

Abstract
Glucagon-like peptide-1 (GLP-1) is an endogenous intestinal peptide that enhances glucose-stimulated insulin secretion. Its natural cleavage product GLP-1(9-36)amide possesses distinct properties and does not affect insulin secretion. Here we report that pretreatment of hippocampal slices with GLP-1(9-36)amide prevented impaired long-term potentiation (LTP) and enhanced long-term depression induced by exogenous amyloid β peptide Aβ(1–42). Similarly, hippocampal LTP impairments in amyloid precursor protein/presenilin 1 (APP/PS1) mutant mice that model Alzheimer's disease (AD) were prevented by GLP-1(9-36)amide. In addition, treatment of APP/PS1 mice with GLP-1(9-36)amide at an age at which they display impaired spatial and contextual fear memory resulted in a reversal of their memory defects. At the molecular level, GLP-1(9-36)amide reduced elevated levels of mitochondrial-derived reactive oxygen species and restored dysregulated Akt–glycogen synthase kinase-3β signaling in the hippocampus of APP/PS1 mice. Our findings suggest that GLP-1(9-36)amide treatment may have therapeutic potential for AD and other diseases associated with cognitive dysfunction.
Introduction
Despite recent impressive progress in basic research, an effective therapy is still lacking for Alzheimer's disease (AD), a devastating neurodegenerative disease that is the most common form of dementia and a leading cause of death in the elderly (Alzheimer's Association, 2011). With the incidence of AD rising as a result of population aging, it is imperative to design and validate disease-modifying treatments targeting multiple cellular and molecular mechanisms (Dyer et al., 2006; Querfurth and LaFerla, 2010; Selkoe, 2011; Ma and Klann, 2012). The amyloid β (Aβ) hypothesis, one of the leading theories for AD etiology, has resulted in current research focusing on lowering levels of brain Aβ, a small peptide derived from amyloid precursor protein (APP) (Haass and Selkoe, 2007). Meanwhile, the deleterious effects of Aβ on downstream signaling also are being vigorously pursued as potential disease-modifying targets (Pimplikar et al., 2010; Selkoe, 2011). One such target is mitochondrial-derived reactive oxygen species (ROS). There is substantial evidence linking ROS with neurodegenerative diseases (Lin and Beal, 2006), and it was reported previously that impairments in hippocampal synaptic plasticity and memory in AD model mice can be alleviated by decreasing mitochondrial ROS (Dumont et al., 2009; Massaad et al., 2009; Ma et al., 2011a).
Glucagon-like peptide-1 (GLP-1) is a primary incretin hormone released from the small intestine in response to nutrient ingestion. The majority of circulating “bioactive GLP-1” is in the form of GLP-1(7-36)amide, which stimulates glucose-dependent insulin secretion (Drucker, 2001; Baggio and Drucker, 2007). GLP-1 has an extremely short half-life (<2 min) and is rapidly cleaved into its “inactive” truncated form, GLP-1(9-36)amide, by the ubiquitous proteolytic enzyme dipeptidyl peptidase-4 (DPP-4) (Baggio and Drucker, 2007). Recent studies have shown that GLP-1(9-36)amide, initially considered to be an inactive degradation product of GLP-1, performs important physiological functions distinct from its precursor (Tomas et al., 2011). For example, GLP-1(9-36)amide does not affect either insulin secretion or glucose homeostasis (Rolin et al., 2004). Furthermore, GLP-1(9-36)amide directly prevents increased mitochondrial production of superoxide induced by either high glucose or high free fatty acids in human arterial endothelial cells, whereas intact GLP-1, in the presence of inhibitors of the GLP-1-degrading proteases DPP-4 and neutral endopeptidase (NEP) 24.11, does not (M.B., unpublished observation). GLP-1(9-36)amide also exerts cytoprotective actions on mouse cardiomyocytes exposed to hydrogen peroxide (Ban et al., 2010). We therefore hypothesized that GLP-1(9-36)amide could reduce hippocampal ROS and their deleterious effects on hippocampal synaptic function and memory in AD model mice.
We investigated the effects of the natural GLP-1 degradation product GLP-1(9-36)amide on AD-associated molecular, synaptic, and memory deficits. AD-associated impairments of hippocampal synaptic plasticity and memory both were improved by GLP-1(9-36)amide treatment. At the molecular level, elevated mitochondrial superoxide and dysregulated Akt–glycogen synthase kinase-3β (GSK3β) signaling in the APP/presenilin 1 (PS1) mice were normalized by GLP-1(9-36)amide. Our findings suggest that GLP-1(9-36)amide has potential as a novel therapeutic for treatment of AD and other disorders associated with cognitive dysfunction.
Materials and Methods
Mice.
All mice were housed in the Transgenic Mouse Facility of New York University, compliant with the National Institutes of Health Guide for Care and Use of Laboratory Animals. The facility is kept on a 12 h light/dark cycle, with a regular feeding and cage-cleaning schedule. APP/PS1 transgenic mice were purchased from The Jackson Laboratory, created via incorporation of a human/murine APP construct bearing the Swedish double mutation and the exon-9-deleted PSEN1 mutation (APPswe + PSEN1/ΔE9) (Jankowsky et al., 2001).
Hippocampal slice preparation and electrophysiology.
Four hundred micrometer hippocampal slices were prepared using a vibratome as described previously (Hu et al., 2006). The slices were maintained at room temperature in artificial CSF for at least 2 h before removal for experiments. For electrophysiology experiments, monophasic, constant-current stimuli (100 μs) were delivered with a bipolar silver electrode placed in the stratum radiatum of area CA3, and the field EPSPs (fEPSPs) were recorded in the stratum radiatum of area CA1. Long-term potentiation (LTP) was induced with a high-frequency stimulation (HFS) protocol consisting of two 1-s-long 100 Hz trains, separated by 60 s, delivered at 70–80% of the intensity that evoked spiked fEPSPs (Tsokas et al., 2007). Long-term depression (LTD) was induced with 300 pulses of low-frequency stimulation (LFS) at 1 Hz (Li et al., 2009).
Drug treatment.
Except for the behavioral tests, drugs were prepared as stock solutions and were diluted to the final concentration before use. The final concentrations and sources of the drugs were as follows: GLP-1(9-36)amide (100 pm; Bachem), MitoQ 10 methanesulfonate (MitoQ; 500 nm; Antipodean Pharmaceuticals), and Aβ(1–42) (100 μm; Bachem). Aβ(1–42) was prepared in water and stored at −20°C for at least 24 h before use at a final concentration of 500 nm. This type of preparation protocol yields ample Aβ oligomers (Ma et al., 2010). For behavioral tests, GLP-1(9-36)amide (500 ng · g−1 · d−1) was freshly made and incorporated into a micro-osmotic pump (model 1002; Alzet).
Detection of mitochondrial superoxide in hippocampal slices.
After completion of either drug or vehicle treatment, hippocampal slices were incubated with 5 μm MitoSOX Red, a mitochondrial superoxide indicator (prepared as 5 mm stock solution immediately before the experiments; Invitrogen) for 10 min. Slices then were fixed with ice-cold 4% paraformaldehyde in PBS overnight at 4°C. Slices were further cut into 40 μm sections and mounted onto presubbed slides with Vectashield mounting medium with DAPI (Vector Laboratories). The sections were imaged using a Leica TCS SP5 confocal microscope at 630×. All parameters (pinhole, contrast, gain, offset) were held constant for all sections from the same experiment.
Behavioral studies.
For all behavioral tasks, APP/PS1 and wild-type littermates (male and female, 10–12 months of age) were used, and the experimenter was blind to the genotypes. Experiments were performed as described previously (Banko et al., 2005). The training paradigm for the hidden platform version of the Morris water maze consisted of four trials (60 s maximum; 15 min interval) each day for 5 consecutive days. The probe trial was performed 2 h after the completion of training on day 5. The visible platform task consisted of four trials each day for 2 consecutive days with the escape platform marked by a visible cue and moved randomly between four locations. The trajectories were recorded with a video tracking system (Ethovision XT).
The training sessions for contextual and cued fear conditioning consisted of a 3 min exploration period, followed by two conditioned stimulus–unconditioned stimulus pairings separated by 1 min (footshock intensity: 0.6 mA, 2 s duration; tone: 85 dB white noise, 30 s duration). Contextual memory tests were performed in the training chamber 24 h after training. Cued memory tests were performed in a distinct chamber 24 h after training.
Western blotting.
Lysates were prepared as described previously (Banko et al., 2005). Sample were loaded on 4–12% Tris–glycine SDS-PAGE (Invitrogen) gels. Membranes were probed overnight at 4°C using primary antibodies for phospho-Akt (Ser473) and Akt (Cell Signaling Technology), phospho-GSK3β (Ser9) and GSK3β (Cell Signaling Technology), Aβ 6E10 (Covance), and Actin (Sigma). Densitometric analysis was performed using Scion Image software (Scion Corporation).
Immunofluorescence and confocal microscopy.
Slices were fixed overnight in ice-cold 4% paraformaldehyde in PBS. Free-floating sections were blocked with 10% normal goat serum, 1% BSA, and 0.1% Na azide in PBS for 2 h and incubated with primary antibodies, including phospho-GSK3β (Ser9) (Cell Signaling Technology) and Aβ(1–42) (Millipore). Alexa Fluor 568 secondary antibodies (Invitrogen) were used. The sections were imaged using a Leica TCS SP5 confocal microscope at 630×. All parameters (pinhole, contrast, gain, offset) were held constant for all sections from the same experiment.
Data analyses.
Data are presented as mean ± SEM. Summary data were presented as group means with SE bars. For comparison between two groups, a two-tailed independent Student's t test was used. For comparisons between multiple groups, ANOVA was used followed by individual post hoc tests when applicable. Error probabilities of p < 0.05 were considered statistically significant.
Results
AD-related alterations in hippocampal synaptic plasticity are prevented and reversed by GLP-1(9-36)amide
Previous studies have established that exogenous Aβ application results in impairment of hippocampal LTP (Shankar et al., 2008; Ma et al., 2011a). We confirmed that LTP induced by HFS was not expressed in hippocampal slices treated with Aβ(1–42) (500 nm) (Fig. 1A,B). Next we pretreated slices with GLP-1(9-36)amide (100 pm) for 30 min and found that HFS now was capable of inducing LTP in the presence of Aβ (Fig. 1A,B). We further observed that GLP-1(9-36)amide by itself did not affect HFS-induced LTP when compared with control slices (Fig. 1A,B). These data indicate that GLP-1(9-36)amide prevents Aβ-induced impairments in LTP.
Figure 1.
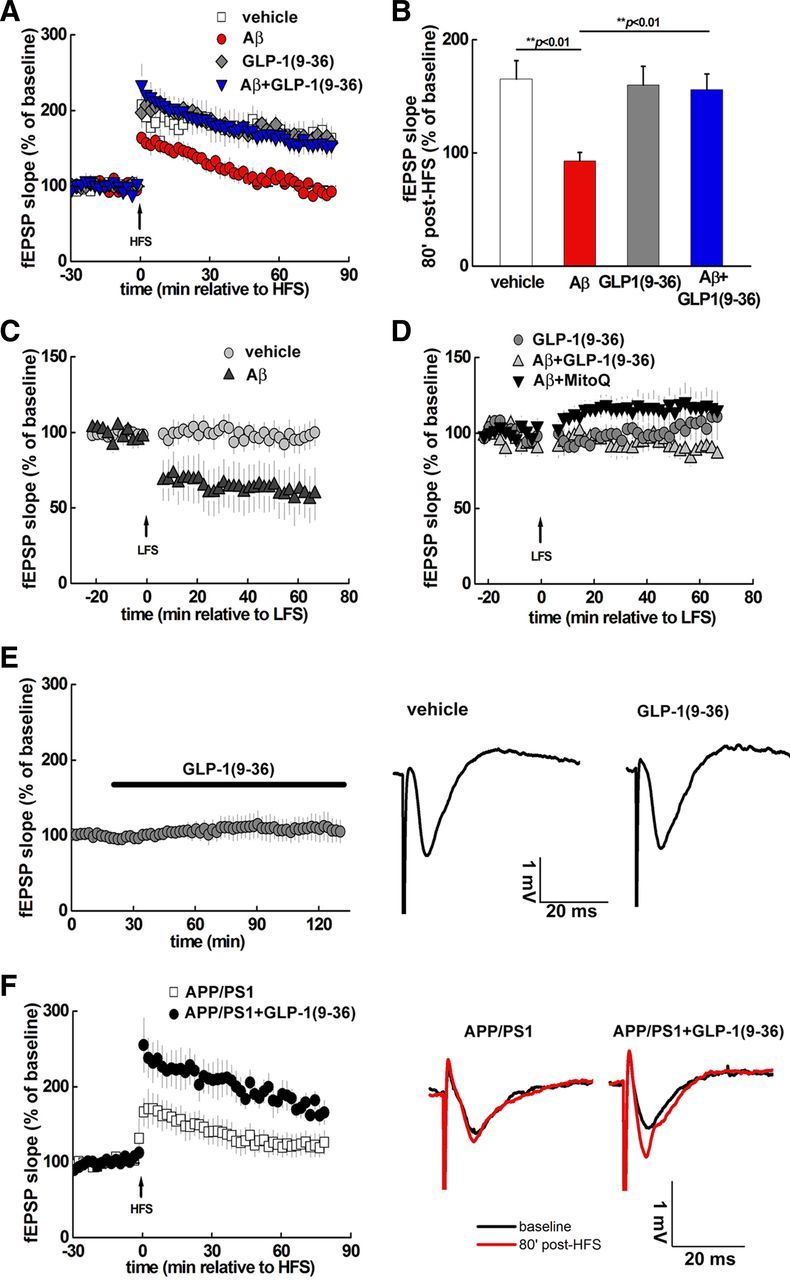
AD-associated aberrant synaptic plasticity is prevented by GLP-1(9-36)amide. A, HFS induced LTP in vehicle-treated hippocampal slices (white squares; n = 5) but not in slices treated with 500 nm Aβ(1–42) (red circles; n = 5). In contrast, 100 pm GLP-1(9-36)amide enabled normal LTP induction in the presence of Aβ(1–42) (blue triangles; n = 6). GLP-1(9-36)amide alone did not alter LTP induction (gray diamonds; n = 5). The dashed line represents the normalized baseline value of 100%. B, Cumulative data showing mean fEPSP slopes 80 min after HFS from the LTP experiments in A. C, Aβ(1–42) facilitated induction of LTD (black triangles; n = 6) by a weak LFS protocol (1 Hz, 300 pulses), which otherwise did not induce LTD (gray circles; n = 5). D, Aβ(1–42) failed to facilitate LTD induction by weak LFS in the presence of either GLP-1(9-36)amide (gray triangles; n = 8) or 500 nm MitoQ (black triangles; n = 7). E, Treatment of slices with GLP-1(9-36)amide did not affect baseline fEPSPs (n = 8). Shown on the right are traces of representative fEPSPs during the baseline recording before and after GLP-1(9-36)amide application. F, HFS-induced LTP was impaired in slices from APP/PS1 mice (white squares; n = 9) and was rescued by GLP-1(9-36)amide treatment (black circles; n = 6). Shown on the right are traces of representative fEPSPs before and after HFS. GLP-1(9-36)amide, Aβ(1–42), and MitoQ were added 30 min before HFS or LFS and left throughout the recording.
Aβ also is known to facilitate LTD, another form of hippocampal synaptic plasticity (Kim et al., 2001; Li et al., 2009). We confirmed that Aβ facilitated induction of LTD with a subthreshold LFS protocol (Fig. 1C). In slices pretreated with GLP-1(9-36)amide, the Aβ-induced facilitation of LTD was absent (Fig. 1D). We recently reported that MitoQ, a mitochondria-targeted antioxidant (Smith and Murphy, 2010), improved hippocampal LTP deficits associated with Aβ (Ma et al., 2011a), which is similar to the effects of GLP-1(9-36)amide on LTP shown here (Fig. 1A,B). Interestingly, Aβ-induced facilitation of LTD also was prevented by MitoQ (500 nm) (Fig. 1D). In addition, MitoQ alone did not alter fEPSPs after the subthreshold LFS protocol (fEPSPs slope 60 min after LFS, 99 ± 14% of baseline, n = 5). Together, these findings indicate that GLP-1(9-36)amide and MitoQ can prevent Aβ-induced facilitation of LTD.
To further explore the effects of GLP-1(9-36)amide on AD-related impairments in hippocampal synaptic plasticity, we examined slices from 10- to 12-month-old APP/PS1 mutant mice, a well-established AD mouse model (Jankowsky et al., 2001). We found that LTP impairments in APP/PS1 mice were reversed by GLP-1(9-36)amide treatment (Fig. 1F). These results are in agreement with the protective effects of GLP-1(9-36)amide on exogenous Aβ-induced alterations in hippocampal synaptic plasticity and suggest that GLP-1(9-36)amide would be effective in reversing memory phenotypes displayed by APP/PS1 mutant mice.
Memory deficits displayed by APP/PS1 mice are rescued by GLP-1(9-36)amide
We next examined whether GLP-1(9-36)amide could rescue AD-associated learning and memory deficits. APP/PS1 mice (10–12 months old) were treated with GLP-1(9-36)amide continuously for 2 weeks (500 ng · g−1 · d−1) and tested on the hidden platform version of the Morris water maze, a hippocampus-dependent learning and memory task. During acquisition, wild-type mice showed a day-to-day decrease in escape latency, whereas APP/PS1 mice displayed significantly higher escape latency (Fig. 2A). Furthermore, APP/PS1 mice spent significantly less time within the target quadrant on probe trial tests (Fig. 2B). Strikingly, the impaired spatial learning and memory deficits exhibited by the APP/PS1 mice were rescued by treatment with GLP-1(9-36)amide, as indicated by reduced escape latency as well as increased target quadrant occupation that were comparable with those displayed by wild-type mice (Fig. 2A,B). To examine the possibility that the effects of GLP-1(9-36)amide on improving spatial learning and memory in the APP/PS1 mice were attributable to effects on vision, motivation, and/or swimming ability, we tested mice on the visible platform task, which is independent of spatial learning and memory (Banko et al., 2005). There were no observable differences between the four groups of mice in the latency to find the visible platform (Fig. 2C).
Figure 2.

Impaired spatial and conditioned fear memory in APP/PS1 mice is rescued by GLP-1(9-36)amide. A, Escape latency in the Morris water maze plotted against the training days. APP/PS1 mice (red circles; n = 7) were slower to learn than wild-type (WT) mice (white squares; n = 7). In contrast, GLP-1(9-36)amide-treated APP/PS1 mice (blue squares; n = 8) exhibited learning curves similar to wild-type mice. Repeated-measures ANOVA were used, p = 0.0001. B, Percentage of time spent in the target quadrant during a 60 s probe trial. APP/PS1 mice spent less time in the target quadrant compared with wild-type mice. GLP-1(9-36)amide-treated APP/PS1 mice demonstrated occupancy time similar to wild-type mice. ANOVA and post hoc Tukey's multiple comparison tests were used with p < 0.05 as significance criteria. C, During the visible platform test, no difference was observed for escape latency among the different groups. Repeated-measures ANOVA test was used, p = 0.24. D, Mean freezing behavior in a contextual fear conditioning test 24 h after training. APP/PS1 mice (n = 7) displayed a significant reduction in freezing compared with wild-type mice (n = 7). In contrast, GLP-1(9-36)amide-treated APP/PS1 mice (n = 7) displayed significantly more freezing behavior than APP/PS1 mice. ANOVA and post hoc Tukey's multiple comparison tests were used with p < 0.05 as significance criteria. E, Mean freezing behavior in cued fear conditioning test 24 h after training. APP/PS1 mice (n = 7) displayed a significant reduction in freezing compared with wild-type mice (n = 7). In contrast, GLP-1(9-36)amide-treated APP/PS1 mice (n = 7) displayed significantly more freezing behavior than APP/PS1 mice. ANOVA and post hoc Tukey's multiple comparison tests were used with p < 0.05 as significance criteria.
We next investigated the effects of GLP-1(9-36)amide on associative learning and memory by training mice with a standard conditioned fear memory paradigm (Banko et al., 2005). For both contextual and cue fear conditioning (tested 24 h after training), APP/PS1 mutant mice exhibited impaired associative memory, as measured by decreased freezing time, compared with wild-type mice (Fig. 2D,E). The conditioned fear memory of GLP-1(9-36)amide-treated APP/PS1 mutant mice was significantly improved as indicated by augmented freezing time in both contextual and cue fear conditioning tests (Fig. 2D,E). Together, these findings indicate that treatment with GLP-1(9-36)amide can reverse memory deficits displayed by APP/PS1 mutant mice.
GLP-1(9-36)amide treatment does not alter the levels of either APP or Aβ in APP/PS1 mice
It was reported that peripheral injection of the diabetes drug liraglutide, a novel, nondegradable GLP-1 analog, has neuroprotective effects and decreases the levels of both APP and Aβ in APP/PS1 mice (McClean et al., 2011). Therefore, we examined whether GLP-1(9-36)amide could affect APP and Aβ levels in APP/PS1 mutant mice. Interestingly, we did not detect any significant difference in the brain levels of either APP or Aβ in APP/PS1 mice treated with GLP-1(9-36)amide (Fig. 3A,B). Using immunofluorescence combined with confocal microscopy, we also examined the effects of GLP-1(9-36)amide on levels of intra-neuronal Aβ. Consistent with the Western blot data, no differences in intra-neuronal Aβ staining was detected in hippocampal area CA1 (Fig. 3C). These findings indicate that treatment of APP/PS1 mutant mice with GLP-1(9-36)amide for 2 weeks reverses memory defects without altering levels of either APP or Aβ in the hippocampus.
Figure 3.

GLP-1(9-36)amide application does not change levels of either APP or Aβ in APP/PS1 mice. A, B, Western blotting was performed on brain extracts of mice from behavioral experiments. No change in the levels of either APP or Aβ was detected between APP/PS1 mice and GLP-1(9-36)amide-treated APP/PS1 mice. n = 5. C, Immunofluorescence confocal microscopy for Aβ in the CA1 region of hippocampal slices harvested from APP/PS1 mice treated with GLP-1(9-36)amide and control group. Scale bar, 50 μm.
GLP-1(9-36)amide decreases elevated levels of mitochondrial superoxide associated with AD
To begin to ascertain the underlying molecular mechanisms responsible for the effects of GLP-1(9-36)amide on AD-associated aberrations in synaptic plasticity and memory, we stained live slices with MitoSOX Red, a fluorogenic dye that is selective for the detection of superoxide in mitochondria. MitoSOX Red is chemically targeted to mitochondria and exhibits red fluorescence when oxidized (Ma et al., 2011a). Both wild-type hippocampal slices treated with Aβ and hippocampal slices from APP/PS1 mice demonstrated a significant enhancement in red fluorescence signal (Fig. 4A,B). This AD-associated increase in mitochondrial superoxide was reversed by GLP-1(9-36)amide, as indicated by the normalizing of the MitoSOX fluorescent signal (Fig. 4A,B). These data provide evidence for a role of GLP-1(9-36)amide as a mitochondrial antioxidant, which may contribute to the correction of aberrant synaptic plasticity induced by Aβ and displayed by APP/PS1 mutant mice.
Figure 4.

GLP-1(9-36)amide normalizes increased mitochondrial superoxide levels associated with AD. A, Treatment of hippocampal slices with 500 nm Aβ(1–42) increased the MitoSOX fluorescent signal (red) in area CA1 compared with control slices. DAPI staining is shown as blue. Pretreatment of slices with 100 pm GLP-1(9-36)amide prevented the Aβ-induced elevation in the MitoSOX fluorescent signal. B, The MitoSOX fluorescent signal was increased in slices from APP/PS1 mice compared with wild-type (WT) slices. Treatment of slices with GLP-1(9-36)amide normalized the increased fluorescent signal. Results are representative of three independent experiments. Scale bar, 50 μm.
GLP-1(9-36)amide treatment reverses activating dephosphorylation of hippocampal GSK3β in APP/PS1 mutant mice
Mounting evidence suggests a critical role of GSK3β in regulating synaptic plasticity (Peineau et al., 2008; Ma et al., 2011b), and dysregulation of GSK3β signaling has been associated with AD (Hooper et al., 2008). Moreover, regulation of GSK3β activity has been linked to mitochondrial function and recently was proposed as a target of Aβ in AD pathogenesis (Jo et al., 2011). We demonstrated that GLP-1(9-36)amide normalized mitochondrial ROS levels in the hippocampus of APP/PS1 mutant mice (Fig. 4B), so we asked whether normalization of hippocampal ROS could affect abnormal activation of GSK3β. Hippocampal slices from in vivo experiments in which APP/PS1 mice were treated with GLP-1(9-36)amide were prepared and stained for the phosphorylation of GSK3β on serine 9, which leads to inhibition of its activity. Using immunofluorescence combined with confocal microscopy, we observed that GSK3β activity was elevated in hippocampal CA1 regions of APP/PS1 mice as measured by decreased immunostaining of phosphorylation on serine 9 and was restored to levels comparable with that of wild-type mice after GLP-1(9-36)amide treatment (Fig. 5). These findings suggest that increased GSK3β signaling in APP/PS1 mutant mice is induced by increased mitochondrial superoxide production, which is reversed by treatment with GLP-1(9-36)amide.
Figure 5.

GLP-1(9-36)amide treatment reverses activating dephosphorylation of GSK3β in APP/PS1 mice. Immunofluorescence for phosphorylated GSK3β on serine 9 (p-GSK3β) in area CA1 of hippocampal slices was reduced in APP/PS1 mice but was restored to wild-type (WT) levels by GLP-1(9-36)amide treatment. Scale bar, 25 μm.
To further explore the molecular signaling mechanisms impacted by GLP-1(9-36)amide, we proceeded to examine hippocampal slices prepared from APP/PS1 mice that had been treated with GLP-1(9-36)amide. In agreement with the immunofluorescence data (Fig. 5), we also found significant reductions of phosphorylated GSK3β levels in slices from APP/PS1 mice that were reversed by treatment with GLP-1(9-36)amide (Fig. 6B). In addition, there was no change in GSK3β phosphorylation in GLP-1(9-36)amide-treated slices from wild-type mice (Fig. 6A).
Figure 6.

Activating dephosphorylation of GSK3β and inactivating dephosphorylation of Akt in hippocampal slices of APP/PS1 mice is reversed by GLP-1(9-36)amide. A, In slices from wild-type (WT) mice, GLP-1(9-36)amide treatment did not change phosphorylated GSK3β levels on serine 9 (p-GSK3β). n = 6. B, Reduced level of phospho-GSK3β in APP/PS1 mice was reversed by GLP-1(9-36)amide. n = 6. C, In slices from wild-type mice, GLP-1(9-36) treatment did not change phosphorylated Akt levels on serine 473 (p-Akt). n = 6. D, Reduced levels of phospho-Akt in APP/PS1 mice was reversed by GLP-1(9-36)amide. n = 6. veh, Vehicle.
An established upstream regulator of GSK3β is Akt, a serine/threonine kinase that inhibits the activity of GSK3β via phosphorylation on serine 9 (Manning and Cantley, 2007). Akt regulation in synaptic plasticity also has been connected to mitochondrial activity (Li et al., 2010). We found that, in slices from APP/PS1 mice, Akt activity was inhibited as indicated by decreased levels of phosphorylation on serine 473 (Fig. 6D), consistent with the observed increase in GSK3β activity. Importantly, the decreased phosphorylation of Akt was reversed by GLP-1(9-36)amide, consistent with its action on reducing GSK3β activity (increasing its phosphorylation). Similar to GSK3β, slices from wild-type mice that had been treated with GLP-1(9-36)amide showed no alteration in Akt phosphorylation (Fig. 6C). A recent study suggested a link between caspase-3 activation and Akt inhibition by Aβ (Jo et al., 2011). Consistent with this report, we found increased levels of active (cleaved) caspase-3 in hippocampal slices from APP/PS1 mice (211 ± 45% of wild-type levels, n = 4, p < 0.05 compared with wild-type mice). However, although there was a trend toward a decrease, the increased levels of active caspase-3 in the APP/PS mice were not altered significantly in slices treated with GLP-1(9-36) (153 ± 22% of wild-type levels, n = 4, p = 0.35 compared with the APP/PS1 group). Together, these findings suggest that GLP-1(9-36)amide reverses abnormal activation levels of GSK3β in APP/PS1 mutant mice by reversing an ROS-induced decrease in Akt activity.
Discussion
In the present study, we show for the first time that GLP-1(9-36)amide, the main endogenously formed cleavage product of the incretin hormone GLP-1, reverses existing impairments in synaptic plasticity and deficits in learning and memory in a mouse model of AD. Hippocampal LTP impairments in APP/PS1 mutant mice were rescued by GLP-1(9-36)amide, and treatment of APP/PS1 mice for 2 weeks with GLP-1(9-36)amide reversed impairments in spatial and conditioned fear memory. At the molecular level, GLP-1(9-36)amide reduced elevated levels of mitochondrial ROS and restored dysregulated Akt–GSK3β signaling in the hippocampus of APP/PS1 mice. These findings suggest that GLP-1(9-36)amide treatment may have therapeutic potential for AD and other diseases associated with cognitive dysfunction. Interestingly, the rescue of synaptic plasticity failure and memory deficits in AD model mice did not correlate with either the reduction of brain Aβ or changes in APP, which is consistent with the recently proposed idea of targeting AD via an Aβ-independent strategy (Pimplikar et al. 2010).
The GLP-1 receptor is expressed widely in the CNS, including the hypothalamus, brainstem, and hippocampus (Alvarez et al., 2005). Thus, it is not surprising that activation of the GLP-1 in brain is associated with a wide variety of physiologic effects, including regulation of feeding, vagus nerve-mediated hormone secretion, hormone action, metabolite flux in peripheral tissues, and decreased arterial blood flow (Turton et al., 1996; Knauf et al., 2005; Cabou et al., 2008; Nogueiras et al., 2009). In normal rodents, signaling through the GLP-1 receptor either maintains or enhances learning and memory. Homozygous GLP-1 receptor knockout mice exhibit learning deficits, which are restored by hippocampal Glp1r gene transfer (During et al., 2003). Central administration of a DPP-4-resistant GLP-1 N-terminal nonapeptide, which activates the GLP-1 receptor, to wild-type rats also enhanced associative and spatial learning (During et al., 2003). The LTP impairments and histological changes that characterize AD were not described in these animal models.
Administration of the GLP-1 receptor agonist liraglutide, an acetylated form of GLP-1(7-36), to APP/PS1 mice produced functional improvements in memory and other behavioral outcomes, increased synaptic plasticity, and reduced evidence of neuronal damage, plaques, and oligomer formation in the CNS (McClean et al., 2011). Although a reduction in Aβ was not observed in our experiments with GLP-1(9-36)amide (Fig. 3), the treatment duration was 25% of that used in the liraglutide study. Liraglutide, unlike peptide GLP-1 receptor agonists such as either exendin-4 or its N-terminal homolog [Ser(2)] exendin (1–9) (During et al., 2003), shares a potential C- terminal mitochondrial-targeting sequence with GLP-1(9-36)amide (Tomas et al., 2011). Furthermore, both GLP-1(9-36)amide and liraglutide share the same potential NEP cleavage site. It has been proposed that the nonapeptide remaining after DPP-4 and NEP cleavage targets mitochondria and suppresses oxidative stress (Tomas et al., 2011). Additional studies will be required to test this proposition.
The overall mechanism by which GLP-1(9-36)amide corrects aberrant synaptic plasticity and memory deficits in AD model mice requires additional elucidation. However, the observation that GLP-1(9-36)amide normalized increased levels of mitochondrial ROS in the APP/PS1 mice (Fig. 4B) is consistent with a body of data linking mitochondrial ROS to synaptic pathology associated with aging and neurodegenerative diseases (Massaad and Klann, 2011). It has been proposed that high levels of ROS produced in pathological situations, such as AD, causes synaptic dysfunction (Balaban et al., 2005; Ma and Klann, 2012). Direct evidence consistent with this idea comes from studies showing that the mitochondria-specific antioxidant MitoQ prevents aberrant LTP (Ma et al., 2011a) and LTD (Fig. 1D) in the APP/PS1 AD model mice. However, MitoQ is poorly absorbed, whereas GLP-1-related peptides administered by nasal inhalation cross the blood–brain barrier easily (During et al., 2003). Thus, GLP-1-related peptides may be easier to deliver as a therapeutic to reduce increased levels of mitochondrial-derived ROS in AD.
Mechanistically, the observation that GLP-1(9-36)amide restored dysregulated Akt–GSK3β signaling in APP/PS1 mice (Figs. 5, 6) is consistent with previous reports in the AD literature. Originally identified as a regulator of glycogen metabolism (Embi et al., 1980), GSK3 is now widely recognized as a critical signaling molecule involved in many cellular processes, and GSK3β dysfunction has been implicated in the pathogenesis of AD (Jope and Johnson, 2004; Hooper et al., 2008; Ma et al., 2011b). Importantly, recent studies show that abnormally high GSK3β activity undermines normal synaptic function, and, conversely, GSK3β inhibition facilitates LTP induction (Peineau et al., 2007; Ma et al., 2010, 2011b; Jo et al., 2011). GLP-1(9-36)amide restored GSK3β inhibitory phosphorylation to normal levels in APP/PS1 mice (Figs. 5, 6A,B), consistent with its efficacy in ameliorating impaired synaptic plasticity associated with AD (Fig. 1). Given the current view that memory deficits in AD are attributable to synaptic failure (Selkoe, 2002), our studies with GLP-1(9-36)amide suggest that this peptide has therapeutic potential for AD and other diseases associated with cognitive dysfunction.
Footnotes
This work was supported by National Institutes of Health Grants NS034007 and NS047834 and Alzheimer's Association Investigator Grant IIRG-09-131756 (E.K.). We thank Dr. Michael P. Murphy for generously providing MitoQ and Dr. Hanoch Kaphzan for excellent technical advice. We thank Alyse J. Wexler for technical assistance on mouse behavioral experiments, Dr. Emanuela Santini for advice concerning statistical analysis of the behavioral experiments, and all other members of the Klann laboratory for critical comments on this manuscript.
The authors declare no competing financial interests.
References
- Alvarez E, Martínez MD, Roncero I, Chowen JA, García-Cuartero B, Gispert JD, Sanz C, Vázquez P, Maldonado A, de Cáceres J, Desco M, Pozo MA, Blázquez E. The expression of GLP-1 receptor mRNA and protein allows the effect of GLP-1 on glucose metabolism in the human hypothalamus and brainstem. J Neurochem. 2005;92:798–806. doi: 10.1111/j.1471-4159.2004.02914.x. [DOI] [PubMed] [Google Scholar]
- Alzheimer's Association. Thies W, Bleiler L. 2011 Alzheimer's disease facts and figures. Alzheimers Dement. 2011;7:208–244. doi: 10.1016/j.jalz.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Ban K, Kim KH, Cho CK, Sauvé M, Diamandis EP, Backx PH, Drucker DJ, Husain M. Glucagon-like peptide (GLP)-1(9-36)amide-mediated cytoprotection is blocked by exendin(9–39) yet does not require the known GLP-1 receptor. Endocrinology. 2010;151:1520–1531. doi: 10.1210/en.2009-1197. [DOI] [PubMed] [Google Scholar]
- Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, Klann E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci. 2005;25:9581–9590. doi: 10.1523/JNEUROSCI.2423-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabou C, Campistron G, Marsollier N, Leloup C, Cruciani-Guglielmacci C, Pénicaud L, Drucker DJ, Magnan C, Burcelin R. Brain glucagon-like peptide-1 regulates arterial blood flow, heart rate, and insulin sensitivity. Diabetes. 2008;57:2577–2587. doi: 10.2337/db08-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drucker DJ. Minireview: the glucagon-like peptides. Endocrinology. 2001;142:521–527. doi: 10.1210/endo.142.2.7983. [DOI] [PubMed] [Google Scholar]
- Dumont M, Wille E, Stack C, Calingasan NY, Beal MF, Lin MT. Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer's disease. FASEB J. 2009;23:2459–2466. doi: 10.1096/fj.09-132928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- During MJ, Cao L, Zuzga DS, Francis JS, Fitzsimons HL, Jiao X, Bland RJ, Klugmann M, Banks WA, Drucker DJ, Haile CN. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med. 2003;9:1173–1179. doi: 10.1038/nm919. [DOI] [PubMed] [Google Scholar]
- Dyer MR, Renner WA, Bachmann MF. A second vaccine revolution for the new epidemics of the 21st century. Drug Discov Today. 2006;11:1028–1033. doi: 10.1016/j.drudis.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem. 1980;107:519–527. [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Serrano F, Oury TD, Klann E. Aging-dependent alterations in synaptic plasticity and memory in mice that overexpress extracellular superoxide dismutase. J Neurosci. 2006;26:3933–3941. doi: 10.1523/JNEUROSCI.5566-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Jo J, Whitcomb DJ, Olsen KM, Kerrigan TL, Lo SC, Bru-Mercier G, Dickinson B, Scullion S, Sheng M, Collingridge G, Cho K. Aβ(1–42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β. Nat Neurosci. 2011;14:545–547. doi: 10.1038/nn.2785. [DOI] [PubMed] [Google Scholar]
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kim JH, Anwyl R, Suh YH, Djamgoz MB, Rowan MJ. Use-dependent effects of amyloidogenic fragments of β-amyloid precursor protein on synaptic plasticity in rat hippocampus in vivo. J Neurosci. 2001;21:1327–1333. doi: 10.1523/JNEUROSCI.21-04-01327.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauf C, Cani PD, Perrin C, Iglesias MA, Maury JF, Bernard E, Benhamed F, Grémeaux T, Drucker DJ, Kahn CR, Girard J, Tanti JF, Delzenne NM, Postic C, Burcelin R. Brain glucagon-like peptide-1 increases insulin secretion and muscle insulin resistance to favor hepatic glycogen storage. J Clin Invest. 2005;115:3554–3563. doi: 10.1172/JCI25764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Jo J, Jia JM, Lo SC, Whitcomb DJ, Jiao S, Cho K, Sheng M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell. 2010;141:859–871. doi: 10.1016/j.cell.2010.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Ma T, Klann E. Amyloid β: linking synaptic plasticity failure to memory disruption in Alzheimer's disease. J Neurochem. 2012;120(Suppl 1):140–148. doi: 10.1111/j.1471-4159.2011.07506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Hoeffer CA, Capetillo-Zarate E, Yu F, Wong H, Lin MT, Tampellini D, Klann E, Blitzer RD, Gouras GK. Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer's disease. PLoS One. 2010;5:pii:e12845. doi: 10.1371/journal.pone.0012845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Hoeffer CA, Wong H, Massaad CA, Zhou P, Iadecola C, Murphy MP, Pautler RG, Klann E. Amyloid β-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J Neurosci. 2011a;31:5589–5595. doi: 10.1523/JNEUROSCI.6566-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Tzavaras N, Tsokas P, Landau EM, Blitzer RD. Synaptic stimulation of mTOR is mediated by Wnt signaling and regulation of glycogen synthetase kinase-3. J Neurosci. 2011b;31:17537–17546. doi: 10.1523/JNEUROSCI.4761-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massaad CA, Klann E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid Redox Signal. 2011;14:2013–2054. doi: 10.1089/ars.2010.3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massaad CA, Washington TM, Pautler RG, Klann E. Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106:13576–13581. doi: 10.1073/pnas.0902714106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClean PL, Parthsarathy V, Faivre E, Hölscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer's disease. J Neurosci. 2011;31:6587–6594. doi: 10.1523/JNEUROSCI.0529-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueiras R, Pérez-Tilve D, Veyrat-Durebex C, Morgan DA, Varela L, Haynes WG, Patterson JT, Disse E, Pfluger PT, López M, Woods SC, DiMarchi R, Diéguez C, Rahmouni K, Rohner-Jeanrenaud F, Tschöp MH. Direct control of peripheral lipid deposition by CNS GLP-1 receptor signaling is mediated by the sympathetic nervous system and blunted in diet-induced obesity. J Neurosci. 2009;29:5916–5925. doi: 10.1523/JNEUROSCI.5977-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Wang YT, Collingridge GL. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Peineau S, Bradley C, Taghibiglou C, Doherty A, Bortolotto ZA, Wang YT, Collingridge GL. The role of GSK-3 in synaptic plasticity. Br J Pharmacol. 2008;153:S428–S437. doi: 10.1038/bjp.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimplikar SW, Nixon RA, Robakis NK, Shen J, Tsai LH. Amyloid-independent mechanisms in Alzheimer's disease pathogenesis. J Neurosci. 2010;30:14946–14954. doi: 10.1523/JNEUROSCI.4305-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Rolin B, Deacon CF, Carr RD, Ahrén B. The major glucagon-like peptide-1 metabolite, GLP-1-(9-36)-amide, does not affect glucose or insulin levels in mice. Eur J Pharmacol. 2004;494:283–288. doi: 10.1016/j.ejphar.2004.05.013. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Resolving controversies on the path to Alzheimer's therapeutics. Nat Med. 2011;17:1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RA, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- Tomas E, Stanojevic V, Habener JF. GLP-1-derived nonapeptide GLP-1(28–36)amide targets to mitochondria and suppresses glucose production and oxidative stress in isolated mouse hepatocytes. Regul Pept. 2011;167:177–184. doi: 10.1016/j.regpep.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Tsokas P, Ma T, Iyengar R, Landau EM, Blitzer RD. Mitogen-activated protein kinase upregulates the dendritic translation machinery in long-term potentiation by controlling the mammalian target of rapamycin pathway. J Neurosci. 2007;27:5885–5894. doi: 10.1523/JNEUROSCI.4548-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turton MD, O'Shea D, Gunn I, Beak SA, Edwards CM, Meeran K, Choi SJ, Taylor GM, Heath MM, Lambert PD, Wilding JP, Smith DM, Ghatei MA, Herbert J, Bloom SR. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature. 1996;379:69–72. doi: 10.1038/379069a0. [DOI] [PubMed] [Google Scholar]