

Abstract
Highly variable (HV) drugs are defined as those for which within-subject variability (%CV) in bioequivalence (BE) measures is 30% or greater. Because of this high variability, studies designed to show whether generic HV drugs are bioequivalent to their corresponding HV reference drugs may need to enroll large numbers of subjects even when the products have no significant mean differences. To avoid unnecessary human testing, the US Food and Drug Administration’s Office of Generic Drugs developed a reference-scaled average bioequivalence (RSABE) approach, whereby the BE acceptance limits are scaled to the variability of the reference product. For an acceptable RSABE study, an HV generic drug product must meet the scaled BE limit and a point estimate constraint. The approach has been implemented successfully. To date, the RSABE approach has supported four full approvals and one tentative approval of HV generic drug products.
KEY WORDS: bioequivalence, generic drugs, highly variable drugs, reference-scaled average bioequivalence, US Food and Drug Administration
INTRODUCTION
Acceptable bioequivalence (BE) between a generic drug product and its corresponding reference product is among the criteria required by the US Food and Drug Administration (FDA) for marketing approval of a new generic drug. A systemically active generic drug is considered to be bioequivalent to the reference-listed drug if the rate and extent of absorption of the two products do not show a significant difference (1). Drug rate and extent of absorption are typically assessed by conducting in vivo BE studies in human subjects in which generic and reference drug plasma pharmacokinetic (PK) profiles are characterized and compared. The PK data are used to obtain peak drug plasma concentration (Cmax) as the BE measure of rate of absorption and area under the drug plasma concentration versus time profile (AUC) as the BE measure of extent of absorption. Most BE studies enroll healthy normal subjects who receive single doses of the generic (test) or reference product via a two-way crossover design. For the most part, the US FDA asks generic drug applicants to statistically compare generic and reference AUC and Cmax values using the two one-sided tests (TOST) procedure (2). Under the TOST, the 90% confidence interval (CI) around the geometric mean ratio (GMR) of the test and reference values of Cmax and AUC is required to fit within BE limits, set from 80% to 125% (3). The width of the 90% CI depends upon the number of subjects in the study and the variability of the BE measure.
Highly variable (HV) drugs are defined as drugs in which the within-subject variability (defined as the %CV) in one or more of the BE measures is 30% or greater (4,5). A survey of generic drug products reviewed at the FDA from 2003 to 2005 suggested that about 20% of the generic drugs evaluated for marketing approval are HV due to their drug substance dispositional characteristics (6). Determining the BE of HV generic drugs is challenging because the high within-subject variability means that large numbers of subjects may be needed to show BE. Figure 1 shows the results of two hypothetical BE studies with GMRs near 1.0. Product A meets the BE limits because within-subject variability is relatively low. Product B fails to meet the BE limits because of high within-subject variability. Thus, although product B, with a GMR near 1.0, appears to be well-designed to perform the same as the reference product in vivo, it will be necessary to increase the number of study subjects—perhaps dramatically—in order for product B to meet the BE limits.
Fig. 1.
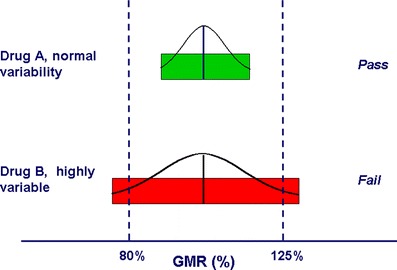
The 80–125% BE limits are represented along the x-axis as two “goal posts.” The BE limits are compared to the hypothetical 90% CIs of the test/reference BE measure GMRs for two drugs, a drug with normal variability (Drug A) and an HV drug (Drug B). The 90% CIs of the two drugs are represented by colored bars. For drug A (normal variability), the 90% CI (green bar) meets the BE limits. For drug B (HV), the 90% CI (red bar) fails to meet the acceptance limits. As the width of the CI is influenced by the number of study subjects, in the hypothetical case of drug B, it is likely that the study would have met the BE limits if more subjects had been used
Several factors influence the sample size needed to meet the regulatory criteria for acceptable BE. First, each one-sided test is carried out at the 5% level of significance, which corresponds to a 90% CI (7). The 5% level of significance represents the type I error rate (α), which is the probability of incorrectly deeming as bioequivalent two formulations whose true (population) GMR fails to meet the BE limits. The second factor influencing sample size is study power, defined as the likelihood or chance of correctly demonstrating BE when it, in fact, exists (8,9). A third factor influencing sample size is the test/reference BE measure ratios. If the true test/reference ratio differs from unity, the overall power to show BE is reduced at any given sample size, resulting in an increase in the number of study subjects needed. Other factors influencing sample size include the study design and the expected within-subject variability. For example, a replicate four-way crossover BE study design, in which each subject receives the test and reference products twice, requires fewer subjects than a two-way crossover BE study design. As within-subject variability increases, the number of subjects needed in a crossover design will also increase, assuming that all other factors remain constant. Thus, BE study sample size is calculated based on a type I error rate of 5% per test, the desired study power, and the best estimates of test/reference ratios and within-subject variability. Table I shows how these factors influence the number of subjects needed to provide an 80% chance of an acceptable BE study.
Table I.
The Number of Study Subjects Required to Show BE with 80% Power is a Function of Within-Subject Variability and GMR (Sample Size Estimations are for the Case σ WT = σ WR and σ D = 0)
| Within-subject %CV | GMR (%) | Sample size for a two-way crossover study | Sample size for a four-way fully replicated crossover |
|---|---|---|---|
| 15 | 100 | 10 | 6 |
| 105 | 12 | 8 | |
| 110 | 20 | 12 | |
| 30 | 100 | 32 | 18 |
| 105 | 38 | 20 | |
| 110 | 68 | 36 | |
| 45 | 100 | 66 | 34 |
| 105 | 80 | 42 | |
| 110 | 142 | 72 | |
| 60 | 100 | 108 | 56 |
| 105 | 132 | 66 | |
| 110 | 236 | 118 | |
| 75 | 100 | 156 | 80 |
| 105 | 190 | 96 | |
| 110 | 340 | 172 |
As shown in Table I, the number of study subjects needed to show BE increases dramatically for HV drugs. The FDA observed in a survey of generic drug product BE studies reviewed from 2003 to 2005 that studies of HV drugs generally used more subjects than studies of lower variability drugs (6).
SHOULD “ONE-SIZE-FITS-ALL” BE LIMITS APPLY TO HV DRUGS
One important observation is that clinical data strongly support a conclusion that HV drugs have wide therapeutic indices. Otherwise, there would have been significant safety issues and lack of efficacy during the pivotal safety and efficacy clinical trials required for initial FDA marketing approval (10). In other words, the reference product, when dosed on different occasions, was safe and efficacious, despite high PK variability. The majority of HV drugs appear to fall into Biopharmaceutics Classification System class II or IV (low aqueous solubility–high intestinal permeability and low aqueous solubility–low intestinal permeability, respectively) (11,12). Dispositional characteristics of HV drugs include extensive presystemic metabolism, low bioavailability, high acid lability, and high lipophilicity (6). Consequently, plasma concentrations of these HV drugs are often very low. In such situations, it may not be possible to accurately characterize PK profiles, with the result that the within-subject variability of BE measures can exceed 30% (13). As the FDA discourages unnecessary human testing, these observations raise questions about whether large numbers of subjects should be used in BE studies of drug products for which high variability does not appear to impact safety and efficacy. It should be stressed that this concern pertains to high PK variability due to drug substance dispositional characteristics and has nothing to do with the formulation performance assessment that is the key question in BE comparisons.
An additional concern is that the large sample sizes needed for BE studies of HV drugs may serve to deter the development of new generic products (14,15). For example, a generic product line may be abandoned because of a high failure rate obtained in in vivo BE studies or it may be necessary to repeat the in vivo studies several times until the outcome of an acceptable BE study is achieved (16). Not only does this situation lead to unnecessary human testing, but it can also serve to increase the prices of generic drugs, conflicting with the principle that generic drugs should be relatively low-cost because they are not subject to extensive and unnecessary clinical testing.
A final concern is that an HV reference product may not be shown to be bioequivalent to itself in a crossover study using a relatively modest number of subjects (e.g., 18–40) (13). This concern is supported by data from in vivo studies of brand-name (reference drug product) formulations of chlorpromazine and verapamil, both of which were characterized by high within-subject PK variability exceeding 30% (17,18). Such findings raise the question of whether the 80–125% BE limits are too stringent for HV drug products.
EVOLUTION OF A NEW BE APPROACH FOR HV GENERIC DRUGS AT THE FDA
The issues surrounding BE evaluation of HV drugs and proposals for modifying the BE approach for such products were discussed over many years within the pharmaceutical sciences community, in the literature, and at various national and international venues (17–21). In 2004, FDA considered seriously whether an alternative BE approach to the “one-size-fits-all” paradigm should be developed for generic HV drugs. In April of that year, FDA’s Office of Generic Drugs (OGD) brought the issue of optimizing BE study designs for HV drugs before its Pharmaceutical Science and Clinical Pharmacology Advisory Committee (hereafter Advisory Committee). Speakers from the FDA, academia, and industry speakers presented the various issues surrounding BE evaluation of HV drugs to the Advisory Committee (22). Among the proposals debated were whether to change the BE limits, for example, using a GMR constraint of 80–125% or expanding the 90% CI limits to 70–143% (23) versus whether to use reference product within-subject PK variability to scale the BE limits (16). Following deliberations, the Advisory Committee made several recommendations (22).
There is a need to understand where the variability originated. Prior knowledge of all biostudies may help set more appropriate specifications to make decisions.
There is an undue reliance on the use of 80–125% BE limits for all drug products. As such, a paradigm shift for HV drugs is in order.
The use of reference scaling should be explored. A limit on the GMR should be used along with reference scaling.
FDA’s OGD agreed to initiate a series of simulation studies to determine whether such an approach was feasible and, if so, how to optimize the approach. Subsequently, the OGD formed an interdisciplinary working group (hereafter Working Group) to investigate the feasibility of applying a reference-scaled average bioequivalence (RSABE) approach to generic HV drugs. The Working Group conducted simulations to investigate expanding BE limits as a function of reference product within-subject variability. Based on the simulation study results, the Working Group members presented a tentative proposal for an RSABE method to the Advisory Committee for Pharmaceutical Science and Clinical Pharmacology in April of 2006. Speakers from academia and from the FDA discussed the advantages and limitations of the following approaches: (a) implementing several different scaling methods (24–27); (b) recommending a minimum number of subjects for RSABE (25,26); and (c) including a constraint on the GMR of the BE study (25–28). The Advisory Committee’s recommendations at the meeting’s conclusion are summarized below (29).
The Advisory Committee was not asked to comment on the merits of RSABE as it had previously advocated investigating the approach in April 2004.
The Advisory Committee agreed that these studies should enroll a minimum number of subjects. The vote was split as to whether this number should be 24 or 36.
The majority of Advisory Committee members either voted against a GMR constraint or abstained from voting on this issue.
FDA’s OGD considered the Advisory Committee’s recommendations in finalizing the RSABE approach for HV drugs (30).
FDA’S RECOMMENDATIONS FOR RSABE STUDIES OF HV DRUGS
Throughout this section, the three basic approaches used for determining if two products are bioequivalent will be illustrated using a series of inequalities based on population parameters.
Average Bioequivalence
The usual manner of statistically analyzing BE study data is by the average bioequivalence (ABE) approach. The acceptance of BE is stated if the difference between the logarithmic means is between preset regulatory limits, as shown below:
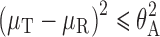 |
where μT is the population average response of the log-transformed measure for the test (T) formulation, μR is the population average response of the log-transformed measure for the reference (R) formulation, and θA is equal to ln(1.25).
As −ln(1.25) = ln(0.8), the BE limits are:
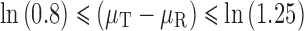 |
Individual Bioequivalence
For a time, the FDA worked toward implementing an individual bioequivalence (IBE) approach for studies submitted to New Drug Applications (NDAs) and Abbreviated New Drug Applications (ANDAs, for generic drugs). It was argued that requiring drug products to meet an IBE rather than an ABE criterion would improve formulation switchability (31,32). The proposed criterion for acceptable IBE included the (a) comparison of test and reference means; (b) comparison of within-subject variances; (c) assessment of subject-by-formulation interaction; and (d) ability to scale the BE limits if within-subject variability following the administration of the reference product exceeded a predetermined value (15). An additional justification offered for implementing IBE was that the ability to scale BE limits would address the concerns associated with BE studies of HV drugs (33–35).
Under IBE, the inequality used to determine if two products are bioequivalent is as follows:
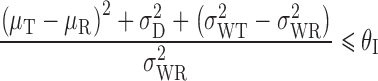 |
where 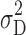 is the population subject-by-formulation interaction variance components,
is the population subject-by-formulation interaction variance components, 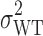 is the population within-subject variance of the test formulation,
is the population within-subject variance of the test formulation, 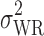 is the population within-subject variance of the reference formulation, and θI is the BE limit for IBE.
is the population within-subject variance of the reference formulation, and θI is the BE limit for IBE.
From 1999 to 2001, at the FDA’s request, the pharmaceutical industry applied the IBE study design and analysis to NDAs and ANDAs for modified-release drug products (36). The IBE was used to evaluate the BE of modified-release drug products because it was thought that, due to the relative complexity of modified-release formulations, the likelihood was greatest of detecting possible subject-by-formulation interactions with these types of drug products. However, analysis of these data failed to detect the presence of clinically significant subject-by-formulation interactions (37). The FDA concluded that expansion of the IBE approach to all BE studies submitted for marketing approval was not warranted and subsequently halted its implementation (38). Notably, during a discussion of IBE at its November 29, 2001 meeting, the Advisory Committee for Pharmaceutical Science and Clinical Pharmacology suggested that the FDA consider applying reference scaling to ABE studies of HV drugs (39).
Reference-Scaled Average Bioequivalence
The RSABE for both AUC and Cmax is evaluated as shown below:
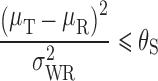 |
where 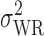 is the population within-subject variance of the reference formulation,
is the population within-subject variance of the reference formulation, 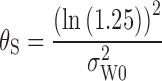 is the BE limit, and
is the BE limit, and 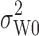 is a predetermined constant set by the regulatory agency.
is a predetermined constant set by the regulatory agency.
Under this model, the implied limits (which represent FDA’s desired consumer risk model) on μT − μR are:
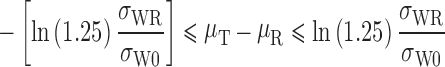 |
If σWR = σW0, the implied limits are equal to the standard unscaled BE limits of ±ln(1.25) (0.80 to 1.25). If σWR > σW0, the implied limits are wider than the standard limits. If σWR < σW0, the implied limits are narrower than the standard limits.
The FDA recommends a mixed scaling approach, as the Agency has determined that it is acceptable for the implied limits to be wider than the standard limits only when σWR is large (as for HV drugs). The mixed scaling model is as shown below.
T and R are considered BE if:
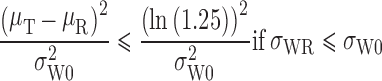 |
and if:
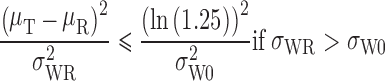 |
FDA sets the value of σW0 at 0.25 (30,40). Under this mixed scaling model and with σW0 = 0.25, the implied limits on μT − μR are as depicted in Fig. 2.
Fig. 2.
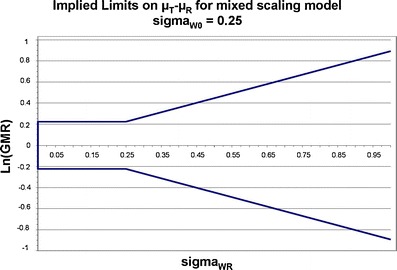
Implied BE limits are plotted as a function of the population reference product within-subject variability of the BE measure. When σ W0 ≤ 0.25, for an acceptable BE study, the 90% CI of the BE measure test/reference GMRs must fall within the 80–125% limits. When σ W0 > 0.25, the implied BE limits scale as reference product within-subject variability increases. The slope of this portion of the curve is determined by the value of σ W0
Direct implementation of FDA’s desired consumer risk model is impossible because σWR is a characteristic of the entire population and thus not directly measured in any particular study. Therefore, FDA has proposed an implementation algorithm (described in the “FDA Draft Guidance for Industry, Bioequivalence Recommendations for Progesterone Oral Capsules”) and discussed in more detail later in this paper. In the FDA-recommended implementation algorithm for mixed scaling studies, the observed within-subject variability of the reference sWR (determined in the BE study) is compared to a cutoff value of 0.294, above which reference scaling is used. This implementation reduces the type I error (defined relative to FDA’s desired consumer risk model) when the within-subject variability is near σW0.
FDA RECOMMENDATIONS FOR DESIGNING RSABE STUDIES
The reference product is administered twice in order to determine its within-subject variability (30). As such, the BE study can use either a partial replicate (three-way crossover: RTR, RRT, or TRR) or full replicate (four-way crossover: RTRT or TRTR) design, but should enroll a minimum of 24 subjects (40,41). The FDA recommends an sWR cutoff value of 0.294, at or above which reference scaling is permitted and below which the unscaled limits of 0.8 to 1.25 are applied (30,40). The selection of 0.294 as the variation at which use of reference scaling of the limits is consistent with the general understanding that drugs may be considered HV if the within-subject coefficient of variation (%CV) observed in the study is ≥30% and, as such, is determined by using the conversion formula of 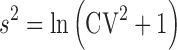 .
.
FDA determined that there are advantages to choosing an sWR cutoff somewhat larger than σW0. The Working Group conducted a series of simulations of BE study results (1,000,000 per condition) to compare the effects of applying different values to σW0. The study results showed that using a σW0 of 0.25 (1) increased the study power compared to ABE, without causing relatively large numbers of studies to pass when the GMR ≥ 1.2 and (2) resulted in a lower inflation of type I error compared to a σW0 of 0.294 (42,43).
The FDA recommends a secondary point estimate constraint criterion of 0.8 to 1.25 on the GMR (30,40). As it is possible that, using RSABE, two products can be shown to be bioequivalent but have an estimated GMR outside of the 0.8 to 1.25 range, it is thought that use of the secondary point estimate constraint will improve the confidence of clinicians and patients(15,28). Several simulation studies investigating the relationship between FDA’s recommended RSABE approach and study power have shown that, when within-subject variability exceeds 50–60%, the point estimate constraint becomes the dominant regulatory criterion rather than the scaling (43–45). It has also been shown that applying the point estimate constraint can increase the sample size needed to show BE for drugs with within-subject variability >50–60% (45). Thus, using a point estimate constraint results in a situation where the benefits of using RSABE are reduced when applied to drugs with very high within-subject variability.
However, it has also been argued that, without a point estimate constraint in effect, the permissiveness of the RSABE approach can become excessively high (42). Another argument favoring incorporating a point estimate constraint is that it has a long history of successful application by regulatory agencies as a BE study acceptance criterion (15). Using a point estimate constraint on the GMR as a criterion for study outcome acceptance is used by the Therapeutic Products Directorate of Health Canada for Cmax in all BE studies (46) and, until 2003, by the FDA for both AUC and Cmax in BE studies that were conducted under fed conditions.
APPLICATION OF RSABE TO REGULATORY REVIEW OF ANDAs FOR GENERIC HV DRUGS
Using the RSABE approach recommended by the FDA, two products are bioequivalent when the 95% upper confidence bound for 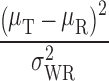 is ≤θS or, equivalently, a 95% upper confidence bound for
is ≤θS or, equivalently, a 95% upper confidence bound for 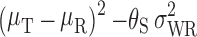 is ≤0. In addition, the GMR of the two products should fall between 0.8 and 1.25.
is ≤0. In addition, the GMR of the two products should fall between 0.8 and 1.25.
Steps for Conducting an RSABE Analysis
It should be noted that we do not know the values of the above population parameters and can never know their values. What we can and do in actuality is calculate CIs around the BE metrics, log(AUC) or log(Cmax).
FDA posted a Guidance for Industry providing step-by-step instructions on how to statistically analyze BE study data using RSABE (40). The intention to use RSABE for an HV drug should be stated a priori in the study protocol. The first step in the analysis is to determine sWR, the within-subject standard deviation (SD) of the reference product estimated from the study, for the BE measures AUC and Cmax. If sWR < 0.294, then the TOST procedure should be used to determine ABE for the BE measure. If sWR ≥ 0.294, then RSABE can be used for the BE measure. Figure 3 illustrates the decision-making process that the FDA uses when determining whether it is suitable to use RSABE for a generic drug.
Fig. 3.
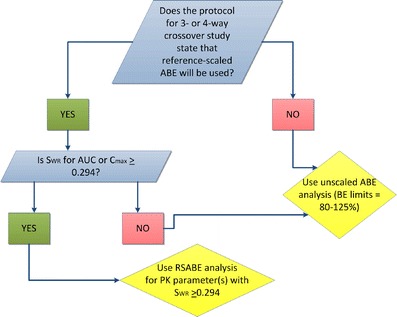
Decision tree showing the process by which FDA’s OGD decide whether it is suitable to use RSABE or unscaled ABE. The first condition to be met is that the study protocol must state a priori that RSABE will be the method of statistical analysis
Once it has been decided that RSABE can be applied to a BE measure, the next step is to determine the 95% upper confidence bound for 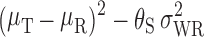 based on Howe’s Approximation I (47). The test and reference product are concluded to be bioequivalent if:
based on Howe’s Approximation I (47). The test and reference product are concluded to be bioequivalent if:
the 95% upper confidence bound for
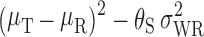 is ≤0 AND
is ≤0 ANDthe test/reference GMR in the study falls within [0.8, 1.25].
Case Studies
Two examples, provided in Table II, show how FDA applies RSABE to studies of HV generic drugs. Both cases are of approved generic drugs, for which the BE studies used three-way partial replicate study designs. As FDA scientific reviewers conduct their own independent calculations in performing BE analysis, the data in Table II are from FDA’s calculations, performed using SAS® Version 9.2.
Table II.
Summary BE Statistics for Two HV Generic Drugs, Analyzed Using RSABE or Unscaled ABE, Depending upon the Value of s WR
| Summary of statistical analysis-scaled data: least-square geometric means, point estimates and 90% confidence intervals | ||||||||
| Parameter | T/R ratio | Lower 90% CL | Upper 90% CL |
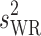
|
s WR | Criteria bound | Method used | Outcome |
| Drug A: fed bioequivalence study, N = 43 | ||||||||
| AUC0 − t | 1.17 | 93.91 | 132.91 | 0.5723 | 0.7565 | −0.2892 | Scaled/PE | Pass |
| AUC∞ | 1.11 | 94.64 | 124.91 | 0.3452 | 0.5875 | −0.1767 | Scaled/PE | Pass |
| C max | 1.19 | 100.47 | 133.26 | 0.3768 | 0.6138 | −0.1684 | Scaled/PE | Pass |
| Drug B: fasting bioequivalence study, N = 36 | ||||||||
| AUC0 − t | 0.99 | 89.82 | 109.53 | 0.07425 | 0.2725 | −0.03914 | Unscaled | Pass |
| AUC∞ | 1.02 | 92.57 | 111.77 | 0.06561 | 0.2561 | −0.03126 | Unscaled | Pass |
| C max | 0.98 | 85.70 | 110.54 | 0.1069 | 0.3270 | −0.05294 | Scaled | Pass |
As shown in Table II, in the case of drug A, the first step of the analysis determined that values of sWR exceeded the regulatory cutoff of 0.294 for AUC0 − t, AUC∞, and Cmax. Thus, the reviewer proceeded to use RSABE for all three BE measures. Table II shows that the calculated 95% upper confidence bound values for all three parameters were <0. Therefore, the product met the first BE acceptance criterion. In addition, the point estimates for all three BE measures are within 0.8 and 1.25. Thus, in this study, drug A met the two RSABE acceptance criteria and was deemed bioequivalent to its corresponding reference.
The analysis of drug B, shown in Table II, was conducted in a different manner because it was not suitable to use RSABE for all BE measures. In the first part of the analysis, sWR for Cmax exceeded 0.294 and values of sWR for AUC0 − t and AUC∞ were <0.294. Thus, ABE with the TOST procedure was used for AUC, for which the 90% CIs met the 80–125% limits. RSABE analysis was only applied to Cmax, which met both acceptance criteria, as the 95% upper confidence bound was <0 and the test/reference GMR point estimate was within 0.8 and 1.25. The case of drug B illustrates the important points that (a) both ABE and RSABE analysis can be used for data from the same study depending on BE measure variability and (b) there is no penalty if the applicant uses a three-way or four-way study design with the intent of using RSABE but sWR fails to make the 0.294 cutoff.
Statistics on BE Study Designs in Recent ANDA Submissions of Generic HV Drugs
Since 2007, the FDA has evaluated 46 ANDAs containing 64 studies in which RSABE was applied. Figure 4 displays the statistics on these studies. Sixty-two of these studies met the RSABE acceptance criteria, although some studies were considered “incomplete” for other reasons, many due to questions relating to the bioanalytical method (48). Only two studies were unacceptable because they did not meet the acceptance criteria. In the first instance, as one BE measure, sWR, was <0.294, the RSABE analysis could not be applied. Thus, it was necessary to apply ABE and the TOST procedure, but the study did not meet the acceptance limits because the upper confidence limit exceeded 125%. In the second instance, the study data met the criteria for applying the RSABE analysis, and the 90% upper confidence bound for all three BE measures was <0, but the GMRs of all BE measures exceeded 1.25. Interestingly, the applicant conducted a second successful study on this product which met the BE limits using RSABE analysis without reformulating or increasing the number of subjects.
Fig. 4.
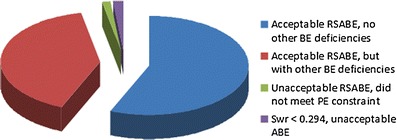
Outcome of the review of 64 RSABE studies reviewed at the OGD. All but two of the studies met the BE acceptance criteria. For 36 of these studies, there were no other deficiencies related to the BE portion of the ANDA submission. For 26 studies, other miscellaneous deficiencies unrelated to RSABE analysis (most often related to the bioanalytical method used) were present in the BE portion of the submission. One study did not meet the point estimate constraint and thus the study did not meet the RSABE acceptance criteria. Another study could not be analyzed using RSABE because the s WR for C max was <0.294. However, when the TOST was applied, the 90% CI for C max did not fall within the 80–125 limits; thus, this study did not meet the ABE acceptance criteria
As of this writing, FDA has fully approved four HV generic drug products for which RSABE was used. In addition, one HV generic drug product for which RSABE was used was tentatively approved (because a patent related to the reference product has not yet expired).
One of the major reasons offered to justify moving away from the “one-size-fits-all” BE limits for HV drugs was concern that BE studies would be unnecessarily repeated because of underpowering. Until the “All Bioequivalence Studies” Final Rule became effective in 2009, it was not possible for the FDA to assess the prevalence of failed and repeated BE studies on HV drugs because generic applicants were not required to submit failed BE studies (49). As a result, FDA’s assessment of the scope of BE problems in regulatory submissions of HV drugs was possibly biased because the Agency did not see the results of failed BE studies. As applicants are now required to submit to ANDAs all BE studies conducted on the same drug product formulation as the one intended for marketing (50,51), it is hoped that such submissions will provide valuable information about the development of HV drug formulations.
To test the hypotheses that a high percentage of BE studies fail because they are of (a) HV drugs and (b) do not enroll enough subjects, we surveyed all failed BE studies of generic drugs reviewed from the time that the Final Rule went into effect until the time of this writing. As shown in Table III, 205 failed BE studies were reviewed during this time period. Of these, 92 of these failed BE studies were of HV drugs, representing 45% of the total. This appears to be a disproportionately high percentage of failed BE studies, as HV drugs are thought to represent about 20% of drug products submitted to FDA in ANDAs.
Table III.
Number of Failed BE Studies Reviewed at FDA’s OGD Since the “All Bioequivalence Studies” Rule Became Effective in July 2009
| Description | BE Studies | ANDAs | ||
|---|---|---|---|---|
| Number | Percent of total | Number | Percent of total | |
| Within-subject variability of AUC and C max ≥ 30% | 92 | 45 | 45 | 37 |
| Within-subject variability of AUC and C max < 30% | 113 | 55 | 76 | 63 |
| Totals | 205 | 100 | 121 | 100 |
In addition, we surveyed the reasons why these 92 BE studies submitted in 45 ANDAs failed. The results confirm that a high percentage of studies of HV drugs appear to fail the BE acceptance criteria due to underpowering. As shown in Table IV, for all but one of 45 different ANDAs, generic products that initially failed one or more two-way crossover studies with TOST analysis met the BE limits in subsequent studies when power was increased, either by enrolling more subjects or by changing to an RSABE design.
Table IV.
Changes Made to the Study Design or Formulation to Achieve Successful Pivotal BE Studies of HV Drugs after the Initial BE Studies Failed to Meet the Acceptance Criteria
| Change made | No. of ANDAs | Percent |
|---|---|---|
| Increase in sample size in two-way crossover BE study | 41 | 91 |
| Changed to three-way study design and RSABE | 3 | 7 |
| Reformulated | 1 | 2 |
| Total | 45 | 100 |
RSABE APPROACHES FOR HV DRUGS IN OTHER JURISDICTIONS
In August 2010, the European Medicines Agency (EMA) issued a new Guideline on the Investigation of Bioequivalence, recommending a scaling approach for BE assessment of HV drugs (15,42,52,53). EMA recommends that BE limits scale with variability only for Cmax and only up to a maximum within-subject variability value of 50% (where %CV is defined as 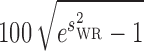 ), after which they remain constant at the static limits of 69.84% to 143.19%. In BE studies, Cmax is usually more variable than AUC (15,54,55). Either a three-way or four-way crossover design, in which the reference product is administered twice, can be used in the BE study, in order to determine the reference product within-subject variability for scaling. The applicant should demonstrate that the within-subject variability of the reference product Cmax is >30%. The extent of the widening of the acceptance limits is defined based upon the within-subject variability seen in the BE study using scaled average BE according to [U, L] = exp [±k × sWR], where U is the upper limit of the acceptance range, L is the lower limit of the acceptance range, k is the regulatory constant set to 0.760, and sWR is the within-subject SD of the log-transformed values of Cmax of the reference product (15,42,52). Using these values, the regulatory limits range from 80% to 125% when the within-subject %CV is 30% to 69.84–143.19% when the within-subject variability is 50%. EMA also recommends a point estimate constraint on the Cmax GMR if scaling is used.
), after which they remain constant at the static limits of 69.84% to 143.19%. In BE studies, Cmax is usually more variable than AUC (15,54,55). Either a three-way or four-way crossover design, in which the reference product is administered twice, can be used in the BE study, in order to determine the reference product within-subject variability for scaling. The applicant should demonstrate that the within-subject variability of the reference product Cmax is >30%. The extent of the widening of the acceptance limits is defined based upon the within-subject variability seen in the BE study using scaled average BE according to [U, L] = exp [±k × sWR], where U is the upper limit of the acceptance range, L is the lower limit of the acceptance range, k is the regulatory constant set to 0.760, and sWR is the within-subject SD of the log-transformed values of Cmax of the reference product (15,42,52). Using these values, the regulatory limits range from 80% to 125% when the within-subject %CV is 30% to 69.84–143.19% when the within-subject variability is 50%. EMA also recommends a point estimate constraint on the Cmax GMR if scaling is used.
The South African Medicines Control Council suggests that the RSABE may be used for AUC and Cmax if specified a priori in the protocol and justified by sound scientific principles; applicants also have the option of applying static BE limits with a wider acceptance interval for HV Cmax (56,57).
CONTROVERSIES
One of the more controversial aspects of the RSABE approach is the selection of the value of σW0, the variability at which the BE limits can potentially begin to scale. The FDA sets σW0 at 0.25, but also proposes an implementation algorithm that does not permit BE limits to scale until the observed within-subject variability reaches the cutoff value of 0.294 for sWR. Endrenyi and Tothfalusi (44) argue that the FDA’s approach to RSABE is discontinuous and conducted simulations claiming that, when σW0 ranges from 0.246 to 0.294, consumer risk is substantially larger than 5% and the regulatory uncertainty about a decision on acceptance or rejection is enhanced. This claim is based on a different definition of consumer risk than FDA uses. Endrenyi and Tothfalusi define consumer risk relative to BE limits of 80–125% (44). FDA’s position is that a true population variance of σWR ≥ 0.25 justifies using BE limits that are wider than 80–125%. The use of sWR ≥0.294 as part of FDA’s implementation method affects the producer risk—there will be a higher probability of failing a product that is acceptable according to FDA’s consumer risk model.
Results of simulations conducted by members of the HV Drug Working Group support the position that using a cutoff value of 0.294 for sWR maintains an acceptable type I error rate relative to FDA’s desired consumer risk model. This can be illustrated by using a value of 0.29399 for σWR. In this case, the acceptable limits on μT − μR are ±ln(1.25) × 0.29399/0.25 = ±0.262408. Results of simulated three-way partial replicate studies of 36 subjects result in 46,434 conclusions of BE out of 1,000,000 simulated studies or an estimated type I error rate of 0.046434 relative to FDA’s desired consumer risk model. It is important to carefully note the distinction between σWR and sWR in the analysis of the scaling method.
Finally, likelihood of regulatory uncertainty in evaluating RSABE study results when within-subject variability is near 30% is substantially reduced by the evaluation approach used by BE review scientists in FDA’s OGD. Although the RSABE analysis is triggered when sWR ≥ 0.294 in a three-way or four-way replicated study, review scientists are encouraged to evaluate the appropriateness of reference scaling in the context of other available information on the PK variability of a given drug. The three-level review process for all BE submissions in the OGD (reviewer, team leader, and division director) also encourages critical evaluation of data and consistent policy application.
FUTURE DIRECTIONS
Lack of harmonization in setting the regulatory constant and cutoff for RSABE could potentially be resolved by using the approach for all generic products, as was formerly proposed for IBE. Such an approach could also be more favorable to ensuring drug product switchability. The FDA recently proposed to adapt RSABE for BE studies of narrow therapeutic index (NTI) drugs (58). Since NTI drugs are characterized by low within-subject variability, this would effectively result in narrower BE limits.
CONCLUSIONS
Figure 5 provides a timeline showing how RSABE evolved at the FDA, from inception to application to ANDAs for generic HV drugs. FDA implemented RSABE for generic HV wide therapeutic index drugs to ease regulatory burden. In the RSABE approach recommended by the FDA, the within-subject variability of the reference product BE measure is determined in a three-way or four-way replicated study which should use at least 24 subjects. The implied BE limits scale to reference within-subject variability once σWR = σW0 = 0.25 or greater. However, to preserve an acceptable (<5%) type I error rate, applicants cannot apply reference scaling to calculate BE limits until the reference product within-subject SD in the BE study is at least 0.294. Acceptance criteria are that the 95% upper confidence bound for the GMR test/reference BE measure ratio is ≤0 and the point estimate of the BE measure GMR is within 0.8 to 1.25. The approach has been applied successfully to ANDAs, with 64 RSABE studies submitted and reviewed since 2007. To date, FDA has granted four full marketing approvals and one tentative approval for several diverse HV generic drug products based on the RSABE analysis. The RSABE is not a “one-size-fits-all” approach, but rather a system that self-adjusts to the characteristics of the reference. As a result, RSABE could be used to potentially set appropriately strict BE limits for all products.
Fig. 5.
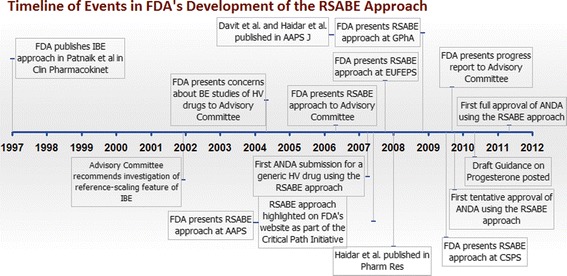
This timeline summarizes the evolution of the RSABE approach at the FDA. The FDA first proposed reference scaling as a component of IBE analysis, although the FDA subsequently decided not to continue implementing IBE. Three years later, in 2004, the FDA began developing RSABE using the scaling approach from IBE. The first ANDA using RSABE was submitted in 2007. The first tentative approval of an HV drug for which RSABE was successfully applied was in 2009. The first full approval of an HV drug supported by acceptable RSABE was in 2011. From 2004 to 2010, FDA scientists published several peer-reviewed scientific articles on RSABE and made a number of presentations at national and international venues. The FDA has also posted a Draft Guidance for Industry with a step-by-step description of how to perform RSABE
ACKNOWLEDGMENTS
The authors thank Hoainhon Nguyen Caramenico, Parthapratim Chandaroy, Svetlana Cherstniakova, Li Gong, Heather Lim, Anitha Palamakula, and Xiaofai Wang for contributing data to the examples described throughout the manuscript. The opinions stated in this article represent those of the authors and do not necessarily represent the official position of the US Food and Drug Administration.
REFERENCES
- 1.Federal Food, Drug, and Cosmetic Act, Chapter V, Subchapter A, Drugs and Devices Section 355(j)(8)(B)(i).
- 2.Schuirmann DJ. A comparison of the two one-sided tests procedure and the power approach for assessing the equivalence of average bioavailability. J Pharmacokinet Biopharm. 1987;15:657–80. doi: 10.1007/BF01068419. [DOI] [PubMed] [Google Scholar]
- 3.Westlake WJ. Bioequivalence testing: a need to rethink. Biometrics. 1981;37:589–94. doi: 10.2307/2530573. [DOI] [Google Scholar]
- 4.Blume HH, Midha KK. Bio-International 92, conference on bioavailability, bioequivalence, and pharmacokinetic studies. J Pharm Sci. 1993;82:1186–9. doi: 10.1002/jps.2600821125. [DOI] [PubMed] [Google Scholar]
- 5.Shah VP, Yacobi A, Barr WH, Benet LZ, Breimer D, et al. Evaluation of orally administered highly variable drugs and drug formulations. Pharm Res. 1996;13:1590–4. doi: 10.1023/A:1016468018478. [DOI] [PubMed] [Google Scholar]
- 6.Davit BM, Conner DP, Fabian-Fritsch B, Haidar SH, Jiang X, et al. Highly variable drugs: observations from bioequivalence data submitted to the FDA for new generic drug applications. AAPSJ. 2008;10:148–56. doi: 10.1208/s12248-008-9015-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.FDA Guidance for Industry, statistical approaches to establishing bioequivalence. US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research. 2001. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070244.pdf. Accessed 23 Jan 2012.
- 8.Patterson SD, Zariffa NMD, Montague TH, Howland K. Non-traditional study designs to demonstrate average bioequivalence for highly variable drug products. Eur J Clin Pharmacol. 2001;57:663–70. doi: 10.1007/s002280100371. [DOI] [PubMed] [Google Scholar]
- 9.Phillips KF. Power of the two one-sided tests procedure in bioequivalence. J Pharmacokinet Biopharm. 1990;18:137–44. doi: 10.1007/BF01063556. [DOI] [PubMed] [Google Scholar]
- 10.Benet L. Why highly variable drugs are safer. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology meeting transcript. US Food and Drug Administration Dockets. Apr 14 2004. http://www.fda.gov/ohrms/dockets/ac/04/transcripts/4034T2.htm. Accessed 23 Jan 2012.
- 11.Cook JA, Davit BM, Polli JE. Impact of Biopharmaceutics Classification System-based biowaivers. Mol Pharmaceutics. 2010;7:1539–44. doi: 10.1021/mp1001747. [DOI] [PubMed] [Google Scholar]
- 12.Amidon GL. Sources of variability: physicochemical and gastrointestinal. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology meeting transcript. US Food and Drug Administration Dockets. Apr 14 2004. http://www.fda.gov/ohrms/dockets/ac/04/transcripts/4034T2.htm. Accessed 23 Jan 2012.
- 13.Conner DP. Bioequivalence methods for highly variable drugs and drug products. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology meeting transcript. US Food and Drug Administration Dockets. Aug 5 2009. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/UCM179891.pdf. Accessed 23 Jan 2012.
- 14.Diliberti CE. Why bioequivalence of highly variable drugs is an issue. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology meeting transcript. US Food and Drug Administration Dockets. Apr 14 2004. http://www.fda.gov/ohrms/dockets/ac/04/transcripts/4034T2.htm. Accessed 23 Jan 2012.
- 15.Tothfalusi L, Endrenyi L, Arieta AG. Evaluation of bioequivalence for highly variable drugs with scaled average bioequivalence. Clin Pharmacokinet. 2009;48:725–43. doi: 10.2165/11318040-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 16.Endrenyi L. Bioequivalence methods for highly variable drugs. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology meeting transcript. US Food and Drug Administration Dockets. Apr 14 2004 http://www.fda.gov/ohrms/dockets/ac/04/transcripts/4034T2.htm. Accessed 23 Jan 2012.
- 17.Midha KK, Rawson MJ, Hubbard JW. The bioequivalence of highly variable drugs and drug products. Int J Clin Pharmacol Ther. 2005;43:485–98. doi: 10.5414/cpp43485. [DOI] [PubMed] [Google Scholar]
- 18.Tsang YC, Pop R, Gordon P, Hems J, Spino M. High variability in drug pharmacokinetic complicates determination of bioequivalence: experience with verapamil. Pharm Res. 1996;13:846–50. doi: 10.1023/A:1016040825844. [DOI] [PubMed] [Google Scholar]
- 19.Blume H, McGilvery I, Midha K. Bio-International 94 conference on bioavailability, bioequivalence and pharmacokinetic studies. Eur J Pharm Sci. 1995;3:113–24. doi: 10.1016/0928-0987(94)00080-J. [DOI] [PubMed] [Google Scholar]
- 20.Blume HH. “One-size-fits-all” in bioavailability and bioequivalence testing. Int J Clin Pharmacol Ther. 2009;47:419–20. doi: 10.5414/cpp47419. [DOI] [PubMed] [Google Scholar]
- 21.Yu LX. Bioequivalence of highly variable drugs: issues and challenges. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology Meeting Transcript. Apr 14 2004. US Food and Drug Administration Dockets. http://www.fda.gov/ohrms/dockets/ac/04/transcripts/4034T2.htm. Accessed 23 Jan 2012.
- 22.Executive Secretary, Advisory Committee for Pharmaceutical Science and Clinical Pharmacology. Summary meeting minutes. In: FDA Advisory Committees meeting materials. US Food and Drug Administration Dockets. Apr 14 2004. http://www.fda.gov/ohrms/dockets/ac/04/minutes/4034M1.htm. Accessed 23 Jan 2012.
- 23.Haidar SH. FDA perspective. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology meeting transcript . US Food and Drug Administration Dockets. Apr 14 2004. http://www.fda.gov/ohrms/dockets/ac/04/transcripts/4034T2.htm. Accessed 23 Jan 2012.
- 24.Midha K. Bioequivalence of highly variable drugs. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology meeting transcript. US Food and Drug Administration Dockets. Apr 6 2006. http://www.fda.gov/ohrms/dockets/ac/06/transcripts/2006-4241t2-01.pdf. Accessed 23 Jan 2012.
- 25.Haidar SH. Evaluation of a scaling approach for highly variable drugs. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology meeting transcript. US Food and Drug Administration Dockets. Apr 6 2006. Accessed 23 Jan 2012.
- 26.Davit BM. FDA’s proposal. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology meeting transcript. US Food and Drug Administration Dockets. Apr 6 2006. http://www.fda.gov/ohrms/dockets/ac/06/transcripts/2006-4241t2-01.pdf. Accessed 23 Jan 2012.
- 27.Endrenyi L. Some issues on the determination of bioequivalence for highly variable drugs. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology meeting transcript. US Food and Drug Administration Dockets. Apr 6 2006. http://www.fda.gov/ohrms/dockets/ac/06/transcripts/2006-4241t2-01.pdf. Accessed 23 Jan 2012.
- 28.Benet L. Therapeutic considerations of highly variable drugs. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology meeting transcript. US Food and Drug Administration Dockets. Apr 6 2006. http://www.fda.gov/ohrms/dockets/ac/06/transcripts/2006-4241t2-01.pdf. Accessed 23 Jan 2012.
- 29.Executive Secretary, Advisory Committee for Pharmaceutical Science and Clinical Pharmacology. Summary meeting minutes. In: FDA Advisory Committees meeting materials. US Food and Drug Administration Dockets. Apr 6 2006. http://www.fda.gov/ohrms/dockets/ac/06/minutes/2006-4241m2.pdf. Accessed 23 Jan 2012.
- 30.Haidar SH, Davit BM, Chen M-L, Conner D, Lee LM, et al. Bioequivalence approaches for highly variable drugs and drug products. Pharm Res. 2008;25:237–41. doi: 10.1007/s11095-007-9434-x. [DOI] [PubMed] [Google Scholar]
- 31.Hauck WH, Hyslop T, Chen M-L, Patnaik R, Williams RL. Subject-by-formulation interaction in bioequivalence: conceptual and statistical terms. Pharm Res. 2000;17:375–80. doi: 10.1023/A:1007508516231. [DOI] [PubMed] [Google Scholar]
- 32.Patnaik RN, Lesko LJ, Chen M-L, Williams RL. Individual bioequivalence: new concepts in the statistical assessment of bioequivalence metrics. Clin Pharmacokinet. 1997;33:1–6. doi: 10.2165/00003088-199733010-00001. [DOI] [PubMed] [Google Scholar]
- 33.Chen M-L. Background and concepts of individual bioequivalence. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology meeting transcript .US Food and Drug Administration Dockets. Nov 29 2001. http://www.fda.gov/ohrms/dockets/ac/01/transcripts/3804t2_03_Afternoon_Session.pdf. Accessed 23 Jan 2012.
- 34.Patnaik R. Results from replicate design studies in ANDAs. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology meeting transcript. US Food and Drug Administration Dockets. Nov 29 2001. http://www.fda.gov/ohrms/dockets/ac/01/transcripts/3804t2_03_Afternoon_Session.pdf. Accessed 23 Jan 2012.
- 35.Benet L. Individual bioequivalence: have the opinions of the scientific community changed? In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology meeting transcript. US Food and Drug Administration Dockets. Nov 29 2001. http://www.fda.gov/ohrms/dockets/ac/01/transcripts/3804t2_03_Afternoon_Session.pdf. Accessed 23 Jan 2012.
- 36.Executive Secretary, Advisory Committee for Pharmaceutical Science and Clinical Pharmacology. Briefing document, bioequivalence criteria research program. In: FDA Advisory Committee Meeting Materials. US Food and Drug Administration Dockets. Nov. 29 2001. http://www.fda.gov/ohrms/dockets/ac/01/briefing/3804b2_08_Bioequiv%20Criteria.pdf. Accessed 23 Jan 2012.
- 37.Chen M-L. Results from replicate design studies in NDAs and FDA database. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology meeting transcript . US Food and Drug Administration Dockets. Nov 29 2001. http://www.fda.gov/ohrms/dockets/ac/01/transcripts/3804t2_03_Afternoon_Session.pdf. Accessed 23 Jan 2012.
- 38.FDA Guidance for Industry, bioavailability and bioequivalence studies for orally administered drug products: general considerations. US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research. 2003. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070124.pdf. Accessed 23 Jan 2012.
- 39.Members of the FDA Advisory Committee for Pharmaceutical Science and Clinical Pharmacology. Discussion of individual bioequivalence issues. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology meeting transcript. US Food and Drug Administration Dockets. Nov 29 2001. http://www.fda.gov/ohrms/dockets/ac/01/transcripts/3804t2_03_Afternoon_Session.pdf. Accessed 23 Jan 2012.
- 40.FDA Draft Guidance for Industry, bioequivalence recommendations for progesterone oral capsules. US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research, Silver Spring. 2011. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM209294.pdf. Accessed 23 Jan 2012.
- 41.Davit BM, Conner DP. The United States of America. In: Kanfer I, Shargel L, editors. Generic drug product development: international regulatory requirements for bioequivalence. New York: Informa Healthcare; 2010. pp. 254–81. [Google Scholar]
- 42.Karalis V, Sylmillides M, Macheras P. Bioequivalence of highly variable drugs: a comparison of the newly-proposed regulatory approaches by FDA and EMA. Pharm Res. 2011. doi:10.1007/s11095-011-0651-y. [DOI] [PubMed]
- 43.Haidar SH, Makhlouf F, Schuirmann DJ, Hyslop T, Davit B, et al. Evaluation of a scaling approach for the bioequivalence of highly variable drugs. AAPSJ. 2008;10:450–4. doi: 10.1208/s12248-008-9053-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Endrenyi L, Tothfalusi L. Regulatory conditions for the determination of bioequivalence of highly variable drugs. J Pharm Pharmaceut Sci. 2009;12:138–49. doi: 10.18433/j3zw2c. [DOI] [PubMed] [Google Scholar]
- 45.Tothfalusi L, Endrenyi L. Sample size for designing bioequivalence studies for highly variable drugs. J Pharm Pharmaceut Sci. 2011;15:73–84. doi: 10.18433/j3z88f. [DOI] [PubMed] [Google Scholar]
- 46.Health Canada Guidance for Industry, conduct and analysis of bioavailability and bioequivalence studies—part A: oral dosage formulations used for systemic effects. Therapeutic Products Directorate, Canada Minister of Health, Ottawa. 1997. http://www.hc-sc.gc.ca/dhp-mps/alt_formats/hpfb-dgpsa/pdf/prodpharma/bio-a-eng.pdf. Accessed 23 Jan 2012.
- 47.Howe WG. Approximate confidence limits on the mean of X + Y where X and Y are two tabled independent variables. J Amer Statist Assoc. 1974;69:789–94. [Google Scholar]
- 48.Liu Q, Davit BM, Cherstniakova SA, Dandamudi S, Walters JF, et al. Common deficiencies with bioequivalence submissions in abbreviated new drug applications assessed by the FDA. AAPSJ. 2011. doi:10.1208/s12248-011-9312-7. [DOI] [PMC free article] [PubMed]
- 49.US Food and Drug Administration, Department of Health and Human Services. Final rule, 21 CFR Parts 314 and 320, Docket No. FDA-2003-N-0209. Fed Regist 2009; 74:2849–62.
- 50.FDA Guidance for Industry, submission of summary bioequivalence data for abbreviated new drug application. US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research, Silver Spring. 2011. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM134846.pdf. Accessed 23 Jan 2012.
- 51.US Food and Drug Administration, Department of Health and Human Services. Bioequivalence. In: Title 21 Code of Federal Regulations, Food and Drugs. Office of the Federal Register National Archives and Records Administration. Washington; 2011. p. 132–3.
- 52.European Medicines Agency. Guideline on the investigation of bioequivalence. 2010. http://www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf. Accessed 23 Jan 2012. [DOI] [PubMed]
- 53.Morais JA, Lobato Mdo R. The new European Medicines Agency Guideline on the investigation of bioequivalence. Basic Clin Pharmacol Toxicol. 2010;106:251–60. doi: 10.1111/j.1742-7843.2009.00518.x. [DOI] [PubMed] [Google Scholar]
- 54.Davit B. Bioequivalence of highly variable drugs case studies. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology Meeting Transcript. US Food and Drug Administration Dockets. Apr 16 2004. http://www.fda.gov/ohrms/dockets/ac/04/transcripts/4034T2.htm. Accessed 23 Jan 2012.
- 55.Davit BM, Nwakama PE, Buehler GJ, Conner DP, Haidar SH, et al. Comparing generic and innovator drugs: a review of 12 years of bioequivalence data from the United States Food and Drug Administration. Ann Pharmacother. 2009;43:1583–97. doi: 10.1345/aph.1M141. [DOI] [PubMed] [Google Scholar]
- 56.Walker RB, Kanfer I, Skinner MF. Bioequivalence assessment of generic products: an innovative South African approach. Clin Res Regul Affair. 2006;23:11–20. doi: 10.1080/10601330500534014. [DOI] [Google Scholar]
- 57.Medicines Control Council. Human medicines guideline, biostudies. MCC Guidelines and Forms. South African Registrar of Medicines. Capetown. 2007. http://www.mccza.com/dynamism/default_dynamic.asp?grpID=30&doc=dynamic_generated_page.asp&categID=177&groupID=30. Accessed 23 Jan 2012.
- 58.Davit B. FDA Proposal for bioequivalence of generic narrow therapeutic index drug products. In: FDA Advisory Committee for Pharmaceutical Sciences and Clinical Pharmacology meeting transcript. US Food and Drug Administration Dockets. Jul 26 2011. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/UCM272112.pdf. Accessed 23 Jan 2012.