from small molecules only to a blended portfolio consisting of both small molecules and biologics. This trend brings many opportunities for scientists working in the drug disposition research field. In anticipation of these opportunities and associated challenges, this review highlights impact of ADME studies on clinical and commercial success of biologics, with a particular focus on emerging applications and technologies and linkage with mechanistic pharmacokinetic/pharmacodynamic modeling and biomarker research.
Key words: ADME, biodistribution, pharmacokinetics, therapeutic biologics
INTRODUCTION
With recent successes in the approval and commercialization of monoclonal antibodies (mAbs), Fc proteins and peptides, drug development for therapeutic biologics has captured substantial attention. Over 20 mAbs have been approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMEA). Since the approval of adalimumab in 2002, human or humanized mAbs are a dominant fast-growing category of targeted therapeutic agents (1). In recent years, other novel modalities and platforms of biologic moieties have emerged rapidly in drug discovery pipelines (Table I) (2–14).
Table I.
Examples of New Modalities and Platforms for Therapeutic Biologics
| Biological modalities and platforms | Examples |
|---|---|
| Synthetic peptides (2) | Hydrocarbon stapled α-helical peptide |
| Bi-functional biologics (3) | IL-2/IL-12 Fc fusion protein |
| Antibody fragments, such as antigen-binding fragment (Fab), single chain variable fragment (scFv), and immunoRNase (4,5) | Ranibizumab |
| Efungumab | |
| Erb-hRNase | |
| Compact antibodies, such as small modular immunopharmaceuticals (SMIP) and nanobodies (6,7) | 2LM20-4 |
| ALX-0081 | |
| Small molecule/peptide and protein conjugates, such as antibody-drug conjugate (ADC) (8,9) | Brentuximab vedotin |
| siRNA, oligonucleotides, and aptamers (10–12) | Mipomersen |
| ALN-VSP02 | |
| Pegaptanib | |
| Biologics with half-life extension approaches, such as PEGylation, HESylation, glycosalation, Fc fusion, IgG binding, enhanced FcRn binding, albumin binding or fusion (13,14) | Certolizumab pegol |
| Ozoralizumab (ATN-103) |
For example, a wave of novel, antigen-specific antibody fragment-based constructs may soon enter clinical evaluation (4). Specifically, several types of antibody fragment technologies are currently employed: antigen-binding fragments (e.g., ranibizumab); single-chain variable fragments (e.g., efungumab); recombinant proteins with size reduction achieved via removal of the domains that are non-essential for function. With the intention to increase potency and prolong half-life, a new class of peptides (or miniproteins) has been synthesized by a ring-closing metathesis reaction to make an all-hydrocarbon “staple” between successive turns of a peptide α-helix (2). It is postulated that stapling of peptides could have an improved resistance to proteolysis compared to traditional linear peptides, while achieving high cell penetration through endocytic vesicle trafficking (2). Other modalities and platforms of biologics that are at various stages of development include multifunctional biologics (3), mAb or Fc protein conjugates with exogenous active moieties (8), and biologics with a variety of half-life extension approaches (e.g., albumin binding, pegylation) (13,14). Selective targeting of tumors by radiation and cytotoxic drugs conjugated to mAbs has been a topic of considerable interest and an area of continued development (8,9). This class of molecules can be illustrated by the recently approved brentuximab vedotin, a CD30-specific antibody–drug conjugate (ADC). In addition to therapeutic proteins, oligonucleotides, small interfering RNAs (siRNA), and aptamers also have been actively pursued for variety of indications (10–12). In parallel with novel modalities and platforms of biologics, new technologies for delivering therapeutic biologics (e.g., needle-free devices, formulation with nanoparticles, intranasal and ocular delivery) and novel approaches to disease treatments (e.g., stem cell therapy, individualized medicine) continue to be developed. These advances will bring new challenges for in vivo disposition assessment of biologics.
Although pharmacokinetics (PK) of therapeutic biologics are routinely determined in nonclinical research and in clinical trials, in-depth assessments of absorption, distribution, metabolism, and excretion (ADME) properties and mechanisms driving those properties have not been the focus of much attention since the 1992 publication of Protein Pharmacokinetics and Metabolism by Ferraiolo et al. (15). Recently, unusual PK profiles of mAbs have been reported for a number of mAbs, such as Anti-Abeta Ab2, a humanized monoclonal antibody against amino acids 3–6 of primate amyloid beta (Abeta), and a humanized antifibroblast growth factor receptor 4 antibody (16,17). To understand possible causes for such unusual PK profiles, mechanistic-based ADME studies are often required. Other important, yet poorly understood ADME issues include the role of neonatal Fc receptor (FcRn) in absorption and tissue distribution, the relative contribution of lymphatic system to subcutaneous (SC) absorption in different species, the influence of net charge and local charge clusters on tissue distribution of therapeutic proteins, and the complex impact of glycosylation on PK profiles (17). As an increasing number of novel therapeutic biologics enter drug discovery pipelines, demands for mechanistic ADME studies of biologics will continue to grow.
Despite the great clinical and commercial success for some biologic drugs, the rate of clinical success across the industry needs to be improved. Recently, significant research efforts have been focused on understanding the correlation between PK, especially drug concentrations at the target site, and pharmacodynamics (PD) in order to improve clinical trial outcomes. As pointed out by van der Graaf, lack of clinical efficacy in phase II trials is considered as the primary reason for drug failure (18). Thus, a concept of three pillars of survival for drug development was proposed by the author, in which three key questions should be addressed before a drug candidate is selected for clinical trials: (1) Does the compound reach the target organ(s) at the concentration that is necessary for the desired target coverage? (2) Does the compound bind to the target(s) in vivo with the coverage required for biological activity? (3) Does the compound exert the functional modulation of the target? In order to answer these key questions, in-depth investigations of ADME properties and relationship between ADME profiles and pharmacological effects (e.g., biomarker activity) are essential in preclinical and clinical studies. The conventional systemic PK assessment strategy for therapeutic biologics needs to be expanded to cover the drug exposure information at the target site and enable the correlation of drug concentrations and in vivo activities by mechanistic PK/PD modeling. This review will identify the challenges related to ADME studies, provide perspectives on scientific and technical approaches to address these challenges during various stages of drug development of therapeutic biologics, and discuss emerging applications and technologies. Due to the increased emphasis on linking ADME studies with mechanistic PK/PD modeling, the recent progress in biodistribution and biomarker assay development will also be highlighted in this review.
CHALLENGES FOR MECHANISTIC ADME STUDIES OF THERAPEUTIC BIOLOGICS
Due to their large molecular size and complicated tertiary structure (Fig. 1), the ADME issues for therapeutic biologics often are different from that for small molecules. The common considerations for the ADME-related issues for therapeutic biologics are summarized in Table II, which include target-mediated clearance, the FcRn recycling for Fc-containing proteins, immunogenicity, isoform heterogeneity, and metabolic stability, especially for relatively small molecular weight (MW) proteins and peptides. Many of the challenges for the mechanistic ADME studies of therapeutic biologics stem from the lack of effective and validated in vitro systems. For example, the metabolic/catabolic stability of mAbs or other large therapeutic proteins cannot be easily assessed in vitro with liver microsomes or cultured hepatocytes, which are commonly used for small molecule studies. In contrast to prediction of oral bioavailability for small molecules, there are no reliable in vitro systems that can help to predict bioavailability of therapeutic biologics after SC administration. Therapeutic biologics generally have limited distribution in tissues. Thus, it is important to understand the tissue penetration mechanism and, consequently, the relationship between tissue concentration and efficacy.
Fig. 1.
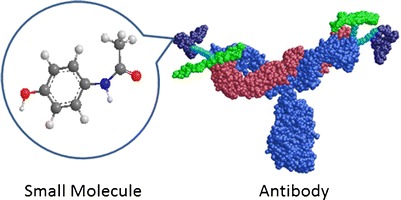
Size comparison of a typical small molecule (illustrated by acetaminophen) and an IgG antibody
Table II.
Key Factors Determining ADME Profiles of Therapeutic Biologics
| ADME-related considerations for biologics | Key contributing factors |
|---|---|
| Physical/chemical properties | Size, shape, charge, stability, heterogeneity in isoforms (including post-translational modifications) |
| Absorption mechanism | Route of administration specific issues (e.g., contribution of lymphatic absorption after subcutaneous injection), formulation, injection site, subject characteristics, FcRn- and target-dependent mechanisms, physical/chemical properties |
| Distribution patterns | Size, shape, charge, target binding, FcRn- and target-dependent mechanisms, route of administration, formulation |
| Elimination pathways | Proteolysis, target-mediated clearance, nonspecific endocytosis and formation of immune-complexes followed by complement- or Fc receptor-mediated clearance, protection from catabolism via FcRn mechanism |
| Nonlinear kinetics | Saturable target mediated clearance, immunogenicity, FcRn (for very high doses or possibly for FcRn mutants) |
| Subject characteristics | Body weight, age, sex, disease status, prior exposure to biologics, concomitant medications |
| Species difference in PK profiles | Target binding affinity, FcRn/IgG interactions, immunogenicity, contribution of lymphatic absorption, off-target effects |
In general, biologics are metabolized/catabolized into small peptide fragments or amino acids that are ready for renal excretion or recycling into protein synthesis. The rate of metabolism for biologics is compound- or modality-dependent. On one end of the spectrum, metabolism of small peptides and recombinant human proteins with a low MW tends to be very rapid. The metabolic stability for this type of molecules can potentially be assessed in vitro, similar to the assays employed for small molecules. On the other end of the spectrum, metabolism of human IgG proteins is very slow, resulting in a long half-life of 7–28 days. As extensively reviewed in literature, antibody molecules with an Fc portion is protected from degradation by binding to FcRn in endothelial cells, explaining the long half-lives of these proteins (19–21). Thus, for mechanistic-based metabolism studies of biologics, validated in vitro systems that could maintain biologic activities (e.g., receptor-mediated uptake, endocytosis, internalization, lysosomal digestion, and/or antigen presentation) for days or weeks would be extremely valuable. To this end, a 3D hollow fiber human hepatocyte bioreactor system has been developed and characterized recently for long-term metabolism and toxicity studies, although further refinement of the system is needed before it can be adapted for the in vitro assessment of biologics.
Another challenge involved in ADME studies of biologics is the species difference in target-binding properties and host immune response to a biologic. It is well established that the target-mediated clearance and antidrug–antibody-mediated clearance for therapeutic biologics are species-dependent. In addition, there are significant species differences in FcRn/IgG interactions (22). Thus, common laboratory animals (e.g., rodents, dogs) may not always be relevant for prediction of ADME profiles of therapeutic biologics in humans. While transgenic animals that express human targets or receptors can be useful for the qualitative assessment of potential clearance mechanisms, a more quantitative approach is needed to predict ADME profiles in humans.
Analytical assay development for therapeutic biologics is another hurdle encountered in ADME studies. Structure similarity between endogenous proteins and therapeutic biologics and the hydrophilic nature of these macromolecules make the extraction and purification of biologic drugs from in vivo samples very difficult. The detection limit of analytical instruments makes the direct measurement (i.e., without prior purification/enrichment step, such as immunocapture) of therapeutic proteins in biologic matrices (plasma or tissue homogenates) a challenging task. Very often, enzymatic digestion may be required prior to the structure identification or quantitation of therapeutic proteins for in vivo samples. In these cases, the recovery of therapeutic biologics during sample treatment and the assay interference of endogenous proteins become major concerns. However, with the improvement in instrumentation and sample handling methodology, significant progress on assay throughput and detection has been made in recent years.
EMERGING TECHNOLOGIES FOR ADME STUDIES OF THERAPEUTIC BIOLOGICS
Quantitative Analytical Method Development
Conventional analytical approaches for pharmacokinetic studies of biologics heavily rely on ligand binding assays, such as the enzyme-linked immunosorbent assay (ELISA) (23,24). Advantages of ligand-binding assays include high specificity and sensitivity, ease of sample handling, and low cost. However, generating high quality and specific reagents for ELISA methods developed to quantify therapeutic biologics and antidrug antibodies (ADAs) could be a time-consuming and labor-intensive process (25,26). While pharmaceutical companies usually are willing to spend resources on lead candidates and focus on resolving complicated issues halting advancement of these candidates, analytical support for early discovery research often is limited. On the other hand, the pace of generating new biotherapeutic candidates has improved dramatically due to the advancement in protein engineering and supporting purification/production technologies. Thus, the demand for supporting discovery activity, such as lead optimization, is paramount. In order to improve the throughput rate, new analytical approaches and instrument platforms are being continuously pursued. One example is the application of “generic ELISA”, which uses a reagent recognizing the Fc portion of a human protein, utilized to support PK studies of human Fc containing biologics in animals. Automation with robotic systems is also highly desirable for the screening of new biologic constructs. One example is the Gyrolab immunoassay platform (Gyros, Uppsala, Sweden), which incorporates the microfluidic concept into a ligand binding assay (27). Applications of the Gyrolab immunoassay workstation for pharmacokinetic studies have been reported recently (28,29). The nanoliter sample requirement for the assay is particularly valuable for pharmacokinetic studies in mice, an animal species commonly used in early efficacy evaluations. With a reduced blood sample size of 10–20 μL for each time point, serial bleeding can be conducted in the same mouse over the time course for a PK study, minimizing the intersubject variability associated with nonserial sampling needed for a mouse PK study that relies on a conventional ELISA for bioanalysis.
As the use of biologics or macromolecules spreads to medical, cosmetic, and food sectors, interference of endogenous proteins similar to therapeutic biologics on an immunoassay developed for therapeutic proteins will become a major issue. Alternative approaches that are less liable to such interference have been actively pursued.
One technology particularly attractive for bioanalysis is mass spectrometry (MS), which has been extensively used in the drug development for small molecules (30,31). Characteristics and performances of commonly used mass spectrometers for proteins have been reviewed by Domon and Aebersold (32). Advantages of MS over ELISA include the improved selectivity between structurally similar peptides and proteins, reduced requirements for specific reagents, improved precision and accuracy, and potentially higher throughput rate (33). To improve the sensitivity and reduce the interference of peptide fragments that originate from endogenous proteins, immunoprecipitation is usually applied in the sample extraction process. This is particularly important for tissue homogenates or plasma samples with low drug concentrations; however, this additional step adds to assay complexity and requires additional reagents. Recent developments in the immunoaffinity purification on magnetic beads coated with antibodies for a rapid and efficient purification of therapeutic proteins from biologic matrices (34), and the invention of commercially available temperature-controlled tissue beaters (e.g., Precellys® with the patented cooling technique that keeps temperature at approximate 4°C during homogenization) are expected to enhance the throughput rate in sample treatment for bioanalysis.
To facilitate the mass fragment detection, isotope-labeled protein standards are often used in the protein quantitation by MS (35,36). A recent strategy called Protein Standard Absolute Quantification has been developed, in which a full-length stable-isotope-labeled protein will be synthesized as a standard marker for the therapeutic protein. During the sample analysis process, the full-length stable-isotope-labeled protein standard will be spiked into biologic fluids (e.g., plasma or urine) containing the therapeutic protein that needs to be quantified. Both the labeled protein standard and the therapeutic protein will undergo the purification and enzymatic digestion simultaneously in the same vial. Thus, it will minimize the difference in the sample treatment associated with other methods (e.g., using a stable-isotope-labeled peptide as the standard) (37).
The MS approach also has been applied to the quantitation of antidrug antibodies. Neubert et al. utilized a magnetic bead-based immunoprecipitation method followed by quantitative liquid chromatography–mass spectrometry (LC-MS) to detect ADA in human and cynomolgus serum in the presence of high-circulating concentrations of the protein therapeutic (38). It is anticipated that the development of quantitative mass spectrometric assays will be a booming area for ADME studies of therapeutic biologics.
Non-Invasive Imaging Technologies for Biodistribution and PK/PD Studies
In order to understand in vivo tissue distribution patterns and address the key “Three Pillar”-related questions described in the “INTRODUCTION” section, investigations of biodistribution and target occupancy have become the main focus of preclinical ADME studies. The application of non-invasive imaging technique in biodistribution studies is rapidly growing in both academic and industrial research. As shown in a recent report issued by the National Heart, Lung, and Blood Institute, publications on the application of molecular imaging for cardiovascular research has increased tenfold in the last 10 years (39).
Common imaging technologies used in animal biodistribution studies include optical imaging with luminescence and fluorescence molecular probes, radiotracer-based single photon emission computed tomography (SPECT) and positron emission tomography (PET) imaging, magnetic resonance imaging (MRI), ultrasound imaging, and X-ray imaging (40,41). In contrast to the conventional cut-and-count approach or whole-body autoradiography with radiolabeled biologics, imaging studies can be conducted in live animals, which provides the advantage of obtaining real-time dynamics on the biodistribution of a test article from the same animal. Information gained from in vitro human cell-based assays and in vivo animal studies with fluorescent- or radiotracer-labeled biologics can be used to guide the dose regimen design for clinical trials. However, it should be pointed out that while in-life imaging can provide an overall picture on biodistribution patterns, its sensitivity may be limited, especially for the in vivo fluorescent imaging (42). In most cases, the imaging approach is considered to be semiquantitative or qualitative. Technologies with relatively high sensitivity and selectivity, such as SPECT and PET, are more attractive for clinical studies. To further enhance the sensitivity, combination imaging systems (e.g., PET/CT, SPECT/CT, PET/MRI) have been developed (43).
While a cutting-edge imaging technique is instrumental to support novel research activities, the conventional radioisotope probe method in combination with tissue counting or whole body autoradiography remains to be the workhorse in laboratory research for biologics. The radioisotopes most commonly used in ADME studies for therapeutic biologics include 18F, 99mTc, 32P, 14C, 111In, 131I, 125I, and 123I. Among that, 125I is usually considered as the first choice in the labeling of biologics in preclinical biodistribution studies owing to the low cost, well-known protein iodination chemistry, and its fit-for-purpose physical properties of gamma emission and 60-day decay half-life. However, there are several concerns associated with using 125I labeled proteins, such as a rapid loss of 125I label for certain types of molecules and the potential for bioactivity change of biologics during the iodination procedure (44). Therefore, caution should be applied in the data interpretation of radiolabeled studies.
Structure Elucidation for In Vivo Degradants and Isoforms of Therapeutic Biologics
Similar to the research for small molecules, structure elucidation of major degradants of a therapeutic protein will help to identify the “metabolic soft spot” for biologics. Such information is valuable to guide the design of new constructs with improved in vivo stability and PK properties. In addition, structure elucidation of in vivo samples also helps the assessment of potential differences in the elimination kinetics of protein product isoforms, as a biologic drug is often a collection of large protein isoforms and not a single molecular entity (45). Information gained from the in vivo measurement of various isoforms (i.e., products of a specific posttranslational modification) may guide selection or design of a new drug product with improved ADME profiles.
Identifying metabolic degradants of therapeutic biologics is a technically challenging task. Lack of an effective extraction and purification method is one of the major hurdles. Although immunoprecipitation with drug specific or “generic” (e.g., anti-IgG) antibodies may help the purification process, such approach is not suitable for degradants that do not bind to capture antibodies. Therefore, separation of proteins and degradants by gel electrophoresis followed by in-gel proteolytic digestion or liquid chromatography is frequently performed.
For structure identification and characterization of degradants, biologic isoforms, and immunocomplexes in biologic matrices, MS approaches developed for proteomic research are commonly used. Two fundamental strategies for MS analysis of protein structures are the “bottom-up” and the “top-down” approaches (46–48). In the “bottom-up” approach, purified proteins or complex protein mixtures are subjected to chemical or enzymatic cleavage, and the peptide products are usually separated by chromatography followed by the tandem mass spectrometry analysis. In the “top-down” approach, intact protein ions or large protein fragments are directly subjected to the gas-phase fragmentation for MS analysis (47). Given the complexity in protein structures, a single approach may not be sufficient to provide comprehensive information for the structure identification purpose. An integrated approach has been applied to identify protein isoforms arising from various amino acid modifications (e.g., acetylation, phosphorylation) and genetic variants (e.g., single nucleotide polymorphic isoforms) (48). This combination strategy overcomes the major limitations of the traditional bottom-up (e.g., inability to characterize multiple, unexpected protein isoforms and genetic variants) or the top-down (e.g., low throughput) approach.
The structures of metabolic degradants, isoforms, or immunocomplexes are determined by a comparison of the peptide mass spectra with theoretical peptide masses calculated from a proteomic or genomic database. However, at present, the de novo peptide identification via tandem MS is a time-consuming process (49). Technical challenges include assembling, analysis, and interpretation of a large volume of data generated from fragments of a macromolecule, requiring a construction of sophisticated computation system with a comprehensive database capability. With the rapid progress in MS technology and bioinformatics, structure identification technique for in vivo degradants is expected to mature with a reasonable throughput rate in the future.
Recent advances in nuclear magnetic resonance (NMR) allow to use this technology to rapidly determine protein and protein–ligand structures, to efficiently screen fragment-based libraries to identify biological relevant ligand interactions, and, ultimately, to identify new therapeutic targets (50). However, the application of NMR can be limited due to the general upper weight limitation of ~25 kDa and concerns about the interference of endogenous proteins. Despite these limitations, NMR technology has significant potential in drug discovery and in the development of therapeutic biologics.
In Vitro Predictive Tools for ADME of Therapeutic Biologics
Despite the urgent need for metabolic stability screening of new biologic constructs in discovery research, there is, to date, no validated in vitro system that can be used for prediction of in vivo clearance (51). For example, optimizing the interactions between IgG and FcRn is becoming a more widespread approach to increase exposure and reduce dosing frequency of an IgG-based biologic (52). In vitro FcRn binding properties (e.g., cell-based binding in FcRn-expressing cell lines) have been used to guide Fc engineering of new mAbs with a desirable half-life (53, 54). However, the correlation between in vitro binding constant (kd) and in vivo clearance is not always straightforward (53). Therefore, quantitative modeling that incorporates the kinetic parameters for FcRn and Fc-containing protein interactions (e.g., kon and koff rates at acidic and neutral pHs) and other determinants of clearance is needed to improve the in vitro and in vivo correlations, since the relative contribution of a given in vitro binding parameter to the overall in vivo clearance may differ among a series of Fc-containing proteins (17).
In vitro predictive tools for tissue penetration are also highly desirable for therapeutic biologics. In recent research on tumor uptake prediction, an in silico model has been proposed by Thurber et al. that could help the design of biologic constructs for solid tumors (55). The authors indicate that theoretical analyses of ADME profiles of a potential therapeutic biologic will provide specific guidance with respect to design of biophysical and biochemical properties of the therapeutic biologic, such as agent size, affinity, and target antigen. The current data analyses suggest that IgG-sized constructs exhibit the most favorable balance between systemic clearance and vascular extravasation, resulting in a maximal tumor uptake. Quantitative predictions of the effect of dose and binding affinity on tumor uptake and penetration can be achieved using the information on antigen expression level and metabolic half-life (56). This quantitative model has been tested in xenografted tumors in mice, and needs to be further verified in cancer patients. The use of computational design to engineer therapeutic biologics based on tissue distribution patterns and target binding properties is an attractive area of future research.
In Vitro Predictive Tools for Immunogenicity
Immunogenicity is often a major concern during the discovery and development of biotherapeutics and may affect ADME profiles in species-dependent manner (57,58). Successful prediction of immunogenicity will help to reduce the risk in drug candidate selection (59). Although immunogenicity of a novel therapeutic protein is usually assessed in animals (most commonly in monkeys) prior to human trials, the primary goal of these assessments is to support preclinical PK, pharmacology, and toxicology data interpretation. Due to species differences in a variety of immune system components involved in an immune response to a particular therapeutic biologic and differences in the “degree of foreignness” of a biologic between animals and humans, predictions of human immunogenicity based on animal data are generally not considered to be reliable, hence the value of mechanistic immunogenicity studies in animals can be restricted (60). Thus, it is desirable to use in silico or in vitro human cell-based assessments for the immunogenicity prediction, including predictions of aggregation prone regions and T and B cell epitopes. To this end, Kumar et al. described the importance of investigating the impact of aggregation on immunogenicity and potential coupling of T and B cell epitopes and aggregation-prone regions (61). Computational tools that can predict aggregation prone regions as well as T- and B-cell immune epitopes from protein sequence and structure have become available recently from several commercial vendors (58, 62–64). However, none of these tools have been thoroughly validated with in vivo human data and, therefore, the standards for prediction tools have not been established (63).
Cell-based assays using human blood are being used in combination with in silico approaches to predict human immune responses for a variety of therapeutic applications, including vaccines, transplantation, and, in recent years, therapeutic biologics. For example, peripheral blood mononuclear cells (PBMC)-derived T cell immunogenicity assays have been used to monitor and predict immune response and outcome in the field of transplantation (65). The assay has the capability to elicit the response from a PBMC population with low frequency of antigen-specific T cells and potentially can be used to distinguish therapeutic proteins with high and low immunogenicity potential. However, depending on assay conditions, different responses may be obtained for a given therapeutic protein in PBMCs, therefore, similar to in silico tools, cell-based assays need to be optimized with respect to predictive value in clinical human studies. The considerations for optimization and validation of an in vitro PBMC-derived T cell assay for immunogenicity prediction of biotherapeutics have been discussed by Wullner et al. (66). To streamline the application of pharmacogenomics to immunogenicity predictions, an individualized T cell epitope measure (iTEM) tool has been developed to estimate an individual’s T cell response to a protein antigen based on HLA-binding predictions. While further refinement of the in vitro conditions is needed, the system is preliminarily validated for prospective iTEM predictions using data from in vitro and in vivo studies (67).
In summary, a collective in silico and in vitro approach in conjunction with in vivo animal models (e.g., genetically modified animals designed to have some aspects of human immune systems) could be used to rank–order protein drug candidates for their immunogenic potential. However, the validation of these predictions is yet to come and will rely on the collection of comprehensive databases and likely collaboration across biopharmaceutical companies. Since immunogenicity impacts both the ADME profile and biological activity of biologics, advances in predictive immunogenicity tools are likely to impact the success rate in biotherapeutic drug development.
Assay Development for Biomarkers
As discussed in the “INTRODUCTION” section, biomarker assay development is a growing area of research and stems from the need to improve the current low success rate of clinical trials, as well as by the increasing emphasis on personalized medicine and targeted therapies. In many cases, the failure of clinical trials is due to the fact that only a subset of patients expresses the responding phenotype. Pharmaceutical companies together with regulatory agencies have made significant investments in biomarker development, with the focus on translation to clinical practice and linkage of PK, PD, and disease biomarkers. Ultimately, it is important to select a “right” biomarker, i.e., to identify key biomarkers related to intended indication(s) (68,69). In addition, developing appropriate biomarkers in animal disease models that can be used in translational research to support human clinical trials will be very valuable. Both ex vivo, in situ, and in vivo biomarkers approaches are being pursued, sometimes in parallel, as discussed below.
Common techniques used in ex vivo biomarker assays include immunoassays, MS, flow cytometry, polymerase chain reaction, and bioactivity assays. The assay development for biomarkers could be a time-consuming process. Hurdles to method development include the requirements on specificity and stability in complex biological matrices, wide dynamic range, and assay sensitivity. Requirements for method validation (e.g., specificity, selectivity, assay acceptance criteria, stability, and reproducibility) and the utility of protein biomarkers have been reviewed by Lee (70). Targeted proteomic strategies have been applied to quantify biomarker candidates at concentrations of nanograms per milliliter or picograms per milliliter by LC-MS (71).
Recently, matrix-assisted laser desorption/ionization imaging MS, a new in situ molecular imaging technology, has been used in the biomarker field (72–74). The imaging MS can conduct a direct in situ analysis of biologics from thin tissue sections and provide important insights into biological processes because the native distributions of molecules and histological features remain intact throughout the analysis. A wide variety of molecules can be imaged, including proteins, peptides, and metabolites. From the perspectives of translational research and clinical proteomics, this method can correlate molecular readouts to histopathological changes found in disease tissues (74,75).
Quantitative or semiquantitative in vivo approaches for directly monitoring pharmacological activities of therapeutic biologics are becoming more widespread in translational research. For example, the putative utility and validity of electroencephalograph (EEG) as a surrogate biomarker for a number of major central nervous system disorders, including depression, schizophrenia, and pain, have been described (76). Similarities in spontaneous EEG and cortical activity potential measures from rodents to humans provide a solid foundation for the use of EEG as a translatable biomarker. EEG monitoring can be performed in a relatively non-invasive, stress- and pain-free manner, and the identical computational algorithms can be used for EEG signal processing and analyses across lab animal species and humans. It should be emphasized that in order to make a link between biomarker data and a clinical outcome (efficacy and/or safety), biomarker data in animals and humans need to be interpreted in an integrated manner together with the corresponding PK/ADME information.
PRACTICAL APPLICATIONS
Case Studies
To demonstrate the importance and practical aspects of mechanistic ADME studies in drug discovery and development research, two case studies are presented in this review. The first case study emphasizes the importance of biodistribution assessment in early phase studies of new biologics to assist in clinical development of ADCs. The second case study illustrates the importance of integrated assessment of presystemic metabolism and absorption rate on the in vivo exposure of a peptide drug after SC injection with a controlled release (CR) formulation.
Case Study 1
ADCs are complex drugs that consist of potent cytotoxic drugs linked to antibodies via chemical linkers. The concept of ADCs has attracted extensive interests due to its promises of potential tumor-specific delivery of cytotoxic drugs, improved safety window compared to the monodrug, and long half-life associated with the antibody portion of the molecule (77).
CMD-193 is an ADC comprised of a humanized antibody directed against the Lewis Y (Ley) antigen conjugated with calicheamicin. CMD-193 binds to the Ley antigen, a tetrasaccharide expressed on the cell surface of many solid tumors (e.g., liver cancer), via its antibody moiety (78). Upon binding to its target on tumor cells, CMD-193 is internalized; the active calicheamicin moiety is released and is able to bind to the minor groove of tumor cell DNA, causing double-strand DNA breaks, the inhibition of DNA synthesis, and apoptosis.
In a phase I study, biodistribution of [111In]CMD-193 was evaluated in patients who had histologically confirmed solid malignancies displaying Ley antigen positive. The PK and tissue distribution pattern of [111In]CMD-193, determined by whole body γ camera scans and SPECT imaging, were compared to those for [111In]Hu3S193, a parental antibody of CMD-193. Following IV infusion, biodistribution and PK of [111In]CMD-193 were found to be significantly different from that for [111In]hu3S193 (78). In contrast to prominent tumor-specific uptake and long half-life in blood observed for [111In]hu3S193, [111In]CMD-193 had fast clearance and short half-life, rapid uptake in liver parenchyma, and lack of tumor uptake (Table III). Although the actual cause for the altered biodistribution of CMD-193 compared to the parent antibody is unknown, it is speculated that a physicochemical change induced by conjugation of the antibody with calicheamicin may lead to the altered biodistribution profile. Thus, understanding the effect of payload on the biodistribution of ADC will help to optimize the tumor uptake and improve the efficacy. In this example, biodistribution data provided a valuable content for the interpretation of the lack of efficacy observed in the clinical trial for CMD-193.
Table III.
Peak Tumor Uptake and Liver Uptake of [111In]CMD-193 and its Parental Antibody [111In]hu3S193
| [111In]CMD-193 | [111In]hu3S193 | |
|---|---|---|
| Peak tumor uptake (μg/g) | 0.15 ± 0.03 | 2.9 ± 1.7 |
| Liver uptake (% injected dose) | 33 ± 6.0 | 7.3 ± 1.5 |
Adapted from Herbertson et al. [78]
Case Study 2
For most therapeutic biologics, SC injection is preferred over the IV administration due to convenience. The SC absorption rate depends on multiple factors, including the size of a molecule and the formulation used. In an effort to develop a SC-delivered peptide therapeutic for weight loss and/or diabetes treatment, a CR formulation of an oxyntomodulin analog peptide (referred to as “Peptide A”) was developed with the intent to prolong the absorption process and reduce the dosing frequency compared to that from the conventional instantaneous released (IR) formulation (79). In rats, the SC absorption rate of peptide A was reduced by the CR formulation, as evidenced by a shift in the time (Tmax) to reach the maximal concentration (Cmax) from 1.3 h for the IR formulation to 10 h for the CR formulation after SC injection of 2 mg/kg. However, the overall systemic exposure, determined by the Cmax and the area under the concentration–time curve (AUC), were decreased significantly in rats treated with the CR formulation compared to that with the IR formulation. The SC bioavailability with the CR formulation was reduced by ~40% compared to that for the IR formulation. Similar findings were observed in monkeys and humans following SC administration with both IR and CR formulations.
To understand the potential cause for the reduced systemic exposure, detailed ADME studies for peptide A were conducted in rats. Biodistribution and excretion studies with [125I]Peptide A indicated that the SC administered radioactive dose was absorbed well from the injection site and the radioactivity was almost completely recovered in urine (as free iodine and/or degradants) over a collection period of 10 days. Further studies by in vitro metabolic stability testing of [125I]Peptide A with fresh rat skin and in vivo metabolic profiling of rat plasma and skin samples collected at the SC injection site suggested that peptide A was metabolically unstable in skin and that degradation of the peptide in skin during its prolonged residence at the injection site could potentially contribute to a decrease in the SC bioavailability for the CR formulation. This example illustrates that in vivo PK profiles depend on the interplay of ADME properties of therapeutic biologics. Knowing the mechanisms that govern PK profiles will help rational optimization in half-life extension and/or delivery strategy.
Hot Topics and Emerging Applications
ADME studies for biologics may bridge into multiple aspects of drug discovery and development, and the applications listed below highlight the emerging focus in the pharmaceutical industry:
PK/PD modeling: Because of the interdependency of PK, biomarkers and efficacy, mechanistic PK/PD modeling is a critical tool in the design of clinical dose regimens, as described in the previous sections. While sophisticated modeling tools and model libraries are already available or continued to be built, validation of these PK/PD models will require extensive experimental data generated from in vitro or in vivo ADME studies.
Blood–brain barrier (BBB) transport: Although BBB transport has been extensively studied for small molecules, this area has not been fully explored for therapeutic proteins. Knowledge of the mechanism of BBB transports for biologics will benefit the drug discovery for central nervous system (CNS) indications, as well as the safety assessment for therapeutic biologics. A recent approach to improve BBB penetration of therapeutic biologics is to engineer a bifunctional IgG-based fusion protein, with one arm binding to a CNS target and the second arm targeting an endogenous BBB receptor, such as the human insulin receptor (HIR) or the transferrin receptor (80). This approach can be illustrated by the brain uptake of the human insulin receptor mAb-human TNF receptor fusion protein (81). However, sufficient delivery of protein drugs to the CNS remains as a major challenge for pharmaceutical research (51).
Drug–drug interactions: As the number of approved therapeutic biologics increases, potential drug–drug interactions (DDI) between concurrently administered drugs (small molecule–biologic drugs or biologic–biologics drugs) become a concern. The importance on therapeutic protein–drug interactions and guidance that recommends how and when to evaluate such interactions have been published recently by FDA (82,83). However, preclinical tools and in vitro test systems for assessing drug interaction potential of therapeutic proteins are limited. Thus, DDI assessment is often evaluated as a part of clinical trials. Knowledge gaps and areas for future research have been summarized in the AAPS Workshop Report on strategies to address DDI for biologics (84). This topic will be monitored closely in the future development of therapeutic biologics. With continuous data collection and knowledge-building process, strategies and methodologies to address DDI potential for new biologics will become mature.
Novel delivery systems: With only a few exceptions, therapeutic biologics are almost exclusively administered by parenteral routes. To increase target-specific delivery, improve safety and convenience for patients, and support for individualized medication, new delivery systems are actively pursued. Some examples include polymeric microstructured arrays for potential delivery of proteins (85), needle-free injection devices for SC injection (86), and nanoparticles to protect siRNA from serum degradation (87). To assess the utility of these new drug delivery systems, PK and ADME properties of biologics delivered by novel technologies often are compared to conventional IV or SC injection. Species selection for nonclinical PK evaluations of new delivery systems can be drug-, device-, and route-dependent, ranging from rodents to monkeys and mini-pigs (88).
-
Biosimilar/comparability assessments: Biosimilars are a new class of drugs intended to offer comparable safety and efficacy to the reference, off-patent biologics. Biosimilars have attracted great attention with the hope that they will allow wide-spread availability of currently expensive biologic products (89). Given their structural complexity, multifaceted manufacturing process, and, as a result, challenges for predicting impact of the manufacturing process changes on immunogenicity, biosimilars are not generic alternatives per se and generally are not interchangeable (90). Thus, unique regulatory pathways are required for biosimilars (91,92). PK and immunogenicity assessments, together with efficacy and toxicology studies, are key components in the development of biosimilars.
Traditionally, comparability assessment has been conducted during the manufacturing process of therapeutic biologics, since it is a complex process that involves continual refinement throughout product development, post-approval, and marketing. Common reasons to alter the manufacturing process include the benefits of better yields, scale-up, new technologies and increased purity, as well as new formulations or product delivery systems (93). Since the definition of biosimilars continues to evolve, comparability assessment and characterization of biologic products during the manufacturing process will remain a “hot” topic. Strategies and methodologies for the comparability assessment, including PK and ADME studies, will continue to emerge, which will bring research in this area to a new horizon.
Placenta transport of therapeutic biologics: Unlike the drug development for small molecules, placenta transport of biologic drugs is not studied extensively, likely because of the common belief that the placental transport is minimal for biologics. However, placental transport of immunoglobulins has been recognized for more than 50 years (94,95). A specific FcRn-mediated binding of IgG at the maternal surface of the placenta has been proposed as the first step in the transport mechanism by which IgG is transferred from the mother to the fetus (94,96,97). The role of the second placental barrier, the fetal capillary endothelium, is not yet clear. The expression of FcRn is known to be relatively low on fetal vessels’ endothelium, suggesting possible other mechanism(s) involved in IgG transport across this placental layer (98). The challenge of drug concentration measurements for fetal tissues limits placental transport studies. Additional research is required to fully understand the mechanism of placental transfer of various biological modalities and its implications in the safety assessment of therapeutic biologics.
SUMMARY
Driven by the need for improved clinical success and by the competitive landscape in pharmaceutical industry, demands for mechanistic ADME studies of novel biologics continue to increase. Despite advancements in the science of ADME processes of biologic drugs, many fundamental questions remain unanswered. ADME studies for therapeutic biologics could be a challenging task due to the complex nature of these novel molecules and to limited bioanalytical methods. Despite significant progress in the technologies pertaining to ADME studies of therapeutic biologics, a breakthrough in bioanalytical methodology and in vitro predictive tools will be required to bring the ADME science of therapeutic biologics to a new horizon. The cost for development, validation, and implementation of these new technologies is significant. Thus, research activities should focus on critical issues that impact the development of drug candidates, and mechanistic ADME studies should be carried out on a case-by-case basis. One of the key objectives of ADME studies is to link PK profiles in circulation and action site to in vivo responses. Therefore, advances, as well as limitations in biomarker technologies, may drive the priorities and design of PK/PD and ADME studies for therapeutic biologics. Over time, knowledge gained from in-depth ADME mechanistic studies in nonclinical and clinical settings will advance our understanding on key factors that govern the in vivo disposition of therapeutic biologics across various modalities. This knowledge is instrumental for optimization of disposition properties, and, ultimately, human efficacy and safety profiles of new therapeutic biologics.
Acknowledgments
The authors would like to thank Jennifer Spencer-Pierce, David DeFranco, Jianqing Chen, Mengmeng Wang, Kathy Laws, Shakey Quazi, Josef Olzer, Lisa Dyleski, Allison Joyce, Rosemery Lawrence-Henderson, Beth Leary, Mania Kavosi, Chris Shea, Jeremy Wellen, Mauricio Leal, Chengjie Ji, Bonnie Rup, Wenwei Huang, and Krishna Balakrishnan for their contributions to this manuscript. The authors also would like to acknowledge the support and contributions of Andover Comparative Medicine and Protein Formulation Group, Pfizer Inc.
Contributor Information
Xin Xu, Phone: +1-301-2174673, FAX: +1-301-2175736, Email: xin.xu3@nih.gov.
Yulia Vugmeyster, Email: yulia.vugmeyster@pfizer.com.
References
- 1.Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–774. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- 2.Verdine GL, Hilinski GJ. Stapled peptides for intracellular drug targets. Methods Enzymol. 2012;503:3–33. doi: 10.1016/B978-0-12-396962-0.00001-X. [DOI] [PubMed] [Google Scholar]
- 3.Gillies SD, Lan Y, Brunkhorst B, Wong WK, Li Y, Lo KM. Bi-functional cytokine fusion proteins for gene therapy and antibody-targeted treatment of cancer. Cancer Immunol, immunother: CII. 2002;51:449–460. doi: 10.1007/s00262-002-0302-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelson AL. Antibody fragments: hope and hype. mAbs. 2010;2:77–83. doi: 10.4161/mabs.2.1.10786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Lorenzo C, D'Alessio G. Human anti-ErbB2 immunoagents–immunoRNases and compact antibodies. FEBS J. 2009;276:1527–1535. doi: 10.1111/j.1742-4658.2009.06896.x. [DOI] [PubMed] [Google Scholar]
- 6.Roovers RC, van Dongen GA, van Bergen en Henegouwen PM. Nanobodies in therapeutic applications. Curr Opin Mol Ther. 2007;9:327–335. [PubMed] [Google Scholar]
- 7.Nickerson-Nutter C, Tchistiakova L, Seth NP, Kasaian M, Sibley B, Olland S, et al. Distinct in vitro binding properties of the anti-CD20 small modular immunopharmaceutical 2LM20-4 result in profound and sustained in vivo potency in cynomolgus monkeys. Rheumatol (Oxford, England) 2011;50:1033–1044. doi: 10.1093/rheumatology/keq423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Younes A, Yasothan U, Kirkpatrick P. Brentuximab vedotin. Nat Rev Drug Discov. 2011;11:19–20. doi: 10.1038/nrd3629. [DOI] [PubMed] [Google Scholar]
- 9.Alley SC, Okeley NM, Senter PD. Antibody-drug conjugates: targeted drug delivery for cancer. Curr Opin Chem Biol. 2010;14:529–537. doi: 10.1016/j.cbpa.2010.06.170. [DOI] [PubMed] [Google Scholar]
- 10.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 11.Bell DA, Hooper AJ, Burnett JR. Mipomersen, an antisense apolipoprotein B synthesis inhibitor. Expert Opin Investig Drugs. 2011;20:265–272. doi: 10.1517/13543784.2011.547471. [DOI] [PubMed] [Google Scholar]
- 12.Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nature Rev Drug Discov. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kratz F, Elsadek B. Clinical impact of serum proteins on drug delivery. J Control Release. 2012;161:429–45. doi:10.1016/j.jconrel.2011.11.028. [DOI] [PubMed]
- 14.Horton S, Walsh C, Emery P. Certolizumab pegol for the treatment of rheumatoid arthritis. Expert Opin Biol Ther. 2012;12:235–249. doi: 10.1517/14712598.2012.645533. [DOI] [PubMed] [Google Scholar]
- 15.Faggioni R. Protein pharmacokinetics and metabolism. New York: Plenum Press; 1992. [Google Scholar]
- 16.Vugmeyster Y, Szklut P, Wensel D, Ross J, Xu X, Awwad M, et al. Complex pharmacokinetics of a humanized antibody against human amyloid beta peptide, anti-abeta Ab2, in nonclinical species. Pharm Res. 2011;28:1696–1706. doi: 10.1007/s11095-011-0405-x. [DOI] [PubMed] [Google Scholar]
- 17.Vugmeyster Y, Xu X, Theil FP, Khawli L, leach MW. Pharmacokinetics and toxicology of therapeutic proteins: advances and challenges. World J Biol Chem. 2012;3:73–92. doi: 10.4331/wjbc.v3.i4.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Graaf P. Influencing early portfolio decision making using preclinical M&S: how early is early and when is it too late? San Francisco: AAPS National Biotechnology Conference; 2010. [Google Scholar]
- 19.Keizer RJ, Huitema ADR, Schellens JHM, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49:493–507. doi: 10.2165/11531280-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 20.Kuo TT, Aveson VG. Neonatal Fc receptor and IgG-based therapeutics. mAbs. 2011;3:422–430. doi: 10.4161/mabs.3.5.16983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 22.Andersen JT, Daba MB, Berntzen G, Michaelsen TE, Sandlie I. Cross-species binding analyses of mouse and human neonatal Fc receptor show dramatic differences in immunoglobulin G and albumin binding. J Biol Chem. 2010;285:4826–4836. doi: 10.1074/jbc.M109.081828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeSilva B, Smith W, Weiner R, Kelley M, Smolec J, Lee B, et al. Recommendations for the bioanalytical method validation of ligand-binding assays to support pharmacokinetic assessments of macromolecules. Pharm Res. 2003;20:1885–1900. doi: 10.1023/B:PHAM.0000003390.51761.3d. [DOI] [PubMed] [Google Scholar]
- 24.Lee JW, Kelley M, King LE, Yang J, Salimi-Moosavi H, Tang MT, et al. Bioanalytical approaches to quantify "total" and "free" therapeutic antibodies and their targets: technical challenges and PK/PD applications over the course of drug development. AAPS J. 2011;13:99–110. doi: 10.1208/s12248-011-9251-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gorovits B. Antidrug antibody assay validation: industry survey results. AAPS J. 2009;11:133–138. doi: 10.1208/s12248-009-9091-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rup B, O'Hara D. Critical ligand binding reagent preparation/selection: when specificity depends on reagents. AAPS J. 2007;9:E148–E155. doi: 10.1208/aapsj0902016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yeo LY, Chang HC, Chan PP, Friend JR. Microfluidic devices for bioapplications. Small. 2011;7:12–48. doi: 10.1002/smll.201000946. [DOI] [PubMed] [Google Scholar]
- 28.Mora JR, Obenauer-Kutner L, Vimal Patel V. Application of the Gyrolab platform to ligand-binding assays: a user's perspective. Bioanalysis. 2010;2:1711–1715. doi: 10.4155/bio.10.122. [DOI] [PubMed] [Google Scholar]
- 29.Roman J, Qiu J, Dornadula G, Hamuro L, Bakhtiar R, Verch T. Application of miniaturized immunoassays to discovery pharmacokinetic bioanalysis. J Pharmacol Toxicol Methods. 2011;63:227–235. doi: 10.1016/j.vascn.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Ezan E, Dubois M, Becher F. Bioanalysis of recombinant proteins and antibodies by mass spectrometry. Analyst. 2009;134:825–834. doi: 10.1039/b819706g. [DOI] [PubMed] [Google Scholar]
- 31.Li F, Fast D, Michael S. Absolute quantitation of protein therapeutics in biological matrices by enzymatic digestion and LC-MS. Bioanalysis. 2011;3:2459–2480. doi: 10.4155/bio.11.237. [DOI] [PubMed] [Google Scholar]
- 32.Domon B, Aebersold R. Mass spectrometry and protein analysis. Science. 2006;312:212–217. doi: 10.1126/science.1124619. [DOI] [PubMed] [Google Scholar]
- 33.Ewles M, Goodwin L. Bioanalytical approaches to analyzing peptides and proteins by LC–MS/MS. Bioanalysis. 2011;3:1379–1397. doi: 10.4155/bio.11.112. [DOI] [PubMed] [Google Scholar]
- 34.Cristea IM, Chait BT. Affinity purification of protein complexes. Cold Spring Harbor Laboratory Protocol; 2011. [DOI] [PMC free article] [PubMed]
- 35.Schoenherr RM, Zhao L, Whiteaker JR, Feng LC, Li L, Liu L, et al. Automated screening of monoclonal antibodies for SISCAPA assays using a magnetic bead processor and liquid chromatography-selected reaction monitoring-mass spectrometry. J Immunol Methods. 2010;353:49–61. doi: 10.1016/j.jim.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW. Mass spectrometric quantitation of peptides and proteins using stable isotope standards and capture by anti-peptide antibodies (SISCAPA) J Proteome Res. 2004;3:235–244. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- 37.Brun V, Dupuis A, Adrait A, Marcellin M, Thomas D, Court M, et al. Isotope-labeled protein standards: toward absolute quantitative proteomics. Molecular & Cell Proteomics: MCP. 2007;6:2139–2149. doi: 10.1074/mcp.M700163-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Neubert H, Grace C, Rumpel K, James I. Assessing immunogenicity in the presence of excess protein therapeutic using immunoprecipitation and quantitative mass spectrometry. Anal Chem. 2008;80:6907–6914. doi: 10.1021/ac8005439. [DOI] [PubMed] [Google Scholar]
- 39.Buxton DB, Antman M, Danthi N, Dilsizian V, Fayad ZA, Garcia MJ, et al. Report of the National Heart, Lung, and Blood Institute working group on the translation of cardiovascular molecular imaging. Circulation. 2011;123:2157–2163. doi: 10.1161/CIRCULATIONAHA.110.000943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vasquez KO, Casavant C, Peterson JD. Quantitative whole body biodistribution of fluorescent-labeled agents by non-invasive tomographic imaging. PLos One. 2011;6e20594. doi:10.1371/journal.pone.0020594. [DOI] [PMC free article] [PubMed]
- 41.Khalil MM, Tremoleda JL, Bayomy TB, Gsell W. Molecular SPECT imaging: an overview. International J Mol Imaging. 2011;2011:796025. doi: 10.1155/2011/796025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hillman EM, Amoozegar CB, Wang T, McCaslin AF, Bouchard MB, Mansfield J, et al. In vivo optical imaging and dynamic contrast methods for biomedical research. Philos Trans Series A, Math, Physical, Eng Sci. 2011;369:4620–4643. doi: 10.1098/rsta.2011.0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.von Schulthess GK, Schlemmer HP. A look ahead: PET/MR versus PET/CT. Eur J Nucl Med Mol Imaging. 2009;36(Suppl 1):S3–S9. doi: 10.1007/s00259-008-0940-9. [DOI] [PubMed] [Google Scholar]
- 44.Vugmeyster Y, DeFranco D, Szklut P, Wang Q, Xu X. Biodistribution of [125I]-labeled therapeutic proteins: application in protein drug development beyond oncology. J Pharm Sci. 2010;99:1028–1045. doi: 10.1002/jps.21855. [DOI] [PubMed] [Google Scholar]
- 45.Katsila T, Siskos AP, Tamvakopoulos C. Peptide and protein drugs: the study of their metabolism and catabolism by mass spectrometry. Mass Spectrom Rev. 2012;31:110–133. doi: 10.1002/mas.20340. [DOI] [PubMed] [Google Scholar]
- 46.Reid GE, Stephenson JL, Jr, McLuckey SA. Tandem mass spectrometry of ribonuclease A and B: N-linked glycosylation site analysis of whole protein ions. Anal Chem. 2002;74:577–583. doi: 10.1021/ac015618l. [DOI] [PubMed] [Google Scholar]
- 47.Chen CH. Review of a current role of mass spectrometry for proteome research. Anal Chim Acta. 2008;624:16–36. doi: 10.1016/j.aca.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 48.Wu S, Lourette NM, Tolic N, Zhao R, Robinson EW, Tolmachev AV, et al. An integrated top-down and bottom-up strategy for broadly characterizing protein isoforms and modifications. J Proteome Res. 2009;8:1347–1357. doi: 10.1021/pr800720d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hunt DF, Yates JR, 3rd, Shabanowitz J, Winston S, Hauer CR. Protein sequencing by tandem mass spectrometry. Proc Natl Acad Sci U S A. 1986;83:6233–6237. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Powers R. Advances in nuclear magnetic resonance for drug discovery. Expert Opin Drug Discov. 2009;4:1077–1098. doi: 10.1517/17460440903232623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin JH. Pharmacokinetics of biotech drugs: peptides, proteins and monoclonal antibodies. Curr Drug Metabol. 2009;10:661–691. doi: 10.2174/138920009789895499. [DOI] [PubMed] [Google Scholar]
- 52.Kuo TT, Baker K, Yoshida M, Qiao SW, Aveson VG, Lencer WI, et al. Neonatal Fc receptor: from immunity to therapeutics. J Clin Immunol. 2010;30:777–789. doi: 10.1007/s10875-010-9468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yeung YA, Leabman MK, Marvin JS, Qiu J, Adams CW, Lien S, et al. Engineering human IgG1 affinity to human neonatal Fc receptor: impact of affinity improvement on pharmacokinetics in primates. J Immunol. 2009;182:7663–7671. doi: 10.4049/jimmunol.0804182. [DOI] [PubMed] [Google Scholar]
- 54.Wang W, Lu P, Fang Y, Hamuro L, Pittman T, Carr B, et al. Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metabol Dispos: Biol Fate Chem. 2011;39:1469–1477. doi: 10.1124/dmd.111.039453. [DOI] [PubMed] [Google Scholar]
- 55.Thurber GM, Schmidt MM, Wittrup KD. Factors determining antibody distribution in tumors. Trends Pharmacol Sci. 2008;29:57–61. doi: 10.1016/j.tips.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wittrup KD, Thurber GM, Schmidt MM, Rhoden JJ. Practical theoretic guidance for the design of tumor-targeting agents. Methods Enzymol. 2012;503:255–268. doi: 10.1016/B978-0-12-396962-0.00010-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ponce R, Abad L, Amaravadi L, Gelzleichter T, Gore E, Green J, et al. Immunogenicity of biologically-derived therapeutics: assessment and interpretation of nonclinical safety studies. Regulatory Toxicol Pharmacol: RTP. 2009;54:164–182. doi: 10.1016/j.yrtph.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 58.De Groot AS, McMurry J, Moise L. Prediction of immunogenicity: in silico paradigms, ex vivo and in vivo correlates. Curr Opin Pharmacol. 2008;8:620–626. doi: 10.1016/j.coph.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 59.Stas P, Lasters I. Strategies for preclinical immunogenicity assessment of protein therapeutics. IDrugs: Investig Drugs J. 2009;12:169–173. [PubMed] [Google Scholar]
- 60.Brinks V, Jiskoot W, Schellekens H. Immunogenicity of therapeutic proteins: the use of animal models. Pharm Res. 2011;28:2379–2385. doi: 10.1007/s11095-011-0523-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kumar S, Singh SK, Wang XL, Rup B, Gill D. Coupling of aggregation and immunogenicity in biotherapeutics: T- and B-cell immune epitopes may contain aggregation-prone regions. Pharm Res. 2011;28:949–961. doi: 10.1007/s11095-011-0414-9. [DOI] [PubMed] [Google Scholar]
- 62.Vita R, Zarebski L, Greenbaum JA, Emami H, Hoof I, Salimi N, et al. The immune epitope database 2.0. Nucleic Acids Res. 2010;38(Database issue):D854–D862. doi: 10.1093/nar/gkp1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gorovits B. Immunogenicity: prediction, detection and effective assay development. Bioanalysis. 2010;2:1539–1545. doi: 10.4155/bio.10.121. [DOI] [PubMed] [Google Scholar]
- 64.Koren E, De Groot AS, Jawa V, Beck KD, Boone T, Rivera D, et al. Clinical validation of the "in silico" prediction of immunogenicity of a human recombinant therapeutic protein. Clin Immunol. 2007;124:26–32. doi: 10.1016/j.clim.2007.03.544. [DOI] [PubMed] [Google Scholar]
- 65.Hernandez-Fuentes MP, Salama A. In vitro assays for immune monitoring in transplantation. Methods Mol Biol. 2006;333:269–290. doi: 10.1385/1-59745-049-9:269. [DOI] [PubMed] [Google Scholar]
- 66.Wullner D, Zhou L, Bramhall E, Kuck A, Goletz TJ, Swanson S, et al. Considerations for optimization and validation of an in vitro PBMC derived T cell assay for immunogenicity prediction of biotherapeutics. Clin Immunol. 2010;137:5–14. doi: 10.1016/j.clim.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 67.Cohen T, Moise L, Ardito M, Martin W, De Groot AS. A method for individualizing the prediction of immunogenicity of protein vaccines and biologic therapeutics: individualized T cell epitope measure (iTEM). J Biomed Biotechnol. 2010. doi:10.1155/2010/961752. [DOI] [PMC free article] [PubMed]
- 68.Frank R, Hargreaves R. Clinical biomarkers in drug discovery and development. Nature Rev Drug Discov. 2003;2:566–580. doi: 10.1038/nrd1130. [DOI] [PubMed] [Google Scholar]
- 69.Taube SE, Clark GM, Dancey JE, McShane LM, Sigman CC, Gutman SI. A perspective on challenges and issues in biomarker development and drug and biomarker codevelopment. J Natl Cancer Inst. 2009;101:1453–1463. doi: 10.1093/jnci/djp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee JW. Method validation and application of protein biomarkers: basic similarities and differences from biotherapeutics. Bioanalysis. 2009;1:1461–1474. doi: 10.4155/bio.09.130. [DOI] [PubMed] [Google Scholar]
- 71.Huttenhain R, Malmstrom J, Picotti P, Aebersold R. Perspectives of targeted mass spectrometry for protein biomarker verification. Curr Opin Chem Biol. 2009;13:518–525. doi: 10.1016/j.cbpa.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Caprioli RM, Farmer TB, Gile J. Molecular imaging of biological samples: localization of peptides and proteins using MALDI-TOF MS. Anal Chem. 1997;69:4751–4760. doi: 10.1021/ac970888i. [DOI] [PubMed] [Google Scholar]
- 73.Seeley EH, Schwamborn K, Caprioli RM. Imaging of intact tissue sections: moving beyond the microscope. J Biol Chem. 2011;286:25459–25466. doi: 10.1074/jbc.R111.225854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cazares LH, Troyer DA, Wang B, Drake RR, Semmes OJ. MALDI tissue imaging: from biomarker discovery to clinical applications. Anal Bioanal Chem. 2011;401:17–27. doi: 10.1007/s00216-011-5003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McDonnell LA, Corthals GL, Willems SM, van Remoortere A, van Zeijl RJ, Deelder AM. Peptide and protein imaging mass spectrometry in cancer research. Journal of proteomics. 2010;73:1921–1944. doi: 10.1016/j.jprot.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 76.Leiser SC, Dunlop J, Bowlby MR, Devilbiss DM. Aligning strategies for using EEG as a surrogate biomarker: a review of preclinical and clinical research. Biochem Pharmacol. 2011;81:1408–1421. doi: 10.1016/j.bcp.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 77.Polson AG, Ho WY, Ramakrishnan V. Investigational antibody–drug conjugates for hematological malignancies. Expert Opin Invest Drugs. 2011;20:75–85. doi: 10.1517/13543784.2011.539557. [DOI] [PubMed] [Google Scholar]
- 78.Herbertson RA, Tebbutt NC, Lee FT, MacFarlane DJ, Chappell B, Micallef N, et al. Phase I biodistribution and pharmacokinetic study of Lewis Y-targeting immunoconjugate CMD-193 in patients with advanced epithelial cancers. Clin Cancer Res. 2009;15:6709–6715. doi: 10.1158/1078-0432.CCR-09-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang MM, Defranco D, Wright K, Quazi S, Spencer-Pierce J, et al. Decreased exposure of Peptide X in Zn formulation after subcutaneous dosing and in vitro metabolism in skin. AAPS J. 2011;13(S1):M1086. [Google Scholar]
- 80.Pardridge WM, Boado RJ. Reengineering biopharmaceuticals for targeted delivery across the blood–brain barrier. Methods Enzymol. 2012;503:269–292. doi: 10.1016/B978-0-12-396962-0.00011-2. [DOI] [PubMed] [Google Scholar]
- 81.Pardridge WM. Biologic TNFalpha-inhibitors that cross the human blood–brain barrier. Bioeng Bugs. 2010;1:231–234. doi: 10.4161/bbug.1.4.12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.U.S. Department of Health and Human Services, Food and Drug Administration. Guidance for industry: nonclinical safety evaluation of drug or biologic combinations. 2006. http://www.fda.gov/OHRMS/DOCKETS/98fr/05d-0004-gdl0002.pdf.
- 83.Lee JI, Zhang L, Men AY, Kenna LA, Huang SM. CYP-mediated therapeutic protein–drug interactions: clinical findings, proposed mechanisms and regulatory implications. Clin Pharmacokinet. 2010;49:295–310. doi: 10.2165/11319980-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 84.Girish S, Martin SW, Peterson MC, Zhang LK, Zhao H, Balthasar J, et al. AAPS workshop report: strategies to address therapeutic protein–drug interactions during clinical development. AAPS J. 2011;13:405–416. doi: 10.1208/s12248-011-9285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Park J-H, Davis S, Yoon Y-K, Prausnitz MR, Allen MG (eds). Micromachined biodegradable microstructures. Proc IEEE Microelectromechanical Systems Conference. 2003; 371–4.
- 86.Webb JL, Cullifer RE, Jr, Lee D. Market trends of injectable drug delivery systems. J Med Mark. 2011;11:237–243. doi: 10.1177/1745790411412241. [DOI] [Google Scholar]
- 87.Shi J, Votruba AR, Farokhzad OC, Langer R. Nanotechnology in drug delivery and tissue engineering: from discovery to applications. Nano Lett. 2010;10:3223–3230. doi: 10.1021/nl102184c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van der Laan JW, Brightwell J, McAnulty P, Ratky J, Stark C. Regulatory acceptability of the minipig in the development of pharmaceuticals, chemicals and other products. J Pharmacol Toxicol Methods. 2010;62:184–195. doi: 10.1016/j.vascn.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 89.Roger SD. Biosimilars: current status and future directions. Expert Opin Biol Ther. 2010;10:1011–1018. doi: 10.1517/14712591003796553. [DOI] [PubMed] [Google Scholar]
- 90.Dranitsaris G, Amir E, Dorward K. Biosimilars of biological drug therapies: regulatory, clinical and commercial considerations. Drugs. 2011;71:1527–1536. doi: 10.2165/11593730-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 91.Minghetti P, Rocco P, Cilurzo F, Del Vecchio L, Locatelli F. The regulatory framework of biosimilars in the European Union. Drug Discov Today. 2012;17:63–70. doi: 10.1016/j.drudis.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 92.Woodcock J, Griffin J, Behrman R, Cherney B, Crescenzi T, Fraser B, et al. The FDA's assessment of follow-on protein products: a historical perspective. Nature Rev Drug Discov. 2007;6:437–442. doi: 10.1038/nrd2307. [DOI] [PubMed] [Google Scholar]
- 93.Chirino AJ, Mire-Sluis A. Characterizing biological products and assessing comparability following manufacturing changes. Nat Biotechnol. 2004;22:1383–1391. doi: 10.1038/nbt1030. [DOI] [PubMed] [Google Scholar]
- 94.Brambell FW. The transmission of immunity from mother to young and the catabolism of immunoglobulins. Lancet. 1966;2:1087–1093. doi: 10.1016/S0140-6736(66)92190-8. [DOI] [PubMed] [Google Scholar]
- 95.Redman DR. Prenatal influence on immunocompetence of the neonate. J Anim Sci. 1979;49:258–267. doi: 10.2527/jas1979.491258x. [DOI] [PubMed] [Google Scholar]
- 96.Kane SV, Acquah LA. Placental transport of immunoglobulins: a clinical review for gastroenterologists who prescribe therapeutic monoclonal antibodies to women during conception and pregnancy. Am J Gastroenterol. 2009;104:228–233. doi: 10.1038/ajg.2008.71. [DOI] [PubMed] [Google Scholar]
- 97.Chaparro M, Gisbert JP. Transplacental transfer of immunosuppressants and biologics used for the treatment of inflammatory bowel disease. Curr Pharm Biotechnol. 2011;12:765–773. doi: 10.2174/138920111795470903. [DOI] [PubMed] [Google Scholar]
- 98.Pentsuk N, van der Laan JW. An interspecies comparison of placental antibody transfer: new insights into developmental toxicity testing of monoclonal antibodies. Birth Defects Res Part B, Dev Reprod Toxicol. 2009;86:328–344. doi: 10.1002/bdrb.20201. [DOI] [PubMed] [Google Scholar]