

Abstract
Plasma cell leukemia (PCL) is a rare hematologic malignancy with aggressive clinical and biologic features. Data regarding its prognosis with the use of the novel agents, i.e., the immunomodulatory drugs thalidomide and lenalidomide, and the proteasome inhibitor bortezomib, are limited. We retrospectively reviewed clinical outcomes, response to therapy, and survival of 17 patients seen at the Penn State Hershey Cancer Institute since the availability of novel agents (2006–2011). Twelve patients had primary PCL (pPCL), and 5 secondary PCL (sPCL). PCL was associated with aggressive clinicobiological features, such as high-risk cytogenetics, elevated serum beta-2-microglobulin and lactate dehydrogenase, International Staging System stage III, and rapid relapse after therapy. With the use of thalidomide, lenalidomide, and bortezomib in 53%, 53%, and 88% patients, respectively, median overall survival (OS) was 18 months in the whole group (95% confidence interval, 11–21 months), and 21 and 4 months in pPCL and sPCL, respectively (P=0.015). OS was inferior to that of 313 consecutive patients with multiple myeloma (MM) treated in the same period, even when compared with a subset of 47 MM with high-risk cytogenetics. Although our data are limited by the small sample size, we conclude that novel agents may modestly improve survival in patients with PCL, when compared to historical controls. Novel therapies do not seem to overcome the negative prognosis of PCL as compared with MM.
Key words: Plasma cell leukemia, multiple myeloma, extramedullary disease.
Introduction
Plasma cell leukemia (PCL) is a very rare malignancy, with an estimated incidence of approximately 4 cases per 10,000,000 persons per year.1 In order to standardize criteria for the diagnosis of PCL, in 1974 Kyle et al. have proposed an absolute plasma cell count >2000/ L or plasma cells comprising >20% of the total white cell count (WBC) in the peripheral blood.2,3 There are two forms of PCL: primary (pPCL), which occurs as a de novo presentation of the leukemia, and secondary (sPCL), which is the leukemic transformation of a previously diagnosed multiple myeloma (MM). pPCL accounts for 2–4% of MM cases.4 The immunophenotype of PCL is typically similar to MM, with cytoplasmic light chain restriction, positivity for CD38 and CD138, and loss of CD19 expression on neoplastic plasma cells. However, in a high proportion of PCLs (80%), plasma cells lack the aberrant CD56 expression.5
Several studies have shown that both pPCL and sPCL are characterized by an aggressive course, and this has been explained by its association with adverse prognostic factors, such as high tumor mass, extramedullary disease, high serum lactate dehydrogenase (LDH), DNA hypodiploidy, and high-risk molecular and cytogenetic abnormalities.3,4,6–8 For example, the loss of the tumor suppressor gene TP53 on chromosome 17p13, is a common event, as it is observed in 50% and 75% of pPCL and sPCL, respectively.9 Overexpression of MYC genes due to copy number alterations in chromosome 8 is also common, and it distinguishes PCL from MM.10 Gene-expression profiling of 27 patients with pPCL demonstrated a high-risk molecular signature. The differentially expressed genes were those involved in the liver X receptor/retinoic X receptor (LXR/RXR) activation, inositol metabolism, hepatic fibrosis/hepatic stellate-cell activation, and lipopolysaccharide/IL-1-mediated inhibition of RXR function pathways.11 The prognosis of PCL is very poor, because median overall survival, according to historical data, is 6–12 months, and less than 3 months in patients with the secondary form.3,12 There are no consensus guidelines for treating PCL, and there is great variability in clinical practice, due to the rarity of the disorder. Therapy is generally based upon expert opinion, extrapolation from results obtained in patients with MM, and single case reports or small retrospective studies. In the last decade, the therapy of MM has been revolutionized by the introduction of novel agents, which have led to improved outcomes: the proteasome inhibitor bortezomib, approved for the treatment of MM by the U.S. Food and Drug Administration in 2003, and the immunomodulatory drugs (IMiDs) thalidomide and lenalidomide, both approved in 2006, have increased therapeutic response rates and prolonged survival in patients with MM.13 Here we review the clinical outcomes of PCL in a period when all three drugs were available for being integrated in the treatment of this disease.
Materials and Methods
We identified and reviewed data of all patients with a diagnosis of PCL followed at the Myeloma Clinic of the Penn State Cancer Hershey Cancer Institute between January 2006 and December 2011. We compared their clinical and laboratory features with those of 313 consecutive MM patients seen in the same period. Response criteria were defined as previously published.14 High-risk cytogenetics were defined as the presence of a complex kariotype or monosomy 13 at metaphase cytogenetics, or the presence of 17p- or translocation t(4;14) at fluorescence in situ hybridization. Statistical analysis was conducted using Minitab 16 Statistical Software 2010 (Minitab, Inc., State College, PA). Event-free survival and overall survival (OS) were calculated from the time of PCL diagnosis, unless otherwise specified. The method of Kaplan and Meier was used to compute the survival curves and to estimate median survival. The differences in survival curves were tested with the log-rank method. P<0.05 was considered statistically significant.
Results
We identified 17 patients with PCL, of whom 12 had sPCL and 5 sPCL. Patient characteristics at baseline are shown in Table 1. The median time between the initial diagnosis of MM and the development of sPCL was 28 months (range, 24–34 months). Median WBC count in the peripheral blood was 12×103/ L (range, 7–48), with a median % of plasma cells 31 (range, 14–89). When defining hypercalcemia as serum calcium level ≥12.0 mg/dL, ane mia as hemoglobin ≤10.0 g/dL, and renal insufficiency as serum creatinine ≥ 2.0 mg/dL, following the thresholds indicated by the Durie-Salmon classification,15 we did not find statistically significant differences in rates of hypercalcemia, anemia, and renal insufficiency between PCL and MM. The paraprotein secreted by the PCL plasma cells was IgG (8 pts), IgG (3 pts), , IgA (1 pt), IgE (1 pt), light chain (2 pts), and light chain (2 pts). High-risk cytogenetics were present in 9 of 16 (56%) evaluable cases (Table 1), and cyclin D1 amplification, assessed by immunohistochemistry staining, was positive in 6 of 9 (67%) evaluable cases.
Table 1. Patient characteristics of plasma cell leukemia and comparison with multiple myeloma.
| PCL n=17 | MM n=313 | P | |
|---|---|---|---|
| Age, median (range), years | 60 (21–92) | 63 (36–92) | 0.39 |
| Gender (male/female), no. of pts | 10/7 | 175/138 | 0.81 |
| EMD, no. of patients | 2 (12%) | 28 (8%) | 0.65 |
| CRAB symptoms | |||
| Hypercalcemia (Ca ≥12 mg/dL), no. of pts | 3/17 (18%) | 35/279 (13%) | 0.47 |
| Renal insufficiency (Cr ≥2.0 mg/dL), no. of pts | 4/17 (24%) | 57/270 (21%) | 0.76 |
| Anemia (Hb <10 g/dL), no. of pts | 9/17 (53%) | 102/287 (36%) | 0.15 |
| Osteolytic lesions at x-rays, no. of pts | 8/17 (47%) | 218/302 (72%) | 0.05* |
| β2-microglobulin, median (range), mg/L | 16.4 (0.1–56) | 5.7 (2.5–76) | <0.01* |
| Elevated serum LDH, no. of pts | 6/13 (46%) | 37/256 (14%) | <0.01* |
| Paraprotein, no. of patients | |||
| IgG, IgA | 12 (71%) | 239 (76%) | 0.57 |
| IgD, IgE, light chain, nonsecretory | 5 (29%) | 74 (24%) | |
| Percent plasma cells in BM aspirate, mean (±SD) | 44 (±29) | 65 (±28) | <0.01* |
| High-risk cytogenetics, no. of pts: | 9/16 (56%) | 47/235 (20%) | <0.01* |
| Complex kariotype or -13 at metaphase cytogenetics | 7/16 (44%) | 28/235 (12%) | |
| 17p- at FISH | 2/16 (12%) | 19/205 (9%) | |
| t(4;14) at FISH | 0/16 (0%) | 10/205 (5%) | |
| ISS stage III, no. of pts | 14/17 (82%) | 83/260 (32%) | <0.01* |
Ca, calcium; Cr, creatinine; dx = diagnosis; EMD, extramedullary disease; FISH, fluorescence in situ hybridization; ISS, International Staging System; LDH, lactate dehydrogenase; MM, multiple myeloma; n/a, not available; PCs, plasma cells; PCL, plasma cell leukemia; pPCL, primary PCL; pts, patients; SCT, stem cell transplantation; SD, standard deviation; sPCL, secondary PCL; WBC, white blood cell.
P<0.01
Treatment included thalidomide-based regimen (9 pts, 53%), lenalidomide-based regimen (9 pts, 53%), bortezomib-based regimen (15 pts, 88%), single autologous stem cell transplantation (SCT) (4 pts, 24%), tandem autologous SCT (4 pts, 24%), and allogeneic SCT (2 pts, 12%). Response rate (RR) to first-line therapy, defined as PR or better, was seen in 14 patients (82%), and nCR/CR was obtained in 4 patients (24%). Median progression-free survival after the initial induction therapy was 10 months (range, 2–63), and 1.2 months (range, 0.5–8.8) in pPCL and sPCL, respectively (P=0.0011). The median OS for all patients was 18 months (95% C.I., 11–21 months), and 21 and 4 months in pPCL and sPCL, respectively (p=0.0154). Median OS in patients with sPCL, when calculated from the initial diagnosis of MM, was 31 months (95% confidence interval, 28–46). Median OS in the whole group of PCL was not only inferior to that observed in 313 consecutive MM patients treated in the same period (91 months, P<0.01), but also inferior to the median OS in a subgroup of 47 consecutive MM patients with high-risk cytogenetics (44 months, P<0.01) (Figure 1A). OS was similar between pPCL and high-risk MM (Figure 1B). After a median follow-up of 25 months, 11 patients with PCL (65%) were dead, and the cause of death was progressive disease in 10 pts, and transplant-related mortality in 1 pt.
Figure 1.
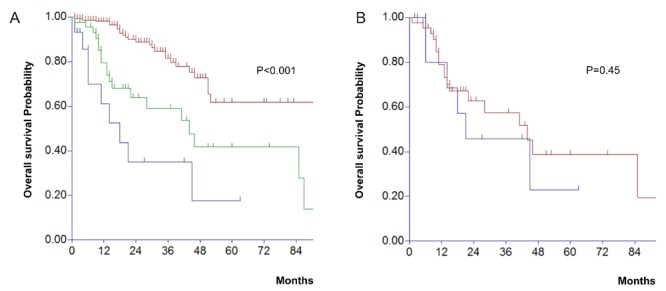
A) Overall survival of plasma cell leukemia (PCL) (n=17, blue line), as compared with high-risk multiple myeloma (MM) (n=47, green line), and standard-risk MM (n=188, brown line) (P<0.001). B) Overall survival of primary PCL (n=12, blue line), as compared with high-risk MM (n=47, red line).
Discussion
Our retrospective analysis of 17 patients with PCL showed that some of the clinical characteristics of PCL are similar to those of MM (e.g., age, gender, incidence of EMD, and type of paraprotein secreted by the neoplastic cells), whereas others have a different distribution. Among the typical CRAB symptoms of MM (i.e., hypercalcemia, renal insufficiency, anemia, and bone lesions), only the presence of osteolytic lesions was different, with a lower prevalence in PCL (P borderline at 0.05). This can be explained by the fact that PCL cells leave the bone marrow and enter the peripheral circulation before forming bone tumors, presumably because of a loss of homing signal in the bone marrow. The higher levels of serum beta-2-microglobulin and LDH seen in PCL are also expected, due to the rapid proliferation of the neoplastic clone, as seen in acute leukemias. Other groups have reported a higher proportion of a paraprotein consisting of light chain or less common isotypes in PCL (e.g., IgD, and IgE) when compared with MM,3,9 but we did not observe this association. We confirmed the higher proportion of high-risk cytogenetics, observed by others.4,9 It seems that this finding alone does not explain by itself the poor prognosis of PCL, because the Kaplan-Meier estimates of PCL vs high-risk MM does show an inferior survival for PCL, as shown in Figure 1A.
Our study is limited by its retrospective nature and the small sample size, which is due to the rarity of PCL. Moreover, the treatment received by our patients was heterogeneous. We report a median OS of 18 months in the era of novel agents, which is higher than that historically seen in the medical literature, and not very different from the 23.6 months reported by the University of South Florida in 25 consecutive PCL patients,16 and from the 22 months seen in 27 patients treated at the University of Arkansas with the Total Therapy approach.11 Obviously, it is difficult to separate the benefit conferred by the use of novel agents over SCT, as many of our patients received both. The outcomes of PCL observed in the past were poor. The combination of melphalan and prednisone, one of the old standards of care, produced a RR of 17% and a median OS of only 3 months in PCL.3 Intravenous chemotherapy with vincristine, adriamycin, and dexamethasone, another old standard of care, or high-dose cyclophosphamide and etoposide, increased RR and prolonged median OS to 15–20 months at the cost of significant toxicity.3,4,9 The novel agent bortezomib has improved RR but not OS: in a study of 12 PCL patients, the use of bortezomib was associated with a very high RR (92%), but median OS was only 12 months.17 As for the IMiDs, the activity of thalidomide has been disappointing, after a series of 5 cases found no responses and a median OS of 2 months,18 whereas the experience with lenalidomide is too premature to draw conclusions.19,20 Regarding SCT, the Mayo Clinic in Florida reported a median OS of 34 months.9 The largest cohort of patients with pPCL treated with autologous SCT was published by the European Group for Blood and Marrow Transplantation.21 This retrospective study showed median PFS and OS of 14.3 and 25.7 months, respectively. As for allogeneic SCT, its role remains controversial, because decreased rates of PCL relapse are paid at the cost of transplant-related mortality and morbidity. A large retrospective series of 50 pts with pPCL treated with allogeneic SCT found a 3-year PFS and OS of 20% and 39%, respectively.22 However, we should interpret all these reports with caution, because they did not include consecutive patients -as in our study - and selection bias is likely present (e.g., patients with better performance status, younger, and with chemotherapy-responsive PCL).
Although outcomes of patients with PCL appear to be improved with the availability of novel agents, the results are not outstanding. These patients respond very well to the initial induction therapy (82% RR in our cohort), but they relapse quickly, and the proportion of long-term survivors and operative cure is negligible. The novel agent do not overcome the negative prognosis of PCL as compared with MM, and more research is need, not only in terms of available therapies, but also for identifying specific molecular pathways responsible for the neoplastic transformation of PCL cells. Even the definition of PCL may need to be revised in the future: we need to recognize that monoclonal PCs circulate in the peripheral blood even in patients without PCL, and 80% of patients with untreated MM harbor circulating PCs, with a median of 6% PCs.23 Therefore, the degree of peripheral blood involvement by monoclonal PCs constitutes a continuous curve, and the current thresholds used to define PCL are purely arbitrary.
Conclusions
We can speculate that PCL and MM are, in fact, the same disease, with PCL being an acute presentation of MM with aggressive biological features.24 According to this view, pPCL resembles those cases of acute leukemias that are found to be blastic crises of chronic myelogenous leukemias presenting de novo. Therefore, in our opinion, instead of differentiating between PCL and MM, it would be more appropriate to distinguish, as in leukemias, chronic myelomas, with relatively indolent course, and acute myelomas (or blastic crisis of MM), characterized by unfavorable biologic features and a more rapidly fatal course.
References
- 1.Sant M, Allemani C, Tereanu C, et al. Incidence of hematologic malignancies in Europe by morphologic subtype: results of the HAEMACARE project. Blood. 2010;116:3724–34. doi: 10.1182/blood-2010-05-282632. [DOI] [PubMed] [Google Scholar]
- 2.Kyle RA, Maldonado JE, Bayrd ED. Plasma cell leukemia. Report on 17 cases. Arch Intern Med. 1974;133:813–8. doi: 10.1001/archinte.133.5.813. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Sanz R, Orfao A, Gonzalez M, et al. Primary plasma cell leukemia: clinical, immunophenotypic, DNA ploidy, and cytogenetic characteristics. Blood. 1999;93:1032–7. [PubMed] [Google Scholar]
- 4.Dimopoulos MA, Palumbo A, Delasalle KB, et al. Primary plasma cell leukaemia. Br J Haematol. 1994;88:754–9. doi: 10.1111/j.1365-2141.1994.tb05114.x. [DOI] [PubMed] [Google Scholar]
- 5.Pellat-Deceunynck C, Barille S, Jego G, et al. The absence of CD56 (NCAM) on malignant plasma cells is a hallmark of plasma cell leukemia and of a special subset of multiple myeloma. Leukemia. 1998;12:1977–82. doi: 10.1038/sj.leu.2401211. [DOI] [PubMed] [Google Scholar]
- 6.Shimazaki C, Gotoh H, Ashihara E, et al. Immunophenotype and DNA content of myeloma cells in primary plasma cell leukemia. Am J Hematol. 1992;39:159–62. doi: 10.1002/ajh.2830390302. [DOI] [PubMed] [Google Scholar]
- 7.Costello R, Sainty D, Bouabdallah R, et al. Primary plasma cell leukaemia: a report of 18 cases. Leuk Res. 2001;25:103–7. doi: 10.1016/s0145-2126(00)00102-8. [DOI] [PubMed] [Google Scholar]
- 8.Jimenez-Zepeda VH, Dominguez-Martinez VJ. Plasma cell leukemia: a highly aggressive monoclonal gammopathy with a very poor prognosis. Int J Hematol. 2009;89:259–68. doi: 10.1007/s12185-009-0288-3. [DOI] [PubMed] [Google Scholar]
- 9.Tiedemann RE, Gonzalez-Paz N, Kyle RA, et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia. 2008;22:1044–52. doi: 10.1038/leu.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiecchio L, Dagrada GP, White HE, et al. Frequent upregulation of MYC in plasma cell leukemia. Genes Chromosomes Cancer. 2009;48:624–36. doi: 10.1002/gcc.20670. [DOI] [PubMed] [Google Scholar]
- 11.Usmani SZ, Nair B, Qu P, et al. Primary plasma cell leukemia: clinical and laboratory presentation, gene-expression profiling and clinical outcome with Total Therapy protocols. Leukemia. 2012 Apr 17; doi: 10.1038/leu.2012.107. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albarracin F, Fonseca R. Plasma cell leukemia. Blood Rev. 2011;25:107–12. doi: 10.1016/j.blre.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516–20. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Durie BG, Harousseau JL, Miguel JS, et al. International uniform response criteria for multiple myeloma. Leukemia. 2006;20:1467–73. doi: 10.1038/sj.leu.2404284. [DOI] [PubMed] [Google Scholar]
- 15.Durie BG, Salmon SE. A clinical staging system for multiple myeloma. Correlation of measured myeloma cell mass with presenting clinical features, response to treatment, and survival. Cancer. 1975;36:842–54. doi: 10.1002/1097-0142(197509)36:3<842::aid-cncr2820360303>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 16.Lebovic D, Zhang L, Alsina M, et al. Clinical outcomes of patients with plasma cell leukemia in the era of novel therapies and hematopoietic stem cell transplantation strategies: a single-institution experience. Clin Lymphoma Myeloma Leuk. 2011;11:507–11. doi: 10.1016/j.clml.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 17.Musto P, Rossini F, Gay F, et al. Efficacy and safety of bortezomib in patients with plasma cell leukemia. Cancer. 2007;109:2285–90. doi: 10.1002/cncr.22700. [DOI] [PubMed] [Google Scholar]
- 18.Petrucci MT, Martini V, Levi A, et al. Thalidomide does not modify the prognosis of plasma cell leukemia patients: experience of a single center. Leuk Lymphoma. 2007;48:180–2. doi: 10.1080/10428190601007570. [DOI] [PubMed] [Google Scholar]
- 19.Benson DM, Jr., Smith MK. Effectiveness of lenalidomide (Revlimid) for the treatment of plasma cell leukemia. Leuk Lymphoma. 2007;48:1423–5. doi: 10.1080/10428190701361851. [DOI] [PubMed] [Google Scholar]
- 20.Musto P, Pietrantuono G, Guariglia R, et al. Salvage therapy with lenalidomide and dexamethasone in relapsed primary plasma cell leukemia. Leuk Res. 2008;32:1637–8. doi: 10.1016/j.leukres.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 21.Drake MB, Iacobelli S, van Biezen A, et al. Primary plasma cell leukemia and autologous stem cell transplantation. Haematologica. 2010;95:804–9. doi: 10.3324/haematol.2009.013334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahindra A, Kalaycio ME, Vela-Ojeda J, et al. Hematopoietic cell transplantation for primary plasma cell leukemia: results from the Center for International Blood and Marrow Transplant Research. Leukemia. 2012;26:1091–7. doi: 10.1038/leu.2011.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Witzig TE, Gertz MA, Lust JA, et al. Peripheral blood monoclonal plasma cells as a predictor of survival in patients with multiple myeloma. Blood. 1996;88:1780–7. [PubMed] [Google Scholar]
- 24.Smadja NV, Bastard C, Brgaudeau C. Primary plasma cell leukemia and multiple myeloma: one or two diseases according to the methodology. Blood. 1999;94:3607–9. [PubMed] [Google Scholar]