

Abstract
The concept of treating cancer with antibody-drug conjugates (ADCs) has gained momentum with the favorable activity and safety of trastuzumab emtansine (T-DM1), SAR3419, and lorvotuzumab mertansine (IMGN901). All three ADCs utilize maytansinoid cell-killing agents which target tubulin and suppress microtubule dynamics. Each ADC utilizes a different optimized chemical linker to attach the maytansinoid to the antibody. Characterizing the absorption, distribution, metabolism, and excretion (ADME) of these ADCs in preclinical animal models is important to understanding their efficacy and safety profiles. The ADME properties of these ADCs in rodents were inferred from studies with radio-labeled ADCs prepared with nonbinding antibodies since T-DM1, SAR3419, IMGN901 all lack cross-reactivity with rodent antigens. For studies exploring tumor localization and activation in tumor-bearing mice, tritium-labeled T-DM1, SAR3419, and IMGN901 were utilized. The chemical nature of the linker was found to have a significant impact on the ADME properties of these ADCs—particularly on the plasma pharmacokinetics and observed catabolites in tumor and liver tissues. Despite these differences, T-DM1, SAR3419, and IMGN901 were all found to facilitate efficient deliveries of active maytansinoid catabolites to the tumor tissue in mouse xenograft models. In addition, all three ADCs were effectively detoxified during hepatobiliary elimination in rodents.
Key words: antibody–drug conjugate, cancer, maytansinoid
INTRODUCTION
Antibody–drug conjugates (ADCs) are targeted anti-cancer agents that utilize the specificity of a monoclonal antibody (Ab) to deliver a cell-killing agent specifically to a cancer cell that expresses the target antigen (1, 2). A design goal of an ADC is to maximize delivery of the cell-killing agent to the tumor tissue while minimizing delivery to normal tissues. The concept of treating cancer with ADCs has gained momentum with the approval by the FDA of brentuximab vedotin (SGN-35, Adcetris®) for the treatment of patients with Hodgkin’s lymphoma and anaplastic large cell lymphoma, and with the favorable activity and safety profile reported in clinical trials of trastuzumab emtansine (T-DM1), SAR3419, and lorvotuzumab mertansine (IMGN901; 3–7). Understanding the absorption, distribution, metabolism, and excretion (ADME) properties of these promising clinical candidates is essential to understanding what attributes may be necessary for clinical success. The ADME properties of T-DM1, SAR3419, and IMGN901 are the focus of this review. All three ADCs utilize maytansinoid cell-killing agents that target tubulin, thus suppressing microtubule dynamics leading to cell cycle arrest in the G2/M phase of the cell cycle, and ultimately, to cell death (8).
ANTIBODY–MAYTANSINOID CONJUGATES
T-DM1, SAR3419, and IMGN901 utilize different chemical linkers to attach the maytansinoid to the antibody (Fig. 1). Similar conjugation strategies are employed for all three ADCs. The selected cross-linking reagent couples the thiol group of the maytansinoid (DM1 or DM4) to an ε-amino group of lysine residues of the antibody (9). Reaction conditions are controlled so that an average of about 3.5 molecules of the maytansinoid are linked per antibody molecule (9). This method of maytansinoid conjugation has been shown to preserve the binding characteristics and activity properties associated with the antibody component (10). The impact of the linker chemistry on the efficacy of an ADC has been found to require empirical evaluations of different linkers (2). Selection of a linker for an ADC typically involves preparing a panel of conjugates with different linkers and evaluating these preclinically for efficacy and safety. The linker that affords the widest margin between the minimally efficacious dose in mouse xenograft models and the best safety profile in an appropriate animal model whose normal tissues react with the ADC similarly to human normal tissues. A thioether-based linker was chosen for T-DM1, while disulfide-based linkers were selected for SAR3419 and IMGN901 (Fig. 1). The disulfide bond of SAR3419 is more sterically hindered as compared to that of IMGN901 (Fig. 1), and thus is less susceptible to cleavage via thiol-disulfide exchange (11, 12). One factor influencing the outcome of such assessments is the effect of linker choice on the pharmacokinetics of the conjugates in vivo (6, 13–16). Another factor is the safety profile: for example, in preclinical rodent models, the trastuzumab–maytansinoid conjugate made with the uncleavable SMCC linker was found to be better tolerated than trastuzumab-SPP-DM1 (17, 18), while, across several antibodies studied, Ab-SPP-DM1 and Ab-SPDB-DM4 were found to have similar tolerability (16). A third factor is the anti-tumor activity of the catabolites generated with the different designs. The catabolites generated from conjugates using thioether-based linkers were shown to have less bystander killing activity than the catabolites generated from ADCs prepared with cleavable disulfide-based linkers (19). In addition, a highly cleavage-resistant linker may slow the rate of release of the active payload at the tumor relative to a more labile disulfide linker (18). Empirical selection in preclinical models allows the relative importance of these factors to be assessed for each antigen–antibody pair in the context of the target disease.
Fig. 1.
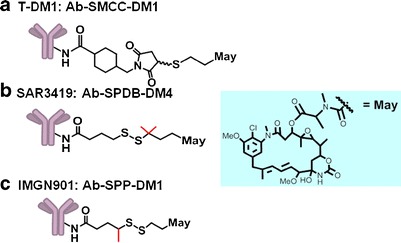
Structure of ADCs
PHARMACOKINETICS
The antibody component of T-DM1, SAR3419, and IMGN901 do not cross-react with rodent antigens. Thus, mice or rats can be used to evaluate the ADME of these ADC compounds without the complication of the additional contribution to clearance and distribution from antigen-mediated effects. Indeed, in general, the ADME parameters of such ADCs may be inferred from the behavior of model ADCs prepared with representative nonbinding antibodies of matched isotype. For simplicity in the following discussion, the conjugates used models where the antigen is not expressed are denoted as Ab-SMCC-DM1, Ab-SDPB-DM4, and Ab-SPP-DM1. In studies where antigen binding is relevant, the specific antibody is noted.
Enzyme-linked immunosorbent assay (ELISA) methods allow for the measurement of conjugate concentrations (concentration of species containing at least one linked maytansinoid) as well as total antibody concentrations in plasma (16). The clearance profile for a panel of Ab–maytansinoid conjugates was assessed using an ELISA method for the detection of conjugate (containing at least one linked maytansinoid) and found to correlate with their relative susceptibility to chemical cleavage via thiol-disulfide exchange of their linker moiety in vitro (11). For example, Ab-SPP-DM1 conjugate can undergo reductive cleavage with dithiolthreitol in vitro and was found to be cleared faster in mice than the uncleavable Ab-SMCC-DM1 conjugate (Fig. 2a). A similar relationship was observed between the clearance of T-DM1 with SMCC and the T-SPP-DM1 design (Fig. 2b; 20). The more sterically hindered disulfide of the Ab-SPDB-DM4 conjugate was more resistant to reductive cleavage than the disulfide of Ab-SPP-DM1 conjugate in vitro (11), and cleared more slowly from circulation (Fig. 2a).
Fig. 2.
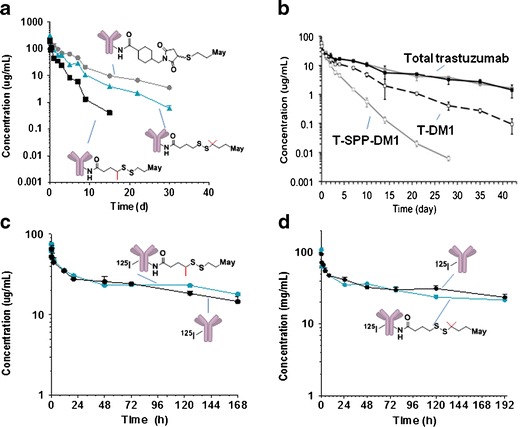
Pharmacokinetics. a Plasma clearance of Ab-SMCC-DM1, Ab-SPDB-DM4, and Ab-SPP-DM1 following a single i.v. bolus administration of 10 mg/kg. The conjugate concentrations were measured using a sandwich ELISA assay in which the conjugate with one or more maytansinoid linked was captured with an anti-maytansinoid antibody and detected with horseradish peroxidase conjugated donkey anti-human IgG antibody. Adapted from (11). b Clearance of T-DM1 and T-SPP-DM1 (T trastuzumab) following a single bolus administration of 3 mg/kg. The conjugate concentrations were measured using a sandwich ELISA assay in which the conjugate with one or more maytansinoid linked was captured with an anti-maytansinoid antibody and detected with biotinylated HER2 extracellular domain and detected with streptavidin horseradish peroxidase. Adapted from (20). c Clearance of 125I-Ab and 125I-Ab-SPP-DM1 following a single bolus i.v. administration of 4.16 mg/kg. Adapted from (26). d Clearance of 125I-Ab and 125I-Ab-SPDB-DM4 following a single bolus i.v. administration of 10 mg/kg. Adapted from (27)
The faster clearance of T-DM1 relative to the clearance of total trastuzumab shown in Fig. 2b suggests that there is another component to the clearance of Ab-maytansinoid conjugates, besides the thiol-disulfide exchange mechanism that likely dominates clearance of conjugates made with relatively labile disulfide linkers. The effect is small, however, and the difference in clearance between total trastuzumab and T-DM1 was barely differentiated with a 7-day observation period (18). The results are not unique to T-DM1 as other Ab-SMCC-DM1 conjugates have similarly shown slightly faster clearance of conjugate versus total antibody in preclinical studies (11, 21). The preclinical observations with T-DM1 appear to translate to the clinic: analysis of data from four clinical studies of single agent T-DM1 administered at 3.6 mg/kg every 3 weeks have shown that the clearance rate of T-DM1 and total trastuzumab in patients ranged from 7 to 13 and 3 to 6 mL/kg/day, respectively, with half-lives of about 4 and 9–11 days for T-DM1 and for total trastuzumab, respectively (13). The mechanism for the faster clearance of T-DM1 as compared to the total trastuzumab is unclear. It has been postulated that it is due to deconjugation (13). Indeed, cleavage of a thioether linkage has been described for cysteine-linked ADCs by thiol-maleimide exchange (22, 23), which has led to speculation that T-DM1 may undergo similar cleavage. However, a recent study has shown that Ab-SMCC-DM1 conjugates are not susceptible to this type of cleavage (24). The superior thioether stability of the Ab-SMCC-DM1 ADCs as compared to the cysteine linked ADCs may stem from inherent differences in the Michael donor reactivity of the sulfhydryl group of DM1 vs. the sulfhydryl group of cysteine side chains (25). Additional work is necessary to understand the mechanism(s) that underlie these pharmacokinetic observations (11, 13, 21). One might speculate that such maytansinoid loss may be accounted for by a low rate of cleavage of the amide bond of the linker-DM1 species from the antibody and/or by a slightly faster rate of clearance from circulation of species with more than the median load of DM1 molecules per antibody versus those species with less than the median number of DM1 molecules per antibody.
TISSUE DISTRIBUTION
The influence of DMx (either DM1 or DM4) conjugation on pharmacokinetics and tissue distribution of the antibody component of Ab-maytansinoid conjugates was assessed by investigating the clearance of 125I-labeled antibody (125I-Ab) and conjugates (125I-Ab-SPP-DM1 and 125I-Ab-SPDB-DM4) in nontumor-bearing mice (26, 27). The clearance of the antibody component of the two conjugates was found to be similar to that of the unmodified antibody (see examples in Fig. 2c and d). In addition, 125I-Ab, 125I-Ab-SPDB-DM4, and 125I-Ab-SPP-DM1 were found to exhibit similar biodistribution profiles in mice following a single bolus injection of 1 mg/kg (125I-Ab and 125I-Ab-SPDB-DM4, data shown in Fig. 3). The conjugates and antibody share the expected distribution profile of an IgG antibody with most of the administered dose confined to the blood, and with minimal accumulation in the brain. The results suggest that conjugation in the range of about 3.5–4.0 DMx molecules per antibody has no detectable impact on the pharmacokinetic or biodistribution properties of the antibody (27). In contrast, conjugation of auristatin through thiol groups of cysteine residues (17, 28) of the antibody (19, 29) was shown to alter the pharmacokinetic properties of the antibody when more than two auristatin molecules were linked per antibody (29), suggesting that not all conjugation strategies may retain the favorable characteristics described here for DMx conjugations.
Fig. 3.
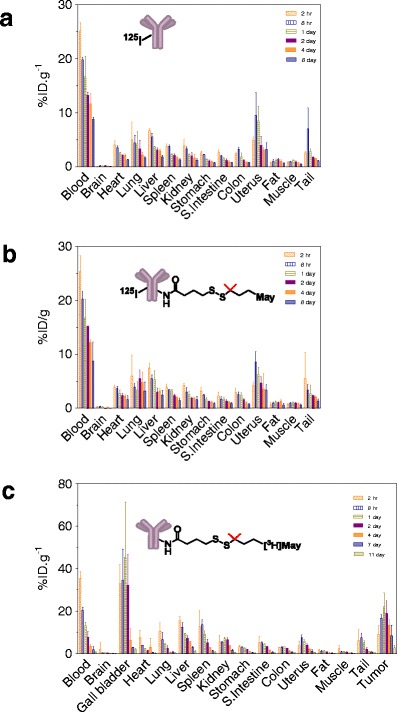
Biodistribution in mice. Tissue distribution of a 125I-Ab and b 125I-Ab-SPDB-DM4 following a single bolus i.v. administration of 10 mg/kg via the tail vein. Adapted from (27). c Tissue distribution of huC242-SPDB-[3H]DM4. Mice bearing established subcutaneous COLO205 xenografts were administered a single bolus i.v. dose of 6.5 mg/kg huC242-SPDB-[3H]DM4. Adapted from (27)
Conjugates prepared with tritium-labeled DMx allow for the distribution and subsequent catabolism of the maytansinoid to be measured (19, 26, 30). Mice bearing COLO 205 tumors that express the CanAg antigen were administered a single bolus dose of anti-CanAg-SPDB-[3H]DM4 and tissues were analyzed for radioactivity to determine the percentage of the injected dose (%ID/g) accumulated in different tissues. At all time points evaluated, the maytansinoid levels in tissues (except tumor and gall bladder) were lower than the corresponding levels in the blood with peak values measured at 2–8 h post-dose, and declining thereafter in concert with declining blood concentrations to nearly undetectable levels by 11 days (Fig. 3c). In tumor tissue, maytansinoid levels peaked at about 20% ID/g between 1 and 2 days post-dose, consistent with antigen-mediated retention and cellular uptake by the tumor. The %ID/g in gall bladder was in the range of 30–50% from 2 h to 2 days post-dose, exceeding the %ID/g of blood after 8 h, an observation consistent with hepatobiliary elimination of the maytansinoid. The anti-CanAg-SMCC-[3H]DM1 conjugate with an uncleavable linker was found to have a similar distribution profile in tumor-bearing mice as shown above (Fig. 3c) for a disulfide-linked conjugate (unpublished data), and was also similar to the tissue distribution reported for labeled maytansinoid in nontumor-bearing rats following administration of a single bolus administration of T-[3H]DM1 (30, 31). In the study with T-DM1 (31), up to 80% of the radioactivity was recovered in the feces over 7 days consistent with the hepatobiliary route for maytansinoid elimination. Similar results were reported for Ab–auristatin conjugates (32).
METABOLISM AND EXCRETION
Given that most of the administered dose of an antibody-based therapeutic appears to be slowly catabolized by the liver and other tissues of the reticuloendothelial system (33), liver tissues of mice were analyzed for maytansinoid metabolites following administration of a single bolus dose of Ab-SMCC-[3H]DM1, Ab-SPDB-[3H]DM4, and Ab-SPP-[3H]DM1 (30). The structures of metabolites isolated from the liver tissues are shown in Fig. 4. A stark difference was noted in the complexity of the liver metabolites obtained for the three conjugates. Lysine-SMCC-DM1 was the major metabolite detected for the Ab-SMCC-DM1 conjugate. Recently, similar studies were performed with T-[3H]DM1 in rats with similar results. Lysine-SMCC-DM1 and MCC-DM1 were observed in the bile, accounting for 72% and 13% of the radiolabel recovered in bile, respectively (31). There was no detectable metabolic modification of the maytansinoid macrocycle or the thioether linker indicating that lysine-SMCC-DM1 resists chemical alteration in the hepatocytes during clearance.
Fig. 4.
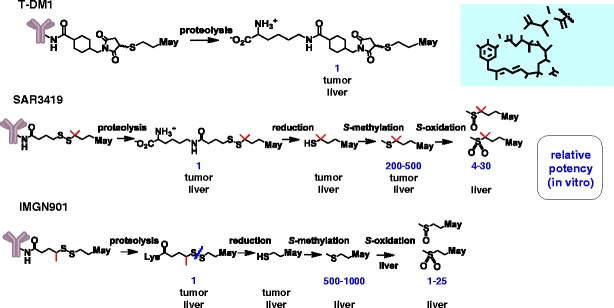
In vivo catabolites and metabolites. The tissue, tumor, or liver, in which the metabolite or catabolite if formed is indicated below each structure. The relative potency of the compounds formed in tumor tissue and/or liver is noted. The free thiol compounds, DM1 and DM4, are not shown since >in vitro IC50 values for these compounds are highly variable due to their rapid oxidation to form mixed disulfides in cell culture medium
The corresponding lysine-linker-DMx species also were detected in the liver extracts of mice treated with the disulfide-linked conjugates, along with several other metabolites. Production of these additional metabolites suggests a degradation path for the conjugates shown in Fig. 4, where initial lysosomal degradation in the liver and the rest of the tissues of the reticuloendothelial system yields the corresponding lysine-linker-DMx. These disulfide-linked species are further cleaved to yield the corresponding free maytansinoid thiols which in turn are S-methylated. All catabolites are ultimately eliminated through the liver where the S-methyl-maytansinoid catabolites are oxidized to the corresponding S-methyl sulfoxide and S-methyl-sulfone derivatives (30). These oxidized metabolites were found to have relatively low cytotoxic potency in cell-based viability assays (see Table I). Efficient S-methylation and oxidation of the catabolites occurs in the liver prior to elimination since only such oxidized species were detected in bile (30). Elimination of the cytotoxic moiety of Ab-maytansinoid conjugates as metabolites of lower potency relative to the “parent” molecule, maytansine, may be an important element in the tolerability of such conjugates. Notably, severe gastrointestinal toxicity was observed in patients treated with maytansine itself (34). However, in general, clinically significant gastrointestinal toxicity has not been observed in patients treated with T-DM1 or with disulfide-linked maytansinoid conjugates (2–4, 6, 7, 15, 35–37). The slower clearance rate for the ADCs as compared to maytansine may also play a role in reducing their gastrointestinal toxicities.
Table I.
Cytotoxic Potencies of Maytansinoids Towards Human Carcinoma Cell Lines in vitro
| Maytansinoid | In vitro IC50 (nM) | ||||
|---|---|---|---|---|---|
| A375 (melanoma) | BJAB (B-cell) | COLO205 (colon) | KB (cervix) | MOLT-4 (T-cell) | |
| Maytansine | 0.05 | 0.03 | 0.08 | 0.05 | 0.09 |
| Lysine-Nε-SPP-DM1 | 10 | 50 | 60 | 10 | >100 |
| Lysine-Nε-SPDB-DM4 | 2 | 7 | 20 | 3 | 16 |
| Lysine-Nε-SMCC-DM1 | 8 | 8 | 17 | 10 | 16 |
| S-methyl-DM1 | 0.02 | 0.01 | 0.03 | 0.02 | 0.03 |
| S-methyl-DM4 | 0.02 | 0.01 | 0.03 | 0.03 | 0.03 |
| S-methyl-DM1 sulfoxide | 9.7 | 7.1 | 17 | 23 | 19 |
| S-methyl-DM1 sulfone | – | 1.7 | 5.9 | 3.5 | – |
| S-methyl-DM4 sulfoxide | 0.55 | 0.55 | 1.3 | 2.0 | 1.9 |
| S-methyl-DM4 sulfone | 0.075 | 0.17 | 0.63 | 0.80 | 1.1 |
All metabolites were chemically synthesized. Cell-killing activities of the metabolites were measured after 5 days using a WST-based cell viability assay. Adapted from (30)
TUMOR LOCALIZATION AND ACTIVATION OF T-DM1
Activation of T-DM1 yields lysine-SMCC-DM1 within HER2-positive breast cancer cells in vitro, following receptor-mediated endocytosis and lysosomal degradation (20). The formation of lysine-SMCC-DM1 within cancer cells in vitro was found to precede the mitotic arrest of cells in G2/M (19). The lysine-SMCC-DM1 catabolite displays poor cytotoxic potency in vitro when added exogenously to cells in cell-based viability assays compared to more lipophilic maytansinoids such as maytansine and S-methyl-DM1 (Table I), likely due to its limited cell permeability as a charged molecule. Thus, Ab-maytansinoid conjugates utilizing the uncleavable SMCC linker will likely not exhibit any bystander killing (38), and so efficient eradication of all tumor cells in vivo will require antibody-mediated delivery of a lethal quantity of lysine-SMCC-DM1 into every cell of a tumor.
The amount of antibody that binds in vivo to the individual tumor cells in a solid tumor is known to be limited by several factors such as stromal and epithelial barriers that limit antibody penetration as well as the number of antigen molecules on the cell surface and the efficiency of internalization of the antibody-antigen complex (39–41). Antibody localization to tumors ranges from 0.003% to 0.08% of the injected dose of antibody per gram (ID/g) of tumor, depending on the tumor type, as measured in human clinical trials (42). An ID/g of 0.01% reaching the tumor translates to a maximal antibody concentration at the tumor of about 200 nM following administration of an antibody dose of 6 mg/kg. Tumor localization values in a mouse xenograft tend to be in the range of 5–20% ID/g (43, 44) which gives a similar antibody concentration of 50–200 nM in the tumor following a dose of 6 mg/kg. The tumor localization values are most relevant for a completely tumor specific target since the Abs do not react with normal tissues in the mouse. For targets with normal tissue expression, the localization data from mice may overestimate the levels that would be expected in patient tumors. Nonetheless, even the lower range of the estimated concentrations should be therapeutically efficacious given that most Ab–maytansinoid conjugates exert their cytotoxic effects towards cancer cells in vitro at picomolar concentrations (45). However, the antibody may not be distributed uniformly, resulting in areas of very high (lethal) concentrations and areas of low (potentially sublethal) concentrations. Indeed, several studies have reported heterogeneous distribution of the antibody in the tumor tissue at doses in this range (46–48). The localization of T-[3H]DM1 to tumor tissue and subsequent activation was assessed in mouse-bearing HER2-positive BT474EEI xenografts (20). Peak uptake of around 9% ID/g was measured between 1 and 2 days post-administration of a single dose of 10 mg/kg. The tumor uptake of the nonspecific IgG1-SMCC-[3H]-DM1 conjugate was also evaluated and found to be about 2.3-fold lower (3.9% ID/g at 24 h). Similar specificity ratios between unconjugated trastuzumab and a nonbinding antibody control have been reported (44).
To determine how much of the localized T-DM1 was subject to intracellular catabolism to yield lysine-SMCC-DM1 in vivo, portions of the tumor homogenates from tumor-bearing mice treated with T-[3H]DM1 were analyzed. Lysine-SMCC-DM1 was the sole catabolite observed in organic extracts of the tumor homogenates, reaching a maximal concentration of nearly 150 nM after about 2 days post-dose (20). The concentrations of lysine-SMCC-DM1 observed in the extracts from the tumor xenografts were similar to the levels that could be achieved in cells exposed to T-DM1 in vitro (20). The concentration of lysine-SMCC-DM1 in the tumors of the control mice treated with a nonbinding control conjugate was fivefold lower—reflecting the difference between the additional benefit of HER2 antigen-mediated binding and internalization for T-DM1 over and above the nonspecific uptake of conjugate (20).
IMPACT OF LINKER ON THE UPTAKE AND CATABOLISM OF T-DM1
To explore the role of the linker on the tumor delivery, the uptake and catabolism of T-SPP-DM1 was compared to that of T-DM1, trastuzumab-SMCC-DM1. As expected from previous reports (11, 18), T-SPP-DM1 had a faster plasma clearance than T-SMCC-DM1 (20). As might be expected, slower T-DM1 clearance translated to higher overall (conjugate plus catabolites) concentrations in the tumor, but, unexpectedly, similar levels of total active catabolites were found in tumors with the two conjugate designs (20). These results indicate that, although, different linkers have a clear impact on the pharmacokinetics and the chemical nature of the catabolites formed, both linkers achieve the same amount of active cell-killing agent at the tumor. The results suggest that more facile release of payload from the disulfide-linked conjugate within tumor cells may compensate for the lower amount of payload reaching the tumor due to its somewhat more rapid clearance.
TUMOR LOCALIZATON AND ACTIVATION OF SAR3419 AND IMGN901
Data to demonstrate the selective tumor uptake and catabolism in mouse xenograft studies have also been described for SAR3419 and IMGN901. The tumor uptake and activation of SAR3419 and IMGN901 in mice were found to be similar to that described for T-DM1 conjugates (49, 50). The major catabolites of IMGN901 were lysine-SPP-DM1 and DM1 (50). Lysine-SPDB-DM4, DM4 and S-methyl DM4 were the major metabolites observed for SAR3419 (51), consistent with the activation pathway described for the CanAg-targeting conjugate, huC242-SPDB-DM4 (19). The observation that unconjugated DM4 was more efficiently methylated than unconjugated DM1, presumably by the S-methyl transferase enzyme(s) endogenous to human carcinoma cells, explains why S-methyl-DM1 is not readily observed in tumor cells as a catabolite of SPP-DM1conjugates (52). Figure 4 summarizes the findings of these studies.
CONCLUSION
The ADME properties for T-DM1, SAR3419, and IMGN901 in preclinical rodent animal models support the favorable efficacy and safety findings for the ADCs in patients. Optimized plasma pharmacokinetics and linker chemistries allow for efficient maytansinoid delivery to tumors for all three ADCs and effective detoxification during hepatobiliary elimination for all three is consistent with their favorable safety profiles. Continued understanding of the ADME properties of antibody–drug conjugates entering clinical evaluations should provide additional insight into what attributes may be necessary for clinical success.
References
- 1.Chari RV. Targeted cancer therapy: conferring specificity to cytotoxic drugs. Acc Chem Res. 2008;41:98–107. doi: 10.1021/ar700108g. [DOI] [PubMed] [Google Scholar]
- 2.Lambert JM. Antibody-maytansinoid conjugates; a new strategy for the treatment of cancer. Drugs Future 2010; 35(6): 471
- 3.Chanan-Khan A, W. J, Gharibo M, Jagannath S, Munshi N, Anderson KC, DePaolo D, Lee K, Miller KC, Guild R, et al. Phase 1 Study of IMGN901, used as a monotherapy, in patients with heavily pre-treated CD56-positive multiple myeloma. A preliminary safety and efficacy analysis. Blood (Ash Annual Meeting Abstracts) abstract 2283. 2009.
- 4.Fossella F, W. P, Lorigan P, Tolcher A, O’Brien M, O’Keeffe J, Zildjian S, Qin A, O’Leary J, Villalona-Calero M. Investigation of IMGN901 in CD56+ solid tumors: results from a phase I/II trial (study 001) and a phase I trial (study 002). 13th World Conference on Lung Cancer. 2009.
- 5.Lambert JM. Drug-conjugated monoclonal antibodies for the treatment of cancer. Curr Opin Pharmacol. 2005;5:543–9. doi: 10.1016/j.coph.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 6.Younes A, G. L, Kim S, Romaguera J, Copeland AR, deCastro Farial S, Kwak L, Fayad L, Hagemeister F, Fanale M, et al. Phase I multi-dose escalation study of the anti-CD19 maytansinoid immunoconjugate SAR3419 administered by IV infusion every 3 weeks to patients with relapsed/refractory B-cell NHL. Blood (Ash Annual Meeting Abstracts) abstract 585. 2009.
- 7.Krop IE, Beeram M, Modi S, Jones SF, Holden SN, Yu W, Girish S, Tibbitts J, Yi JH, Sliwkowski MX, Jacobson F, Lutzker SG, Burris HA. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol. 2010;28:2698–704. doi: 10.1200/JCO.2009.26.2071. [DOI] [PubMed] [Google Scholar]
- 8.Bai R, Friedman SJ, Pettit GR, Hamel E. Dolastatin 15, a potent antimitotic depsipeptide derived from Dolabella auricularia. Interaction with tubulin and effects of cellular microtubules. Biochem Pharmacol. 1992;43:2637–45. doi: 10.1016/0006-2952(92)90153-A. [DOI] [PubMed] [Google Scholar]
- 9.Singh R, Erickson HK. Antibody–cytotoxic agent conjugates: preparation and characterization. Methods Mol Biol. 2009;525:445–67. doi: 10.1007/978-1-59745-554-1_23. [DOI] [PubMed] [Google Scholar]
- 10.Junttila TT, Li G, Parsons K, Phillips GL, Sliwkowski MX. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res Treat. 2011;128:347–56. doi: 10.1007/s10549-010-1090-x. [DOI] [PubMed] [Google Scholar]
- 11.Kellogg BA, Garrett L, Kovtun Y, Lai KC, Leece B, Miller M, Payne G, Steeves R, Whiteman KR, Widdison W, Xie H, Singh R, Chari RV, Lambert JM, Lutz RJ. Disulfide-linked antibody–maytansinoid conjugates: optimization of in vivo activity by varying the steric hindrance at carbon atoms adjacent to the disulfide linkage. Bioconjug Chem. 2011;22:717–27. doi: 10.1021/bc100480a. [DOI] [PubMed] [Google Scholar]
- 12.Widdison WC, Wilhelm SD, Cavanagh EE, Whiteman KR, Leece BA, Kovtun Y, Goldmacher VS, Xie H, Steeves RM, Lutz RJ, Zhao R, Wang L, Blattler WA, Chari RV. Semisynthetic maytansine analogues for the targeted treatment of cancer. J Med Chem. 2006;49:4392–408. doi: 10.1021/jm060319f. [DOI] [PubMed] [Google Scholar]
- 13.Girish S, Gupta M, Wang B, Lu D, Krop IE, Vogel CL, Burris Iii HA, Lorusso PM, Yi JH, Saad O, Tong B, Chu YW, Holden S, Joshi A. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother Pharmacol. 2012;69(5):1229–40. doi: 10.1007/s00280-011-1817-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.LoRusso PM, Weiss D, Guardino E, Girish S, Sliwkowski MX. Trastuzumab emtansine: a unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin Cancer Res. 2011;17:6437–47. doi: 10.1158/1078-0432.CCR-11-0762. [DOI] [PubMed] [Google Scholar]
- 15.Tolcher AW, Ochoa L, Hammond LA, Patnaik A, Edwards T, Takimoto C, Smith L, de Bono J, Schwartz G, Mays T, Jonak ZL, Johnson R, DeWitte M, Martino H, Audette C, Maes K, Chari RV, Lambert JM, Rowinsky EK. Cantuzumab mertansine, a maytansinoid immunoconjugate directed to the CanAg antigen: a phase I, pharmacokinetic, and biologic correlative study. J Clin Oncol. 2003;21:211–22. doi: 10.1200/JCO.2003.05.137. [DOI] [PubMed] [Google Scholar]
- 16.Xie H, Blattler WA. In vivo behaviour of antibody-drug conjugates for the targeted treatment of cancer. Expert Opin Biol Ther. 2006;6:281–91. doi: 10.1517/14712598.6.3.281. [DOI] [PubMed] [Google Scholar]
- 17.Junutula JR, Flagella KM, Graham RA, Parsons KL, Ha E, Raab H, Bhakta S, Nguyen T, Dugger DL, Li G, Mai E, Lewis Phillips GD, Hiraragi H, Fuji RN, Tibbitts J, Vandlen R, Spencer SD, Scheller RH, Polakis P, Sliwkowski MX. Engineered thio-trastuzumab-DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2-positive breast cancer. Clin Cancer Res. 2010;16:4769–78. doi: 10.1158/1078-0432.CCR-10-0987. [DOI] [PubMed] [Google Scholar]
- 18.Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, Blattler WA, Lambert JM, Chari RV, Lutz RJ, Wong WL, Jacobson FS, Koeppen H, Schwall RH, Kenkare-Mitra SR, Spencer SD, Sliwkowski MX. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68:9280–90. doi: 10.1158/0008-5472.CAN-08-1776. [DOI] [PubMed] [Google Scholar]
- 19.Erickson HK, Park PU, Widdison WC, Kovtun YV, Garrett LM, Hoffman K, Lutz RJ, Goldmacher VS, Blattler WA. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006;66:4426–33. doi: 10.1158/0008-5472.CAN-05-4489. [DOI] [PubMed] [Google Scholar]
- 20.Erickson HK, Lewis Phillips GD, Leipold DD, Provenzano CA, Mai E, Johnson HA, Gunter B, Audette CA, Audette CA, Gupta M, Pinkas J, Tibbitts J. The effect of different linkers on target cell catabolism and pharmacokinetics/pharmacodynamics of trastuzumab maytansinoid conjugates. Mol Cancer Ther. 2012;11(5):1133–1142. doi: 10.1158/1535-7163.MCT-11-0727. [DOI] [PubMed] [Google Scholar]
- 21.Stephan JP, Chan P, Lee C, Nelson C, Elliott JM, Bechtel C, Raab H, Xie D, Akutagawa J, Baudys J, Saad O, Prabhu S, Wong WL, Vandlen R, Jacobson F, Ebens A. Anti-CD22-MCC-DM1 and MC-MMAF conjugates: impact of assay format on pharmacokinetic parameters determination. Bioconjug Chem. 2008;19:1673–83. doi: 10.1021/bc800059t. [DOI] [PubMed] [Google Scholar]
- 22.Alley SC, Benjamin DR, Jeffrey SC, Okeley NM, Meyer DL, Sanderson RJ, Senter PD. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug Chem. 2008;19:759–65. doi: 10.1021/bc7004329. [DOI] [PubMed] [Google Scholar]
- 23.Shen BQ, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu SF, Mai E, Li D, Tibbitts J, Baudys J, Saad OM, Scales SJ, McDonald PJ, Hass PE, Eigenbrot C, Nguyen T, Solis WA, Fuji RN, Flagella KM, Patel D, Spencer SD, Khawli LA, Ebens A, Wong WL, Vandlen R, Kaur S, Sliwkowski MX, Scheller RH, Polakis P, Junutula JR. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol. 2012;30:184–9. doi: 10.1038/nbt.2108. [DOI] [PubMed] [Google Scholar]
- 24.Fishkin N, Maloney EK, Chari RV, Singh R. A novel pathway for maytansinoid release from thioether linked antibody-drug conjugates (ADCs) under oxidative conditions. Chem Commun (Camb) 2011;47:10752–4. doi: 10.1039/c1cc14164c. [DOI] [PubMed] [Google Scholar]
- 25.Baldwin AD, Kiick KL. Tunable degradation of maleimide-thiol adducts in reducing environments. Bioconjug Chem. 2011;22:1946–53. doi: 10.1021/bc200148v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie H, Audette C, Hoffee M, Lambert JM, Blattler WA. Pharmacokinetics and biodistribution of the antitumor immunoconjugate, cantuzumab mertansine (huC242-DM1), and its two components in mice. J Pharmacol Exp Ther. 2004;308:1073–82. doi: 10.1124/jpet.103.060533. [DOI] [PubMed] [Google Scholar]
- 27.Mayo F, X. H, Erickson H, Wunderli P, Garrett L, Whitman K, Leece B, Lutz RJ. Pharmacokinetics and Biodistribution of huC242-DM4, an Antibody-Maytansinoid Conjugate That Tartgets CanAg-Postive Tumors, In AACR Meeting. 2005.
- 28.Boswell CA, Mundo EE, Zhang C, Bumbaca D, Valle NR, Kozak KR, Fourie A, Chuh J, Koppada N, Saad O, Gill H, Shen BQ, Rubinfeld B, Tibbitts J, Kaur S, Theil FP, Fielder PJ, Khawli LA, Lin K. Impact of drug conjugation on pharmacokinetics and tissue distribution of anti-STEAP1 antibody-drug conjugates in rats. Bioconjug Chem. 2011;22:1994–2004. doi: 10.1021/bc200212a. [DOI] [PubMed] [Google Scholar]
- 29.Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, Meyer DL, Francisco JA. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10:7063–70. doi: 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- 30.Sun X, Widdison W, Mayo M, Wilhelm S, Leece B, Chari R, Singh R, Erickson H. Design of antibody-maytansinoid conjugates allows for efficient detoxification via liver metabolism. Bioconjug Chem. 2011;22:728–35. doi: 10.1021/bc100498q. [DOI] [PubMed] [Google Scholar]
- 31.Shen BQ, Bumbaca D, Saad O, Yue Q, Pastuskovas CV, Khojasteh SC, Tibbitts J, Kaur S, Wang B, Chu YW, Lorusso PM, Girish S. Catabolic fate and pharmacokinetic characterization of trastuzumab emtansine T-DM1: an emphasis on preclinical and clinical catabolism. Curr Drug Metab. 2012 (in press) [DOI] [PubMed]
- 32.Alley SC, Zhang X, Okeley NM, Anderson M, Law CL, Senter PD, Benjamin DR. The pharmacologic basis for antibody–auristatin conjugate activity. J Pharmacol Exp Ther. 2009;330:932–8. doi: 10.1124/jpet.109.155549. [DOI] [PubMed] [Google Scholar]
- 33.Wright A, Sato Y, Okada T, Chang K, Endo T, Morrison S. In vivo trafficking and catabolism of IgG1 antibodies with Fc associated carbohydrates of differing structure. Glycobiology. 2000;10:1347–55. doi: 10.1093/glycob/10.12.1347. [DOI] [PubMed] [Google Scholar]
- 34.Issell BF, Crooke ST. Maytansine. Cancer Treat Rev. 1978;5:199–207. doi: 10.1016/S0305-7372(78)80014-0. [DOI] [PubMed] [Google Scholar]
- 35.Chanan-Khan A, J. S, Heffner T, Avigan D, Lee K, Lutz RJ, Haeder T, Ruehle M, Uherek C, Wartenberg-Demand A, et al. Phase I Study of BT062 given as repeated single dose once every 3 weeks in patients with relapsed or relapsed/refractory multiple myeloma. Blood (Ash Annual Meeting Abstracts) abstract 1862. 2009.
- 36.Smith SV. Technology evaluation: huN901-DM1, ImmunoGen. Curr Opin Mol Ther. 2005;7:394–401. [PubMed] [Google Scholar]
- 37.Younes A, F-T A, Bartlett NL, Leonard JP, Lynch C, Kennedy DA, Sievers EL. Multiple complete responses in a Phase 1 dose-escalation study of the antibody-drug conjugate SGN-35 in patients with relapsed or refractory CD30-positive lymphomas. Blood (Ash Annual Meeting Abstracts) abstract 2731. 2008.
- 38.Kovtun YV, Audette CA, Ye Y, Xie H, Ruberti MF, Phinney SJ, Leece BA, Chittenden T, Blattler WA, Goldmacher VS. Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 2006;66:3214–21. doi: 10.1158/0008-5472.CAN-05-3973. [DOI] [PubMed] [Google Scholar]
- 39.Jain RK. Transport of molecules, particles, and cells in solid tumors. Annu Rev Biomed Eng. 1999;1:241–63. doi: 10.1146/annurev.bioeng.1.1.241. [DOI] [PubMed] [Google Scholar]
- 40.Thurber GM, Schmidt MM, Wittrup KD. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv Drug Deliv Rev. 2008;60:1421–34. doi: 10.1016/j.addr.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Christiansen J, Rajasekaran AK. Biological impediments to monoclonal antibody-based cancer immunotherapy. Mol Cancer Ther. 2004;3:1493–501. [PubMed] [Google Scholar]
- 42.Sedlacek H-H, Seeman G, Hoffman D, Czech J, Lorenz P, Kolar C, Bosslet K. Antibodies as carriers of cytotoxicity. Marburg: Karger; 1992. [Google Scholar]
- 43.Cai W, Ebrahimnejad A, Chen K, Cao Q, Li ZB, Tice DA, Chen X. Quantitative radioimmunoPET imaging of EphA2 in tumor-bearing mice. Eur J Nucl Med Mol Imaging. 2007;34:2024–36. doi: 10.1007/s00259-007-0503-5. [DOI] [PubMed] [Google Scholar]
- 44.McLarty K, Cornelissen B, Scollard DA, Done SJ, Chun K, Reilly RM. Associations between the uptake of 111In-DTPA-trastuzumab, HER2 density and response to trastuzumab (Herceptin) in athymic mice bearing subcutaneous human tumour xenografts. Eur J Nucl Med Mol Imaging. 2009;36:81–93. doi: 10.1007/s00259-008-0923-x. [DOI] [PubMed] [Google Scholar]
- 45.Kovtun YV, Goldmacher VS. Cell killing by antibody-drug conjugates. Cancer Lett. 2007;255:232–40. doi: 10.1016/j.canlet.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 46.Juweid M, Neumann R, Paik C, Perez-Bacete MJ, Sato J, van Osdol W, Weinstein JN. Micropharmacology of monoclonal antibodies in solid tumors: direct experimental evidence for a binding site barrier. Cancer Res. 1992;52:5144–53. [PubMed] [Google Scholar]
- 47.Blumenthal RD, Fand I, Sharkey RM, Boerman OC, Kashi R, Goldenberg DM. The effect of antibody protein dose on the uniformity of tumor distribution of radioantibodies: an autoradiographic study. Cancer Immunol Immunother. 1991;33:351–8. doi: 10.1007/BF01741594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baker JH, Lindquist KE, Huxham LA, Kyle AH, Sy JT, Minchinton AI. Direct visualization of heterogeneous extravascular distribution of trastuzumab in human epidermal growth factor receptor type 2 overexpressing xenografts. Clin Cancer Res. 2008;14:2171–9. doi: 10.1158/1078-0432.CCR-07-4465. [DOI] [PubMed] [Google Scholar]
- 49.Blanc V, Bousseau A, Caron A, Carrez C, Lutz RJ, Lambert JM. SAR3419: an anti-CD19-maytansinoid immunoconjugate for the treatment of B-cell malignancies. Clin Cancer Res. 2011;17:6448–58. doi: 10.1158/1078-0432.CCR-11-0485. [DOI] [PubMed] [Google Scholar]
- 50.Sun X, C. L, Ellis M, Whiteman KR, Pinkas J, Lazar A, Erickson H. Lorvotuzumab mertansine displays favorable pharmacokinetics and tumor delivery in mouse models. AACR-NCI-EORTC Abstract # B195. 2011.
- 51.Erickson HK, P. C, Mayo MF, Widdison WC, Audette C, et al. Target-cell processing of the anti-CD19 antibody maytansinoid conjugate SAR3419 in preclinical models. AACR Abstract 5473. 2009.
- 52.Erickson HK, M. M, Widdison W, Audette C, Kovtun Y, Chari R, Lutz RJ, Singh R. Linker selection in antibody-maytansinoid conjugates impacts bystander killing in tumor xenograft mouse models. AACR-NCI-EORTC abstract #A86. 2007.