

Abstract
We report a case of 41-year-old lady who presented with chronic headache of 6-month duration and a sellar mass with a suprasellar extension on imaging, which was interpreted as pituitary macroadenoma. She had normal pituitary function and visual perimetry. On clinical examination and imaging it was provisionally diagnosed as pituitary incidentaloma due to hypophysitis and she was advised steroid therapy, but underwent transnasal resection of the tumor against suggestion. Histopathological examination revealed combined granulomatous and lymphocytic hypophysitis most likely of autoimmune in origin. Definitive diagnosis of hypophysitis can be made only on histopathological examination. As most cases of autoimmune hypophysitis are surgically treated, patients should be assessed on individual basis for requirement of steroids in postoperative period.
Keywords: Granulomatous hypophysitis, incidentaloma, lymphocytic hypophysitis
INTRODUCTION
A pituitary incidentaloma that is discovered on an imaging study can be found in 10% of population and is due to varied etiology.[1] Primary granulomatous hypophysitis is a rare disease and its coexistence with lymphocytic hypophysitis is known, and it has been proposed that both of them may be spectrum of same autoimmune disease with lymphocytic hypophysitis occurring during early course and granulomatous hypophysitis developing later on.[2,3] Here we report a case of combined lymphocytic and granulomatous hypophysitis clinically suspected and confirmed on histopathology.
CASE REPORT
A 41-year-old female presented with insidious onset headache of 6-month duration, which was diffuse, dull aching, at times exacerbated to severe throbbing headache. There were no aggravating and relieving factors, no association with visual symptoms, vomiting, fever or any other neurologic symptoms. During evaluation by her neurologist, computerized tomography (CT) of brain was performed, which showed a sellar mass. She was further evaluated by neurosurgeon and a magnetic resonant imaging (MRI) was performed which confirmed a sellar mass with a probable diagnosis of pituitary adenoma. She was referred to us for endocrinological work-up. On further questioning she did not have any symptoms related to pituitary dysfunction. Her past history was significant for being empirically treated for tubercular pericardial effusion 10 years back. She had two children, with the second delivery 8 years back. She had an uneventful postpartum period. General and physical examination was essentially normal. Visual perimetry showed normal visual fields in both eyes.
Her hematological and biochemical work-up was normal barring erythrocyte sedimentation rate (ESR) which was 54 mm at the end of first hour. Her hemoglobin was 14 g/ dl; total leucocyte count was 6700/μl, neutrophil count of 64%, lymphocyte count of 30%, monocyte 4%, eosinophil 2%; fasting plasma glucose 92 mg/dl, blood urea 22 mg/ dl, serum creatinine 0.6 mg/dl, serum bilirubin 0.5 mg/dl, ALT-6IU/l, ALP 32 IU/l, serum alkaline phosphatase 86 U/l, serum calcium 8.5 mg/dl, serum phosphates 3.4 mg/dl. She had normal hormonal work-up (fT3 – 3.4 pg/ ml [2.4-4.2], fT4 – 1.4 ng/ ml [0.8-1.7], TSH – 3.56 [0.5- 6.5] μIU/ml; LH/FSH – 7.8/9.4 IU/l; S. estradiol– 75 pg/ml; S. prolactin – 14 ng/ml [< 25]; S. Cortisol basal/ACTH stimulated – 14 /23.6 μg/dl). The Mantoux test was strongly positive (20×55mm). Chest radiography, 2D echocardiogram, ultrasound abdomen were normal. Serum angiotensin convertase enzyme was 39.0 U/l (8-65). Autoimmune workup did not reveal any abnormality with CRP – 1.2 mg/l (normal < 7 mg/l); antinuclear antibodies (< 1:40), and antineutrophilic cytoplasmic antibodies (< 1:16), anti-TPO Ab - 4 IU/l (< 9), RA factor 4 IU/l (< 20), and anti-TTG Ab- 3.54 IU/ ml (< 15). CT scan of chest and abdomen was normal. A review of MRI brain favored hypophysitis more than pituitary adenoma in view of homogeneity of the mass, thickened and midline pituitary stalk, strong and diffuse enhancement after contrast, and loss of posterior pituitary hyper intensity and maintenance of sellar floor [Figure 1]. The possibility was discussed with the patient and close observation with steroid therapy was contemplated.
Figure 1.
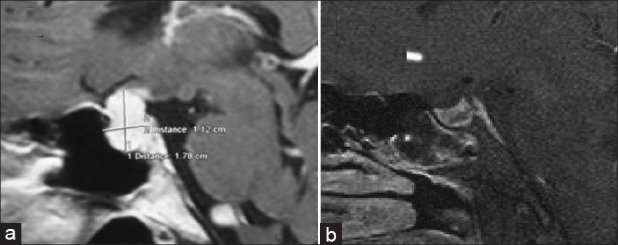
(a) Preoperative image showing 1.78 × 1.12 × 2.2 cm lesion, homogenously hyperintense on T1W postcontrast image, showing thickening of pituitary stalk and dural tail. (b) Postoperatively normal sella with no residual lesion
However, before initiation of steroids she had an episode of severe headache with vomiting without any neurological deficits. The episode made the patient apprehensive of her underlying sellar mass and she favored surgery. She underwent successful transnasal transsphenoidal excision of the mass. Postoperatively she had transient diabetes insipidus on the second and third postoperative days, revealed by marked polyuria and raised serum sodium [154 meq/l (135-145 meq/l)]. It was managed with subcutaneous injection vasopressin, followed by no further requirement of vasopressin from the fourth postoperative day onward. The histopathological examination of the tissue showed the lesion to be filled with well-defined epitheloid granulomas, lymphocytic infiltration, and histiocytes [Figure 2]. Immunohistochemistry stained positive for CD3 (T-lymphocytes), CD20 (B-lymphocytes), and CD68 (histiocytes) [Figure 3]. In view of these findings a diagnosis of combined granulomatous and lymphocytic hypophysitis was made. There were no acid-fast bacilli seen. Tissue PCR could not be done due to financial constraints. Postoperatively she was asymptomatic, had normal pituitary functions, and there was no residual lesion on MRI.
Figure 2.
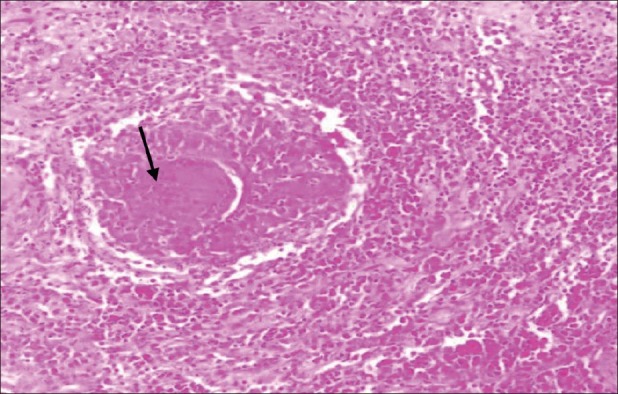
H and E, ×10 showing an effacement of alveolar architecture of pituitary gland by well-defined epitheloid granulomas (center) composed of multinucleate giant cells (arrow) and lymphocytes. Inflammatory infiltrate comprising predominately lymphocytes and histiocytes and occasional eosinophils is noted surrounding the granulomas and pituicytes
Figure 3.
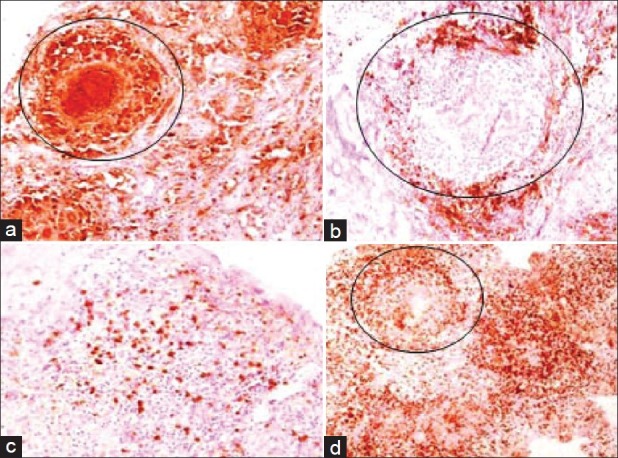
IHC showing (a) CD68 positive histiocytes in granulomas and in surrounding inflammatory infiltrate (10×) (b) CD20 positive lymphocytes around granulomas (10×) (c) CD 20 positive lymphocytes scattered amidst pituicytes (10×) (d) extensive CD3 positive lymphocytic infiltrate around granulomas (circled) (4×)
DISCUSSION
A pituitary incidentaloma is a previously unsuspected pituitary lesion that is discovered on an imaging study performed for an unrelated reason. The imaging study should not be done for a symptom specifically related to the lesion, such as visual loss, or a clinical manifestation of pituitary dysfunction, but rather for the evaluation of symptoms such as headache, or other head or neck neurological or central nervous system complaints or head trauma.[1] In our case imaging was done for headache without any clinical suspicion of pituitary, hence justifies diagnosis of pituitary incidentaloma, though headache is a known feature of pituitary lesions.
Preoperative diagnosis of primary granulomatous hypophysitis (PGH) is very uncommon as it is pathological diagnosis and mostly diagnosed postoperatively. However, on radiological examination features of all subtypes of primary hypophysitis are quite similar. MRI features which favor hypophysitis include diffuse, ill-defined, symmetrical enlargement of pituitary tissue, thickened but undeviated pituitary stalk, intact sellar floor, absence of mucosal thickening in sphenoid sinus, isointensity with gray matter on T1-weighted image, marked homogeneous or heterogeneous enhancement by gadolinium, and a strip of enhanced tissue along the duramater (the so-called “dural tail”), delayed complete contrast enhancement of the whole pituitary in dynamic MRI.[4,5] Our case had typical MRI findings as described; hence we made a clinical diagnosis of hypophysitis. However, despite these differentiating features it is not always possible to distinguish it from pituitary adenoma and final diagnosis in most cases is achieved on histopathological examination.
Hypophysitis is classified into primary and secondary forms. Primary hypophysitis can be autoimmune, granulomatous, necrotizing or xanthomatous.[6] Hypophysitis can also be secondary to local lesions like germinomas. Rathke's cyst, craniopharyngioma or pituitary adenomas or secondary to systemic diseases like tuberculosis, syphilis, sarcoidosis, and Wegener's granulomatosis.[4] In our case, there was past history of adequately treated tuberculosis 10 years back with raised ESR and a strongly positive Mantoux test. However, due to a time leg of 10 years we kept this possibility at low. Moreover, the Mantoux test can remain positive for a long time after treatment and does not indicate active disease.[7] Other secondary causes were excluded from investigation. With this background we made a provisional diagnosis of primary hypophysitis.
Current recommendation for pituitary incidentaloma is observation for patients without visual abnormalities and evidence of pituitary hypo- or hyperfunction. Glucocorticoids have been tried for treatment of PGH in view of good response of PAH to steroids; however, as the number of cases of PGH is so rare, and it is a pathological diagnosis, it has been difficult to assess response of PGH to steroids. Whatever data are there they show partial response to steroids and later recurrence in more cases compared to PAH.[8] However, spontaneous recovery, benign and transient course, and response to steroids have led to suggestions that conservative management with close clinical observation as the primary therapy.[9] In fact steroid therapy with successful outcome has been reported.[10] Trans-sphenoidal surgery should only be performed in cases with progressive compression, especially for those whose clinical and radiological presentation is not typical and diagnosis is in doubt.[11] Surgery is effective in achieving decompression of the sellar mass and thereby resolving headache and visual deficits. Most patients require long-term replacement with one or more hormones. However, surgery has a poor effect on improving the preexisting endocrine defects.[4,11] Thus, we decided to monitor our patient conservatively with steroid therapy in view of previously reported steroid therapy response in autoimmune hypophysitis. However, our patient had one episode of severe headache. This caused distress to the patient and she opted for surgery.
Primary hypophysitis is a rare entity and out of it PGH is even rarer so much so that some authors believe that its incidence rate is 1 in 10 million per annum.[11] Granulomatous hypophysitis was first described in 1917 by Simmond's in postmortem studies.[12] The first antemortem case was reported in 1980.[13] The pituitary in PGH shows diffuse collections of multinucleated giant cells and histiocytes, with surrounding lymphocytes and plasma cells. Lymphocytes are mainly of the T lineage.[4] Clinically PGH seems to be quite different from primary lymphocytic hypophysitis (PLH) as PLH occurs in peripartum women, occasional spontaneous remission, and association with other autoimmune disease which is not the case with PGH.[4] However, PGH coexistence with PLH is known and it has been proposed by Mckeel that they are in continuum with each other with PLH occurring in early course and PGH later on during the course.[2,3] Histologically PGH is characterized by necrotizing granulomas that are formed by collection of histiocytes, multinucleated giant cell, and variable numbers of lymphocytes and plasma cells.[11,14] Our case also had both granulomatous and lymphocytic inflammation as evidenced by well-defined epitheloid granulomas, lymphocytic infiltration and histiocytes on histopathological examination [Figure 2], and immunohistochemistry stained positive for CD3 (T-lymphocytes), CD20 (B-lymphocytes), and CD68 (histiocytes). Combined PGH and PAH are even rare and till now only four cases have been reported.[3,15,16] This further supports the hypothesis that PGH is a continuum of PLH and our case has been picked up at the stage of transition from PLH to PGH. We also speculate that this may further lead to fibrosis of pituitary and atrophic hypophysitis akin to atrophic thyroiditis, leading to hypopituitarism with empty sella of which many cases have been reported in the literature.
Till now 39 cases of PGH have been reported in the literature. It has slight female predilection (male/female ratio – 1:2), mean age of presentation of around 46 years. Clinically it can present with symptoms and signs of sellar compression, hypopituitarism, diabetes insipidus, and hyperprolactinemia. Headache is most common symptom followed by diabetes insipidus, visual compression, vomiting, nausea, extraocular muscle paralysis, and fever in this order.[11] Though autoimmune hypophysitis has been reported to present as pituitary incidentaloma[4] but the literature search performed by us did not reveal any case of combined PAH and PGH or otherwise isolated PGH presenting as incidentaloma. Another feature, which differed from literature in our case, was that there were no sign or symptoms suggestive of compression or pituitary dysfunction making it first time reported presentation of an extremely rare disease.
Thus here we report an uncommon case of PGH combined with lymphocytic hypophysitis with uncommon presentation as pituitary incidentaloma and pre- and postoperative normal pituitary functions.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Freda PU, Beckers AM, Katznelson L, Molit ME, Montori VE, Post KD, et al. Pituitary Incidentaloma: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96:894–904. doi: 10.1210/jc.2010-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKeel DW. Primary hypothyroidism and hypopituitarism in a young woman: Pathological discussion. Am J Med. 1984;77:326–9. doi: 10.1016/0002-9343(84)90710-1. [DOI] [PubMed] [Google Scholar]
- 3.Miyamoto M, Sugawa H, Mori T, Hashimoto N, Imura H. A case of hypopituitarism due to granulomatous and lymphocytic adenohypophysitis with minimal pituitary enlargement: A possible variant of lymphocytic adenohypophysitis. Endocrinol Jpn. 1988;35:607–16. doi: 10.1507/endocrj1954.35.607. [DOI] [PubMed] [Google Scholar]
- 4.Caturegli P, Newschaffer C, Olivi A, Pomper MG, Burger PC, Rose NR. Autoimmune hypophysitis. Endocr Rev. 2005;26:599–614. doi: 10.1210/er.2004-0011. [DOI] [PubMed] [Google Scholar]
- 5.Gutenberg A, Larsen J, Lupi J, Rohde V, Caturegli P. A radiologic score to distinguish autoimmune hypophysitis from nonsecreting pituitary adenoma preoperatively. AJNR Am J Neuroradiol. 2009;30:1766–72. doi: 10.3174/ajnr.A1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi J, Zhang JM, Wu Q, Chen G, Zhang H, Bo WL. Granulomatous hypophysitis. J Zhejiang Univ Sci B. 2009;10:552–8. doi: 10.1631/jzus.B0820355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pottumarthy S, Morris AJ, Harrison AC, Wells VC. Evaluation of the Tuberculin Gamma Interferon Assay: Potential To Replace the Mantoux Skin Test. J Clin Microbiol. 1999;37:3229–32. doi: 10.1128/jcm.37.10.3229-3232.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheung CC, Ezzat S, Smyth HS, Asa SL. The spectrum and significance of primary hypophysitis. J Clin Endocrinol Metab. 2001;86:1048–53. doi: 10.1210/jcem.86.3.7265. [DOI] [PubMed] [Google Scholar]
- 9.Ezzat S, Josse RG. Autoimmune hypophysitis. Trends Endocrinol Metab. 1997;8:74–80. doi: 10.1016/s1043-2760(96)00270-6. [DOI] [PubMed] [Google Scholar]
- 10.Beressi N, Cohan R, Beressi JP, Dumas JL, Legrand M, Iba-Zizen MT, et al. Pseudotumoral lymphocytic hypophysitis successfully treated by corticosteroid alone: First case report. Neurosurgery. 1994;35:505–8. doi: 10.1227/00006123-199409000-00020. [DOI] [PubMed] [Google Scholar]
- 11.Su S, Zhang D, Yue S, Zhang J. Primary granulomatous hypophysitis: A case report and literature review. Endocr J. 2011;58:467–73. doi: 10.1507/endocrj.k10e-357. [DOI] [PubMed] [Google Scholar]
- 12.Simmonds M. Uber das Vorkommen von Riesenzelle in der Hypophyse. Virchows Arch. 1917;223:281–90. [Google Scholar]
- 13.Taylon C, Duff TA. Giant cell granuloma involving the pituitary gland. J Neurosurg. 1980;52:584–857. doi: 10.3171/jns.1980.52.4.0584. [DOI] [PubMed] [Google Scholar]
- 14.Gutenberg A, Hans V, Puchner MJ, Kreutzer J, Bruck W, Caturegli P, et al. Primary hypophysitis: Clinical-pathological correlations. Eur J Endocrinol. 2006;155:101–7. doi: 10.1530/eje.1.02183. [DOI] [PubMed] [Google Scholar]
- 15.Honegger J, Fahlbusch R, Bornemann A, Hensen J, Buchfelder M, Müller M, et al. Lymphocytic and granulomatous hypophysitis: Experience with nine cases. Neurosurgery. 1997;40:713–23. doi: 10.1097/00006123-199704000-00010. [DOI] [PubMed] [Google Scholar]
- 16.Madsen JR, Karluk D. A 71-year-old woman with an enlarging pituitary mass. N Engl J Med. 2000;343:1399–406. doi: 10.1056/NEJM200011093431909. [DOI] [PubMed] [Google Scholar]