

Abstract
Quality population modeling and simulation analyses and reports are something every modeler desires. However, little attention in the literature has been paid to what constitutes quality regarding population analyses. Very rarely do published manuscripts contain any statement about quality assurance of the modeling results contained therein. The purpose of this manuscript is to present guidelines for the quality assurance of population analyses, particularly with regards to the use of NONMEM from an industrial perspective. Quality guidelines are developed for the NONMEM installation itself, NONMEM data sets, control streams, output listings, output data files and resultant post-processing, reporting of results, and the review processes. These guidelines were developed to be thorough yet practical, though are not meant to be completely comprehensive. It is our desire to ensure that what is reported accurately reflects the collected data, the modeling process, and model outputs for a modeling project.
Key words: modeling, NONMEM, quality assurance
INTRODUCTION
Mistakes in quality regularly appear in the news, often with disastrous consequences. In 1999, the Mars Climate Orbiter crashed due to a mix-up in measurement units. Scientists at Lockheed Martin built the orbiter assuming English units while NASA's Jet Propulsion Laboratory used metric units in their calculation of the orbiting process; this conflict in units resulted in an improper entry into the Mars atmosphere with the result being the loss of a $125 million orbiter. In relation to drug development, in 2010 Johnson and Johnson was forced to recall many different consumer products resulting in over $100 million in manufacturing plant remediation and loss of brand respectability by consumers and physicians alike when the Food and Drug administration reported various “inspectional deficiencies of varying degrees of seriousness at all of […their manufacturing] facilities”. Fortunately, no deaths occurred. The same cannot be said when in 2007, five people died from eating accidentally contaminated spinach processed during a single shift in one food processing plant.
While there is no evidence that improper population pharmacokinetic–pharmacodynamic analyses have resulted in death or severe injury, these anecdotes were used to highlight the importance of quality and results that could occur when quality is ignored because as systems and processes become more and more complex, the potential for errors, mistakes, and oversights increases. Increased complexity in the presence of low quality leads to increased probability for failure. Because of this potential, organizations have been working to improve quality in recent decades. But defining quality is difficult. Quality standards may differ from individual to individual and from company to company. Quality control (QC) is thus an elusive concept. QC is sometimes used interchangeably with quality assurance (QA), but this is a mistake. QA is process oriented while QC is product oriented. QC does not ensure QA. For purposes of this manuscript, the focus will be on QC but even with this distinction, what consitutes quality will still be difficult to define.
Quality in relation to drug development has likewise played an increasing role in the last few decades to ensure data accuracy, data and study consistency, and fraud prevention. Quality standards are in place throughout the entire drug development process from formulation consistency in chemistry, manufacturing, and controls to clinical studies through good clinical practices (GCPs). In the data analytic fields, like statistics, formalized QC guidances or guidelines have not been issued by regulatory authorities although all companies put in place their own internal guidelines for QC through the issuance of standard operating procedures (SOPs).
Population analyses of pharmacokinetic and often pharmacodynamic data (PopPK-PD) in support of new drug applications and regulatory submissions are commonplace. The most commonly used program for such analyses is NONMEM, a Fortran-compiled program that reads ASCII files and performs modeling and simulation via a “control stream”, which is a command line-like ASCII file of NONMEM-specific commands. On rare occasions, a small snippet of Fortran code is linked to the control stream which allows NONMEM to calculate some function it has not otherwise defined or some user-defined algorithm. After executing a control stream file, NONMEM generates a list file which summarizes the run and can optionally produce any number of ASCII tab-delimited output files that can be subsequently analyzed by a post-processing program like R, SAS, MATLAB, S-Plus, or Sigmaplot (Fig. 1). NONMEM is either executed from the command line/terminal emulator through DOS/Unix or via a so-called “front end” (like PDx-POP or Pirana (1)) that uses a graphical user interface and runs NONMEM in the background. Another commonly used program that runs from the command line is Perl Speaks NONMEM (PsN), which was designed to automate certain tasks (like bootstrapping and influence analysis) and perform computer-intensive statistical methods operations that early NONMEM versions do not perform (like calculation of conditional weighted residuals).
Fig. 1.
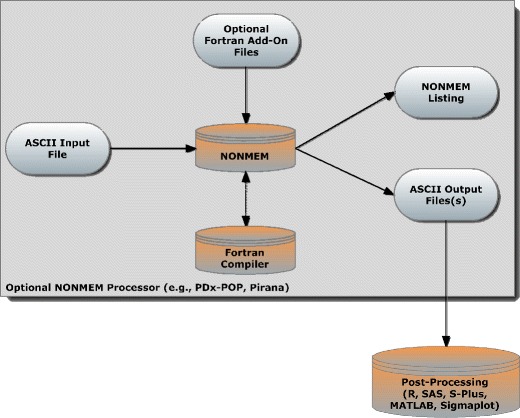
Data flow chart for a NONMEM analysis
QC of data analytic processes encompasses two types of activities that are complementary to each other: verification and validation. Verification tests whether the model is programmed correctly and contains no errors, oversights, or bugs. Verification also ensures that the input data, the NONMEM control stream, and the output data are all grouped together as a unit. Validation relates to whether the model adequately reflects the observed data. The concept of model validation has been discussed in many papers (2–5) and will not be a focus of the QC process herein since validation is a matter of scientific review and opinion. The QC process presented in this manuscript will focus on model verification to the exclusion of model validation, although it must be understood that a credible model requires both validation and verification.
Different types of verification can be performed as part of the QC process and should be done at different times during the execution of the NONMEM analysis. This process is illustrated in Fig. 2. Briefly, the first QC check should occur on the original source data, which should be checked not only for errors but also in terms of completeness of information required for the analysis. Upon fulfillment of the data requirements, the original source data can be used for the first transformation task (transformation task 1) which comprises the data management in order to create NONMEM input data files. A QC check here should also occur to determine whether the input data set is of the appropriate format and contains no errors. After development of the NONMEM model and/or performance of the simulations (transformation task 2), a QC check should occur on the NONMEM control streams, outputs, and listings. At this stage, a content review will take place to ensure that all activities have been performed according to the data analysis plan and that the end user, usually the project team, accepts the performed analysis. Upon acceptance, transformation task 3 occurs in order to generate a report which should be reviewed for quality and content. The QC check should include all NONMEM post-processing used to generate plots, tables, and listings added to the report. Finally, if data transfer to a regulatory body is expected to be necessary a last transformation function (transformation task 4) and subsequent QC check should occur in order to make sure that the data and reports fulfill regulatory requirements (6).
Fig. 2.
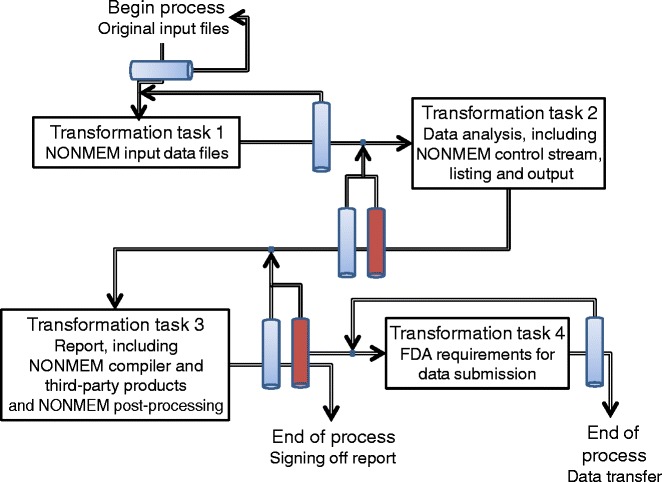
Schematic description of the transformation tasks related to the NONMEM analysis, including all steps that should be reviewed for quality (blue bars) and content (red bars). Every review step has its own checklist as described in the text
If an error is found during any of the QC checks, the related transformation functions should be (partially or fully) repeated after the error is corrected, and a new QC check should be performed. This process should be repeated until all QC checks are passed. Ideally, all QC checks should be performed by a different pharmacometrician than the one performing the analysis. The first three tasks and the related quality checklists will now be discussed in turn. Transformation task 4 will not be discussed herein since this issue is discussed in regulatory guidances. Suffice it to say that it would be useful for sponsors to run the to-be-submitted dataset against the to-be-submitted NONMEM control stream to ensure that the same results are generated as previously reported.
TRANSFORMATION TASK 1
Data Analysis Plan
The data analysis plan (DAP) is a prospectively defined document that is a comprehensive and detailed description of the methods of pharmacokinetic–pharmacodynamic analysis (7). The DAP should include a description of the data that will be used in an analysis, how the data will be handled (e.g., handling of missing data, handling of censored data, definition of outliers, etc.), the modeling methodology that will be used, and the reporting structure with mock tables, listings, and figures whenever possible. The DAP is similar in nature to a statistical analysis plan defined in the ICH E9 guidance that defines the objectives and methods of the statistical analysis of a clinical trial (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, (8)). The same principles apply for the DAP—the objectives and methods of the modeling and simulation project should be clearly stated. The covariates that will be examined in the analysis should be explicitly detailed in the DAP, as well as the rationale for each covariate in the analysis. Model discrimination criteria should also be explicitly stated. For example, a forwards—backwards approach may be taken for covariate selection in which case the P value for covariate inclusion in the model and covariate exclusion in the model should be explicitly defined, usually 0.05 for inclusion and 0.01 for exclusion.
DAPs can be either add-on documents to protocols or standalone documents. Add-on DAPs are usually not as detailed as standalone documents as some parts of the DAP can be omitted, like a description of the study design. Standalone DAPs are usually multi-study analysis plans, but can be single study analysis plans as well. It is recognized that DAPs cannot fully envision every scenario, but they can be of sufficient detail to provide guidance and provide a reasonable level of assurance when followed appropriately. DAPs should be peer reviewed prior to any data analysis and should be reviewed with an eye towards the QC that will occur with the actual modeling.
NONMEM Input Data Files
Confirming that a NONMEM dataset is accurate is intimidating and some might say it's an impossible task for some multi-study phase 2/3 datasets with hundreds of subjects and thousands of observations. However, inaccurate data may lead to incorrect modeling results (as described by the cliche: “garbage in = garbage out”); thus, QC of the data that will be modeled may be the single most critical step in the modeling process. Ideally one would confirm the dataset quality prior to any modeling but this may significantly delay the modeling process. Invariably there will be a period of analysis/modeling and correction of the NONMEM dataset as mistakes are identified and rectified during the initial parts of the modeling process. For this reason, data QC is usually done during or after completion of the analysis, which is far from ideal as errors identified in the dataset may require reworking of part or all of the model development process. Hence, it may be useful to break the QC process into pieces: data specific processes could be tested during data merging, e.g., checking for units, consistency of dates and times, etc., then NONMEM-specific items can be tested, e.g., is the appropriate CMT value used for a compartment.
Errors in the dataset can range from formatting, e.g., decimal numbers reported as integers, to dire errors where the wrong data are captured, e.g., the age of a patient from one study is merged with the identity of another patient from another study having the same patient identifier. NONMEM does some data checking as it reads the data file, e.g., it ensures that all dates and times are sorted within an ID, that a non-missing AMT is associated with a nonzero EVID, etc., but there are still many checks that it fails to do.
Some specific items that can be tested as part of the review process of the NONMEM input dataset are:
- Error checking
- Does the dosing event date occur before the date of the first positive concentration value in each subject?
- Are there any alphabetical or non-numeric values anywhere for any cell (with the exception of any comment columns)?
- Are there any empty cells in the dataset?
- Are there any impossible values in the dataset (such as a negative value for age or weight)?
- Are time-invariant values (ID, gender, certain covariates) consistent within a subject and between subjects?
- Format and units
- Do the variables have the appropriate number of significant digits?
- Are units consistent for each column? If data are merged from multiple studies or multiple sites, are the same formats and units used for concentrations, doses, and covariates in the source datasets? If not, have the data been properly converted to the default units specified in data analysis plan?
- Do the dose (AMT) units correspond to units of concentrations? For example, if concentrations are in nanograms per milliliter, are doses in nanograms? If not, does the scale factor for the observation compartment in the control stream handle the scaling appropriately?
- Is the AMT column the molecular weight-corrected dose if the drug is a salt?
- Was the same assay method for concentration or other laboratory values used across multiple merged studies? If not, are known assay differences accounted for by adjusting the values?
- Dependent variable (DV)
- Are all the dependent variable (DV) values of one type, e.g., drug concentration? If not, is an indicator variable(s) present in the dataset that specifically associates with a DV data type and is that variable used in the NONMEM control file to control the Y function? In cases where DV is of different types, e.g., pharmacokinetic and pharmacodynamic data, an easy check for indicator discreteness is by plotting DV versus the indicator. An overlap would then show that the indicator has not been consistently assigned.
- Are there any DV outliers? If so, are the data real or a data error? What constitutes an outlier should be explicitly defined in the data analysis plan. Trellis plots of DV conditioned on categorical covariates or binned continuous variables can be very helpful to assist in outlier identification.
- Covariates
- What is the maximum and minimum value for each covariate? Are the values in the dataset physically possible?
- Is the covariate time-varying or does it use the baseline or other value throughout the study? Is this rule used consistently across patients?
- Is there a drastic but intermittent shift in the time-varying covariates that may identify an erroneous value or outlier measurement?
- Do any time-invariant covariates change over time? This could be checked by trellis plotting each covariate over time by ID and checking to see if there are any “jumps” in the series plot from one time to another.
- Do histograms of the covariates show any multimodalities? If so, are these real or unit-related errors, e.g., height is in centimeters for some subjects and in meters for others?
- Do categorical variables have the appropriate number of levels? For example, a variable with four levels should have a minimum of four distinct values in the data set (either through dummy coding or direct coding)
- Dates
- Are dates within a subject consistent with the study design? For example, in a single dose phase 1 study, if the dose is given on 1/1/2010 it does not make sense to have an observation 24 h later to be 1/2/2011. Check dates and times for consistency.
- Are all dates and times in consistent format, e.g., 12/10/2010 14:32? The US and EU may store dates differently. For example, the US may store April 3rd 2012 as 4-3-2012 while the EU may store it as 3-4-2012. Users of Microsoft Excel® need to be especially aware of this issue when merging datasets.
- Are elapsed times properly calculated based on the first dose or measurement, and in the appropriate units (hours, days, etc.)?
- Missing and derived data
- Are any zero value covariates present? Are these values real?
- Are missing data handled as planned? (e.g., set to 0, imputed, etc.)
- Are data records to be omitted (outliers, etc.) appropriately commented out and are they described in the report with the reason for omission?
- Are concentrations below the limit of quantification appropriately handled (e.g., set to 0, set to half of LLOQ, imputed, etc.) as planned?
- Are derived variables properly calculated using the appropriate relationships as planned?
- NONMEM specific
- Are dosing records appropriately flagged in the EVID column (e.g., EVID = 1)?
- Are non-dosing events appropriately flagged in the EVID column (e.g., EVID = 2)?
- Are missing DV values appropriately handled? In the simplest case, are missing values coded as MDV = 1 in the MDV column?
- Do the dosing records reflect the study design? If ADDL and II are used, are they correctly indicated and in the correct units? (i.e., II should be in the same units as the TIME column)
- Does the CMT variable in each record correspond to the correct compartment (e.g., for first-order absorption, CMT = 1 for dosing, 2 for concentrations in the central compartment)
It is important to distinguish between the properties a variable actually has and the properties it has in the NONMEM file (9), e.g., decimals are reported in fewer significant digits in the NONMEM dataset. The value of graphical analysis of data columns cannot be overstated. A histogram of a covariate data column can be used to detect outliers and groups with possibly inconsistent units.
For many NONMEM datasets a complete 100% QC of the dataset is an impossible task given available time and resources. A risk assessment should be undertaken to determine whether a complete data QC should be undertaken on some fraction of the data. For example, it may be decided that a complete data QC for a minimum number of randomly selected subjects per study in the dataset of the final model is sufficient.
TRANSFORMATION TASK 2
A framework for the QC of NONMEM analyses will now be presented. Because during the model development process, hundreds, if not thousands, of models can be tested and explored, it will be impossible to QC test each model due to time constraints. Therefore, QC will focus only on those models specifically discussed in the body of a report. Typically these include only two models: the base model without covariates and the final model with covariates. Additional models may sometimes be presented in the body of the report, but these can often be checked with an abbreviated QC checklist.
NONMEM Compiler and Third-Party Products
The type of NONMEM compiler that is used can impact the results of the analysis (10, 11). Small differences in standard errors (related to the matrix inversion process) and errors/warnings reported by NONMEM may result if a GFORTRAN compiler is used as compared to an Intel Fortran compiler, for example.
What type of NONMEM compiler was used?
- In the NONMEM directory tree is the file CONTROL5. Run the CONTROL5 file and examine the output in the listing.
- Did CONTROL5 run without errors?
- Was the minimum value of the Objective Function Value 104.561?
- Were the values of TH(1), TH(2), and TH(3) 2.77, 0.0781, and 0.0363, respectively?
- Were the values of OM(11), OM(22), and OM(33) 5.55, 2.4E−4, and 0.515, respectively?
- Was the value of SIGMA(1) 0.388?
Are any third-party preprocessor software products used in the analysis, e.g., PDx-POP, PSN, etc. and are they installed properly?
NONMEM Control Stream
The NONMEM control stream reads the NONMEM ASCII dataset and then either estimates a model's parameters or simulates a dependent variable conditional on a model. NONMEM generates a listing file based on the control stream and input dataset. From the control file:
Are the number of data columns in the NONMEM $DATA statement equal to the number of data items in the NONMEM input dataset? Is the order of the variables in the $INPUT statement the same as the order of the variables in the dataset?
Does the choice of ADVAN and TRANS used in the control stream reflect the desired model and parameterization? ADVAN1 for 1-compartment model with IV administration, ADVAN2 for 1-compartment model with oral absorption, etc.?
Does the scale value correspond to the sampling compartment volume, e.g., S2 = V2?
Alternatively, does the scale value require a transformation for consistency between the AMT variable and DV variable? For example, if dose is in milligrams, V is in liters, and concentration is in nanograms per milliliter, then S2 must be written as S2 = V2/1,000 for ADVAN2.
Is the INTERACTION option in the $EST statement used in the presence of ETA–epsilon interaction in the error model?
If differential equations are used, does the CMT variable correspond to the correct compartment (e.g., 1 for dosing, 2 for observation for an oral dosing model)? Is TOL in the $SUB statement of greater value than NSIG in the $EST statement?
If the error mode is a transform-both-side (TBS) approach, do both the dependent variable and error model have the same transformation? For example, if a log TBS approach were used, is the dependent variable log-transformed and is the error model of the form Y = f(LOG(F), EPS(n)), e.g., Y = LOG(F) + EPS(1).
If multiple $EST statements are used, is the appropriate estimation method and corresponding options presented in the report?
- If simulation is being performed:
- Is an MSF file being used to input parameters? Does the control stream use the TRUE = FINAL option? Or are the parameters fixed from the final model and are these correct?
- Is the number of simulation subproblems defined?
- Is the simulation seed defined?
- Is the appropriate probability density function being used, e.g., normal or uniform?
Are all parameters that are required for post-processing listed in the $TABLE statement?
Like the NONMEM Input QC process, review of the NONMEM control streams can be saved for specific points during the analysis. For example after identification of the base model, all models used to develop the base model can be reviewed for correctness. Similarly, after covariate identification, all covariate models could be identified, and the same after identification of the final model.
NONMEM Listing
After executing a control stream, NONMEM generates a listing file summarizing its results. If $EST is used, the file will contain model parameter estimates and if $SIM is used, NONMEM will present a summary of the simulation. Results of a model specifically identified in a report should be compared to the listing for accuracy. Specific questions to be addressed include:
Were the total number of individuals and total number of observations reported in the listing file consistent with the values in the dataset?
Were any warning or errors present in the listing?
Do the number of observations reported in the listing correspond to the number of non-commented out observations?
Did the $COV step implement without error? If not, is the model still acceptable?
How many significant digits were used in the final parameter estimates?
Are all the values for THETA, OMEGA, and SIGMA plausible?
Were the standard errors estimable?
Is the degree of shrinkage acceptable?
Are all ETABAR values non-significant?
Was the condition number of the final model (ratio of largest to smallest eigenvalue) < 1,000 (which is one measure that indicates a high degree of collinearity among covariates)?
Do start and stop time in listing file correspond to date and time of output files?
NONMEM Output Files
NONMEM can output and generate a number of different ASCII datasets based on the NONMEM input file and control stream. For each of the NONMEM output datasets generated from a particular control stream:
If the FIRSTONLY option is not used, are the number of data rows of the output dataset the same as the number of rows of the input dataset minus any rows that were removed using the IGNORE or ACCEPT fields in the $INPUT statement?
Is the date and time stamp of the output file consistent with the date and time stamp of the control stream?
Does the NONMEM output file have the same numerical precision as the NONMEM input file?
NONMEM Post-Processing
NONMEM output is often read into a post-processing program (PPP), such as R, Matlab, SAS, SigmaPlot, or S-Plus, for graphical and statistical analysis.
- Have the data been read in to the PPP correctly?
- Does the number of read rows in the PPP correspond to the number of rows in the NONMEM output dataset?
- Does the number of variables in the PPP correspond to the number of columns in the output dataset?
- Does the PPP data have the same numerical precision as the NONMEM output file?
- Do the variables correspond, e.g., CWRES in the NONMEM output dataset is read into the CWRES variable in the PPP program (note that the names do not have to match, simply the data does)?
- If possible, verify that the plots presented in a report use the appropriate NONMEM output data set associated with a specific control stream.
- If the PPP is script based, does the script have appropriate annotation, including the run date and control stream name, and show the path for the NONMEM output file that was input to create the plots and tables?
- If the PPP is GUI-based, is there an audit trail that can be examined to ensure that the appropriate NONMEM output file was input to create the plots and tables?
NONMEM sets DV, PRED, and all computed residuals equal to 0 when MDV = 1. Have these values been reset to missing prior to post-processing?
Are the residuals appropriately plotted, e.g., is CWRES reported as CWRES or as WRES?
TRANSFORMATION TASK 3
Structure of the PopPK-PD Report
Both the Food and Drug Administration (7) and the European Medicines Agency (12) have guidelines for the overall report format of population analyses. The reader is referred to the original documents for details. Briefly, the final report subsections should include a summary, introduction, objectives, data, methods, results, and discussion section. There are no recommendations for the type of plots presented in the report using the FDA's guidelines, but there are recommendations in the EMA document. The report should comprehensively detail the analysis from data collection, database creation, model development, covariate selection, and model validation/evaluation. These results should then be placed in context in the discussion section of the report.
In a recent presentation, Edholm (13) presented on what constitutes regulatory expectations regarding reporting population analysis results. Edholm points out that:
The report should be detailed sufficiently to enable a secondary evaluation by a regulator.
Every assumption and decision made during modeling should be documented, discussed, and justified.
The report should be of sufficient quality such that “the final model can be judged to be a good description of the data in that the results and conclusions… can be considered valid.”
Sponsors are expected to critically evaluate the quality of their model with regards to how well the model describes the observed data, what the limitations of the model are, what is the clinical relevance of the model covariates in their biological plausibility, how the results compared to previous analyses, and how will the results be used, e.g., to support labeling. Edholm also presented reasons for failing expectations which related to the quality of the report (primarily insufficient report detail, missing information, presentation of detailed but irrelevant information, analysis quality, and underlying data used to build the model) and to criticisms related to conclusions. A number of different case studies of reports of low quality were presented. In one case study, the assessor criticized the report for having no information on the number of samples excluded from the analysis for their concentrations being below the limit of quantification of the assay. Also criticized were the use of plots that relied on the Empirical Bayes Estimates of pharmacokinetic parameters when the degree of shrinkage was large (29% to 65%), that high concentrations were not captured in the visual predictive checks suggesting model misspecification, and that weight was used as a covariate in the model even though its inclusion did not result in a decrease of the objective function value. In another report from a different company, the model was criticized for not adequately capturing high concentrations and the report was criticized for not providing shrinkage estimates, for not showing the 5th, 50th, and 95th percentiles of the visual predictive check and for not stratifying by important covariates. Criticisms of the other analyses had to do with shrinkage, visual predictive checks, and insufficient information and detail.
Peer Review for Quality
Peer review of PopPK-PD reports is designed to guarantee that the displayed information is in agreement with the model listing and to ensure that the quality of the NONMEM analysis itself is satisfactory in terms of contents. Specific questions to be answered in relation to a specific model report are:
If possible, rerun reported control streams in NONMEM and compare results to verify accuracy of results in report and NONMEM listing file.
Is the version of NONMEM used reported? Is the version of Fortran Compiler reported?
If any third-party preprocessor software products used in the analysis, e.g., PDx-POP, PSN, etc. are the use of these products with version number documented in the report?
Are any outliers identified outside the Data Analysis Plan specifically addressed in the body of the report?
Does the choice of ADVAN used in NONMEM correspond to the model stated in the report, e.g., ADVAN for 1-compartment model or ADVAN2 for 1-compartment model with oral absorption?
Does the functional form of the covariate model correspond to the functional form stated in the report, i.e., is the covariate submodel stated in the control stream the model stated in the report?
Does the estimation method in the control stream correspond to the estimation method stated in the report?
Was minimization successful? Were any warnings or errors reported?
Are the shrinkage estimates reported? Were there any larger than predefined critical values (e.g., 30% on parameters with random effects) and is this noted in the report?
Are the ETABAR statistics reported? Were any p values less than 0.05 noted?
Are all estimable model parameters and their standard errors reported?
Are the variance components consistently reported as percent coefficient of variation or log-scale variance?
Is the condition number (ratio of largest to smallest eigenvalue) of the final model reported?
Do all model control stream and outputs in the report appendix correspond to the model run mentioned in the body of the report?
Do the diagnostic plots correspond to the model?
In the case of simulation, was the seed and number of simulation replicates defined in the body of the report?
Do all tables providing a overview of the model runs contain correct information when checked against the NONMEM model listings?
Peer Review for Content
Peer review of population-related reports is a necessity as it helps the writer focus on strategy as well as the technical correctness of the document. Professional QC individuals and medical writers, while useful for inspecting the document for style and accuracy, are simply unqualified to evaluate the technical aspects of a population report. Peers with modeling experience, particularly with NONMEM-related experience and who are independent of the project, are required for part of the QC process. Peer review should focus on technical aspects of the document keeping in mind who the ultimate end users are—regulatory authorities. Management should recognize that document review is an important and legitimate process, not something that needs to be tacked on top of one's other duties. The goal of a well-written and well-designed report is to put information in prominent locations, clearly address the issues, and to help reviewers quickly find the information they need.
NONMEM-based modeling reports are large and complex documents, sometimes as large as thousands of pages in length. The document, which is usually written by a single individual, may contain many tables, many figures, and can be verbose particularly with regards to the model development process. Document review in pharmaceutical companies typically takes the form of team review followed by managerial review. Because of the technical complexity of the modeling process, most team members and many managers are unqualified to evaluate the technical aspects related to the report. Although style and accuracy issues can be addressed, issues related to model development, evaluation, and analysis may be beyond their skills. Unfortunately, even technically qualified reviewers may not know how to properly review a document, nor have they ever been trained in that regard.
Document review at the project team and managerial level tends to be informal. Individual document reviewers and teams tend to start at the beginning of the document and work their way towards the end. It is not uncommon to see far more suggestions, recommendations, and wordsmithing at the beginning of a document compared to the end of the document because by the time a reader gets to the end, they are often tired and want to finish. The same thing happens during roundtables where project teams focus so extensively at the beginning of the document that at the end of the meeting they realize that they never really reached the meat of the document and the meeting needs to be rescheduled.
McCulley/Cuppan LLC (14), a medical writing and communication consulting company, has presented some guidelines related to strategic review of documents that can be applied to modeling reports. Basic questions that McCulley/Cuppan say should be addressed during early review of a report include:
“Is there sufficient content to support and resolve issues?”
“Is the required regulatory content in place?”
“Is the document well argued and logical?”
“Is there sufficient context to clarify content?”
“Is the key information presented in prominent locations?”
“Is the document well designed and all visuals clear?”
Basic questions that McCulley/Cuppan say should be addressed during late stage review of a report include:
“Are all data accurate, complete, and consistent?”
“Are gaps and contradictions resolved?”
“Are all visuals well labeled, legible, and interpretable?”
“Is the document consistently formatted?”
“Is the language clear and correct?”
McCulley/Cuppan (14) and Bernhardt (15) also provide additional guidelines for effective reviews. Peers should review reports from a technical point of view with the reader in mind and leave wordsmithing for medical writing professionals (though this may be necessary in some cases with difficult to read reports). Other guidelines include, but are not limited to:
Once a section of a report has been reviewed (and subsequent corrections or improvements are made and approved), in future reviews make that section off-limits so that a “clean” section does not require further revision.
Instead of starting a review at the beginning of a document, start at the methods section, or start at the results section to avoid comment condensation near the beginning of the document.
Reports are often done after completion of the analysis. Trying to write up the results sections days, weeks or sometimes months after completion is difficult. Authors should consider writing modeling reports in real time, even before the analysis is complete; objectives, study descriptions, demographics, basic methodology, etc., can all be prepared and placeholders for predefined tables and figures can be created. Upon the final analysis, the author simply needs to provide any additions to the pre-written sections, and insert the results, interpretation, and conclusions.
Similarly, authors could consider having reviews done as report sections are completed, called staged review, rather than overwhelming reviewers with a single document at the end. Alternatively if modelers are unable to write reports in real time, a suggestion is to have a primary meeting with the team to simply discuss the final results and agree upon the message, conclusions, and implications.
It is not uncommon to see a request from an author to reviewers that simply state something like “Please review this document by the end of the week”. Rather than present reviewers with no guidelines for review except a due date, it may be possible to provide specific instructions for reviewers. For example, reviewers may be told to not focus on the mathematics or methodology but to focus on the document flow or interpretation. They may also be told that specific sections are still in draft form, i.e., not read for team review, or that specific sections do not require review.
Reviewers should be trained in how to review and annotate documents and review with the user in mind. Comments, however, should be made with the author in mind. Simply putting comments like “please revise”, “???”, and “unclear” are in themselves unclear. Reviewers should add sufficient and specific detail to their comments such that the author will not have to come back to ask for clarification.
Consider attaching a checklist to a review that asks for holistic comments. Comments made in the margins of a document are specific and local to a section. Too many reviewers focus on grammar and spelling. Reviewers should learn to make global comments and critiques that improve the overall quality of the document. An example of a global comment might be something like “The model showed that age affected clearance. There are not any graphs that really support this relationship.”
Instead of sending a copy of the document to everyone in a distribution list via e-mail, consider putting a single copy of the document out on a network drive or, ideally, in a version controlled system in which users must check out the document to edit it, then check it back in. In this manner, comments are entered in a cumulative manner with less likelihood for repetition, and version control is maintained. If this is not possible, to control having reviewers working on different versions of the document, each reviewer should send their version of the document with tracked changes to a single recipient (generally the author), and not to the entire team of reviewers. The author would then compile all comments, address what can be modified or clarified at that time, and then send out an updated version to all recipients at prespecified times.
In summary, strategic review asks whether the document makes the right arguments in the right place, whether its arguments are logically sound and well supported by the data, and whether a reviewer can quickly find such information. The goal is to develop a usable document that facilitates understanding.
Identified Errors
Errors are likely to be encountered in every NONMEM-related project simply because of the complexity related to data merging, model development, reporting, and analysis. Errors can be of 3 types: errors related to models and model development, errors related to model analysis, and errors related to reporting of model results. Errors related to model development can range from the trivial like forgetting to include a $COV step to major, such as failure to identify a covariate associated with a pharmacokinetic parameter. Errors related to model development are difficult to spot particularly when modeling itself is such a subjective process; one modeler may choose one model over another but does this mean it was an error or a choice? Do differences in model parameters translate into a model error? Errors related to model analysis could be something like plotting PRED versus DV but labeling the plot as IPRED versus DV. Errors related to reporting model results can be inaccurate reporting of a model parameter value.
Errors identified during the QC process do not necessarily invalidate the results of a population analysis. Errors should be evaluated by the modeler as to the potential impact for affecting the final model and the model development process. Errors related to model development that are identified as minor (e.g., a single concentration value that is reported incorrectly) and unlikely to affect model results can be included in the body of the report as an addendum. Errors identified as major, e.g., a significant covariate having the wrong units for one study in a multi-study dataset, may necessitate rerunning some or all models in a modeling project. The modeler should identify the point in model development after which models should be rerun after error correction. Errors related to model analysis and reporting may require post-processing tables and figures be redone and the report updated accordingly. Again, the modeler's judgment should be exercised as to the degree to which analyses need to be repeated.
Regulatory Review of Population-Based Reports
The Food and Drug Administration (FDA) uses what is called a Question-Based Review (QBR) in their review of New Drug Applications (16). FDA reviewers in the Office of Clinical Pharmacology are presented with a list of questions that they must answer as part of the review process. Questions include “What are the major intrinsic factors responsible for the inter-subject variability in exposure (AUC, Cmax, Cmin) in patients with the target disease and how much of the variability is explained by the identified covariates?” or “Based upon what is known about [exposure–response] relationships in the target population and their variability, what dosage regimen adjustments are recommended for each group?” While there is no specific QBR template for review of population reports, guidances are available from regulatory authorities and within those guidances are recommendations for report format and structure. Authors should write their reports with an eye towards aiding the reviewer to answer the questions they must answer as part of their review and should include report elements as recommended in the regulatory guidances.
CONCLUSIONS
For the results of an analysis to be credible in the eyes of a reviewer, certain criteria need to be met. One of these criteria is report quality. Low report quality is associated with low model credibility. Formatting errors, typographical errors, poor grammar, reporting of wrong model file results, incorrect parameterization, or use of wrong data and graphics can consequently decrease the credibility of the model (and the modeler) in the eyes of the reviewer. A rigorous QC process will increase the readability of a report and allow the reviewer to focus on the details, issues, messages, and conclusions. Companies need to place greater emphasis on the review process as part of the clinical study and drug development process. Efficient QC and document review by companies will allow regulatory reviewers to more efficiently review regulatory submissions. While this paper has focused on the what and when to QC a population analysis, companies will have to decide for themselves how to implement these recommendations.
References
- 1.Keizer RJ, van Benten M, Beijnen JH, Schellens JH, Huitema AD. Piraña and PCluster: a modeling environment and cluster infrastructure for NONMEM. Computer Methods and Programs in Biomedicine. 2011;101:72–9. doi: 10.1016/j.cmpb.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 2.Beal SL. Validation of a population model. NONMEM Topic 006: NONMEM Users Group e-mail discussion. 1994.
- 3.Bruno R, Vivier N, Vergniol JC, De Phillips SL, Montay G, Shiener LB. A population pharmacokinetic model for docetaxel (Taxotere): model building and validation. J Pharmacokinet Biopharm. 1996;24:153–72. doi: 10.1007/BF02353487. [DOI] [PubMed] [Google Scholar]
- 4.Chen C. Validation of a population pharmacokinetic model for adjunctive lamotrigine therapy in children. Br J Clin Pharmacol. 2000;50:135–45. doi: 10.1046/j.1365-2125.2000.00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cobelli C, Carson ER, Finkelstein L, Leaning MS. Validation of simple and complex models in physiology and medicine. Am J Physiol. 1984;246:R259–66. doi: 10.1152/ajpregu.1984.246.2.R259. [DOI] [PubMed] [Google Scholar]
- 6.U.S. Department of Health and Human Services, Division of Drug Information, Center for Drug Evaluation and Research, and Food and Drug Administration. Guidance for Industry—Providing Regulatory Submissions in Electronic Format—Human Pharmaceutical Product Applications and Related Submissions Using the eCTD Specifications. http://www.fda.gov/RegulatoryInformation/Guidances/ucm126959.htm; 2005. Accessed 1 July 2012.
- 7.United States Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, and Center for Biologics Evaluation and Research. Guidance for industry: population pharmacokinetics. 1999.
- 8.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Statistical principles for clinical trials (E9). 1998. [DOI] [PMC free article] [PubMed]
- 9.Gorrell P. A SAS programmers guide to project and program-level quality control. Northeast SAS Users Group (NESUG) 18; 2005.
- 10.Bonate PL. Consistency of NONMEM parameter estimates across platforms and compilers. Presented at the Annual Meeting of the American Association of Pharmaceutical Scientists, Toronto, Canada; 2002.
- 11.Frame B, Koup J, Miller R, Lalonde R. Population pharmacokinetics of clinafloxacin in healthy volunteers and patients with infections: experience with heterogeneous pharmacokinetic data. Clin Pharmacokinet. 2001;40:307–15. doi: 10.2165/00003088-200140040-00006. [DOI] [PubMed] [Google Scholar]
- 12.European Medicines Agency (EMEA) and Committee for Medicinal Products for Human Use (CHMP). Guideline on the reporting the results of population pharmacokinetic analyses. http://www.emea.europa.eu/pdfs/human/ewp/18599006enfin.pdf; 2007.
- 13.Edholm M. Modeling and simulation examples that failed to meet regulator's expectations. Presented at European Medicines Agency-European Federation of Pharmaceutical Industries and Associations Modelling and Simulation Workshop, November 2011; 2011.
- 14.McCulley/Cuppan. Improving the review process and review outcomes; 2011.
- 15.Bernhardt S. Improving document review processes in pharmaceutical companies. J Bus Tech Commun. 2003;17:439–73. doi: 10.1177/1050651903255345. [DOI] [Google Scholar]
- 16.Lesko L, Williams R. The question based review. Applied Clinical Trials. 1999;8:56–62. [Google Scholar]