

Abstract
Patients with Growth Hormone Insensitivity have characteristic phenotypic features and severe short stature. The underlying basis are mutations in the growth hormone receptor gene which gives rise to a characteristic hormonal profile. Although a scoring system has been devised for the diagnosis of this disorder, it has not been indisputably validated. The massive expense incurred in the diagnosis and treatment of this condition with suboptimal therapeutic response necessitates a judicious approach in this regard in our country.
Keywords: Growth hormone insensitivity, IGF-1, Laron syndrome
The syndrome of growth hormone (GH) insensitivity was first described by Laron in three siblings of Israeli origin.[1] These patients have a characteristic phenotype and are associated with severe postnatal growth failure and marked reduction in adult height.
Several abnormalities occur in growth hormone insensitivity (GHI). Some may have intra-uterine abnormalities, including weaker fetal movements, reduced birth length/Intra-uterine growth retardation (IUGR). Rarely congenital abnormalities like strabismus, cataract, aortic stenosis, undescended testis, and congenital dislocation of hip joint may occur. The child may also have sparse hair, frontal bossing, hypoplastic nose, shallow orbit, blue sclera, high-pitched voice, delayed dentition, genital abnormalities, sleep disorder, severe short stature, preserved sub-cutaneous fat, and obesity. There is often delay in puberty.[2]
The hormonal features of growth hormone insensitivity (GHI) consists of normal or elevated serum GH levels, combined with the inability to generate normal/functional quantities of insulin-like growth factor-1 (IGF-1). Circulating concentrations of IGF-1, IGF-II and IGF-binding protein (BP)-3 are reduced.[3] Serum GH-BP, which is identical to the extracellular domain of the GH receptor, was reported to be absent in this condition.[4] It is now reported that in some cases there may be detectable GH-binding activity. This finding led to more studies which have finally led to the conclusion that GHI is a hetergenous group of disorders caused by homozygous or compound heterozygous mutations in the GH receptor gene. A variety of such mutations have been identified and most of which affect the extracellular GH binding domain of the receptor.[5] Research has demonstrated that post GH receptor mechanisms like any abnormality of GH receptor signal transduction, IGF-1 gene mutation, defective stabilisation of circulating IGF-1, IGF-1 receptor mutation can all give rise to disorders similar to that which was classically described by Laron.[6,7]
Hormonal evaluation is essential to confirm the diagnosis and to help prognosticate response to therapy. A scoring system to identify patients with GHI has been suggested. This scoring includes basal GH, IGF-1, IGF-1 Generation, Height SDS (standard deviation scores) GH-BP [Table 1].[8] Maximum possible score is 7 points. Patients with a score of 5 or more are considered to have significant growth hormone insensitivity.
Table 1.
Scoring system for the diagnosis of growth hormone insensitivity
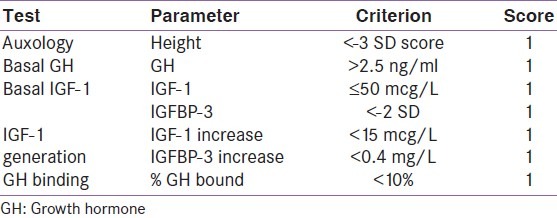
The criteria can be further strengthened by including other criteria like GH secretory profiles rather than isolated basal levels, using age dependent cut-offs of IGF-1 and better IGF-1 assays.[9]
There have been several potential problems with making a diagnosis of GHI. The IGF-1 generation test has been criticised to have not been adequately characterised and normative data is inadequate.[10] In addition, the availability and costs of the tests for diagnosis of GHI in India is a hurdle, which has been well discussed by Balaji et al. in this issue of the journal.
Patients with GHI are not responsive to GH therapy, but usually responsive to treatment with IGF-1. However, the response of patient of GHI to IGF-1 therapy is variable and substantially less than that of a GH deficient patient treated with GH. Recombinant human IGF-1 (rh IGF-1) (Mecasermin, Increlex) has been used in children with GHI. In one study the height velocity increased from 2.8 cm/year at baseline to a mean of 8 cms/year during the first year of treatment and the height velocity remained above baseline for up to eight years following initiation of rh IGF-1 therapy.[11,12]
In view of the costs of diagnosis and treatment, (as mentioned by Balaji et al. in this issue of the journal) and less than optimal outcome to therapy , one might argue that in a country like India it might be a futile effort to actively search for cases of GHI. We agree with that in patients in whom we cannot afford treatment with IGF-1, it is not cost effective to investigate for primary GHI. However, as clinicians we need to have a pragmatic view and bank on phenotypic appearance in making a possible diagnosis of GHI in such cases. It has to be realised that India is a land of diversity (including financial). In the affordable few, the approach should be that which has been advocated by medical literature.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Laron Z, Pertzelan A, Mannheimer S. Genetic pituitary dwarfism with high serum concentation of growth hormone-a new inborn error of metabolism? Isr J Med Sci. 1966;2:152–5. [PubMed] [Google Scholar]
- 2.Laron Z. Laron syndrome (primary growth hormone resistance or insensitivity): The personal experience 1958-2003. J Clin Endocrinol Metab. 2004;89:1031–44. doi: 10.1210/jc.2003-031033. [DOI] [PubMed] [Google Scholar]
- 3.Laron Z, Pertzelan A, Karp M, Kowadlo-Silbergeld A, Daughaday WH. Administration of growth hormone to patients with familial dwarfism with high plasma immunoreactive growth hormone: Measurement of sulfation factor, metabolic and linear growth responses. J Clin Endocrinol Metab. 1971;33:332–42. doi: 10.1210/jcem-33-2-332. [DOI] [PubMed] [Google Scholar]
- 4.Rosenbloom AL, Guevara AJ, Rosenfeld RG, Fielder PJ. The little women of Loja-growth hormone-receptor deficiency in an inbred population of southern Ecuador. N Engl J Med. 1990;323:1367–74. doi: 10.1056/NEJM199011153232002. [DOI] [PubMed] [Google Scholar]
- 5.Daughaday WH, Trivedi B. Absence of serum growth hormone binding protein in patients with growth hormone receptor deficiency (Laron dwarfism) Proc Natl Acad Sci U S A. 1987;84:4636–40. doi: 10.1073/pnas.84.13.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amselem S, Duquesnoy P, Attree O, Novelli G, Bousnina S, Postel-Vinay MC, et al. Laron dwarfism and mutations of the growth hormone-receptor gene. N Engl J Med. 1989;321:989–95. doi: 10.1056/NEJM198910123211501. [DOI] [PubMed] [Google Scholar]
- 7.Bonapace G, Concolino D, Formicola S, Strisciuglio P. A novel mutation in a patient with insulin-like growth factor 1 (IGF1) deficiency. J Med Genet. 2003;40:913–7. doi: 10.1136/jmg.40.12.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woods KA, Dastot F, Preece MA, Clark AJ, Postel-Vinay MC, Chatelain PG, et al. Phenotype: Genotype relationships in growth hormone insensitivity syndrome. J Clin Endocrinol Metab. 1997;82:3529–35. doi: 10.1210/jcem.82.11.4389. [DOI] [PubMed] [Google Scholar]
- 9.Blum WF, Ranke MB, Savage MO, Hall K. Insulin-like growth factors and their binding proteins in patients with growth hormone receptor deficiency: Suggestions for new diagnostic criteria. The Kabi Pharmacia Study Group on Insulin-like Growth Factor I Treatment in Growth Hormone Insensitivity Syndromes. Acta Paediatr Suppl. 1992;383:125–6. [PubMed] [Google Scholar]
- 10.Buckway CK, Guevara-Aguirre J, Pratt KL, Burren CP, Rosenfeld RG. The IGF-I generation test revisited: A marker of GH sensitivity. J Clin Endocrinol Metab. 2001;86:5176–83. doi: 10.1210/jcem.86.11.8019. [DOI] [PubMed] [Google Scholar]
- 11.Chernausek SD, Backeljauw PF, Frane J, Kuntze J, Underwood LE. Long-term treatment with recombinant insulin-like growth factor (IGF)-I in children with severe IGF-I deficiency due to growth hormone insensitivity. J Clin Endocrinol Metab. 2007;92:902–10. doi: 10.1210/jc.2006-1610. [DOI] [PubMed] [Google Scholar]
- 12.Collett-Solberg PF, Misra M. The role of recombinant human insulin-like growth factor-I in treating children with short stature. J Clin Endocrinol Metab. 2008;93:10–8. doi: 10.1210/jc.2007-1534. [DOI] [PubMed] [Google Scholar]