

Abstract
Facing the increasing number of poorly water-soluble drugs, pharmaceutical scientists are required to break new grounds for the delivery of these pharmaceutically problematic drugs. Lipid-based drug delivery systems (LBDDS) have received increased interest as a novel drug delivery platform during the last decades and several successfully marketed products have shown the potential for LBDDS. However, there exists a discrepancy between the clear need for innovative delivery forms and their rational design. In the case of LBDDS, this can be attributed to the complexity of LBDDS after administration. Unlike conventional formulations, LBDDS are susceptible to digestion in the gastrointestinal tract, the interplay of delivery system, drug and physiology ultimately effecting drug disposition. In vitro lipolysis has become an important technique to mimic the enzymatic degradation. For the better understanding of how LBDDS promote drug delivery, in vitro lipolysis requires advanced characterisation methods. In this review, the physiological background of lipid digestion is followed by a thorough summary of the techniques that are currently used to characterise in vitro lipolysis. It would be desirable that the increasing knowledge about LBDDS will foster their rationale development thereby increasing their broader application.
KEY WORDS: in vitro digestion, in vitro lipolysis models, lipid-based drug delivery systems, poorly soluble drugs, self-nanoemulsifying drug delivery systems (SNEDDS)
INTRODUCTION
It has long been observed that the intake of food, notably lipids, can have profound effects on the absorption and bioavailability of drugs (1). In particular, the bioavailability of poorly water-soluble, lipophilic drugs has been shown to benefit from the concomitant ingestion of lipids, sparking the interest in the use of lipids as potential drug delivery systems (2,3). As a result some compounds formulated as lipid-based drug delivery systems (LBDDS) have entered the market successfully (4). However, there appears some reluctance in the broader utilisation of LBDDS, indicated by the still-limited number of products commercially available (4,5). This might be attributed to the intricate interplay of LBDDS and the digestive system. After administration, LBDDS are processed in a complex physiological sequence which is not completely understood. It is this inherent alteration of the drug carrier during its transit through the gastrointestinal tract (GIT) which distinguishes LBDDS from conventional formulations such as tablets. Consequently this attribute asks for protocols capable of assessing and predicting the quality and performance of LBDDS. However, standardised protocols have not been established yet, which can be considered a major obstacle for the broader use of these delivery systems. This review sets out to summarise the recent developments in the characterisation of LBDDS. In the following sections, an overview of the physiological processes in the GIT necessary for the understanding of the fate of LBDDS after oral administration, is presented. Thereafter, the evolution of meaningful in vitro methods necessary for the development and rational testing of LBDDS is highlighted.
LIPID-BASED DRUG DELIVERY
LBDDS are a heterogeneous group of delivery systems comprising solutions of oil, emulsions, suspensions, liposomes and self-emulsifying drug delivery systems (SEDDS) (3,5). The latter can be considered the most complex formulations consisting of oil, surfactant, co-surfactant, cosolvent, and drug (3,4). Upon contact with aqueous medium these isotropic preconcentrates spontaneously generate coarse emulsions, or fine nano-emulsions, referred to as self-nanoemulsifying drug delivery systems (SNEDDS). In the literature, the term SNEDDS is often used in parallel with the term self-microemulsifying drug delivery system (SMEDDS) without further distinction of the true nature of the resulting dispersion. SMEDDS are defined as thermodynamically stable systems whereas SNEDDS are kinetically stable. According to a recent review by Anton, most of the studies that describe SMEDDS are, in fact SNEDDS (6). Therefore, throughout this work the term SNEDDS is used and reference is made to the terminology of other authors where applicable. SNEDDS and SMEDDS have gained considerable attention since the large surface area associated with the fine dispersion of these systems (droplet size, <<100 nm) is believed to foster rapid digestion followed by a more reproducible drug release and absorption compared with non-dispersing LBDDS (2).
Similar to food-derived lipids, the digestive system metabolises LBDDS in a complex interplay with the GIT fluids resulting in the solubilisation and distribution of the co-administered drug between mixed micelles or other colloidal phases potentially facilitating drug absorption. Other mechanisms than enhanced solubilisation might also contribute to improved bioavailability: it is well documented that the lymphatic uptake of very lipophilic compounds can be an alternative route of drug absorption which can be stimulated by long-chain lipids commonly used in LBDDS (7–10). Furthermore, some excipients have been shown to impede the activity of P-glycoproteins and intestinal cytochrome P450 and to enhance the permeability of the intestinal epithelium potentially giving rise to improved absorption (11,12). Finally, gastric emptying can be protracted in response to lipids allowing more time for drug dissolution (13).
LIPID DIGESTION
Gastric Digestion
Lipids are an essential part of the nutrition constituting of up to 170 g of the daily diet in contemporary Europe (14,15). Notwithstanding the problems associated with such a high-energy intake, the physiology of the body is well equipped for the assimilation of lipids, starting in the stomach. Here, lingual (in some species (16)) and gastric lipases, secreted by the salivary glands and the gastric mucosa, respectively, are the first enzymes starting hydrolysis of the lipids (17). Both enzymes have similar preferences regarding the substrate: lipids consisting of medium-chain triglycerides are hydrolysed faster compared with long-chain triglycerides, both reactions producing diglycerides, monoglycerides and (partially unionised) fatty acids. The surface activity of the digestion products together with the dietary phospholipids and the powerful shear forces of the stomach promote the first crude emulsification of the lipids (17). After emulsification in the stomach, the lipid droplets are propelled into the duodenum by the contractions of the pylorus for further processing. On a quantitative basis, the gastric lipolysis step accounts for up to approximately 10–25% of the total lipolysis, whereas the rest of the lipids are hydrolysed by pancreatic enzymes (18,19).
Intestinal Digestion
The arrival of the acidic lipid emulsion in the duodenum triggers several events crucial for the effective and complete hydrolysis of the remaining lipids (17). The pancreas secretes a mixture of fluids containing enzymes of which pancreatic lipase and co-lipase are most important. The secretion of bicarbonate elevates the pH to approximately 6–8, reflecting the pH optimum for pancreatic lipase (20). Pancreatic lipase selectively hydrolyses triglycerides at the Sn1 and Sn3 position, producing 1 mol 2-monoglycerides and 2 mol fatty acids for each mole triglyceride. Similar to gastric lipase the efficiency of pancreatic lipase towards hydrolysis of medium-chain triglycerides is enhanced compared with long-chain triglycerides (20). However, the hydrolytic activity of pancreatic lipase highly depends on a complex interplay with co-lipase, bile acids and calcium.
One of the prominent properties of pancreatic lipase is that the enzyme is active only at the oil/water interface (21). Therefore, its activity is highly influenced by any processes affecting the surface of the substrate. This is particularly important during the progress of lipolysis where the surface of the substrate accumulates amphiphilic digestion products, including fatty acids and 2-monoglycerides that form liquid crystalline and ‘viscous isotropic’ phases (15,22,23). The accumulation of these compounds restricts the access of pancreatic lipase to the oil/water interface. It has been shown that bile acids can displace these amphiphiles from the substrate interface. However, bile acids can also inhibit pancreatic lipase (24). Co-lipase binds to pancreatic lipase in a 1:1 complex reconstituting the activity of pancreatic lipase and serving as an anchor for pancreatic lipase at the substrate interphase (20,25). Furthermore, co-lipase stabilises pancreatic lipase in an ‘open-lid’ conformation (26). The lid comprises a surface loop covering the catalytic centre in the inactive conformation of the enzyme, thereby restricting access to its catalytic centre (21). Stabilisation of the open lid, therefore, increases the likelihood of substrate hydrolysis (18,27).
For an optimal activity of the pancreatic lipase/co-lipase complex, the presence of bile salts is required (28). The most important bile acids in humans are cholic acid, deoxycholic acid and chenodeoxycholic acid. The conjugation with taurine and glycine renders the bile acids more surface active. This facilitates the displacement of digestion products and formulation-derived surfactants from the interface. The accumulation of these compounds would result in the detachment of the pancreatic lipase from the interface thereby preventing further lipolysis. Furthermore, in vivo, bile salts aid in the removal of digestion products from the interface by solubilisation into mixed micelles facilitating the transport across the unstirred water layer and the intestinal mucus into the enterocyte by a mechanism still unknown. Medium-chain fatty acids diffuse directly across the enterocyte accessing the systemic circulation via the portal vein, whereas long-chain fatty acids require re-esterification to triglycerides (29,30). This is accomplished at the endoplasmic reticulum of the enterocyte, followed by the incorporation of the newly synthesised triglycerides into lipoproteins and their subsequent release into the lymphatic system.
Calcium ions have been described as an additional factor regulating lipolysis in vivo (15,22,31). The calcium concentration changes considerably from approximately 3–4 mM in the fasted to approximately 15 mM in the fed state (32). An increase in the calcium concentration has been associated with a decline in the lag phase commonly preceding maximum lipase activity (33). It is assumed that calcium improves the penetration of the pancreatic lipase into the substrate surface by the screening of negative charges from the surface decreasing the electrostatic repulsion of the enzyme (33). Other authors suggested a highly enzymatically active complex of enzyme, phospholipid, mixed micelle and calcium (34). Furthermore, calcium is known to generate liquid crystals with bile salts (22). Free fatty acids are solubilised in this liquid crystalline phase followed by the absorption of the fatty acids by the intestinal epithelium. This mechanism is important as it presents the removal of digestion products from the substrate surface.
The preceding discussion illustrates the dynamics and complexity of lipid digestion. Generally, similar considerations apply to the digestion of lipids originating from LBDDS although the lipid amounts are generally smaller compared with those deriving from food products. Furthermore, excipients (e.g. surfactants) that are not present in food, might be used in LBDDS. Therefore, LBDDS pose particular challenges in regard to their quality assessment and a priori prediction of their performance.
DISPERSION TESTING OF LBDDS
Standardised techniques such as compendial dissolutions studies used for the quality assessment of conventional formulations do not apply to LBDDS as the co-administered drug is commonly presented in solution. However, the same equipment used for compendial dissolution testing (e.g. USP paddle 2) has been used frequently to study the dispersion characteristics of some LBDDS (35–37). With regard to SNEDDS which have attracted most attention amongst LBDDS, compendial media have been used mainly for the assessment of the dispersion characteristics, i.e. the dispersion time, droplet size or zeta potential of dispersed formulations (35,38,39). Interestingly, despite the commonly employed dispersion assessment during the development of SNEDDS, the target values for dispersion have not been defined. The rationale behind the assessment of droplet size is the observation that an increased surface area, as generated by SNEDDS, allows rapid and reliable digestion by lipases facilitating drug partitioning in the solubilising phases (40). In this context two products of the immunosuppressant drug cyclosporine A may be mentioned: Neoral®, forming a microemulsion upon dispersion, and its predecessor Sandimmun® which produces a coarse emulsion. Clinically, Neoral® has shown improved bioavailability and dose linearity compared with the coarse emulsion (40,41). Although this finding stimulated the development of similar SNEDDS, the question whether dispersion alone can determine the performance of LBDDS, has not been sufficiently answered yet. It should be noted that Neoral® in addition contains excipients which have been shown to interact with P-glycoprotein and cytochrome P450. Therefore, factors other than dispersion related, cannot be ruled out (42).
Furthermore, dispersion cannot be assessed for non-dispersing LBDDS such as for formulations based on pure triglycerides. In this case the drug transfer from the lipid into the test medium rather than the dispersion characteristics might be more relevant to assess, which requires the choice of an appropriate test medium. During the last decades, the thorough physicochemical characterisation of human gastrointestinal fluids allowed the development of biorelevant media closer matching the in vivo conditions compared with the rather simple compendial media (43–45). The proposed biorelevant media contain physiological relevant amounts of buffer, lecithin and bile salts mimicking either the fasted or fed state (Table I) and have been subject of recent reviews (46,47).
Table I.
Composition of the Fasted- and Fed-State Simulated Intestinal Fluids Proposed for Biorelevant Dissolution Testing (FaSSIF-V2 and FeSSIF-V2)
| Composition/property | FaSSIF-V2 | FeSSIF-V2 |
|---|---|---|
| Sodium taurocholate (mM) | 3 | 10 |
| Lecithin (mM) | 0.2 | 2 |
| Maleic acid (mM) | 19.12 | 55.02 |
| Sodium chloride (mM) | 68.62 | 125.5 |
| Sodium hydroxide | 34.8 | 81.65 |
| Glyceryl monooleate (mM) | – | 5 |
| Sodium oleate | – | 0.8 |
| Osmolality (mOsm kg−1) | 180 ± 10 | 390 ± 10 |
| Buffer capacity (mmol l−1∆pH) | 10 | 25 |
| pH | 6.5 | 5.8 |
Adapted from Jantratid et al. (49)
Despite the progress in the development of dissolution media the major drawback of most dissolution media is the fact that they are rather static systems that have not been designed to reflect the dynamic changes in the GIT occurring after ingestion of a meal. In contrast, lipid digestion is a highly dynamic process during which amphiphilic species are generated, colloidal structures appear and change over time, which will in turn affect drug distribution and, finally, drug absorption. In the case of LBDDS that are often susceptible to digestion before they deliver the co-administered drug, a dynamic model appears therefore more appropriate.
IN VITRO LIPOLYSIS
In vitro lipolysis is typically carried out in a thermo-controlled reaction vessel containing digestion medium representative of either the fasted or the fed state (Fig. 1).
Fig. 1.
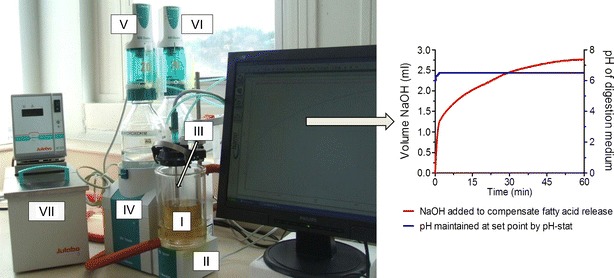
The left panel shows the typical lipolysis set up. The thermo-jacketed reaction vessel (I) contains the digestion medium and is stirred constantly by the magnetic stirrer (II). The pH-electrode (III) measures the pH and temperature of the lipolysis medium. The pH-stat unit (IV) regulates the dispensing of sodium chloride (V) and calcium chloride (VI). The water bath (VII) maintains the temperature of the lipolysis medium at 37°C. The right panel depicts the typical profile of a lipolysis experiment where the pH of the lipolysis medium is maintained constant (blue line). The red line represents the cumulative consumption of NaOH to titrate the liberated fatty acids during in vitro lipolysis
Although the composition varies between different laboratories (Table II), the digestion medium commonly comprises an aqueous buffer solution, supplemented by bile salts, phospholipids and NaCl (48). The concentrations of these compounds are defined by the physiological conditions prevailing in the fasted or fed state. The pH of the medium is monitored by a pH-sensitive electrode connected to a computer-controlled pH-stat device capable of maintaining a predefined pH value via titration with NaOH. Upon addition of pancreatic lipase and co-lipase the hydrolysis of triglycerides and other digestible excipients present in the formulations is initiated. As a result of the enzymatic hydrolysis, free fatty acids are released into the digestion medium leading to a drop in the pH. The change in the pH is immediately compensated by the titration with NaOH. In order to reduce the experimental error and to obtain reproducible results, the concentration of the NaOH solution needs to be adequate to compensate for the release of fatty acids expected to be generated from the amount of lipids during lipolysis. In case of a NaOH concentration chosen too low relative to the amount of substrate, this will result in the dilution of the digestion medium. Since the solubilisation capacity for a drug is a function of the bile salt concentration in the medium, dilution will lead to an underestimation of drug solubilisation. On the other hand, too-concentrated NaOH solutions bear the risk of over titration and the erroneous reading of small volumes of the titrant.
Table II.
Examples of In Vitro Lipolysis Conditions
| Volume (ml) | Duration (min) | pH | Buffer | Lipase | Bile/phospholipid | Calcium | Substrate | References |
|---|---|---|---|---|---|---|---|---|
| 9 | 12 | 6.5 | Tris-maleate (2 mM) | Pancreatin (24 TBU/ml) | NaTDC (8 mM) | 10 mM | 0.45 ml TG emulsion | (50) |
| 5 | 8.5 | Tris-maleate (50 mM) | Porcine pancreas (168–280 TBU/ml) | NaTDC (0–30 mM/lecithin) | 0–30 mM | 0–5% (w/v) of TG emulsion (10%) emulsified with PL | (34) | |
| 10 | 6–9 | Tris-maleate (2 mM) | Purified from human pancreatic fluid (1–8 TBU/ml) | NaTC NaTDC or NaCDC (6 mM) | 10 mM | 0.5 ml olive oil emulsified with gum Arabic | (24) | |
| 40 | 30 | 6.8–7.4 | Tris-maleate (50 mM) | Porcine pancreatin ((8× USP) 1,000 IU/ml) | NaTC (5 mM l-α-PC) | 5 mM | 0.2–1 g TG | (51,52) |
| 37.5 | 20–120 | 7.0 | Phosphate (5 mM) | Porcine pancreas type II (L3126, Sigma; (100–400 U/mg, 2.4 mg/ml) | Porcine bile extract (20 mM) | 10 mM | 0.5% TG emulsion,different emulsifiers,1% | (53) |
| 300 | 40–90 | 6.5 | Tris-maleate (2 mM) | Porcine pancreatin (3× USP; 300–800 USP units/ml) | Porcine bile extract (5–30 mM)/1–5 mM PC | Continuous addition (0.045–0.181 mmol/min) | 15 mM -30 mM TG,1-3 g SNEDDS, | (37,48,54,55,56) |
| 10–40 | 30–60 | 7.5 | Tris-maleate (50 mM) | Porcine pancreatin (8× USP; 1,000 TBU/ml) | NaTDC (5–20 mM)/1.25–5 mM lecithin (60% PC) | 5 mM | 1 g SEDDS , TG | (57,58) |
| 100 | 40 | 6.5 | Tris-maleate (50 mM) | Pancreatin extract (8 TBU/mg) | Bile salts (5 mM)/1.25 mM lecithin | 5 mM | 1 g SEDDS | (59) |
| 20 | 35 | 7.5 | Tris-maleate 50 mM | Porcine pancreatic lipase 40,000 IU/g 4,000 TBU/ml | NaTDC 5 mM/1.25 mM lecithin (92% PC) | 5 mM | 0.21 g TG, sub-microemulsion | (60) |
FFA free fatty acids, NaTC sodium taurocholate, NaTDC sodium deoxytaurocholate, NaCDC sodium chenodeoxycholate, PC phosphatidylcholine, PL phospholipid, TBU Tributyrin unit (1 TBU equals the amount of enzyme that can liberate 1 mol of fatty acid from tributyrin/min) (69)
Due to the stoichiometry of the hydrolysis reaction, 2 mol NaOH are needed for the hydrolysis of 1 mol triglyceride. Complete hydrolysis of the 2-monoglycerides to glycerol and a third mole fatty acid can be mediated either by isomerisation of 2-monoglycerides to 1-monoglycerides in alkaline medium (61) or by hydrolysis directly at the Sn2 position by the less specific carboxyl ester hydrolase (cholesterol esterase) (62). As the velocity of 2-monoglyceride absorption by the intestine is faster compared with the isomerisation reaction (17), the relative generation of glycerol during triglyceride hydrolysis is rather limited, accounting for approximately 22% of the overall digestion products (20).
During in vitro lipolysis samples are withdrawn, followed by the addition of a lipase inhibitor (commonly 4-bromobenzeneboronic acid) to stop further lipase activity within the sample. Thereafter, the samples are analysed either directly or, more frequently, after a centrifugation or ultracentrifugation step separating the sample in two or more phases depending on the sampling time point and on the composition of the formulation (Fig. 2) (63–65). At the beginning of in vitro lipolysis, when more undigested lipids are present, it is more likely to obtain an oil phase as the top layer. However, the presence of a lipid layer also depends on the initial composition of the investigated LBDDS. SNEDDS and SMEDDS commonly produce no lipid layer due to their fine dispersion and reduced lipid content. In contrast, non-dispersing LBDDS (e.g. pure soy bean oil) generate a lipid layer on top of the lipolysis medium. Provided the lipids employed are digestible by the lipases the oil layer diminishes with the progress of in vitro lipolysis. At the same time, the amount of pellet is increasing due to the precipitation of fatty acid as calcium soaps (see below).
Fig. 2.
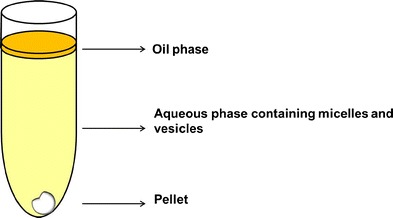
Schematic of the lipolysis medium after ultracentrifugation. The lipolysis medium separated into at least two phases: the pellet, containing mostly calcium soaps of fatty acids and precipitated drug; the aqueous phase containing the drug solubilised in mixed micelles and vesicles; the oil phase containing undigested lipids. Redrawn and adapted from reference (70)
The physiological pH range of the intestine (pH 6.1–7.3 in the fasted and pH 5–6.6 in the fed state, respectively) (47) includes the pH optimum of the pancreatic lipase/co-lipase complex (reported between pH 6 and 9) (20,31). Consequently, in vitro lipolysis should be conducted closely to these values to reflect the in vivo conditions. From an experimental point of view however, the chosen pH needs to be high enough to present the fatty acids in a titratable ionised form (24). The apparent pKa of long-chain fatty acids in lipolysis medium is lower compared with that in water (e.g. approximately pKa 6.5 compared with pKa 8 for oleic acid) probably due to the presence of mixed micelles and Ca2+ ions (48,66). Thus, choosing a physiological pH between 6 and 7, the free fatty acids can—at least to some extent—be titrated (48,54). It has been recommended to increase the pH of the lipolysis medium after the actual lipolysis experiment to values above the pKa of the fatty acids (e.g. to pH 9) to titrate all the fatty acids liberated during in vitro lipolysis which might have been obscured at lower pH (67).
The titration of fatty acids with NaOH is an indirect and unspecific method to monitor lipid hydrolysis. It is noteworthy that a drop in the pH (resulting in an increased NaOH consumption) can partly originate from the absorption of CO2 by the lipolysis medium (68). Other sources of fatty acids might derive from the degradation of proteins, the hydrolysis of impurities present in the crude extracts of bile and pancreas, and the hydrolysis of phospholipids present in the lipolysis medium (63,68). In order to compensate for this ‘background lipolysis’, blank experiments are carried out that involve the ‘digestion’ of pure (i.e. formulation free) lipolysis medium and subsequent subtraction of the recorded NaOH volumes from the formulation-containing lipolysis experiments (55,57).
Since in vitro lipolysis generates fatty acids that potentially inhibit further enzymatic activity of the pancreatic lipase by accumulation at the lipid surface, the end products of in vitro lipolysis have to be removed. In vivo this removal is accomplished by the absorption of fatty acids by the intestinal wall. In lieu of this physiological mechanism, the addition of Ca2+-ions precipitates the fatty acids as calcium soaps during in vitro lipolysis (22). For the addition of Ca2+-ions, two main strategies are followed: either the addition as a bolus at the start of lipolysis (68,51,69), or the continuous Ca2+-addition by a peristaltic pump, also referred to as the ‘dynamic’ in vitro lipolysis model (48,54). The bolus Ca2+-addition (commonly 5 mM) results in a very fast hydrolysis rate with lipolysis being almost complete within the initial 10 min (57) while the continuous Ca2+-addition (e.g. 0.181 mmol/min) allows a controlled lipolysis rate (48,54).
Another difference between the currently employed digestion models arises from the source of bile acids. While the concentration of bile is similar (5–30 mM) throughout the employed lipolysis models, some groups utilise pure taurocholic acid (51,52,71) or taurodeoxycholic acid (57,58,72), whereas others employ a crude (porcine) bile extract (47,73,56). The utilisation of pure bile acids renders the composition of the lipolysis medium more reproducible compared with the more variable crude extract, but lacks the complex composition of human bile (63). Recent studies have shown that the bile acid concentration rather than the structure of the bile acids affects the drug solubilisation in artificial intestinal fluids suggesting that the use of a single bile acid could reflect the physiological situation (74,75). However, these studies were not carried out during in vitro lipolysis where the phenomena at the lipid/water interface have been shown to be sensitive to the different structures and hydrophobicities of a range of bile acids potentially interfering with the lipase activity (33). Therefore, compared with pure bile salts, crude porcine bile extract might provide some advantage, including economic considerations.
The ultimate objective of the in vitro lipolysis model is to predict the in vivo performance of LBDDS a priori from the corresponding in vitro design parameters, hence to achieve in vitro/in vivo correlations. So far, the development of LBDDS has been based on a trial and error approach rather than on rational design. Probably the two most important reasons for this are the heterogeneity of LBDDS, comprising of up to five and more excipients (4) and the intricate interactions of the formulation components with the complex endogenous digestion apparatus. Although the knowledge of the physico-chemical processes underlying lipolysis has grown during the last decades, one of the most probing questions remains yet unanswered: what are the determinants of successful and predictable drug absorption from LBDDS and their digestion products? Several in vivo studies have been able to demonstrate rank order in vitro/in vivo correlations (52,76,77). However, in vivo studies are time consuming and costly. Therefore, it would be desirable to apply predictable in vitro models to the design and assessment of LBDDS.
The currently employed lipolysis models emphasise the intestinal digestion while giving only little attention to the pre-intestinal lipolysis. As mentioned above the activity of gastric lipase can account for approximately 10–25% of the total lipid digestion and the enzyme remains active in the intestine (20). Since the products of gastric lipolysis are surface active, facilitating the emulsification of the lipid before entering the intestine for further lipolysis (17), the gastric lipolysis step might be of greater importance for the assessment of LBDDS than previously assumed. The impact of gastric lipase activity is confirmed further by the observation of sustained lipid absorption in patients suffering from cystic fibrosis despite the incompetence of the patient’s pancreas to secrete lipase (78,79). Further studies will help elucidate the importance of pre-pancreatic lipolysis as part of in vitro digestion models.
PERMEABILITY STUDIES WITH LBDDS
So far none of the current in vitro lipolysis models has routinely accommodated an absorption model following the in vitro digestion step. The literature offers only very limited information regarding in vitro drug permeability and absorption following the digestion of LBDDS. The effects of some formulation parameters (oil structure, surfactant HLB, lipid/surfactant ratio) on the intestinal permeability and drug release kinetics from LBDDS were investigated by Buyukozturk et al. (80) and the effect of SMEDDS on tight junctions was evaluated by Sha et al. (81). Using Caco-2 cell line monolayers the studies revealed useful considerations for the design of LBDDS, including the potential toxicity of these formulations towards Caco2-cell lines, as evident from the observed disruption of the tight junctions. However, in these studies the investigated formulations were diluted with rather simple buffer solutions devoid of bile salts and lipases, making the extrapolation to the complex intestinal environment difficult. In a study by Dahan et al. (52), the in vitro lipolysis model was able to predict the bioavailability of dexamethason and griseofulvin. On the other hand, when the intestinal permeability of the drugs was assessed from samples obtained during in vitro lipolysis in an Ussing chamber, the ex vivo model failed to predict the performance seen in vivo and during in vitro lipolysis (52). It appears that suitable in vitro models capable of withstanding the harsh conditions of representative intestinal conditions have yet to be established. This will ultimately provide a tool for the assessment of LBDDS needed to create a guide for a more rational design of LBDDS.
CHARACTERISATION TECHNIQUES DURING IN VITRO LIPOLYSIS
The rationale of LBDDS is to increase the absorption of poorly water-soluble, lipophilic compounds by improved solubilisation mediated by the interaction of the drug with endogenous and exogenous surfactants, lipids and their digestion products. Although research in lipid digestion is well established (22,61,66), the interest of pharmaceutical scientists in this area is relatively new. The solubility of a drug is intrinsically linked to the fate of the formulation and drug distribution is dictated by the type of colloidal phases generated during digestion (56,82–85). The characterisation techniques (Table III) have been focussed either on the evolution of the colloidal digestion products or on the solubility and distribution of the co-administered drug. The following section will therefore summarise the employed methods using the same differentiation.
Table III.
Recent Techniques Employed for the Characterisation of In Vitro Lipolysis
| In vitro lipolysis | Substrate/drug | Technique | Comment | References |
|---|---|---|---|---|
| Phosphate buffer at pH 6.8, 10 mM bile salts from bile extract 10 mM Ca2+ , porcine pancreatin extract and 150 U/ml lipase activity | 1.5% olive oil emulsion (15 mM triolein)/1 mM tempol benzoate | Drug—EPR spectroscopy, lipids and digestion; products—HPTLC | Quantification of spin label in digestion phases; quantification of lipids by HPTLC | (87) |
| Tris buffer at pH 7.4, 13 mM NaTDC, 8 mM Ca2+, porcine pancreatin extract and 20 U/mg lipase activity | 5% oil emulsion (glyceryl trioctanoate and glyceryl trioleate)/ergosterol, progesterone and vitamin D3 | Multiplex CARS microspectroscopy | Label-free imaging and quantification of drug and lipid digestion products | (88) |
| Tris maleate at pH 6.5, 5 mM bile salts from porcine bile extract, continuous Ca2+ addition (0.045 mM/min), porcine pancreatin extract, 350 U/ml lipase activity | 1 g SNEDDS—55% medium-chain and long-chain lipids, 35% CrRH40 and 10% ethanol | Cryo SEM | Structure of dispersed SNEDDS | (37) |
| tris maleate pH 6.5, 5 mM bile salts from bile extract, continuous Ca2+ addition (0.045 mM/min), porcine pancreatin extract, 800 U/ml lipase activity | 3 g SNEDDS—30% sesame oil, 30% mixed LC glycerides, 30% CrRH40 and 10% ethanol | Cryo TEM | Structures of dispersed SNEDDS and digestion products | (56) |
| Tris maleate pH 7.5, 5 mM/20 mM NaTDC, 5 mM Ca2+, porcine pancreatin ( 8× USP) and 50 TBU/ml | 0.25 g medium-chain or long-chain lipid | HPTLC | Quantification of mono-, di- and triglycerides and fatty acids during digestion | (86) |
| Tris maleate at pH 6.5, 5 mM bile salts from bile extract, continuous Ca2+ addition (0.045 mM/min), porcine pancreatin extract and 800 U/ml lipase activity | 3 g SNEDDS—30% sesame oil, 30% mixed LC glycerides, 30% CrRH40, and 10% ethanol | Bench-top SAXS and cryo TEM | Structures of dispersed SNEDDS and digestion products | (82) |
| Tris maleate pH 7.5, 5 mM NaTDC, 10 mM Ca2+, porcine pancreatin (8× USP), 20,000 TBU | Phytantriol, glyceryl monooleate, SNEDDS—30% sesame oil, 30% mixed LC glycerides, 30% CrRH40 and 10% ethanol | Synchroton SAXS | Real-time observation of dispersed SNEDDS and digestion products | (89) |
Cryo SEM/TEM cryogenic scanning/transmission electron microscopy, EPR electron paramagnetic resonance, HPTLC high-performance thin layer chromatography, NaTDC sodium taurodeoxycholate, SAXS small angle X ray scattering, SNEDDS self-nanoemulsifying drug delivery system, TBU Tributyrin unit (1 TBU equals the amount of enzyme that can liberate 1 mol of fatty acid from tributyrin/min (69))
Methods for the Characterisation of the Lipids and Colloidal Phases
Quantification of the different digestion products, including mono-, di-, triglycerides, fatty acids, bile salts and phospholipids can be carried out by use of HPLC, GC or HPTLC (69,86). This provides an alternative to the indirect measure of digestion by NaOH consumption (69,86).
The bulk structure of diluted SNEDDS has commonly been investigated by dynamic light scattering (DLS) (56,90). In DLS an incident laser beam is scattered by dispersed particles in a sample. The fluctuation in the scattering signal, caused by Brownian motion of the particles, is measured as a function of time. From the correlation function, the particle size is calculated as an approximation from the diffusion coefficient. Although DLS does not allow a direct size measurement, it is a convenient and fast screening tool for particle size measurements including SNEDDS. However, other methods such as electron microscopy and X-ray scattering techniques (discussed below) might provide complementary information.
The rapid immobilisation of aqueous dispersions of SNEDDS by plunge-freezing in liquid nitrogen allowed the preservation of the dispersed particle in the bulk. The subsequent cryogenic electron micrographs of the frozen samples revealed the spherical shape of the dispersed SNEDDS and particle sizes between 30 and 40 nm (37).
Similarly, the utilisation of cryogenic transmission electron microscopy (cryo-TEM) with its superior resolution compared with cryo-SEM allowed the observation of morphological changes of SNEDDS during in vitro lipolysis (56,82). Before lipolysis was initiated, the medium was dominated by the presence of oil droplets (approximately 50 nm) and micelles formed by bile salts and phospholipids (approximately 5–10 nm). With progressing lipid digestion, unilamellar and multilamellar colloidal structures emerged at the expense of oil droplets while micelles were constantly observed. However, after 30 min in vitro lipolysis the number of the unilamellar vesicles declined and multilamellar vesicles disappeared. This behaviour was explained by the likely incorporation of the generated digestion products in micelles, thus forming swollen mixed micelles.
In order to elucidate the structures of the phases observed with cryo-TEM, small-angle X-ray scattering (SAXS) has been used during in vitro lipolysis of SNEDDS. SAXS is a method taking advantage of the scattering of X-rays impinging on macromolecules or self-assembled structures in the nanometre to micrometre range using scattering angles smaller than 10° (2Θ) for the detection of the scattered X-rays (91). This method revealed the steady development of a lamellar phase followed by the co-existence of lamellar and inverse hexagonal phases, the latter prevailing at the end of in vitro lipolysis (Fig. 3).
Fig. 3.
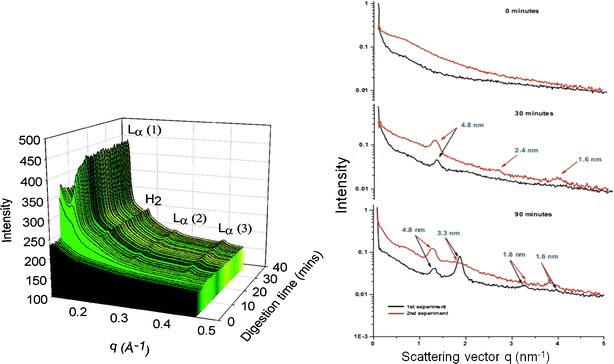
SAXS data using either synchrotron (left) or bench-top SAXS (right). The intensity of the scattered electrons is plotted versus the scattering vector (q), for synchrotron also versus time. The spectra illustrate the evolution of lamellar (L α) and hexagonal (H 2) phases during in vitro lipolysis. The arrows on the right panel denote the Bragg spacing for the lamellar phases at day = 1.6, 2.4, and 4.8 nm and at day = 3.3 nm for the hexagonal phase. At time points 0 min, no liquid crystals are visible, after 30 min a lamellar phase exists, and 90 min hexagonal and lamellar phases co-exist. Reprinted with permission from references (82,89)
It should be noted that for the described cryo-TEM and SAXS experiments, samples were withdrawn during lipolysis, providing snapshots of the dynamic process. Recently, online monitoring of the lyotropic phases generated during lipolysis of the same SNEDDS as described above was reported (92). Using synchrotron radiation the phases identified with bench-top SAXS were confirmed. Differences were observed in the temporal appearance of the phases which could be traced back to the slightly different lipolysis protocols: with the bolus addition of calcium a hexagonal phase was detected by synchrotron SAXS within 5–10 min whereas the same phase was observed from 60 min using continuous calcium addition and bench-top SAXS (Fig. 3).
Methods for the Characterisation of the Drug
With regard to the drug, the analytics carried out during in vitro lipolysis comprises the quantification by HPLC in the supernatant and pellet obtained after a centrifugation step (63,65). Traditionall,y samples of the lipolysis medium have been subjected to ultracentrifugation. In a recent systematic study, the separation of digestion phases of less lipophilic formulation by ultracentrifugation was comparable to conventional bench-top centrifugation enabling a higher sample turn-over for laboratories (93). However, the presence of an oil layer in case of more lipophilic LBDDS required ultracentrifugation for the effective separation of oil, aqueous phase, and pellet.
It is assumed that the drug solubilised in the aqueous phase is the fraction available for absorption, whereas precipitated drug has been considered undesirable since the solid drug requires a dissolution step potentially reducing bioavailability (2,57).
Recently, the solid state properties of the precipitates generated during in vitro lipolysis have gained increased interest. When the pellet obtained after ultracentrifugation of a SMEDDS subjected to in vitro lipolysis was investigated using XRPD and polarising light microscopy, the results indicated that the model drug cinnarizine precipitated in an amorphous form showing a much faster dissolution rate compared with the crystalline drug (94). In a similar study, amorphous halofantrine precipitated during in vitro lipolysis of two supersaturated SNEDDS based on either medium- or long-chain lipids (95). Despite substantial precipitation during in vitro lipolysis the bioavailability of halofantrine was not negatively affected after the administration of the same formulations to beagle dogs. Amorphous drug precipitation was recently shown for ketoconazole in intestinal aspirates (96). Assuming halofantrine precipitated also in an amorphous form in vivo, the rapid dissolution rate of the amorphous drug (as seen in vitro) could explain the retained bioavailability. From these initial studies, it appears that the propensity of drug to precipitate in vitro could be a misleading parameter for in vivo predictions of LBDDS without considering the solid state properties of the precipitate. Therefore, well-established solid state characterisation techniques might find increasing application for the assessment of lipid-based formulations providing valuable information supplementing in vitro lipolysis.
During in vitro digestion the co-administered drug partitions between the continuously changing colloidal phases which have different solubilisation capacity for the drug (84,97). The current standard method employed for the separation of the digestion phases are conventional centrifugation and ultracentrifugation which provide only an unrefined picture of drug distribution. The spatio-temporal patterns of drug distribution in the lipolysis medium might be more accessible by complimentary methods.
One of these methods is electron paramagnetic resonance (EPR) spectroscopy, also known as electron spin resonance spectroscopy. Similar to nuclear magnetic resonance, a magnetic moment (‘spin’) is mandatory for EPR spectroscopy. In the case of EPR, unpaired electrons are excited by a micro- or radio-wave source in a changing magnetic field (98). When in resonance the electron spin reverses, causing some of the electromagnetic radiation to be reflected which corresponds to the EPR signal. The signal is sensitive to alterations in the microenvironment (such as in the polarity) of the paramagnetic compound. As the polarity in the direct vicinity of a compound changes during in vitro lipolysis, EPR spectroscopy was successfully employed to monitor the distribution of the lipophilic, paramagnetic compound tempol benzoate (TB) between undigested oil, aqueous phase and mixed micelles (87). The dynamics of drug distribution was most evident during the initial 20 min of in vitro lipolysis (Fig. 4), corresponding to the digestion of approximately 30% of the oil (as determined by HPTLC). Although a promising, non-invasive technique, a more widespread utilisation of EPR might be restricted due to the requirement of a paramagnetic compound or the need for adequate labelling.
Fig. 4.
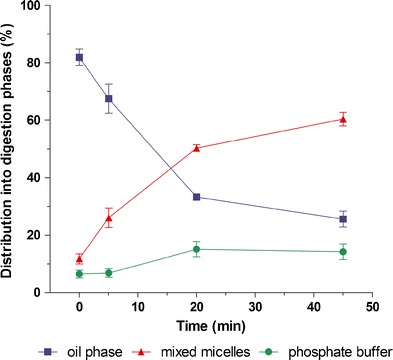
Distribution of the spin probe tempol benzoate during in vitro lipolysis of 1.5% (v/v) olive oil as determined by electron paramagnetic spin resonance spectroscopy, EPR, in the presence of 10 mM bile acids. Redrawn and adapted from Rübe et al. (87)
In contrast, coherent anti-Stokes Raman scattering (CARS) microspectroscopy does not rely on a specific label (99). Images are obtained from molecular Raman vibrations which render the method highly chemical-specific. CARS spectroscopy is a coherent method resulting in signal strengths several orders of magnitude greater than spontaneous Raman scattering (100). The advantage of high sensitivity and spatial resolution has sparked the interest in CARS as a non-invasive tool to probe biological samples such as living cells in real time (99). Recently, multiplex CARS microspectroscopy has been employed to characterise in vitro lipolysis (88). The method was suitable to locate and quantify several lipophilic drugs during progressing lipolysis. As an example, from the increasing concentration of progesterone in the diminishing oil droplets (Fig. 5c), the authors concluded that the investigated vehicle was ill-suited as a delivery system for progesterone since the drug did not distribute into the digestion products, probably due better solubility in the triglyceride.
Fig. 5.

The progress of triglyceryl trioctanoate digestion by pancreatic lipase observed over time by bright field microscopy (a) and CARS (b,c). False colour images of triglyceryl trioctanoate (red) and progesterone (green) revealed diminishing triglyceride content and increasing progesterone concentration. The concentration of progesterone (blue) is provided in the colour bar, corresponding to the false colour images of progesterone within the lipid droplet (c). The initial progesterone concentration was 95 mM, the scale bar equals 10 μm. Reprinted with permission from Day et al. (93). Copyright (2010) American Chemical Society
An additional benefit of CARS arises from its ability to visualise and quantify the digestion of the lipids (88) (Fig. 5b). Further improvements might enable this promising technique to be conducted also under more realistic hydrodynamic conditions and in vivo. This would eventually provide insights into the mechanisms of drug and lipid absorption on a cellular level.
CONCLUSIONS
Lipid-based drug delivery system has become increasingly popular for the delivery of poorly water-soluble, lipophilic compounds due their potential to improve the bioavailability of these drugs. So far, a rational design for these delivery systems has not yet been established. The current work tried to summarise the established methods and show the trends and pitfalls in the characterisation of in vitro lipolysis indicating the need for a multi-disciplinary approach which seems to be the key to further our knowledge about these promising delivery systems.
REFERENCES
- 1.Melander A. Influence of food on the bioavailability of drugs. Clin Pharmacokinet. 1978;3(5):337–351. doi: 10.2165/00003088-197803050-00001. [DOI] [PubMed] [Google Scholar]
- 2.Humberstone AJ, Charman WN. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv Drug Deliv Rev. 1997;25(1):103–128. doi: 10.1016/S0169-409X(96)00494-2. [DOI] [Google Scholar]
- 3.Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58(3):173–182. doi: 10.1016/j.biopha.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Strickley RG. Solubilizing excipients in oral and injectable formulations. Pharm Res. 2004;21(2):201–230. doi: 10.1023/B:PHAM.0000016235.32639.23. [DOI] [PubMed] [Google Scholar]
- 5.Hauss DH. Oral lipid-based formulations. Adv Drug Deliv Rev. 2007;59(7):667–676. doi: 10.1016/j.addr.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 6.Anton N, Vandamme T. Nano-emulsions and micro-emulsions: clarifications of the critical differences. Pharm Res. 2011;28(5):978–985. doi: 10.1007/s11095-010-0309-1. [DOI] [PubMed] [Google Scholar]
- 7.Grove M, Nielsen J, Pedersen G, Müllertz A. Bioavailability of seocalcitol (IV): evaluation of lymphatic transport in conscious rats. Pharm Res. 2006;23(11):2681–2688. doi: 10.1007/s11095-006-9109-z. [DOI] [PubMed] [Google Scholar]
- 8.Holm R, Müllertz A, Pedersen GP, Kristensen HG. Comparison of the lymphatic transport of halofantrine administered in disperse systems containing three different unsaturated fatty acids. Pharm Res. 2001;18(9):1299–1304. doi: 10.1023/A:1013037927882. [DOI] [PubMed] [Google Scholar]
- 9.Khoo S-M, Prankerd RJ, Edwards GA, Porter CJH, Charman WN. A physicochemical basis for the extensive intestinal lymphatic transport of a poorly lipid soluble antimalarial, halofantrine hydrochloride, after postprandial administration to dogs. J Pharm Sci. 2002;91(3):647–659. doi: 10.1002/jps.10045. [DOI] [PubMed] [Google Scholar]
- 10.Lind ML, Jacobsen J, Holm R, Müllertz A. Intestinal lymphatic transport of halofantrine in rats assessed using a chylomicron flow blocking approach: the influence of polysorbate 60 and 80. Eur J Pharm Sci. 2008;35(3):211–218. doi: 10.1016/j.ejps.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 11.Constantinides PP, Wasan KM. Lipid formulation strategies for enhancing intestinal transport and absorption of P-glycoprotein (P-gp) substrate drugs: in vitro/in vivo case studies. J Pharm Sci. 2007;96(2):235–248. doi: 10.1002/jps.20780. [DOI] [PubMed] [Google Scholar]
- 12.Tompkins L, Lynch C, Haidar S, Polli J, Wang H. Effects of commonly used excipients on the expression of CYP 3A4 in colon and liver cells. Pharm Res. 2010;27(8):1703–1712. doi: 10.1007/s11095-010-0170-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kossena G, Charman W, Wilson C, O’Mahony B, Lindsay B, Hempenstall J, et al. Low dose lipid formulations: effects on gastric emptying and biliary secretion. Pharm Res. 2007;24(11):2084–2096. doi: 10.1007/s11095-007-9363-8. [DOI] [PubMed] [Google Scholar]
- 14.Branca F, Nikogosian H, Lobstein T, editors. The challenge of obesity in the WHP European region and the strategies for response. Copenhagen, Denmark: WHO; 2007. [Google Scholar]
- 15.Carey MC, Small DM, Bliss CM. Lipid digestion and absorption. Annu Rev Physiol. 1983;45(1):651–677. doi: 10.1146/annurev.ph.45.030183.003251. [DOI] [PubMed] [Google Scholar]
- 16.DeNigris SJ, Hamosh M, Kasbekar DK, Lee TC, Hamosh P. Lingual and gastric lipases: species differences in the origin of prepancreatic digestive lipases and in the localization of gastric lipase. Biochim Biophys Acta. 1988;959(1):38–45. doi: 10.1016/0005-2760(88)90147-6. [DOI] [PubMed] [Google Scholar]
- 17.Phan CT, Tso P. Intestinal lipid absorption and transport. Front Biosci. 2001;6:d299–d319. doi: 10.2741/Phan. [DOI] [PubMed] [Google Scholar]
- 18.Golding M, Wooster TJ. The influence of emulsion structure and stability on lipid digestion. Curr Opin Colloid Interface Sci. 2010;15(1–2):90–101. doi: 10.1016/j.cocis.2009.11.006. [DOI] [Google Scholar]
- 19.Miled N, Canaan S, Dupuis L, Roussel A, Rivière M, Carrière F, et al. Digestive lipases: from three-dimensional structure to physiology. Biochimie. 2000;82(11):973–986. doi: 10.1016/S0300-9084(00)01179-2. [DOI] [PubMed] [Google Scholar]
- 20.Embleton JK, Pouton CW. Structure and function of gastro-intestinal lipases. Advanced Drug Delivery Reviews. 1997;25(1):15–32. doi: 10.1016/S0169-409X(96)00488-7. [DOI] [Google Scholar]
- 21.Lowe ME. Structure and function of pancreatic lipase and colipase. Annu Rev Nutr. 1997;17:141–158. doi: 10.1146/annurev.nutr.17.1.141. [DOI] [PubMed] [Google Scholar]
- 22.Patton JS, Carey MC. Watching fat digestion. Science. 1979;204(4389):145–148. doi: 10.1126/science.432636. [DOI] [PubMed] [Google Scholar]
- 23.Porter CJH, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(March):231–248. doi: 10.1038/nrd2197. [DOI] [PubMed] [Google Scholar]
- 24.Patton JS, Carey MC. Inhibition of human pancreatic lipase–colipase activity by mixed bile salt-phospholipid micelles. Am J Physiol Gastrointest Liver Physiol. 1981;241(4):G328–G336. doi: 10.1152/ajpgi.1981.241.4.G328. [DOI] [PubMed] [Google Scholar]
- 25.Erlanson-Albertsson C. Pancreatic colipase. Structural and physiological aspects. Biochim Biophys Acta. 1992;1125(1):1–7. doi: 10.1016/0005-2760(92)90147-N. [DOI] [PubMed] [Google Scholar]
- 26.Bezzine S, Ferrato F, Ivanova MG, Lopez V, Verger R, Carrière F. Human pancreatic lipase: colipase dependence and interfacial binding of lid domain mutants. Biochemistry (Mosc) 1999;38(17):5499–5510. doi: 10.1021/bi982601x. [DOI] [PubMed] [Google Scholar]
- 27.Delorme V, Dhouib R, Canaan S, Fotiadu F, Carrière F, Cavalier J-F. Effects of surfactants on lipase structure, activity, and inhibition. Pharm Res. 2011;28(8):1831–1842. doi: 10.1007/s11095-010-0362-9. [DOI] [PubMed] [Google Scholar]
- 28.Larsson A, Erlanson-Albertsson C. The importance of bile salt for the reactivation of pancreatic lipase by colipase. Biochim Biophys Acta. 1983;750(1):171–177. doi: 10.1016/0005-2760(83)90217-5. [DOI] [PubMed] [Google Scholar]
- 29.Bloom B, Chaikoff IL, Reinhardt WO. Intestinal lymph as a pathway for transport of absorbed fatty acids of different chain lengths. Am J Physiol Gastrointest Liver Physiol. 1951;166:451–455. doi: 10.1152/ajplegacy.1951.166.2.451. [DOI] [PubMed] [Google Scholar]
- 30.Bergström S, Blomstrand R, Borgström B. Route of absorption and distribution of oleic acid and triolein in the rat. Biochem J. 1954;58(4):600–604. doi: 10.1042/bj0580600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Armand M, Borel P, Ythier P, Dutot G, Melin C, Senft M, et al. Effects of droplet size, triacylglycerol composition, and calcium on the hydrolysis of complex emulsions by pancreatic lipase: an in vitro study. J Nutr Biochem. 1992;3(7):333–341. doi: 10.1016/0955-2863(92)90024-D. [DOI] [Google Scholar]
- 32.Mansbach CM, Cohen RS, Leff PB. Isolation and properties of the mixed lipid micelles present in intestinal content during fat digestion in man. J Clin Invest. 1975;56(4):781–791. doi: 10.1172/JCI108156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wickham M, Garrood M, Leney J, Wilson PDG, Fillery-Travis A. Modification of a phospholipid stabilized emulsion interface by bile salt: effect on pancreatic lipase activity. J Lipid Res. 1998;39(3):623–632. [PubMed] [Google Scholar]
- 34.Alvarez FJ, Stella VJ. The role of calcium ions and bile salts on the pancreatic lipase-catalyzed hydrolysis of triglyceride emulsions stabilized with lecithin. Pharm Res. 1989;6(6):449–457. doi: 10.1023/A:1015956104500. [DOI] [PubMed] [Google Scholar]
- 35.Khoo S-M, Humberstone AJ, Porter CJH, Edwards GA, Charman WN. Formulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrine. Int J Pharm. 1998;167(1–2):155–164. doi: 10.1016/S0378-5173(98)00054-4. [DOI] [Google Scholar]
- 36.Nielsen FS, Petersen KB, Müllertz A. Bioavailability of probucol from lipid and surfactant based formulations in minipigs: influence of droplet size and dietary state. Eur J Pharm Biopharm. 2008;69(2):553–562. doi: 10.1016/j.ejpb.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 37.Thomas N, Müllertz A, Graf A, Rades T. Influence of lipid composition and drug load on the in vitro performance of self-nanoemulsifying drug delivery systems. J Pharm Sci. 2012;101(5):1721–1731. doi: 10.1002/jps.23054. [DOI] [PubMed] [Google Scholar]
- 38.Hong J-Y, Kim J-K, Song Y-K, Park J-S, Kim C-K. A new self-emulsifying formulation of itraconazole with improved dissolution and oral absorption. J Control Release. 2006;110(2):332–338. doi: 10.1016/j.jconrel.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 39.Li XR, Yuan QA, Huang YQ, Zhou YX, Liu Y. Development of silymarin self-microemulsifying drug delivery system with enhanced oral bioavailability. AAPS PharmSciTech. 2010;11(2):672–678. doi: 10.1208/s12249-010-9432-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mueller EA, Kovarik JM, van Bree JB, Tetzloff W, Grevel J, Kutz K. Improved dose linearity of cyclosporine pharmacokinetics from a microemulsion formulation. Pharm Res. 1994;11(2):301–304. doi: 10.1023/A:1018923912135. [DOI] [PubMed] [Google Scholar]
- 41.Kovarik JM, Mueller EA, Van Bree JB, Tetzloff W, Kutz K. Reduced inter- and intraindividual variability in cyclosporine pharmacokinetics from a microemulsion formulation. J Pharm Sci. 1994;83(3):444–446. doi: 10.1002/jps.2600830336. [DOI] [PubMed] [Google Scholar]
- 42.Wandel C, Kim RB, Stein CM. “Inactive” excipients such as Cremophor® can affect in vivo drug disposition. Clin Pharmacol Ther. 2003;73(5):394–396. doi: 10.1016/S0009-9236(03)00010-9. [DOI] [PubMed] [Google Scholar]
- 43.Dressman JB, Berardi RR, Dermentzoglou LC, Russell TL, Schmaltz SP, Barnett JL, et al. Upper gastrointestinal (GI) pH in young, healthy men and women. Pharm Res. 1990;7(7):756–761. doi: 10.1023/A:1015827908309. [DOI] [PubMed] [Google Scholar]
- 44.Kleberg K, Jacobsen J, Müllertz A. Characterising the behaviour of poorly water soluble drugs in the intestine: application of biorelevant media for solubility, dissolution and transport studies. J Pharm Pharmacol. 2010;62(11):1656–1668. doi: 10.1111/j.2042-7158.2010.01023.x. [DOI] [PubMed] [Google Scholar]
- 45.Klein S. The use of biorelevant dissolution media to forecast the in vitro/in vivo performance of a drug. AAPS J. 2010;12(3):397–406. doi: 10.1208/s12248-010-9203-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Di Maio S, Carrier RL. Gastrointestinal contents in fasted state and post-lipid ingestion: in vivo measurements and in vitro models for studying oral drug delivery. J Control Release. 2011;151(2):110–122. doi: 10.1016/j.jconrel.2010.11.034. [DOI] [PubMed] [Google Scholar]
- 47.Kleberg K, Jacobsen F, Fatouros DG, Müllertz A. Biorelevant media simulating fed state intestinal fluids: colloid phase characterization and impact on solubilization capacity. J Pharm Sci. 2010;99(8):3522–3532. doi: 10.1002/jps.22122. [DOI] [PubMed] [Google Scholar]
- 48.Zangenberg NH, Müllertz A, Kristensen HG, Hovgaard L. A dynamic in vitro lipolysis model: I. Controlling the rate of lipolysis by continuous addition of calcium. Eur J Pharm Sci. 2001;14(2):115–122. doi: 10.1016/S0928-0987(01)00169-5. [DOI] [PubMed] [Google Scholar]
- 49.Jantratid E, Janssen N, Reppas C, Dressman JD. Dissolution media simulating conditions in the proximal human gastrointestinal tract: An update. Pharm Res. 2008;25(7):1663–1676. doi: 10.1007/s11095-008-9569-4. [DOI] [PubMed] [Google Scholar]
- 50.Reymond J-P, Sucker H, Vonderscher J. In vivo model for ciclosporin intestinal absorption in lipid vehicles. Pharm Res. 1988;5(10):677–679. doi: 10.1023/A:1015939307478. [DOI] [PubMed] [Google Scholar]
- 51.Dahan A, Hoffmann A. Use of a dynamic in vitro lipolysis model to rationalize oral formulation development for poor water soluble drugs: correlation with in vivo data and the relationship to intra-enterocyte processes in rats. Pharm Res. 2006;23(9):2165–2174. doi: 10.1007/s11095-006-9054-x. [DOI] [PubMed] [Google Scholar]
- 52.Dahan A, Hoffmann A. The effect of different lipid based formulations on the oral absorption of lipophilic drugs: the ability of in vitro lipolysis and consecutive ex vivo intestinal permeability data to predict in vivo bioavailability in rats. Eur J Pharm Biopharm. 2007;67(1):96–105. doi: 10.1016/j.ejpb.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 53.Li Y, McClements DJ. New mathematical model for interpreting pH-stat digestion profiles: Impact of lipid droplet characteristics on in vitro digestibility. J Agric Food Chem. 2010;58(13):8085–8092. doi: 10.1021/jf101325m. [DOI] [PubMed] [Google Scholar]
- 54.Zangenberg NH, Müllertz A, Kristensen HG, Hovgaard L. A dynamic in vitro lipolysis model: II: evaluation of the model. Eur J Pharm Sci. 2001;14(3):237–244. doi: 10.1016/S0928-0987(01)00182-8. [DOI] [PubMed] [Google Scholar]
- 55.Larsen A, Holm R, Pedersen M, Müllertz A. Lipid-based formulations for danazol containing a digestible surfactant, Labrafil® M2125CS: in vivo bioavailability and dynamic in vitro lipolysis. Pharm Res. 2008;25(12):2769–2777. doi: 10.1007/s11095-008-9641-0. [DOI] [PubMed] [Google Scholar]
- 56.Fatouros DG, Bergenstahl B, Müllertz A. Morphological observations on a lipid-based drug delivery system during in vitro digestion. Eur J Pharm Sci. 2007;31(2):85–94. doi: 10.1016/j.ejps.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 57.Cuiné JF, Claire L, McEvoy CL, Charman WN, Pouton CW, Edwards GA, Benameur H, et al. Evaluation of the impact of surfactant digestion on the bioavailability of danazol after oral administration of lipidic self-emulsifying formulations to dogs. J Pharm Sci. 2008;97(2):995–1012. doi: 10.1002/jps.21246. [DOI] [PubMed] [Google Scholar]
- 58.Kaukonen AM, Boyd BJ, Charman WN, Porter CJH. Drug solubilization behavior during in vitro digestion of suspension formulations of poorly water-soluble drugs in triglyceride lipids. Pharm Res. 2004;21(2):254–260. doi: 10.1023/B:PHAM.0000016283.87709.a9. [DOI] [PubMed] [Google Scholar]
- 59.Ali H, Nazzal M, Zaghloul A-AA, Nazzal S. Comparison between lipolysis and compendial dissolution as alternative techniques for the in vitro characterization of α-tocopherol self-emulsified drug delivery systems (SEDDS) Int J Pharm. 2008;352(1–2):104–114. doi: 10.1016/j.ijpharm.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 60.Han SF, Yao TT, Zhang XX, Gan L, Zhu C, Yu H-Z, et al. Lipid-based formulations to enhance oral bioavailability of the poorly water-soluble drug anethol trithione: effects of lipid composition and formulation. Int J Pharm. 2009;379:18–24. doi: 10.1016/j.ijpharm.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 61.Borgström B. Influence of bile salt, pH, and time on the action of pancreatic lipase: physiological implications. J Lipid Res. 1964;5(4):522–531. [PubMed] [Google Scholar]
- 62.Lombardo D, Guy O. Studies on the substrate specificity of a carboxyl ester hydrolase from human pancreatic juice. II. Action on cholesterol esters and lipid-soluble vitamin esters. BBA Enzym. 1980;611(1):147–155. doi: 10.1016/0005-2744(80)90050-9. [DOI] [PubMed] [Google Scholar]
- 63.Larsen AT, Sassene P, Müllertz A. In vitro lipolysis models as a tool for the characterization of oral lipid and surfactant based drug delivery systems. Int J Pharm. 2011;417(1–2):245–255. doi: 10.1016/j.ijpharm.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 64.FDA. Guidance for Industry: waiver of in vivo bioavailabilty and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutical classification system. Rockville, MD, USA: Food and Drug Administration, Center for Drug Evaluation and Research (CDER); 2000 [cited 2012 15.05.]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070246.pdf.
- 65.Porter CJH, Charman WN. In vitro assessment of oral lipid based formulations. Adv Drug Deliv Rev. 2001;50(Supplement 1):S127–S147. doi: 10.1016/S0169-409X(01)00182-X. [DOI] [PubMed] [Google Scholar]
- 66.Staggers JE, Hernell O, Stafford RJ, Carey MC. Physical–chemical behavior of dietary and biliary lipids during intestinal digestion and absorption. 1. Phase behavior and aggregation states of model lipid systems patterned after aqueous duodenal contents of healthy adult human beings. Biochemistry (Mosc) 1990;29(8):2028–2040. doi: 10.1021/bi00460a011. [DOI] [PubMed] [Google Scholar]
- 67.Fernandez S, Jannin V, Rodier JD, Ritter N, Mahler B, Carrière F. Comparative study on digestive lipase activities on the self emulsifying excipient Labrasol®, medium chain glycerides and PEG esters. BBA Mol Cell Biol Lipids. 2007;1771(5):633–640. doi: 10.1016/j.bbalip.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 68.MacGregor KJ, Embletona JK, Lacy JE, Perry EA, Solomon LJ, Seager H, et al. Influence of lipolysis on drug absorption from the gastro-intestinal tract. Adv Drug Deliv Rev. 1997;25(1):33–46. doi: 10.1016/S0169-409X(96)00489-9. [DOI] [Google Scholar]
- 69.Sek L, Pouton CW, Charman WN. Characterisation and quantification of medium chain and long chain triglycerides and their in vitro digestion products, by HPTLC coupled with in situ densitometric analysis. J Pharm Biomed Anal. 2001;25(3–4):651–661. doi: 10.1016/S0731-7085(00)00528-8. [DOI] [PubMed] [Google Scholar]
- 70.Fatouros DG, Müllertz A. In vitro lipid digestion models in design of drug delivery systems for enhancing oral bioavailability. Expert Opin Drug Metab Toxicol. 2008;4(1):65–76. doi: 10.1517/17425255.4.1.65. [DOI] [PubMed] [Google Scholar]
- 71.Stillhart C, Kuentz M. Comparison of high-resolution ultrasonic resonator technology and Raman spectroscopy as novel process analytical tools for drug quantification in self-emulsifying drug delivery systems. J Pharm Biomed Anal. 2012;59:29–37. doi: 10.1016/j.jpba.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 72.Kaukonen AM, Boyd BJ, Porter CJH, Charman WN. Drug solubilization behavior during in vitro digestion of simple triglyceride lipid solution formulations. Pharm Res. 2004;21(2):245–253. doi: 10.1023/B:PHAM.0000016282.77887.1f. [DOI] [PubMed] [Google Scholar]
- 73.Christensen J, Schultz K, Mollgard B, Kristensen HG, Müllertz A. Solubilisation of poorly water-soluble drugs during in vitro lipolysis of medium- and long-chain triacylglycerols. Eur J Pharm Sci. 2004;23(3):287–296. doi: 10.1016/j.ejps.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 74.Söderlind E, Karlsson E, Carlsson A, Kong R, Lenz A, Lindborg S, et al. Simulating fasted human intestinal fluids: understanding the roles of lecithin and bile acids. Mol Pharm. 2010;7(5):1498–1507. doi: 10.1021/mp100144v. [DOI] [PubMed] [Google Scholar]
- 75.Zughaid H, Forbes B, Martin GP, Patel N. Bile salt composition is secondary to bile salt concentration in determining hydrocortisone and progesterone solubility in intestinal mimetic fluids. Int J Pharm. 2012;422(1–2):295–301. doi: 10.1016/j.ijpharm.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 76.Porter CJH, Kaukonen AM, Boyd BJ, Edwards GA, Charman WN. Susceptibility to lipase-mediated digestion reduces the oral bioavailability of danazol after administration as a medium-chain lipid-based microemulsion formulation. Pharm Res. 2004;21(8):1405–1412. doi: 10.1023/B:PHAM.0000036914.22132.cc. [DOI] [PubMed] [Google Scholar]
- 77.Fatouros DG, Nielsen FS, Douroumis D, Hadjileontiadis LJ, Müllertz A. In vitro-in vivo correlations of self-emulsifying drug delivery systems combining the dynamic lipolysis model and neuro-fuzzy networks. Eur J Pharm Biopharm. 2008;69(3):887–898. doi: 10.1016/j.ejpb.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 78.Ross CAC. Fat absorption studies in the diagnosis and treatment of pancreatic fibrosis. Arch Dis Child. 1955;30(152):316–321. doi: 10.1136/adc.30.152.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abrams CK, Hamosh M, Hubbard VS, Dutta SK, Hamosh P. Lingual lipase in cystic fibrosis—quantitation of enzyme activity in the upper small intestine of patients with exocrine pancreatic insufficiency. J Clin Invest. 1984;73(2):374–382. doi: 10.1172/JCI111222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Buyukozturk F, Benneyan JC, Carrier RL. Impact of emulsion-based drug delivery systems on intestinal permeability and drug release kinetics. J Control Release. 2010;142(1):22–30. doi: 10.1016/j.jconrel.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 81.Sha X, Yan G, Wu Y, Li J, Fang X. Effect of self-microemulsifying drug delivery systems containing Labrasol® on tight junctions in Caco-2 cells. Eur J Pharm Sci. 2005;24(5):477–486. doi: 10.1016/j.ejps.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 82.Fatouros DG, Deen GR, Arleth L, Bergenstahl B, Nielsen FS, Pedersen JS, et al. Structural development of self nanoemulsifying drug delivery systems (SNEDDS) during in vitro lipid digestion monitored by small-angle X-ray scattering. Pharm Res. 2007;24(10):1844–1853. doi: 10.1007/s11095-007-9304-6. [DOI] [PubMed] [Google Scholar]
- 83.Fatouros DG, Walrand I, Bergenstahl B, Müllertz A. Colloidal structures in media simulating intestinal fed state conditions with and without lipolysis products. Pharm Res. 2008;26(2):361–374. doi: 10.1007/s11095-008-9750-9. [DOI] [PubMed] [Google Scholar]
- 84.Kossena GA, Charman WN, Boyd BJ, Dunstan DE, Porter CJH. Probing drug solubilization patterns in the gastrointestinal tract after administration of lipid-based delivery systems: a phase diagram approach. J Pharm Sci. 2004;92(2):332–348. doi: 10.1002/jps.10554. [DOI] [PubMed] [Google Scholar]
- 85.Kossena GA, Charman WN, Boyd BJ, Porter CJH. Influence of the intermediate digestion phases of common formulation lipids on the absorption of a poorly water-soluble drug. J Pharm Sci. 2005;94(3):481–492. doi: 10.1002/jps.20260. [DOI] [PubMed] [Google Scholar]
- 86.Sek L, Pouton CW, Kaukonen AM, Charman WN. Evaluation of the in-vitro digestion profiles of long and medium chain glycerides and the phase behaviour of their lipolytic products. J Pharm Pharmacol. 2002;54(1):29–41. doi: 10.1211/0022357021771896. [DOI] [PubMed] [Google Scholar]
- 87.Rübe A, Klein S, Mäder K. Monitoring of in vitro fat digestion by electron paramagnetic resonance spectroscopy. Pharm Res. 2006;23(9):2024–2029. doi: 10.1007/s11095-006-9069-3. [DOI] [PubMed] [Google Scholar]
- 88.Day JPR, Rago G, Domke KF, Velikov KP, Bonn M. Label-free imaging of lipophilic bioactive molecules during lipid digestion by multiplex coherent anti-Stokes Raman scattering microspectroscopy. J Am Chem Soc. 2010;132(24):8433–8439. doi: 10.1021/ja102069d. [DOI] [PubMed] [Google Scholar]
- 89.Warren DB, Anby MU, Hawley A, Boyd BJ. Real time evolution of liquid crystalline nanostructure during the digestion of formulation lipids using synchrotron small-angle X-ray scattering. Langmuir. 2011;27(15):9528–9534. doi: 10.1021/la2011937. [DOI] [PubMed] [Google Scholar]
- 90.Nielsen FS, Gibault E, Ljusberg-Wahren H, Arleth L, Pedersen JS, Müllertz A. Characterization of prototype self-nanoemulsifying formulations of lipophilic compounds. J Pharm Sci. 2007;96(4):876–892. doi: 10.1002/jps.20673. [DOI] [PubMed] [Google Scholar]
- 91.Dong Y-D, Boyd BJ. Applications of X-ray scattering in pharmaceutical science. Int J Pharm. 2011;417(1–2):101–111. doi: 10.1016/j.ijpharm.2011.01.022. [DOI] [PubMed] [Google Scholar]
- 92.Roshan Deen G, Aagaard AE, Pedersen JS, Fatouros D, Müllertz A, Experimental set-up with flow-through cell for SAXS studies of in-situ degradation of drug formulations under gastro-intestinal mimicking conditions. 13th International Conference on Small-Angle Scattering; 2006; Kyoto, Japan.
- 93.Williams HD, Sassene P, Kleberg K, Bakala-N’Goma J-C, Calderone M, Jannin V, et al. Toward the establishment of standardized in vitro tests for lipid-based formulations, part 1: method parameterization and comparison of in vitro digestion profiles across a range of representative formulations. J Pharm Sci. 2012. doi:10.1002/jps.23205 (online first). [DOI] [PubMed]
- 94.Sassene PJ, Knopp MM, Hesselkilde JZ, Koradia V, Larsen A, Rades T, et al. Precipitation of a poorly soluble model drug during in vitro lipolysis: characterization and dissolution of the precipitate. J Pharm Sci. 2010;99(12):4982–4991. doi: 10.1002/jps.22226. [DOI] [PubMed] [Google Scholar]
- 95.Thomas N, Holm R, Müllertz A, Rades T. In vitro and in vivo performance of novel supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS) J Control Release. 2012;160(1):25–32. doi: 10.1016/j.jconrel.2012.02.027. [DOI] [PubMed] [Google Scholar]
- 96.Psachoulias D, Vertzoni M, Goumas K, Kalioras V, Beato S, Butler J, et al. Precipitation in and supersaturation of contents of the upper small intestine after administration of two weak bases to fasted adults. Pharm Res. 2011;28(12):3145–3158. doi: 10.1007/s11095-011-0506-6. [DOI] [PubMed] [Google Scholar]
- 97.Boyd BJ, Porter CJH, Charman WN. Using the polymer partitioning method to probe the thermodynamic activity of poorly water-soluble drugs solubilized in model lipid digestion products. J Pharm Sci. 2003;92(6):1262–1271. doi: 10.1002/jps.10390. [DOI] [PubMed] [Google Scholar]
- 98.Lurie DJ, Mäder K. Monitoring drug delivery processes by EPR and related techniques—principles and applications. Adv Drug Deliv Rev. 2005;57(8):1171–1190. doi: 10.1016/j.addr.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 99.Schie IW, Wu J, Weeks T, Zern MA, Rutledge JC, Huser T. Label-free imaging and analysis of the effects of lipolysis products on primary hepatocytes. J Biophotonics. 2011;4(6):425–434. doi: 10.1002/jbio.201000086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Day JPR, Domke KF, Rago G, Kano H, Hamaguchi H-O, Vartiainen EM, et al. Quantitative coherent anti-Stokes Raman scattering (CARS) microscopy. J Phys Chem B. 2011;115(24):7713–7725. doi: 10.1021/jp200606e. [DOI] [PubMed] [Google Scholar]