

Abstract
Biotherapeutics are becoming an increasingly common drug class used to treat autoimmune and other inflammatory conditions. Optimization of absorption, distribution, metabolism, and excretion (ADME) profiles of biotherapeutics is crucial for clinical, as well as commercial, success of these drugs. This review focuses on the common questions and challenges in ADME optimization of biotherapeutics for inflammatory conditions. For these immunomodulatory and/or immunosuppressive biotherapeutics, special consideration should be given to the assessment of the interdependency of ADME profiles, pharmacokinetic/pharmacodynamic (PK/PD) relationships, and immunogenicity profiles across various preclinical species and humans, including the interdependencies both in biology and in assay readouts. The context of usage, such as dosing regimens, extent of disease, concomitant medications, and drug product characteristics may have a direct or indirect (via modulation of immunogenicity) impact on ADME profiles of biotherapeutics. Along these lines, emerging topics include assessments of preexisting reactivity to a biotherapeutic agent, impact of immunogenicity on tissue exposure, and analysis of penetration to normal versus inflamed tissues. Because of the above complexities and interdependences, it is essential to interpret PK, PD, and anti-drug antibody results in an integrated manner. In addition, because of the competitive landscape in autoimmune and inflammatory markets, many pioneering ADME-centric protein engineering and subsequent in vivo testing (such as optimization of novel modalities to extend serum and tissue exposures and to improve bioavailability) are being conducted with biotherapeutics in this therapeutic area. However, the ultimate challenge is demonstration of the clinical relevance (or lack thereof) of modified ADME and immunogenicity profiles.
KEY WORDS: ADME, autoimmune disease, immunogenicity, pharmacokinetics, therapeutic proteins
INTRODUCTION
Therapeutic proteins (also referred to as biotherapeutics) are becoming an increasingly common class of drugs to treat autoimmune and other inflammatory conditions (e.g., sepsis, osteoarthritis, and chronic viral infections) in the USA, Europe, as well as in Canada and Japan. Examples of therapeutic proteins that are approved for inflammatory conditions are shown in Table I. There are more than 70 different autoimmune diseases, with rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), plaque psoriasis, inflammatory bowel disease (IBD, includes two different diseases: ulcerative colitis [UC] and Crohn's disease [CD]), asthma, type 1 diabetes mellitus, multiple sclerosis (MS), immune thrombocytopenia being some of the most prevalent ones. The rationale behind the use of biotherapeutics for inflammatory conditions is the potential for a gentler and more targeted immunomodulation compared to conventional small-molecule compounds, such that only a subset of the inflammatory or immunoregulatory pathways are targeted.
Table I.
Examples of Therapeutic Proteins Approved for Autoimmune and Other Inflammatory Conditions
| Therapeutic | Trade name | Modality, target(s), and/or mode of action | Inflammatory indication(s)a | Dosing regimenb |
|---|---|---|---|---|
| Type I alpha-interferon (IFN-α), interferon alfacon 1, consensus interferon | Infergen | Recombinant (non-naturally) occurring type-I IFN-α; immunoregulator | Chronic hepatitis C infection | 15 μg daily SC |
| Interferon-α2a (IFN-α2a) | Roferon-A | Recombinant IFN-α2a; immunoregulator | Chronic hepatitis C infection | 3 MIU three times a week SC |
| PegInterferon-α2a | Pegasys | Recombinant IFN-α2a conjugated to PEG (40 kDa); immunoregulator | Chronic hepatitis C infection | 180 μg QW SC |
| Interferon-α2b (IFN-α2b) | Intron A | Recombinant IFN-α2b; immunoregulator | Chronic hepatitis C, hepatitis B infections | Hepatitis C:3 MIU TIW SC or IM |
| Hepatitis B: 5 MIU QD or 10 MIU TIW SC or IM | ||||
| PegInterferon-α2b | Peg-Intron | Recombinant IFNα2b conjugated to PEG (12 kDa); immunoregulator | Chronic hepatitis C infection | 1.0 μg/kg QW SC |
| Interferon-αn3 (IFN-αn3) | Alferon N | Nonrecombinant human IFN-α-n3 purified from pooled human leukocytes; immunoregulator | Condylomata acuminata (genital warts) | 250,000 IU per wart, twice weekly |
| Interferon-β1a (rIFN-β1a) | Avonex | Recombinant rIFN-β1a; antiviral and immunoregulator | MS | 30 μg QW IM |
| Interferon-β1b (rIFN-β) | Betaseron | Recombinant rIFN-β1b; antiviral and immunoregulator | MS | Initial: titration 0.0625 to 0.25 mg Q2D SC |
| Maintenance: 0.25 mg Q2D SC | ||||
| Omalizumab | Xolair | Humanized IgG1 directed against IgE and inhibits IgE binding to the high-affinity IgE receptor on mast cells and basophils | Asthma | Titrated to total serum IgE levels: 150–375 mg Q2W or Q4W SC |
| Rituximab | Rituxan | Chimeric (human/mouse) IgG1 that binds CD20, a cell surface protein found on normal and some malignant B cells, resulting in selective depletion of CD20+ B cells | RA | Two 1-g IV infusions separated by 2 weeks repeated every 24 weeks |
| Antithymocyte globulin (rabbit) | Thymoglobulin | Purified, pasteurized, IgG, obtained by immunization of rabbits with human thymocytes; immunosuppressant that selectively depletes T cells | Acute kidney transplant rejection, aplastic anemia | 1.5 mg/kg IV QD |
| Basiliximab | Simulect | Chimeric (human/mouse) IgG1 that binds to IL-2Rα chain, expressed on the surface of activated T lymphocytes, inhibiting the IL2-mediated activation of lymphocytes | Prophylaxis against allograft rejection in renal transplant patients | Two 20 mg IV doses |
| Daclizumab | Zenapax | Humanized IgG1 that binds to IL-2Rα chain, inhibiting the IL2-mediated activation of lymphocytes | Prophylaxis against acute allograft rejection in patients receiving renal transplants | 1 mg/kg IV Q2W transplant |
| Muromonab-CD3 | Orthoclone, OKT3 | Mouse mAb that binds CD3 and results in selective depletion of CD3+ T cells | Acute renal allograft rejection, steroid-resistant cardiac or hepatic allograft rejection | 5 mg/day IV for 10–14 days |
| Abatacept | Orencia | Recombinant human fusion protein consisting of the ECD of CTLA4 and the Fc (IgG1 modified) that inhibits T cell activation by binding to CD80 and CD86, thereby blocking interaction with CD28 | JIA, RA | JIA and RA “IV regimen”: |
| 500–1,000 mg or 10 mg/kg (based on body weight) Q2W 3 doses, followed by maintenance Q4W | ||||
| RA “SC regimen”: initial dose given by IV as above followed by 125 mg SC weekly | ||||
| Anakinra | Antril, Kineret | Recombinant form of IL-1R antagonist (IL-1Ra) that binds to IL-1 and inhibits it binding to IL-1 type 1 receptor (IL-1RI) | RA | 100 mg SC daily |
| Rilonacept | Arcalyst | Recombinant human fusion protein consisting of ECD of IL-1RI, IL-1 receptor accessory protein, and Fc (IgG1) that binds to IL-1 and inhibits its binding to the IL-1Rs | Cryopyrin-associated periodic syndromes | 160 mg SC QW, with 320 mg SC loading dose |
| Canakinumab | Llaris | Humanized IgG1 directed against IL-1β inhibits its binding to the IL-1Rs | Cryopyrin-associated periodic syndromes | 150 mg SC Q8W |
| Adalimumab | Humira | Human IgG1 that binds to soluble and membrane TNF-α and inhibits its binding to TNFRs | AS, CD, JIA plaque psoriasis, psoriatic arthritis, RA | RA, AS psoriatic arthritis: 40 mg SC Q2W |
| JIA: (15–30 kg) 20 mg SC Q2W, (≥30 kg), 60 mg SC Q2W | ||||
| CD: Initial: 160 mg SC, 80 mg 1 week later | ||||
| Maintenance: 40 mg SC Q2W | ||||
| Plaque psoriasis: initial: 80 mg SC | ||||
| Maintenance: 40 mg SC Q2W (starting the week following the initial dose) | ||||
| Etanercept | Enbrel | Recombinant soluble human TNF-R1 and Fc (IgG1) fusion protein that binds to soluble and membrane TNF-α and inhibits its binding to TNFRs | AS, JIA, plaque psoriasis, psoriatic arthritis, RA | RA, psoriatic arthritis: 50 mg SC QW |
| JIA: 0.8 mg/kg (max, 50 mg) SC QW | ||||
| Psoriasis: 50 mg SC twice weekly every 3 months | ||||
| Infliximab | Remicade | Chimeric (mouse/human) IgG1 that binds to soluble and membrane TNF-α and inhibits its binding to TNFRs; induction of activated T cell and macrophage apoptosis | AS, CD, plaque psoriasis, psoriatic arthritis, RA, UC | CD, UC, psoriatic arthritis, and plaque psoriasis: Initial: 5 mg/kg IV 0, 2, and 6 weeks |
| Maintenance: 5 mg/kg IV Q8W | ||||
| RA: 3 mg/kg IV at weeks 0, 2, and 6 | ||||
| Maintenance: 3 mg/kg IV Q8W | ||||
| AS: 3 mg/kg IV at weeks 0, 2, and 6 | ||||
| Maintenance: 3 mg/kg IV Q6W | ||||
| Certolizumab pegol | Cimzia | Humanized Fab-PEG conjugate that binds to soluble and membrane TNF-α and inhibits its binding to TNFRs | CD, RA | CD: initial: 400 mg SC (two 200-mg injections) at weeks 0, 2, and 4 |
| Maintenance: 400 mg Q4W | ||||
| RA: initial: 400 mg SC (two 200-mg injections) at weeks 0, 2, and 4 | ||||
| Maintenance: 200 mg Q2W | ||||
| Golimumab (Simponi) | Simponi | Human IgG1 Ab that binds to soluble and membrane TNF-α and inhibits its binding to TNFRs | AS, psoriatic arthritis, RA | 50 mg SC Q4W |
| Alefacept | Amevive | Recombinant human LFA3 ECD and Fc (IgG1) fusion protein that inhibits LFA3–CD2 interaction and blocks lymphocyte activation | Plaque psoriasis | 7.5 mg Q1W IV or 15 mg Q1W IM |
| Efalizumab | Raptiva | Humanized IgG1 that binds CD11a, a subunit of LFA-1 (expressed on all leukocytes), decreases cell surface expression of CD11a and inhibits its binding to ICAM-1, preventing T cell activation, adhesion, and migration to site of inflammation | Plaque psoriasis (withdrawn) | 1 mg/kg Q1W (with 0.7 mg/kg SC first conditioning dose) |
| Natalizumab | Tysabri | Humanized IgG4 that binds to the α4-subunit of α4β1 and α4β7 integrins, blocking their interactions with VCAM1 and MadCAM1 and preventing transmigration of leukocytes into inflamed tissues | CD, relapsing MS | 300 mg IV, Q4W |
| Belimumab | Benlysta | Human IgG1 that binds BLyS, a B cell survival factor, and prevents binding to its receptors on B cells, thereby inhibiting the survival of B cells, (including autoreactive B cells) and differentiation of B cells into Ig-producing plasma cells | SLE | 10 mg/kg IV Q2W for the first 3 doses and Q4W thereafter |
| Ustekinumab | Stelara | Humanized IgG1 directed against p40 subunit of IL-23/IL-12 and prevents interaction between receptors, inhibiting IL-12 and IL-23-mediated immune responses | Plaque psoriasis | 45 or 90 mg (based on body weight) Q4W for the first 2 doses and Q12W SC thereafter |
| Eculizumab | Soliris | Humanized mAb that binds complement protein C5 and inhibits its cleavage to C5a and C5b, preventing the formation of the terminal complement complex and reducing hemolysis | Paroxysmal nocturnal hemoglobinuria | Initial: 600 mg QW IV for 4 weeks, followed by 900 mg IV 1 week later. Maintenance: 900 mg Q2W |
| Tocilizumab | Actemra | Humanized IgG1 directed against soluble and membrane and inhibits IL-6R-mediated signaling through these receptors | JIA, RA | RA: 4 mg/kg or increased to 8 mg/kg (based on clinical experience) IV Q4W |
| JIA: 8 or 12 mg/kg (based on child's body weight) IV Q2W |
IV intravenous; SC subcutaneous; IM intramuscular; PEG polyethylene glycol; IL interleukin; Ig immunoglobulin; R receptor; Fab fragment, antigen binding; Fc fragment, crystallizable; ECD extracellular domain; LFA3 lymphocyte function-associated antigen; TNF tumor necrosis factor α; CTLA4 cytotoxic T lymphocyte antigen 4; BLyS B lymphocyte stimulator; MIU million international units; TIW three times a week; QD daily, QW weekly; QXD every X days; QXW every X weeks; RA rheumatoid arthritis; JIA juvenile idiopathic arthritis; MS multiple sclerosis; SLE systemic lupus erythematosus; CD Chron's disease; UC ulcerative colitis; AS ankylosing spondylitis
aOther indications for the same drug not listed
bDosing regimen is shown for listed indication(s) only. Some drugs are approved in combinations with compounds, which are not specified here. The dosing information is from the package insert
These therapeutic proteins could be grouped into several subclasses, based on the point of intervention within an inflammatory cascade that is being modulated and the mode of action (Fig. 1): (1) cytokine/cytokine receptor antagonists, with multiple tumor necrosis factor-α (TNF) inhibitors approved for a variety of autoimmune indications and, recently, anti-B lymphocyte stimulator (BLys) Ab for SLE being the most famous examples; (2) lymphocyte-depleting agents, such as anti-CD20 and anti-CD4 monoclonal antibodies (mAbs) for RA; (3) agents that interfere with T cell receptor complex-mediated signal transduction and costimulatory signaling in T cell activation, including tolerance-inducing agents, such as CTLA4-Ig (a fusion protein of the external domain of cytotoxic T lymphocyte-associated antigen 4 and the Fc region of human IgG1) and anti-CD3 nonactivating mAb for RA or transplant rejection; (4) agents that interfere with cell trafficking or adhesion of T cells to antigen-presenting cells (APC), such as the mAb against the α4-subunit of α4β1 and α4β7 integrins for CD and MS; (5) agents that target components of the innate immune system, such as an anti-C5 complement subunit mAb for paroxysmal nocturnal hemoglobinuria; and (6) immunomodulatory agents, such as interferon beta or interferon alpha (for MS or as an antiviral treatment, with yet to be clarified mode of action). Note that therapeutic proteins in subclasses 1, 3, 4, and 5 may exert their function by blocking the ligand–receptor interactions and/or by downmodulation of the expression of the cell surface molecules. In addition, some agents have multiple modes of action and thus may be included in more than one subclass.
Fig. 1.

Common subclasses of therapeutic proteins for treatment of autoimmune and other inflammatory conditions based on points of intervention and mode of action. 1 cytokine/cytokine receptor antagonists; 2 lymphocyte-depleting agents; 3 agents that interfere with T cell–receptor (TCR) complex-mediated signal transduction and costimulatory signaling in T cell activation, including tolerance-inducing agents; 4 agents that interfere with cell trafficking or adhesion of T cells to APCs (such as dendritic cells, macrophages, B cells) and/or endothelium (including integrin antagonists); 5 agents that target components of the innate immune system, including anti-complement (5a), anti-chemokine drugs (5b), or drugs targeting cellular components of the innate immune system (5c). Agents in subclasses 1, 3, 4, and 5c may exert their function by blocking the ligand–receptor interactions and/or by downmodulation of the expression of cell surface molecules. Some agents have multiple modes of actions and thus may be included in more than one subclass. Additional therapeutic proteins that do not directly fall in the above subclasses, as described in the text. TCR-associated complex includes TCR, CD3, CD8, or CD4 on T cells and MHC presenting an antigenic peptide on APCs. Costimulatory factors include CD28 and lymphocyte function-associated antigen-1 (LFA) on T cells, CD80, CD86, and intercellular adhesion molecule 1 (ICAM1) on APCs. Phagocytes = dendritic cells, macrophages, and/or neutrophils
Some therapeutic proteins do not directly fall in any of the above categories, for example intravenous immunoglobulin products (used to treat a number of primary immunodeficiencies and autoimmune diseases), anti-IgE for asthma, as well as some locally delivered agents for osteoarthritis. Chan and Carter have recently reviewed the key insights learned from the development of therapeutic antibodies for autoimmunity and inflammation, the most common class of therapeutic proteins (1). This review focuses on the absorption, distribution, metabolism, and excretion (ADME) of antibodies, as well as other therapeutic proteins from the point of view of drug development in this therapeutic area.
In general, there are multiple practical and commercial constraints for delivering an effective and safe dose of a therapeutic protein, including dosing volume, dosing frequency, cost of goods, target properties (expression profile/turnover rate), as well as a range of modalities available for a particular target. Many of the above constraints are constantly changing with the commercial landscape and advances in protein engineering, pharmaceutical, and ADME sciences. These constraints are used to guide the design of an ideal target product profile, which, in turn, evolves itself, as a therapeutic protein moves along the pipeline. When considering optimization of ADME profiles, several factors—some of which are uniquely affected by inflammation and autoimmune indications—should be considered. The purpose of this review is to highlight common questions and challenges encountered during the optimization of ADME and pharmacokinetic/pharmacodynamic (PK/PD) profiles of therapeutic proteins for treating these disorders.
INTERDEPENDENCY OF PK, PD, AND IMMUNOGENICITY PROFILES
Humanization of mouse- or rat-derived antibodies has significantly improved the immunogenicity profiles of therapeutic antibodies (1–4). In addition, advances in protein engineering tools, such as phage display or mice expressing human immunoglobulin genes, have enabled production of fully human mAbs (reviewed in (1)). However, even fully human mAbs contain unique sequences in their CDRs, and it has become clear that nearly all therapeutic proteins, including fully human ones, may be immunogenic, depending on the context of usage (1–4). The context of usage includes patient/subject population (species, disease status, age, and sex), dosing regimen (dose level, route, and duration of treatment), concomitant medications, as well as the manufacturing process and formulation (affecting post-translational modifications, impurities, and aggregation).
Immunogenicity may affect both the PK and PD profiles and sometimes the efficacy and safety of therapeutic proteins (Fig. 2). Specifically, anti-drug antibody (ADA) responses may introduce additional clearance and distribution pathways, dependent on the formation of drug/ADA complexes. When a drug/ADA immune complex (IC) is formed, the clearance of a drug within the IC may be much faster compared to that for a drug not bound to ADA, leading to a rapid concentration drop in a concentration-time profile. Because the extent and rate of IC formation vary among human subjects, the IC-related clearance could be considered a major contributor to the intersubject variability in clinical and nonclinical PK profiles for therapeutic proteins. The alternate scenario is also possible, especially for biotherapeutics with relatively fast clearance, in which there is a time-dependent decrease in the apparent elimination rate of a drug caused by formation of drug/ADA complexes that are cleared at a slower rate compare to the drug not in complex with ADA. In addition to the “real impact” of ADA on PK, ADA may interfere in the PK assay, such that an apparent rapid concentration drop may be a consequence of this interference. A “real PK impact” of ADA usually correlates with a biological effect (PD, efficacy, and/or toxicity), while an apparent PK impact caused by assay interference may have a lower impact on PD, except in cases where there is a strong neutralizing component in ADA and drug concentrations are relatively low. ADA impact on PD of a therapeutic protein includes neutralization of biological activity or introduction of new biological activity. New biological activity in some cases also may lead to toxicity: for example, IC deposition in the kidney, crosslinking-dependent signaling (leading to agonistic effects and/or cytokine storm), effector function mediated by ADA in complex with a therapeutic protein (leading to hypersensitivity reactions or depletion of target-expressing cells), and immune reaction to or neutralization of an endogenous protein (especially if a biotherapeutic is constructed using moieties/sequences that are expressed in the study population) (5).
Fig. 2.
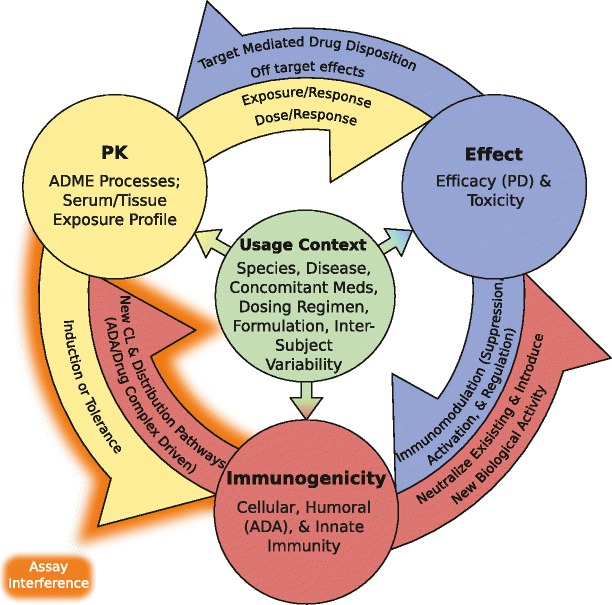
The relationship between PK, effect, and the host immune response to a therapeutic protein. Immunogenicity may affect both the PK and biological activity and ultimately the efficacy and safety of therapeutic proteins. Specifically, ADAs may introduce additional clearance and distribution pathways, dependent on formation of drug–ADA complexes. ADAs may neutralize existing biological activity and/or introduce new biological activity, in some cases causing toxicity: for example, IC deposition in the kidney, crosslinking, and subsequent signaling/agonistic activity; ADA-mediated effector functions (which may lead to depletion of target-expressing cells and hypersensitivity reactions), as well as immune reaction to or neutralization of an endogenous protein. In turn, both the PK and the biological activity of therapeutic proteins impact the host immune response to the drug. Exposure profiles (magnitude and duration) of a biotherapeutic in serum and in immune organs may shift the balance between the induction of an immune response and tolerance. Because of the intended biological activity of immune modulators, the biological activity is directly linked to immunogenicity, such that ADA formation or lack thereof (a consequence of an immune response or tolerance, respectively) may be viewed as a PD readout. Special considerations for interdependencies in PK–effect relationships for biotherapeutics include disease-dependent changes in expression of the intended (or unintended) target that impact contribution of TMDD to the ADME profile. The context of usage impacts PK, effect, and the host immune response and adds another layer of complexity in their inexpediencies. The context of usage includes subject population (for example species, disease status, age, and sex), dosing regimen (dose level, route, duration of treatment), concomitant medications, as well as the manufacturing process and formulation. Impact of potential reciprocal assay interferences in PK and ADA readouts should also be taken into account for data interpretation. PK pharmacokinetics; PD pharmacodynamics; ADA anti-drug antibody; TMDD target-mediated drug disposition; ADME absorption, distribution, metabolism, excretion; CL clearance; IC immune complex
PK and PD are also likely to affect ADA profiles, due to either the actual biology of the system (“real” impact of PK or PD on ADA, Fig. 2) or an interference caused by the drug in ADA assay readouts (or both). In particular, for immunomodulatory drugs (including both immunosuppressants and immunoactivators), the ADA formation (or lack thereof) may be viewed as a PD readout. In cases where the drug interferes in the ADA assay (such that there is a false-negative result in the ADA assay), changes in PK and PD may be used as alternative indicators of ADA formation. Optimization of assay conditions to minimize drug interference or employment of orthogonal methods may be used to confirm the presence of ADAs (4,6). For immunosuppressants, there may be an inverse dose response for the induction of ADAs, which may be related to suboptimal pharmacological activity at lower dose levels (providing a more permissive environment for ADA development) or more potent pharmacological activity (i.e., immunosuppression, resulting in the downregulation of the immune response against the drug itself) and/or development of tolerance at higher dose levels. Furthermore, some therapeutic proteins for the treatment of autoimmune conditions are administered in combination with small molecule immunomodulators (such as azathioprine, 6-mercaptopurine, methotrexate, cyclosporin A), which can introduce even more complexity in the dose–response relationship for the immunogenicity response and its subsequent effect on the PK/PD profile, as well as issues concerning translation from animals to humans. Below, we provide examples of nonclinical and clinical studies of therapeutic proteins for the treatment of autoimmune conditions that illustrate the interdependencies and challenges described above.
Case Study 1: Anti-IL-21R Abs
An example of the interdependency between PK, PD, and immunogenicity profiles, as well as inverse dose response for induction of ADAs, is described for the neutralizing anti-IL-21R antibodies that are being investigated for the treatment of autoimmune conditions (7). The pharmacological activities and PK profiles of two different human anti-IL-21R antibodies (referred to as Ab-01 and Ab-02) that differ in their in vitro potency and PK were examined in the MRL-Faslpr mouse model of lupus. The results indicated that only the more potent anti-IL-21R antibody (Ab-01) was able to elicit a full pharmacological effect, and this effect was also associated with lower or delayed ADA levels and slower clearance, compared to Ab-02 and also compared to the same antibody (Ab-01) administered at suboptimal doses.
Specifically, when Ab-01 was administered at 10 mg/kg three times per week (TIW) for 12 weeks to MRL-Faslpr mice, significant reductions in both a biomarker of disease (anti-double-stranded DNA antibodies) and IgG deposits in the kidney were observed. At this pharmacologically active dose level of 10 mg/kg, trough median serum Ab-01 concentrations were higher than predicted based on a single-dose PK profile (assuming linearity), and ADA titers appeared lower compared to those in the isotype control group (Fig. 3); however, it should be noted that circulating Ab-01 might have interfered in the ADA assay. When Ab-01 was administered at 2.5 or 5 mg/kg TIW for 12 weeks, only a partial pharmacological response was observed (with no reduction in IgG deposits in the kidney). At these sub-efficacious dose levels, median trough serum Ab-01 concentrations were either below the limit of quantitation (LOQ) or relatively low (i.e., decreased in more than a dose-proportional manner compared to the corresponding values in the 10 mg/kg group). Specifically, in the 5-mg/kg group, median trough Ab-01 concentrations were above the LOQ only for the first 4 weeks of treatment, while in the 2.5-mg/kg group, these values were below the LOQ at all time points tested (Fig. 3). All trough samples in the 2.5- and 5-mg/kg group tested positive for ADA in a screening assay (titers were not determined for these dose levels; Vugmeyster et al., unpublished observations). Overall, the more pharmacologically effective dose of Ab-01 was also associated with lower or delayed ADA levels and slower clearance.
Fig. 3.
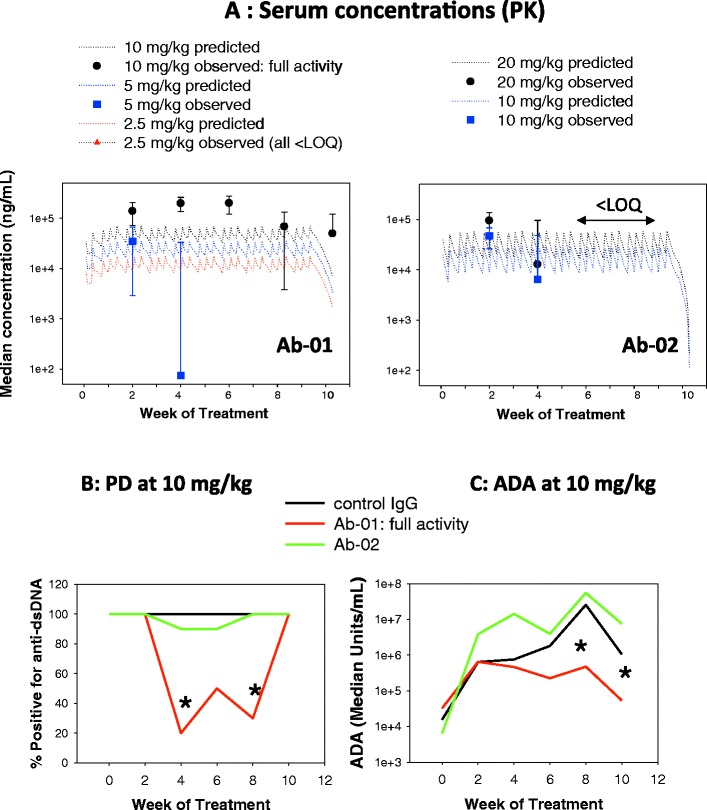
Example of interdependencies in PK, PD, and ADA data in preclinical studies of a biotherapeutic. Twelve-week-old MRL-Fas lpr mice were administered anti-IL-21R antibody Ab-01 (10, 5, or 2.5 mg/kg), anti-IL-21R antibody Ab-02 (20 or 10 mg/kg), or an isotype control antibody (10 mg/kg) by intraperitoneal (IP) injection three times per week over 10 weeks and sacrificed ~3 days after the last dose. Serum was collected prior to dosing (week 0) and then at 2, 4, 6, 8, and 10 weeks post-dose. Sera were analyzed for test article concentration (panel A), anti-dsDNA biomarker (panel B), and ADAs (panel C) by immunoassays. Panel A shows observed median ± standard deviations (symbols with error bars) and predicted concentrations (dotted lines; based on simulations using serum concentration data of Ab-01 and Ab-02 observed after a single IP dose to MRL-Fas lpr mice). Asterisks indicate a significant difference in anti-dsDNA biomarker (B) and ADA (C), as compared to the isotype control group. IgG deposits in the kidney sections were assessed by immunohistochemistry at sacrifice (not shown), and only the 10-mg/kg Ab-01 dose group showed significant reduction in IgG deposits (referred to as “full activity”). These data were compiled based on the results reported by Vugmeyster et al. (7) PK pharmacokinetics, PD pharmacodynamics, ADA anti-drug antibodies, LOQ limit of quantification, anti-dsDNA anti-double-stranded DNA antibodies
Although both Ab-01 and Ab-02 had biological activity in mouse and human IL-21-dependent cell-based assays, Ab-02 had lower potency in mouse cell-based assays. After a single 10-mg/kg IP dose to MRL-Faslpr mice, Ab-02 had ~2–3-fold lower exposure compared to Ab-01, and both antibodies had a sharp decline in the terminal phase, coincidental with the development of ADA. In contrast to Ab-01, Ab-02 administered at 20 mg/kg (or 10 mg/kg) TIW for 12 weeks had no significant pharmacological activity in the MRL-Faslpr mouse model of lupus. Trough median serum concentrations of Ab-02 given at 20 mg/kg were relatively low (compared to those of Ab-01 given at the pharmacologically active dose of 10 mg/kg) for the first 4 weeks and below the LOQ after 4 weeks (Fig. 3). ADA titers in mice treated with 10 or 20 mg/kg of anti-Ab-02 TIW for 12 weeks (or with a control human IgG) were 10–100-fold higher than those measured in animals treated with 10 mg/kg Ab-01 for 12 weeks.
The absence of pharmacological activity of Ab-02 in the MRL-Faslpr model may be a consequence of both rapid elimination (likely driven by both ADA-dependent and ADA-independent clearance mechanisms) and insufficient potency in blocking the effects of mouse IL-21R. Because IL-21 has a critical role in primary antibody responses (8), it is plausible that an antibody that neutralizes IL-21R effectively upon repeated dosing also reduces or delays the development of ADA against itself, which in turn would lead to slower elimination of this antibody compared to a less potent anti-IL-21 Ab or to suboptimal dose regimens for the same antibody. This effect may be enhanced in a mouse strain such as MRL-Faslpr, which is prone to autoimmune disease and polyclonal B cell expansion, and also has fast elimination of normal IgG, possibly due to the disease-induced impairment of the function of the neonatal Fc receptor (FcRn) (9).
Case Study 2: Anti-TNF Antibody CDP571
Another example of the complex interdependency between PK and immunogenicity profiles (including an inverse dose response for induction of ADA, species differences in these relationships, and an impact of concomitant medications) and ultimately the translation challenges are described for a humanized anti-TNF antibody CDP571. In single-dose studies of CDP571 in normal human volunteers (NHVs), ADAs exhibited an inverse dose response and had no pronounced impact on PK, and based on further characterization, the risk of increased ADA response upon chronic dosing was deemed low. In repeat dosing studies in autoimmune patients, ADA frequency was relatively low. However, since the patients were on concomitant immunosuppressive medications that affected multiple study endpoints including incidence of ADA, it was not possible to conclude that the prediction of low risk of increased ADA response was confirmed. Species differences are highlighted by the finding that, in contrast to NHVs, in single-dose monkey studies of CDP571, ADAs had a significant impact on PK, and an isotype class switch was observed, despite the fact that a major ADA epitope appeared the same in NHVs and monkeys.
Specifically, during preclinical and early clinical development, CDP571 was administered via a single IV infusion to NHVs and cynomolgus monkeys at doses ranging from 0.1 to 10 mg/kg (10). ADAs were detectable in all NHVs receiving 0.1 mg/kg CDP571, but the frequency and titer decreased with increasing dose (i.e., an inverse dose response for induction of ADAs). ADA response in NHVs was characterized as weak, transient, mostly of IgM isotype, non-neutralizing, and, in general, specific for a certain epitope (with no ADA response detectable to the constant or framework regions of CDP571). The authors used these ADA characterization data, as well as the observation of the decreasing ADA response at higher (therapeutically relevant) doses, as an argument of potential feasibility of chronic therapy with CDP571, since the risk of increased ADA response upon repeated dosing was deemed relatively low. In contrast to NHVs, after a single dose of CDP571 to cynomolgus monkeys, IgM to IgG isotype class switch for ADAs was detected in a majority of the animals, and ADAs appeared to have a more pronounced impact on PK profiles, compared to NHVs. However, as in humans, ADAs were, in general, specific for the same conformational epitope. In both monkeys and NHVs, the authors attributed the reduced anti-CDP571 response at higher dosages to induction of tolerance rather than sustained immunosuppression. These CDP571 studies in monkeys and NHVs highlight the difficulty in extrapolating the impact of ADAs on PK profiles from monkeys to humans, even in the cases when the majority of ADA response is considered to be directed to the same epitope.
In the subsequent repeated dosing studies of CDP571 in humans with CD (two separate clinical trials), a relatively low percentage of patients were ADA positive (~6–11%) (11,12). However, it is not possible to conclude that the prediction of a low risk of increased ADA response (made based on the single-dose data in NHVs) was confirmed, because an interpretation of repeated dosing data may have been confounded by the concomitant immunosuppressive medications that affected the ADA profile (as well as efficacy, safety, and PK). While the co-administration of immunosuppressive medications appeared to correlate with lower ADA frequency within each clinical trial, the overall and subgroup-specific frequencies of ADA response were variable across the two trials. These studies illustrate additional complexities in interpretation of PK, PD, and ADA data of immunosuppressive proteins that are co-administered with other immunosuppressive medications and the need for an integrated interpretation of ADA impact on study results.
Case Study 3: Anti-CD4 Antibody MTRX1011A
Preexisting antibodies to some therapeutic proteins, which tend to be more prevalent in the disease population than in NHVs, are an emerging issue that contributes to potential ADA impact on PK/PD profiles and may complicate data interpretation and translation of PK/PD and ADA profiles from NHVs to patients with an autoimmune disease. Preexisting antibody reactivity may be caused by exposure to related biotherapeutics or environmental antigens or may be naturally occurring antibodies. An example of the confounding impact of preexisting reactivity and disease status on interpretation of PK/PD data is presented for a humanized nondepleting anti-CD4 antibody MTRX1011A.
MTRX1011A contains a mutation in the Fc region (N434H) designed to improve MTRX1011A binding to FcRn and thereby decreases the nonspecific systemic elimination rate (Kel) and increases exposure (13). Preexisting (IgM) antibodies that recognize N434H in MTRX1011A were detected in approximately 70% of RA patients but not in NHVs or baboons. In baboons, the benefit of incorporating N434H mutation on serum PK was clearly demonstrated compared to the parent chimeric anti-CD4 antibody TRX1 that had no FcRn modulating mutation (13). However, the impact of N434H mutation on human Kel could not be demonstrated. Authors indicate that comparison of PK/PD data for TRX1 and MTRX1011A in humans was confounded by the differences in populations used for the first-in-human studies, which were RA for MTRX1011A and of NHVs for TRX. These differences in study subjects are especially important because of the potential impact of disease status on the PK/PD properties of these antibodies: in part because of the significant contribution of target-mediated disposition, as well as because of the high prevalence of preexisting ADAs for MTRX1011A in RA populations but not for TRX1 in NHVs. While very few patients developed non-IgM anti-MTRX1011A antibodies post MTRX1011A treatment and there was no apparent relationship between ADA status (both IgM and non-IgM) and PK of MTRX1011A, the authors suggested that some of the anti-MTRX1011A reactivity might be partially neutralizing with respect to the expected beneficial attenuation of FcRn binding. The authors indicated that the cross-population comparison and test article choice were not optimal for testing the benefits of the FcRn modulating mutation in humans, and therefore, further studies would be required to test the benefit of N434H in humans.
In summary, for immunomodulatory/immunosuppressive biotherapeutics, special consideration should be given to assessment of the interdependency of ADME, PK/PD, and immunogenicity (including preexisting reactivity) profiles across various preclinical species and humans, including both the interdependencies in biology and assay readouts. The differences in the context of usage (such as dosing regimens, disease status, and concomitant medications) may affect immunogenicity profiles and its impact on ADME and PK/PD profiles. Because of the above complexities and interdependences, it is essential to interpret PK, PD, and ADA readouts and ultimately human efficacy and safety data in an integrated manner.
DISTRIBUTION TO NORMAL VERSUS DISEASE TISSUE
Several recent studies report differences in tissue penetration of macromolecules in normal versus inflamed tissues and exemplify the potential importance and challenges of addressing tissue penetration of different biotherapeutic modalities for treatment of inflammatory conditions in the disease (inflamed) settings. Palframan et al. examined uptake of several TNF inhibitors into the paws of healthy mice and inflamed paws of mice with collagen-induced arthritis (CIA), a mouse model of RA (14). Specifically, healthy and CIA mice were injected with a single 2 mg/kg IV dose of fluorescently labeled certolizumab pegol (PEGylated recombinant, humanized anti-TNF Fab′), adalimumab (a recombinant human anti-TNF mAb), or infliximab (a chimeric anti-TNF mAb), and paw uptake was quantified using a noninvasive biofluorescence imaging method. All three TNF inhibitors distributed more effectively into the inflamed tissue than the noninflamed tissues in this animal model of arthritis. Similar differences in tissue penetration of these TNF inhibitors into inflamed versus noninflamed mouse tissues were also obtained in the DSS-induced animal model of IBD relative to the control healthy mice (15).
It is likely that in many cases, the relative impact of ADAs on tissue distribution and resulting tissue exposure will be different between the healthy and disease subjects and that these relative differences cannot be directly extrapolated from serum profiles. As an example, for the anti-IL-21R antibody Ab-01 (mentioned in Case Study 1), biodistribution studies were conducted in wild type and MRL-FASlpr lupus-prone mice following a single dose of 125I-labeled drug (16). Although both the serum and tissue Ab-01 concentrations were lower in MRL-FASlpr mice than in healthy mice, the difference between serum and tissue drug concentrations was more pronounced in the lupus-prone animals. Although ADA likely was a major cause of these differences, other strain and disease-specific factors have likely contributed including the increased receptor-mediated clearance and/or tissue uptake (due to expansion of IL-21R-expressing lymphoid cells in MRL-FASlpr mice), impaired FcRn function, and/or increased vascular permeability (9,17). It should be noted that similar to the impact of ADAs, effects of altered target expression and/or increased vascular permeability may not always be evident from serum PK profiles, even in cases where there is a significant impact of these factors on distribution, as exemplified by the biodistribution study of anti-RAGE Ab in target-expressing and knockout mice (18).
Similar considerations related to disease status apply for characterization of retention profiles of locally administered biotherapeutics, for example intra-articular (IA) injections for various arthritic conditions or intradermal injection for psoriasis or atopic dermatitis. In a recent study, disposition of 125I-labeled recombinant human lubricin ([125I]LUB:1) was examined in normal rats and in the rat meniscal tear model of post-traumatic arthritis following a single IA injection (19). Micro-autoradiography analysis of tissue sections suggested that [125I]LUB:1 tended to localize to damaged joint surfaces in rats with meniscal tear; however, no differences in total knee counts between healthy and operated rats were noted.
In humans, due to ethical considerations, side-by-side comparisons of tissue penetration of a therapeutic protein in healthy versus inflamed tissues are usually not available. In addition, there is a paucity of data on tissue penetration in inflammatory/autoimmune patients, because of both the logistics of sample availability and limitations in bioanalytical methodologies. In one study conducted nearly 40 years ago, catabolism and serum-synovial transport of a purified rheumatoid factor were examined in patients with inflammatory joint disease, and this study suggested that in this patient population, the synovial membrane offers little barrier to antibody transport (20). Further studies with several TNF inhibitors indicated that concentrations of these therapeutic proteins in synovial fluid of RA patients is 30–100% of that in serum (21, 22). Penetration into the brain is an especially challenging but highly relevant problem when considering the number of indications with the site of action in the central nervous system. For example, rituximab (an anti-CD20 Ab) and alemtuzumab (an anti-CD52 antibody) are currently being developed for the treatment of MS (23). However, in the case of rituximab, the steady-state cerebrospinal fluid levels in MS patients are reported as <0.25% to <0.1% of serum levels (23). It should be noted that for mAbs that are being explored for neurological indications, it is not known whether brain penetration is required for efficacy, since pharmacological activity in the periphery (e.g., B cell depletion by rituximab) may be the main driver for the clinical endpoints.
In general, several factors may explain the differences in tissue penetration of therapeutic proteins in normal versus inflamed tissues (Box 1). Inflamed tissues tend to have a disrupted endothelium (24), which means there is less of a barrier to passive diffusion compared to normal tissue. In addition, in autoimmune conditions such as RA, new vessel formation at the site of inflammation has been reported, providing additional mechanistic basis for the apparent increased vascular permeability of macromolecules in RA (reviewed in (25)). Also, for biotherapeutics that target various components of the immune system, the target expression may be higher in inflamed tissues, including both an upregulation of target expression per cell and an increase in the number of target-expressing cells (related to increased vascular permeability and chronic inflammatory cell infiltration, as well as to chronic lymphoproliferative responses), which means that there may be a target-dependent tissue retention or uptake of an immunomodulatory therapeutic. The differences in lymphatic drainage and in the impact of ADA on tissue profiles (as mentioned above) (16) in normal versus inflamed settings have also been reported (26,27). Thus, the mechanistic basis for differences in penetration between the normal and inflamed tissues is likely to vary with the biotherapeutic modality being employed, and an understanding of these mechanisms is crucial for engineering of the next generation of biotherapeutics with improved tissue penetration to the site of action. It is expected that the application of emerging noninvasive quantitative imaging technologies would be instrumental in advancing our knowledge in tissue penetration of biotherapeutics in humans, including addressing differences between healthy and inflamed states. The follow-up challenge would be investigating and establishing the clinical relevance (including impact on dosing regimens) of these potential tissue penetration differences.
QUEST FOR HALF-LIFE EXTENSION
The autoimmune and inflammatory disease market for biological therapies is one of the most competitive in the industry, with more than 30 proteins approved and hundreds of therapeutic proteins in various stages of development (Table I and (1,28)). Because of the persistent nature of autoimmune and inflammatory conditions, chronic treatments are needed to adequately manage these diseases. Furthermore, identical or similar targets and/or active moieties are often being exploited by different pharmaceutical companies (Table I). Therefore, the PK/PD profile, target affinities, and formulations that are compatible with infrequent dosing delivered by routes amenable to self-administration (most commonly subcutaneous [SC] but also intramuscular [IM]) provide a clear commercial advantage and are used to define target product profiles during development. In general, there are multiple approaches to attenuate the half-life of a therapeutic protein, and the state of the art in half-life extension (HLE) of therapeutic proteins has been recently extensively reviewed (29). A significant amount of ADME-centric protein engineering and subsequent in vivo validations and investigations are being conducted for biotherapeutics intended for autoimmune or other inflammatory conditions. Below, we focus on the classic examples and emerging science in this field.
Fc Engineering
One of the earliest and most common approaches for HLE is the attachment of an Fc domain to gain protection from endosomal degradation by taking advantage of FcRn recycling (reviewed in (30–32)). Enbrel, comprised of a soluble TNF receptor (p75) fused to the Fc domain of a human IgG1 and developed to treat RA was the first Fc-fusion protein to be marketed (33). The modern version of this approach is mutating the FcRn binding site to gain half-life extension beyond that for wild-type IgGs (reviewed in (30–32)). As mentioned above (Case Study 3), the first FcRn-engineered antibody reported to be administered to humans is an anti-CD4 antibody MTRX1011A for RA (13).
While significant knowledge has been acquired on the impact of the FcRn-site engineering on serum half-lives of Fc-containing biotherapeutics, the impact of these mutations on biodistribution and SC absorption is an emerging research field. Wang et al. provided initial evidence for the role of FcRn on SC absorption of an antibody: for a model antiplatelet antibody 7E3, the systemic bioavailability after a SC dose was ∼3-fold higher in WT mice than in FcRn-deficient mice (34). Deng et al. compared SC absorption profiles of a wild-type anti-TNF Ab IgG and two FcRn-engineered variants (N434A and N434H) in both mice and monkeys (35). While some differences in SC absorption parameters between the wild-type and FcRn-engineered variants of an anti-TNF Ab were noted and hypotheses to explain these differences were proposed, mechanistic studies to understand the role of FcRn in SC absorption of antibodies remain to be conducted. Comprehensive evaluation of the contribution of FcRn to transport of IgGs, likewise, needs to be carried out. It is likely that biotherapeutic candidates for the treatment of autoimmune conditions that have been engineered to contain FcRn mutations, such as anti-TNF and anti-CD4 mAbs described above, will be used as model compounds for these studies (13,35).
PEGylation
Attachment of a polyethylene glycol (PEG) polymer is commonly used to prolong in vivo exposure of therapeutic proteins. Different PEGs ranging in size from 1 up to 50 kDa and also differing in site and chemistry of attachment are available, and all result in some improvement in serum half-life, ranging from a 4-fold to a nearly 1,000-fold longer half-life compared to the unconjugated protein (reviewed in (36–39)). Two basic mechanisms are likely to contribute to the effect of PEGylation on PK profiles of therapeutic proteins: reduction in renal clearance through increased overall size/molecular weight and masking of the proteolytic sites on the therapeutic protein (reviewed in (37,38,40)). It is believed that protein–PEG conjugates with larger size and/or hydrodynamic volume of PEG (and in some but not all cases, PEGs with a higher degree of branching) are more likely to have higher serum exposures (39,41–43). However, since the loss of biological activity may be an unwanted consequence of PEGylation, the optimal PEG structure may vary for different therapeutic proteins (40).
PEGylated interferon-α (IFN-α) for treatment of chronic hepatitis C, Peg-Intron, was one of the first approved PEGylated products. Currently, there are two different FDA-approved PEGylated interferon alpha treatments: Peg-Intron, a conjugate of recombinant IFN-α2b and a linear 12-kDa PEG, and Pegasys, a conjugate of IFN-α2a and a 40-kDa PEG. Both Peg-Intron and Pegasys are administered via weekly SC injections and are recommended to be used in combination with ribavirin (reviewed in (44) and Table I). The mean terminal half-life after SC dosing in patients with chronic hepatitis C appears shorter for PEG-Intron (~40 h) compared to Pegasys (~80 h), which is consistent with the difference in PEG size between the two conjugates (45,46). It should be noted that in comparative clinical trials at the approved dose regimens, Pegasys and PEG-Intron had comparable efficacy and safety profiles (47).
Similar to the case for FcRn engineering, the impact of PEGylation on ADME properties of biotherapeutics beyond serum half-life extension is not well understood. The pioneering studies on the impact of PEGylation on biodistribution were performed for certolizumab pegol (Cimzia), which is the PEGylated recombinant humanized anti-TNF Fab′ and is the first antibody fragment approved for clinical use (indicated for CD and RA, Table I). The fluorescent imaging study in mice by Palframan et al. to address biodistribution of certolizumab pegol and anti-TNF IgGs is outlined in the previous section (14). In this study, the ratio of penetration of certolizumab pegol into arthritic paws compared with normal paws was greater than that observed with the two different anti-TNF IgGs, although all three anti-TNF inhibitors had more efficient penetration into inflamed paws. Furthermore, the accumulation of certolizumab pegol in inflamed paws was more responsive to the severity of inflammation when compared with the anti-TNF IgGs. The authors suggested that these differences in tissue penetration of certolizumab pegol compared to anti-TNF IgGs were conferred on the molecule by PEGylation. However, since different active anti-TNF moieties were used for the Fc-based versus the PEG-based HLE approaches, this hypothesis needs to be confirmed by additional experiments. Nevertheless, this study suggested that the differences in tissue penetration between various HLE approaches may be more pronounced in the disease settings.
The studies that address the impact of PEG characteristics (such as size, degree of branching, and attachment site) on tissue penetration of protein–PEG conjugates are sparse. Yamoka et al. showed differences in tissue uptake for naked (non-protein attached) PEG molecules of different molecular weight after IV administration in mice (42,43). A recent case study with a PEGylated TNF-binding Nanobody suggested that the impact of the degree of PEG branching on tissue distribution may not necessarily parallel the impact on serum PK (48). In this study, mice were given a single IV injection of 125I-labeled anti-TNF Nanobody conjugated to either linear or branched 40-kDa PEG and serum PK, and tissue penetration in normal mice was examined. While serum exposure was higher for the branched TNF Nanobody–PEG conjugate, the tissue exposure in some organs (e.g., spleen) was similar between these two constructs.
Hu et al. studied the differences in absorption kinetics of PEG–IFN β-1a, following IM versus SC administration to rhesus monkeys (49). PEG–IFN β-1a is a second-generation IFN β-1a–20-kDa PEG conjugate and is currently in phase III studies for the treatment of MS, with the goal of decreasing the frequency of administration compared to the approved unconjugated IFN β-1a (Anovex, Table I). In rhesus monkeys, PEG–IFN β-1a was absorbed more quickly after a single IM injection (evident by earlier Tmax and higher Cmax), such that the absorption rate constant (ka) for the SC route was 5-fold lower than that for the IM route. The difference in ka between IM and SC routes in rhesus monkeys appears to be unique to the PEGylated IFN β-1a, since the reported Tmax for non-PEGylated IFN β-1a in rhesus monkeys was similar between the IM and SC routes. However, the route differences between the absorption kinetics of PEGylated IFN β-1a in rhesus monkeys did not translate to humans (50), again highlighting the challenges in translating ADME profiles from animals to humans, especially for novel modalities.
Albumin Binding
Attachment of albumin/albumin-binding domains (such as nanobodies, albumin-binding domain derived from streptococcal protein G [ABD], and other albumin-binding peptides) is another HLE approach, which provides protection from degradation through FcRn recycling, conceptually similar to the Fc-fusion approach (reviewed in (31)). Zablin, the human albumin fusion protein that has advanced the farthest clinically, is a genetic fusion of human albumin and interferon-α2b developed for the treatment of chronic hepatitis C. The desired product profile for Zalbin included requirement for less frequent injections (administered every 2 weeks (Q2W) in clinical trials (51)) compared to the previously discussed competitor products Peg-Intron and Pegasys (approved for weekly dosing (QW (45,46)). Zablin submission was not approved by the FDA (because of the risk/benefit profile), and development was stopped.
As noted above for Fc engineering and PEGylation, there is little knowledge on the impact of fusing albumin or an albumin-binding domain to a biotherapeutic on the absorption kinetics or biodistribution beyond the expected decrease in systemic elimination. Andersen et al. reported that in healthy rats, the ABD molecule fused to epidermal growth factor 2 (HER2)-targeting affibody had a serum half-life and biodistribution profile similar to that of rat serum albumin (RSA) alone (52). Interestingly, these data also suggested that distribution of this ABD fusion or RSA shows a more rapid and wider distribution compared to that of a typical IgG. Coppieters et al. assessed distribution of a 125I-labeled Nanobody fusion construct comprised of anti-mouse TNF and anti-albumin Nanobodies in arthritic mice (CIA mouse model or RA) (53). In this study, accumulation of radioactivity in inflamed paws was detected (using a gamma camera); however, no suitable control groups were included to assess the specific contribution of the albumin-binding nanobody. Studies by Bocci et al. showed that when albumin is added to an IFN-α formulation, it acts as an interstitial fluid expander, thus favoring interferon absorption through lymphatics rather than blood capillaries, suggesting that albumin may influence SC absorption pathways of macromolecules (54,55).
In summary, because of the chronic nature of the diseases and the competitive landscape in autoimmune and inflammatory markets, many pioneering investigations on ADME-driven protein engineering approaches are being conducted with biotherapeutics in this therapeutic area. While significant knowledge has been acquired on the HLE capabilities of various modalities with respect to the serum PK profile, the impact on tissue distribution and absorption, as well as translation of ADME properties of novel modalities from animals to humans, is an emerging area of research.
CONCLUSION
Optimization of ADME and PK/PD profiles of protein therapeutics intended for the treatment of autoimmune and other inflammatory conditions often requires creative approaches to protein engineering, ADME study designs, and bioanalysis. Special consideration should be given to the assessment of the interdependency of PK, PD, distribution, and immunogenicity profiles across various preclinical species and humans, including animal and human subjects with different status of disease, inflammation, and concomitant medications. Along these lines, emerging questions include assessments of a preexisting reactivity to a biotherapeutic in disease populations and its impact on PK and PD profiles, as well as analysis of distribution to normal versus inflamed tissue, including contributions of target-mediated mechanisms, increased vascular permeability, differences in lymphatic drainage, and impact of ADA. Due to complexities and interdependences in underlying biology and assay readouts for PK, PD, and ADA profiles of biotherapeutics intended for treatment of autoimmune or inflammatory conditions, these data need to be interpreted in an integrated manner.
In addition, many pioneering ADME investigations on new approaches to extend the serum and the site-of-action exposures are being conducted for biotherapeutics in this therapeutic area, in part because treatment of inflammatory conditions requires chronic treatments and drug penetration to the tissues and, in part, due to strong competitive pressure in this market. To date, significant knowledge has been acquired on serum half-life extension capabilities of various approaches, including Fc engineering, PEGylation, and fusion to albumin-binding domains. On the other hand, characterization and reliable prediction (based on preclinical and/or in vitro data) of absorption and tissue penetration profiles of these new constructs are an emerging science, which holds great promise for optimizing efficacy and providing commercial advantage in the field of autoimmune and other inflammatory diseases.
However, similar to any therapeutic area in which biotherapeutic modalities are pursued, one of the most challenging tasks ahead is linking novel methodologies, investigations, and/or data analysis strategy to a clinically relevant endpoint.
ACKNOWLEDGMENTS
We thank Bonnie Rup for critical review of the manuscript.
REFERENCES
- 1.Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. 2010;10(5):301–316. doi: 10.1038/nri2761. [DOI] [PubMed] [Google Scholar]
- 2.Koren E, Zuckerman LA, Mire-Sluis AR. Immune responses to therapeutic proteins in humans–clinical significance, assessment and prediction. Curr Pharm Biotechnol. 2002;3(4):349–360. doi: 10.2174/1389201023378175. [DOI] [PubMed] [Google Scholar]
- 3.Sauerborn M, Brinks V, Jiskoot W, Schellekens H. Immunological mechanism underlying the immune response to recombinant human protein therapeutics. Trends Pharmacol Sci. 2009;31(2):53–59. doi: 10.1016/j.tips.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 4.Buttel IC, Chamberlain P, Chowers Y, Ehmann F, Greinacher A, Jefferis R, et al. Taking immunogenicity assessment of therapeutic proteins to the next level. Biologicals. 2011;39(2):100–109. doi: 10.1016/j.biologicals.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 5.Vugmeyster Y, Xu X, Theil FP, Khawli LA, Leach MW. Pharmacokinetics and toxicology of therapeutic proteins: advances and challenges. World J Biol Chem. 2012;3:73–92. doi: 10.4331/wjbc.v3.i4.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.EMEA/CHMP. Guideline on immunogenicity assessment of biotechnology derived therapeutic proteins. 2007.
- 7.Vugmeyster Y, Guay H, Szklut P, Qian MD, Jin M, Widom A, et al. In vitro potency, pharmacokinetic profiles, and pharmacological activity of optimized anti-IL-21R antibodies in a mouse model of lupus. MAbs. 2010;2(3):335–346. doi: 10.4161/mabs.2.3.11850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ozaki K, Spolski R, Feng CG, Qi C-F, Cheng J, Sher A, et al. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298:1630–1634. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- 9.Zhou J, Pop LM, Ghetie V. Hypercatabolism of IgG in mice with lupus-like syndrome. Lupus. 2005;14(6):458–466. doi: 10.1191/0961203305lu2129oa. [DOI] [PubMed] [Google Scholar]
- 10.Stephens S, Emtage S, Vetterlein O, Chaplin L, Bebbington C, Nesbitt A, et al. Comprehensive pharmacokinetics of a humanized antibody and analysis of residual anti-idiotypic responses. Immunology. 1995;85:668–674. [PMC free article] [PubMed] [Google Scholar]
- 11.Feagan BG, Sandborn WJ, Lichtenstein G, Radford-Smith G, Patel J, Innes A. CDP571, a humanized monoclonal antibody to tumour necrosis factor-alpha, for steroid-dependent Crohn's disease: a randomized, double-blind, placebo-controlled trial. Aliment Pharmacol Ther. 2006;23:617–628. doi: 10.1111/j.1365-2036.2006.02791.x. [DOI] [PubMed] [Google Scholar]
- 12.Sandborn WJ, Feagan BG, Radford-Smith G, Kovacs A, Enns R, Innes A, et al. CDP571, a humanised monoclonal antibody to tumour necrosis factor alpha, for moderate to severe Crohn's disease: a randomised, double blind, placebo controlled trial. Gut. 2004;53:1485–1493. doi: 10.1136/gut.2003.035253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng Y, Scheerens H, Davis JC, Jr, Deng R, Fischer SK, Woods C, et al. Translational pharmacokinetics and pharmacodynamics of an FcRn-variant anti-CD4 monoclonal antibody from preclinical model to phase I study. Clin Pharmacol Ther. 2011;89(2):283–290. doi: 10.1038/clpt.2010.311. [DOI] [PubMed] [Google Scholar]
- 14.Palframan R, Airey M, Moore A, Vugler A, Nesbitt A. Use of biofluorescence imaging to compare the distribution of certolizumab pegol, adalimumab, and infliximab in the inflamed paws of mice with collagen-induced arthritis. J Immunol Methods. 2009;348(1–2):36–41. doi: 10.1016/j.jim.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 15.Marenzana M, Eddleston A, Vugler A, Nesbitt A. Differential distribution of a PEGylated Fab′ into inflamed versus normal tissue compared to an IgG in arthritis and colitis models [abstract] Arthritis Rheum. 2009;60(Suppl 10):771. [Google Scholar]
- 16.Vugmeyster Y, DeFranco D, Szklut P, Wang Q, Xu X. Biodistribution of [125I]-labeled therapeutic proteins: application in protein drug development beyond oncology. J Pharm Sci. 2010;99(2):1028–1045. doi: 10.1002/jps.21855. [DOI] [PubMed] [Google Scholar]
- 17.Newkirk MM, Novick J, Stevenson MM, Fournier MJ, Apostolakos P. Differential clearance of glycoforms of IgG in normal and autoimmune-prone mice. Clin Exp Immunol. 1996;106(2):259–264. doi: 10.1046/j.1365-2249.1996.d01-847.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vugmeyster Y, DeFranco D, Pittman DD, Xu X. Pharmacokinetics and lung distribution of a humanized anti-RAGE antibody in wild-type and RAGE-/- mice. MAbs. 2010;2(5):571–575. doi: 10.4161/mabs.2.5.13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vugmeyster Y, Wang Q, Xu X, Harrold J, Daugusta D, Li J, et al. Disposition of human recombinant lubricin in naive rats and in a rat model of post-traumatic arthritis after intra-articular or intravenous administration. AAPS J. 2012;14:97–104. doi: 10.1208/s12248-011-9315-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bluestone R, Cracchiolo A, 3rd, Goldberg LS, Pearson CM. Catabolism and synovial transport of rheumatoid factor. Ann Rheum Dis. 1970;29(1):47–55. doi: 10.1136/ard.29.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou H. Clinical pharmacokinetics of etanercept: a fully humanized soluble recombinant tumor necrosis factor receptor fusion protein. J Clin Pharmacol. 2005;45(5):490–497. doi: 10.1177/0091270004273321. [DOI] [PubMed] [Google Scholar]
- 22.Abbott Laboratories. Humira [package insert]. Abbott Laboratories; 2011.
- 23.Petereit HF, Rubbert-Roth A. Rituximab levels in cerebrospinal fluid of patients with neurological autoimmune disorders. Mult Scler. 2009;15(2):189–192. doi: 10.1177/1352458508098268. [DOI] [PubMed] [Google Scholar]
- 24.Walsh DA, Wade M, Mapp PI, Blake DR. Focally regulated endothelial proliferation and cell death in human synovium. Am J Pathol. 1998;152(3):691–702. [PMC free article] [PubMed] [Google Scholar]
- 25.Koch AE. Angiogenesis as a target in rheumatoid arthritis. Ann Rheum Dis. 2003;62(Suppl 2):ii60–ii67. doi: 10.1136/ard.62.suppl_2.ii60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Semaeva E, Tenstad O, Skavland J, Enger M, Iversen PO, Gjertsen BT, et al. Access to the spleen microenvironment through lymph shows local cytokine production, increased cell flux, and altered signaling of immune cells during lipopolysaccharide-induced acute inflammation. J Immunol. 2010;184(8):4547–4556. doi: 10.4049/jimmunol.0902049. [DOI] [PubMed] [Google Scholar]
- 27.Zhou Q, Guo R, Wood R, Boyce BF, Liang Q, Wang YJ, et al. Vascular endothelial growth factor C attenuates joint damage in chronic inflammatory arthritis by accelerating local lymphatic drainage in mice. Arthritis Rheum. 2011;63(8):2318–2328. doi: 10.1002/art.30421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bell GM, Reynolds G, Isaacs JD. Biologic therapies in non-rheumatic diseases: lessons for rheumatologists? Nat Rev. 2011;7(9):507–516. doi: 10.1038/nrrheum.2011.106. [DOI] [PubMed] [Google Scholar]
- 29.Kontermann RE. Strategies to extend plasma half-lives of recombinant antibodies. BioDrugs. 2009;23(2):93–109. doi: 10.2165/00063030-200923020-00003. [DOI] [PubMed] [Google Scholar]
- 30.Roopenian DC, Sun VZ. Clinical ramifications of the MHC family Fc receptor FcRn. J Clin Immunol. 2010;30(6):790–797. doi: 10.1007/s10875-010-9458-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuo TT, Baker K, Yoshida M, Qiao SW, Aveson VG, Lencer WI, et al. Neonatal Fc receptor: from immunity to therapeutics. J Clin Immunol. 2010;30(6):777–789. doi: 10.1007/s10875-010-9468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaneko E, Niwa R. Optimizing therapeutic antibody function: progress with Fc domain engineering. BioDrugs. 2011;25(1):1–11. doi: 10.2165/11537830-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 33.Amgen Inc. and Pfizer Inc. Enbrel [package insert]. 2011;1–29.
- 34.Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548–558. doi: 10.1038/clpt.2008.170. [DOI] [PubMed] [Google Scholar]
- 35.Deng R, Loyet KM, Lien S, Iyer S, DeForge LE, Theil FP, et al. Pharmacokinetics of humanized monoclonal anti-tumor necrosis factor-{alpha} antibody and its neonatal Fc receptor variants in mice and cynomolgus monkeys. Drug Metab Dispos. 2010;38(4):600–605. doi: 10.1124/dmd.109.031310. [DOI] [PubMed] [Google Scholar]
- 36.Bailon P, Won CY. PEG-modified biopharmaceuticals. Expert Opin Drug Deliv. 2009;6(1):1–16. doi: 10.1517/17425240802650568. [DOI] [PubMed] [Google Scholar]
- 37.Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nat Rev Drug Discov. 2003;2(3):214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 38.Caliceti P, Veronese FM. Pharmacokinetic and biodistribution properties of poly(ethylene glycol)-protein conjugates. Adv Drug Deliv Rev. 2003;55(10):1261–1277. doi: 10.1016/S0169-409X(03)00108-X. [DOI] [PubMed] [Google Scholar]
- 39.Pasut G, Veronese FM. State of the art in PEGylation: The great versatility achieved after forty years of research. J Control Release. 2011 (in press) [DOI] [PubMed]
- 40.Fishburn CS. The pharmacology of PEGylation: balancing PD with PK to generate novel therapeutics. J Pharm Sci. 2008;97(10):4167–4183. doi: 10.1002/jps.21278. [DOI] [PubMed] [Google Scholar]
- 41.Fee CJ. Size comparison between proteins PEGylated with branched and linear poly(ethylene glycol) molecules. Biotechnol Bioeng. 2007;98(4):725–731. doi: 10.1002/bit.21482. [DOI] [PubMed] [Google Scholar]
- 42.Yamaoka T, Tabata Y, Ikada Y. Comparison of body distribution of poly(vinyl alcohol) with other water-soluble polymers after intravenous administration. J Pharm Pharmacol. 1995;47(6):479–486. doi: 10.1111/j.2042-7158.1995.tb05835.x. [DOI] [PubMed] [Google Scholar]
- 43.Yamaoka T, Tabata Y, Ikada Y. Distribution and tissue uptake of poly(ethylene glycol) with different molecular weights after intravenous administration to mice. J Pharm Sci. 1994;83(4):601–606. doi: 10.1002/jps.2600830432. [DOI] [PubMed] [Google Scholar]
- 44.Rosen HR. Clinical practice. Chronic hepatitis C infection. N Engl J Med. 2011;364(25):2429–2438. doi: 10.1056/NEJMcp1006613. [DOI] [PubMed] [Google Scholar]
- 45.Genentech. Pegasys [package insert]. Genentech, 1 DNA Way, South San Francisco, CA 9400–4990.
- 46.Genentech. Pegasys [package insert]. 2011;1–65.
- 47.McHutchison JG, Lawitz EJ, Shiffman ML, Muir AJ, Galler GW, McCone J, et al. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infection. N Engl J Med. 2009;361(6):580–593. doi: 10.1056/NEJMoa0808010. [DOI] [PubMed] [Google Scholar]
- 48.Vugmeyster Y, Entrican CA, Joyce AP, Lawrence-Henderson RF, Leary BA, Mahoney CS, et al. Pharmacokinetic, biodistribution, and biophysical profiles of TNF Nanobodies conjugated to linear or branched Poly(ethylene glycol). Bioconjugate Chem. 2012. doi:10.1021/bc300066a. [DOI] [PubMed]
- 49.Hu X, Miller L, Richman S, Hitchman S, Glick G, Liu S, et al. A novel PEGylated interferon beta-1a for multiple sclerosis: safety, pharmacology, and biology. J Clin Pharmacol. 2011;52:798–808. doi: 10.1177/0091270011407068. [DOI] [PubMed] [Google Scholar]
- 50.Hu X, Olivier K, Polack E, Crossman M, Zokowski K, Gronke RS, et al. In vivo pharmacology and toxicology evaluation of polyethylene glycol-conjugated interferon beta-1a. J Pharmacol Exp Ther. 2011;338(3):984–996. doi: 10.1124/jpet.111.180661. [DOI] [PubMed] [Google Scholar]
- 51.HGS. Zalbin. 2012. Available from: http://www.hgsi.com/albuferona.html. Accessed Feb 10, 2012
- 52.Andersen JT, Pehrson R, Tolmachev V, Daba MB, Abrahmsen L, Ekblad C. Extending half-life by indirect targeting of the neonatal Fc receptor (FcRn) using a minimal albumin binding domain. J Biol Chem. 2011;286(7):5234–5241. doi: 10.1074/jbc.M110.164848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coppieters K, Dreier T, Silence K, de Haard H, Lauwereys M, Casteels P, et al. Formatted anti-tumor necrosis factor alpha VHH proteins derived from camelids show superior potency and targeting to inflamed joints in a murine model of collagen-induced arthritis. Arthritis Rheum. 2006;54(6):1856–1866. doi: 10.1002/art.21827. [DOI] [PubMed] [Google Scholar]
- 54.Bocci V, Muscettola M, Grasso G, Magyar Z, Naldini A, Szabo G. The lymphatic route. 1) Albumin and hyaluronidase modify the normal distribution of interferon in lymph and plasma. Experientia. 1986;42(4):432–433. doi: 10.1007/BF02118644. [DOI] [PubMed] [Google Scholar]
- 55.Bocci V, Muscettola M, Naldini A, Bianchi E, Segre G. The lymphatic route–II. Pharmacokinetics of human recombinant interferon-alpha 2 injected with albumin as a retarder in rabbits. Gen Pharmacol. 1986;17(1):93–96. doi: 10.1016/0306-3623(86)90017-0. [DOI] [PubMed] [Google Scholar]