

Abstract
Fragment-based drug discovery (FBDD) has become a new strategy for drug discovery where lead compounds are evolved from small molecules. These fragments form low affinity interactions (dissociation constant (KD) = mM − μM) with protein targets, which require fragment screening methods of sufficient sensitivity. Weak affinity chromatography (WAC) is a promising new technology for fragment screening based on selective retention of fragments by a drug target. Kinases are a major pharmaceutical target, and FBDD has been successfully applied to several of these targets. In this work, we have demonstrated the potential to use WAC in combination with mass spectrometry (MS) detection for fragment screening of a kinase target—cyclin G-associated kinase (GAK). One hundred seventy fragments were selected for WAC screening by virtual screening of a commercial fragment library against the ATP-binding site of five different proteins. GAK protein was immobilized on a capillary HPLC column, and compound binding was characterized by frontal affinity chromatography. Compounds were screened in sets of 13 or 14, in combination with MS detection for enhanced throughput. Seventy-eight fragments (46 %) with KD < 200 μM were detected, including a few highly efficient GAK binders (KD of 2 μM; ligand efficiency = 0.51). Of special interest is that chiral screening by WAC may be possible, as two stereoisomeric fragments, which both contained one chiral center, demonstrated twin peaks. This ability, in combination with the robustness, sensitivity, and simplicity of WAC makes it a new method for fragment screening of considerable potential.
Keywords: Fragment screening, Fragment-based drug discovery, Cyclin G-associated kinase, Weak affinity chromatography
Introduction
During the last 10 years, the fragment-based drug discovery (FBDD) paradigm, where a lead compound is evolved from small molecular fragments that bind to a biological target, has to an increasing extent been implemented both within academia and the pharmaceutical industry [1–3]. Problems presented by high-throughput screening (HTS) such as low hit rates and difficulties in optimizing hits into lead compounds with adequate ADME properties [4] has led to the use of FBDD either as a complement or replacement to HTS. It is believed that FBDD enables the development of inhibitors against difficult targets where HTS has failed [5].
Originally, the concept of FBDD was presented by Jencks in 1981 [6]. In 1996, Shuker et al. presented the “SAR-by-NMR” approach where nuclear magnetic resonance (NMR) was used to find two different fragments that bound to various locations of the binding site of a target protein. Subsequent linking of the two fragments to each other resulted in a compound with nanomolar affinity to the target, demonstrating the concept of FBDD in practice [7].
In 2011, the FDA approved the first drug that had been developed by FBDD—the oncogenic B-Raf inhibitor Zelboraf as a therapy against malignant melanoma [8]. Several other candidate drugs that have been developed by FBDD are currently in clinical trials, and many of these are kinase inhibitors. Kinases are involved in a range of different biological processes such as signaling, proliferation, apoptosis, and differentiation [9]. As kinases have been related to many different types of pathological states, such as cardiovascular diseases, cancer, and neurodegenerative diseases [9, 10], a considerable amount of research is now devoted to finding drugs that target kinases [11]. FBDD has proven successful when applied to kinases as drug targets and numerous examples exist of FBDD projects targeting kinases, such as PIM [12], PI3K [13], and PDK1 [14]. The majority of kinase inhibitors target the ATP-binding site which, due to the conserved nature of this site among kinases, often leads to difficulties obtaining specificity [11]. However, it has also been proposed that promiscuity of kinase inhibitors can be beneficial in some instances [15, 16].
As the number of possible combinations of a molecule increases exponentially with the number of heavy atoms in a molecule, screening of molecules that are smaller in size enables coverage of a greater portion of chemical space [4, 17]. As a result, fragment libraries are much smaller than HTS libraries, consisting of only hundreds to a few thousand compounds [18, 19].
Due to the small size of fragments (<300 Da [20]), fragments form weak although high-quality interactions to the target with dissociation constants (KD) in the micromolar to millimolar range [2, 19]. This puts great demands on technologies used for fragment screening, for example in terms of sensitivity and throughput. Surface plasmon resonance (SPR), NMR, X-ray crystallography, and isothermal titration calorimetry are techniques that are frequently employed today for fragment screening [19, 21]. Weak affinity chromatography (WAC) [22] has in a number of studies been applied to analytical affinity separations including carbohydrate [23, 24] and chiral analyses [25]. In recent studies, it has also been applied to fragment screening on proteases as model target proteins [26]. This is a technique which carries all the inherent properties of HPLC combined with the selectivity of affinity. Moreover, analysis of sample mixtures combined with MS detection further enables high throughput and sensitivity of detection.
In this study, fragment screening by WAC with MS detection is demonstrated for the first time against a kinase target by screening a larger set of substances. The cyclin G-associated kinase (GAK) was used as a model protein target for kinase, which also has been suggested as a potential drug target for Parkinson’s disease [27, 28]. GAK was immobilized on a capillary column for minimal protein consumption, and characterization was performed by frontal affinity chromatography to assess the number of active sites. Screening by WAC was preceded by virtual screening of a 3,200-compound commercial fragment library against the ATP-binding site of five different protein structures. One hundred seventy fragments with the highest average rank were subject to fragment screening by WAC. WAC showed promise as a technology for fragment screening against kinases including the potential to select stereoisomeric fragments.
Materials and methods
Chemicals
A fragment-based library of 3,200 fragments was obtained from TimTec, Newark, DE. The molecular weights of the fragments were between 75 and 289 Da. All fragments had a cLogP, ≤2.5; hydrogen bond acceptors, ≤5; hydrogen bond donors, ≤3; rotatable bonds, ≤3; and heavy atoms (nonhydrogen), ≤20. The fragments were dissolved in dimethyl sulfoxide (DMSO; Sigma Aldrich, St Louis, MO) at 1.5 mM and were further diluted in water to working concentrations. Periodic acid, adenosine, sodium cyanoborohydride, and ethanolamine were purchased from Sigma Aldrich. LB Media was purchased from Novagen (Darmstadt, Germany).
Production of GAK
DNA (residues 20–347) of human GAK was PCR amplified from synthetic DNA and inserted into vector pNIC28-Bsa4 (GI 124015065) [29] by ligation-independent cloning. The plasmid DNA was transformed into Escherichia coli Rosetta (DE3) for protein expression. From an overnight culture grown in LB (Miller, Novagen) media containing 50 μg/mL kanamycin and 34 μg/mL chloramphenicol, 6 × 10 mL were used to inoculate 6 × 1 L cultures of LB media containing 50 μg/mL kanamycin, in baffled shaker flasks. Cultures were grown at 37 °C until an OD600 of ∼0.7. The temperature was reduced to 18 °C for 1 h, and the cultures were then induced with 0.5 mM isopropyl-β-thio galactopyranoside and left shaking overnight. Cells were harvested by centrifugation.
The cells were resuspended in 50 mM Hepes at pH 7.5, 500 mM sodium chloride, 5 % glycerol, 5 mM imidazole, and 0.5 mM tris(2-carboxyethyl)phosphine (TCEP) (binding buffer) and lysed by sonication. Polyethyleneimine was added to a final concentration of 0.25 %, and the cell debris and precipitated DNA were spun down. The supernatant was passed through a 3-mL nickel-nitriloacetic acid (Ni-NTA) column, which was washed with binding buffer (25 mL) and binding buffer containing 25 mM imidazole (25 mL). The protein was eluted with binding buffer containing 250 mM imidazole (15 mL). The eluted fraction was concentrated to 4 mL and injected onto an S200 16/60 gel filtration column (GE Healthcare, Waukesha, WI) in 25 mM Hepes at pH 7.4, 150 mM sodium chloride, 0.5 mM TCEP (GF buffer). Fractions containing GAK were pooled, and the protein was digested with tobacco etch virus protease. The sample was passed through 2 mL of Ni-NTA, eluting with GF buffer containing 20 mM imidazole. The sample was concentrated to 13 mg/mL and frozen at −80 °C. Typical yield of purified GAK protein is about 10 mg/L culture media.
Preparation of affinity capillary columns
GAK was thawed on ice and prepared for immobilization by dialysis 1/10,000 in 0.1 M sodium phosphate buffer at pH 7.0 in a Slide-A-Lyser® (Pierce, Rockford, IL) at 4 °C. The buffer was exchanged three times during dialysis of the protein. The GAK concentration was estimated by absorbance readings at 280 nm (NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE)).
Immobilization of GAK and ethanolamine was performed in situ on two stainless-steel capillary columns (100 × 0.5 mm) packed with spherical silica particles (5 μm in diameter, 300 Å pore size; Kromasil, EKA Chemicals, Bohus, Sweden) which had been silanized into diol-substituted silica according to standard procedures. Immobilization was performed on an Agilent 1200 HPLC system (Agilent Technologies, Waldbronn, Germany) by reductive amination of mainly lysine side chains of the protein and aldehyde groups of the silica surface essentially as described earlier [26]. The diol-silica column was rinsed with water (20 column volumes) (immobilization of ethanolamine) or was rinsed with isopropanol (20 column volumes) and then with water (20 column volumes; immobilization of GAK). For immobilization of GAK and ethanolamine, the diol-silica columns were oxidized into aldehyde-silica by 10 × 40-μL injections of 0.13 g/mL periodic acid at a flow rate of 20 μL/min for 2 min. The flow rate was then stopped for 11 (GAK) or 20 min (ethanolamine) between injections. The total reaction time was 2 (GAK) or 3.3 h (ethanolamine), and it was performed at 12 (GAK) or 22 °C (ethanolamine). The column was then rinsed with water and with 0.1 M sodium phosphate buffer at pH 7.0 (>20 column volumes). For the coupling procedure of GAK, 9 × 40-μL injections of GAK (2.24 mg/mL) and sodium cyanoborohydride (9 mg/mL) and 1 × 40-μL injection of sodium cyanoborohydride (9 mg/mL) were performed at a flow rate of 20 μL/min for 2 min. The flow was stopped for 2 h between injections. The total reaction time was 20 h, and it was performed at 12 °C. Remaining aldehyde-silica groups present on the column were encapsulated by ethanolamine. This was achieved by 6 × 40-μL injections of ethanolamine (6.15 mg/mL) and sodium cyanoborohydride (9 mg/mL) dissolved in 0.1 M sodium phosphate buffer at pH 7.0 at a flow rate of 20 μL/min for 2 min. The flow was stopped for 1 h between injections. The reaction was performed at 12 °C and the total reaction time was 6 h. For immobilization of ethanolamine as a reference column, 10 × 20-μL injections of ethanolamine (6.15 mg/mL) and sodium cyanoborohydride (9 mg/mL) dissolved in 0.1 M sodium phosphate buffer at pH 7.0 were performed with a flow rate of 20 μL/min for 1 min. The flow was stopped for 75 min between injections. The total reaction time was 12.5 h and it was performed at 12 °C. Both columns were rinsed with 0.1 M sodium phosphate at pH 7.0 (>20 column volumes). The eluate (including washings) from the immobilization of GAK was collected and the yield was determined indirectly from the absorbance at 280 nm of the eluate and applied GAK sample.
Selection of fragments for screening
All calculations were performed using the Schrödinger Suite 2010 (Portland, OR) and with default settings. Five 3D structures of proteins with an ATP-binding site to which a ligand was docked was downloaded from the protein data bank (PDB): Mammalian STE20-like protein kinase 3 (MST3; PDB ID 3CKX), heat shock protein 90 (Hsp90; PDB ID 2XDK), c-Jun N-terminal kinase 3 (JNK3; PDB ID 3G90), protein kinase A (PKA; PDB ID 1Q8T), and vascular endothelial growth factor (VEGFR; PDB ID 3BE2). The protein 3D structures were prepared in the Protein Preparation Wizard. Any missing side chains in the structures were predicted by Prime (Prime version 2.2.108). The water present in the structures was deleted and a receptor grid was prepared of the ATP-binding site. The 3,200 structures of the fragments were prepared by LigPrep (Ligprep version 2.4.107) at pH 7.4 ± 2.0 using the ionizer and a maximum of 32 stereoisomers per ligand. LigPrep generated 5,636 3D structures with variations in, e.g., tautomeric and ionic form, which were docked flexibly in standard precision-mode in Glide (Glide version 5.6.107) to the generated receptor grid of each of the five-protein 3D structures.
The obtained Glide gscores, which is related to the affinity of a fragment to a protein, were used to rank the 3,200 fragments internally with regard to each of the five-protein 3D structures. For each fragment, the average rank with regard to all five-protein 3D structures was calculated and the 170 fragments with the highest average rank were selected for further studies.
Column characterization by frontal affinity chromatography
Frontal affinity chromatography was performed essentially as described by Kasai et al. [31]. An Agilent 1200 HPLC system equipped with a diode array multiple wavelength detector was used for the frontal analysis. As mobile phase, 20 mM ammonium acetate at pH 6.8 was used. The flow rate was 18 μL/min, and the column temperature was 22 °C. Adenosine was dissolved in the mobile phase and injected onto the GAK capillary column at 100 μL at concentrations of 0.025, 0.050, 0.100, 0.200, 0.300, 0.400, 0.500, and 0.750 mM. The breakthrough curve was monitored by UV at 214 nm. The void time of the column was determined with DMSO (0.1 % in water) by an injection volume of 0.4 μL. Chromatograms were analyzed by the Agilent ChemStation version B.04.01 chromatography data system, and the front time was assessed from the local maximum of the first derivative of the front curve. The volume for column saturation V´R was calculated by adjusting for the void volume of the column.
The number of moles of adenosine required for saturation of all active sites present on the column was calculated from V´R and the concentration of adenosine for each injection. These values were plotted against the concentrations of adenosine in each injection in order to generate a one-site total binding hyperbola using GraphPad Prism 5.0c (San Diego, CA). Here, the contribution from nonspecific binding is defined as a linear relation between analyte concentration and amount bound. By nonlinear regression analysis, the number of binding sites on the column (Btot) and the KD of adenosine to GAK were determined (Eq. 1).
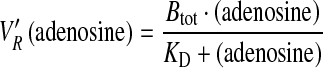 |
1 |
Zonal affinity chromatography screening of the fragment library
Fragment screening was performed on an Agilent 1200 series capillary HPLC system equipped with a single quadropole mass spectrometry (MS) detector. On MS detection, fragments were ionized by electrospray at atmospheric pressure in positive and negative mode alternatively. Drying nitrogen gas flow was 7 L/min at 350 °C. The nebulizer pressure was 15 psig, and the capillary voltage was 3,000 V in positive mode and −2,500 V in negative mode. MS signal acquisition was set at selected ion monitoring (SIM) mode on sample target masses. Both the [M+1]+ and the [M−1]− ion was monitored for each analyte. The fragmentor was set to 100 V in positive SIM mode and to 173 V in negative SIM mode. Retention times were based on peak apexes of the extracted ion chromatogram (EIC). Chromatograms were analyzed with the Agilent ChemStation version B.04.01 chromatography data system.
Ammonium acetate buffer (20 mM) at pH 6.8 was used as mobile phase throughout the studies and the analyses were performed isocratically. Fragments were analyzed in mixtures with 13 or 14 fragments in each mixture, resulting in 13 different mixtures. The concentration was 5.0 or 5.3 μM of each fragment and a DMSO concentration of 4.6 or 4.7 %. Adenosine was also present as a control in each mixture at an equal concentration as the other analytes present in the mixture. The retention time of adenosine was monitored continuously and was used as a measure of the activity of the column. Each mixture was analyzed in duplicate samples on both the GAK column and the reference (ethanolamine) column. Screening was performed with an injection volume of 0.4 μL and a flow rate of 20 μL/min. The column temperature was set to 22 °C.
Data analysis
The KD of each fragment with regard to binding to GAK was approximated [23, 30] according to:
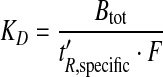 |
2 |
where F is the flow rate during screening and t´R, specific is the adjusted retention time on the GAK column (t’R, GAK), corrected for the adjusted retention time on the reference column (t´R, ref):
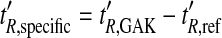 |
3 |
The adjusted retention times were calculated by subtraction of the retention time of the void marker DMSO from the retention times of the analytes. Equation 2 is valid under a linear isotherm, i.e. when the concentration of the analyte during analysis on the column is much smaller than the analyte’s KD value.
Results and discussion
Selection of fragments by virtual screening
Through virtual screening by Glide, 170 fragments were selected from a fragment based library of 3,200 substances on the basis of their average internal rank towards the ATP-binding site of five crystal structures of four kinases of different types and one chaperone (MST3, Hsp90, JNK3, PKA, and VEGFR). The structures of these randomly selected proteins with an ATP-binding site were used for virtual screening as there were no crystal structures available for GAK with a docked inhibitor that could be used for ligand docking by Glide. The object was to find fragments with affinity to the ATP-binding site, which would produce a set of fragments that were likely to bind to GAK or other kinases due to the conserved nature of the ATP-binding site. The object with ligand docking to more than one-protein 3D structure was to overcome inherent limitations of the computational modeling method, as well as to minor flaws that may be present in the individual crystal structures.
As water present in the protein crystal structures was deleted prior to virtual screening, ligands that use waters to form interactions with the ATP-binding site may have been missed. In addition, as the virtual screening was performed using one single conformation of each protein structure, ligands that utilize the inherent plasticity of the kinase for their binding may have been excluded. The molecular weights of the 170 selected substances were between 147 and 284 Da. The fragments contained ≤20 heavy (nonhydrogen) atoms, ≤3 hydrogen bond donors, and ≤5 hydrogen bond acceptors and had a cLogP between −1.87 and 2.52.
Production and characterization of GAK capillary column
GAK was immobilized onto a diol-silanized silica capillary column to prevent nonspecific interactions with the naked silica surface and to introduce functional groups for protein coupling. The diol silica was oxidized into aldehyde silica and GAK was immobilized by reductive amination (0.78 mg (20.8 nmol); 39.8 mg/mL) at a high coupling yield (90 %). The GAK column was characterized by frontal affinity chromatography and the number of active enzyme sites on the capillary column was determined to be 4.6 ± 0.7 nmol (R2 = 0.988). Thus, the ratio of active sites versus protein content on the column was 22 %. This value is typically lower than was experienced in previous studies with other ligands [23, 26]. The reason for this may be a decrease in kinase activity during storage prior to immobilization or inactivation by coupling to lysine residues present in the ATP-binding site of GAK. However, the concentration of active sites in the capillary column (0.23 mM) still makes it theoretically possible to study weak binding in the low millimolar range. By miniaturization of column size into capillary columns, consumption of target protein was well below 1 mg which could be further minimized by an order of magnitude by going into chip format or shorter capillary columns.
Fragment screening by zonal affinity chromatography
Performance of screening
Figure 1 demonstrates typical chromatograms from analysis of one mixture of fragments on the GAK column with MS detection, both the total ion chromatograms (TICs) of SIM-positive and SIM-negative mode, and EICs for individual analytes. Adenosine was present in each mixture during analysis. The KD value for adenosine, as measured by zonal chromatography (41 μM), was in good agreement with the KD obtained from frontal affinity chromatography (63 μM). There was no trend towards shorter retention times of adenosine throughout the screening study (a few weeks) indicating no loss of activity of the GAK column. Out of the 170 selected fragments that were screened by WAC, 145 (85 %) were detected by MS during analysis on the GAK column and 141 (83 %) on the reference column. One hundred forty (82 %) of the fragments were detected on both columns. The distribution in estimated KD values for these fragments is shown in Fig. 2. For 78 (46 %) of the screened fragments, the estimated KD was 200 μM or lower, which represents a difference in retention time between the GAK column and the reference column of 1× the void time of the GAK column (1.13 min; Eq. 2). This number constitutes 2.4 % of the 3,200 compounds in the original Timtec library. The high number of fragment hits of relatively high affinity indicates that the selection of fragments for screening that was performed in silico was successful. A number of false positives may however be included as fragments binding to parts of the protein other than the ATP site would also have been detected.
Fig. 1.

Chromatograms of fragments of mixture 4. (a) TIC of SIM-positive mode, (c) TIC of SIM-negative mode, (b) EICs of individual analytes in SIM-positive mode (1, ST025584; 2, ST055793; 3, ST090280; 4, ST079160; and 5, adenosine (reference); 6, ST059588; and 8, ST082975), and (d) EICs on individual analytes in SIM negative mode (1, ST056929; 2, ST066868; 3, ST069077; 4, ST059588; and 5, ST066881). Spikes are artifacts of the MS detector. Some fragments were detectable both in SIM-positive and SIM-negative mode
Fig. 2.

Distribution of dissociation constants (KD) for fragments as screened by WAC
In Fig. 3, some of the best hits from screening of the 170 fragments towards GAK are presented. Fragment ST014664 and ST034758 were the highest ranked hits with adjusted retention times of 117 and 108 min on the GAK column and 1.14 and 7.39 min on the ethanolamine column, respectively. Fragment ST059935, which is structurally similar to ST014664 and ST034758 had an adjusted retention time of 47 min on the GAK column and consequently a very high ligand efficiency (0.60). Fragments ST057236 and ST057640 each contain a chiral center, resulting in the presence of two enantiomers of each compound which exhibited different affinities to GAK, as shown by double peaks in the chromatograms (see discussion below) (Fig. 4). These two fragments hold together with fragment ST048901 a structural resemblance to the favonoid Quercetin, which caused an increase of melting temperature (ΔTm) of GAK of 9.4 °C during a thermal shift assay performed as described in [32]. Fragment ST088036, which constitutes a part of adenosine, was also identified as one of the highest ranked fragments and the measured affinity was interestingly higher than that of adenosine to GAK (Fig. 3).
Fig. 3.

Quercetin, reference compound adenosine, and examples of fragment hits from screening against GAK
Fig. 4.

Structure of fragment ST057236 and EIC from analysis on the GAK column (solid line) and on the reference column (dotted line). The chiral center of fragment ST057236 is indicated by a black circle. Detection was performed in SIM negative mode of the [M−1]− ion m/z 271. The KD values of the enantiomeres were estimated to 2.5 and 4.9 μM. Spikes are artifacts of the MS detector
The use of MS detection enables analysis of mixtures by WAC, increasing the throughput considerably. However, screening of a selected library such as the one used in this study requires relatively long elution times; in these studies each sample was eluted for 140 min. Thus, analysis of the 170 substances as mixtures of 13 compounds would require 33 h. Throughput could be enhanced by screening of mixtures containing more analytes. In this study, the number of compounds in each mixture was limited due to the fragment concentration in the original library. Using a library at higher concentrations or in another organic solvent than DMSO would enable up to 100 analytes in each mixture and as a result would lead to significantly higher throughput.
Fragment screening by WAC requires very low sample concentrations (<0.1 mM), which distinguishes the method from other methods used in fragment screening, making issues such as analyte solubility, nonspecific binding and aggregation less of a problem. Thus, the sample consumption was only 2 pmol of each analyte (one injection).
Chiral separation
Out of the substances that were selected for screening towards GAK, 11 contained chiral centers. During analysis of two of these substances, double peaks of equal area were detected for the same m/z (Fig. 4). Analysis of these substances as single injections verified that both peaks belong to the same analyte. Meanwhile, analysis of these compounds on the ethanolamine column only resulted in one peak. All these results taken together indicate that the fragment existed in a racemic mixture and that the enantiomers of these two fragments are retarded differently due to different affinities in the micrometer range (see Fig. 3). The occurrence of only one single peak during analysis of the remaining chiral fragments can be explained by several factors: (1) the chiral centers of these fragments are not important for binding; (2) only one enantiomer is present in the sample; or (3) the second enantiomer of the racemic mixture was not detected due to carrying an affinity that was out of the range of detection of this method.
The ability of WAC to perform a chiral separation and hence determine affinity constants for individual enantiomers presents a significant advantage for WAC for fragment screening as compared with other technologies for fragment screening such as SPR.
Mass Spectrometry detection
Some fragments (approximately 15 %) were not detected during screening in this study. Most of the nondetected fragments were flavones. No extensive optimization study of the spray chamber settings was conducted in order to be able to detect all fragments at the low sample concentration that was used and the parameters applied were probably not optimal for all fragments. It cannot be excluded that some of the fragments were nonionizable and consequently unsuitable for MS detection by ESI. Some fragments were detected on the reference column, but not on the GAK column. Some of them had structural similarities with other fragments of rather high affinity, such as the flavones. Therefore, it may be assumed that the affinity of these fragments was above the range of this screening method. Analytes with KD < 1 μM are not suitable for this method as they are not eluted within a reasonable time. In addition, their slow kinetics may lead to nondetectable peaks due to considerable peak broadening. However, highly retarded fragments may be detectable by forced elution by such means as changes in temperature, pH, ionic strength or hydrophobicity of mobile phases. However, such elution methods have to be compatible with the immobilized protein, the support material and MS detection.
Analytes eluted in the void volume were generally more difficult to detect due to signal suppression effects from DMSO and other analytes competing for surface area of the electrospray droplets. To reduce these effects, the amount of DMSO present in the samples had to be kept to a minimum. In this study, the number of fragments in each mixture was limited to 14 in order to keep the concentration of DMSO in each mixture to below 5 %. Using a fragment library where the original samples are provided at higher concentrations would enable preparation of mixtures containing a higher number of analytes and consequently higher throughput, while the lower DMSO concentration in each mixture would limit the influence of DMSO on MS detection. As DMSO is not an ideal organic solvent to be used in combination with MS, a library that is dissolved in another more volatile solvent would be more suitable for this technique and enable higher throughput. Nevertheless we have demonstrated that DMSO, which is commonly used to dissolve compound libraries, can be used with this technique.
The use of reference column
For 23 (16 %) of the 140 fragments that were detected during screening on both columns, the adjusted retention time on the reference column was longer than on the GAK column. These fragments were between 0.03 and 2.90 column volumes longer retarded (0.2–18 min) on the ethanolamine column than on the GAK column. On the GAK column a portion of the column surface is shielded by immobilized protein, but it is difficult to determine to what extent this changed the binding to the naked matrix. Estimation of affinity constants is difficult for these fragments with considerable affinity to the derivatized silica matrix and they are often regarded as nonhits. For screening of such troublesome fragments, alternative reference columns could be used based on other matrices or other surface chemistries.
In theory, the GAK column used in this study could be used to detect millimolar-binding events, as the affinity range is dependent on the amount of immobilized active protein. However, the contribution from nonspecific interactions to the overall retention time on the column could be considerable in the millimolar affinity range [26]. The cut-off for accurate KD determination was therefore set to <200 μM in this study. To determine accurate affinity constants of weaker binding events, a reference column with immobilized, inactivated, target protein would be preferred. This could be achieved either by a covalent inhibitor or a high affinity noncovalent inhibitor present in the mobile phase [26]. This would also enable identification of fragments binding to the target protein outside the active site(s) which could be of potential interest for developing allosteric inhibitors.
Conclusions
This paper presents an alternative method of fragment screening of kinases by introducing affinity LC/MS. One hundred seventy fragments, selected by virtual screening of a 3,200-member fragment library, were subjected to WAC on a capillary column with the immobilized kinase GAK where some highly efficient binders were defined. WAC also enabled resolution of racemic mixtures and determination of affinity constants for individual enantiomers which represents a unique benefit of WAC for fragment screening. Combining MS detection with WAC is highly advantageous in terms of throughput, identification and detection capability which could be further optimized by the use of fragment libraries especially designed for this technology.
Fragment screening by WAC has now been demonstrated on a few targets including proteases and kinases and it has been proven to be labor and time efficient. Furthermore, screening can be performed using standard HPLC equipment. Very little sample is consumed; only nanograms of each analyte are required. Low fragment concentrations in the high μM range are used which minimize problems with sample solubility and aggregation. Another major benefit is that only one run with a fragment is required to give an estimate of its affinity and theoretically of its kinetics. The use of capillary columns limits protein consumption and further miniaturization is possible. Gentle immobilization of a protein in porous gels frequently enhances its stability and as a result affinity columns can be used for months and withstand hundreds of injections of sample mixtures.
We conclude that WAC is a complementary method showing great potential for fragment screening in drug discovery. The method facilitates detection of weak binding events in a broad affinity range and could be used as a primary screening tool where ranking of binders in terms of affinity constants can be obtained by a single sample injection. Comparison and correlation of the results to data from other existing fragment screening techniques such as NMR and SPR will be the subject of future investigations.
References
- 1.Sun C, Petros AM, Hajduk PJ. Fragment-based lead discovery: challenges and opportunities. J Comput Aided Mol Des. 2011;25:607–610. doi: 10.1007/s10822-011-9451-z. [DOI] [PubMed] [Google Scholar]
- 2.Murray CW, Rees DC. The rise of fragment-based drug discovery. Nat Chem. 2009;1:187–192. doi: 10.1038/nchem.217. [DOI] [PubMed] [Google Scholar]
- 3.Kuo LC. Fragment-based drug design: tools, practical approaches and examples. San Diego: Elsevier; 2011. [DOI] [PubMed] [Google Scholar]
- 4.Bembenek SD, Tounge BA, Reynolds CH. Ligand efficiency and fragment-based drug discovery. Drug Discov Today. 2009;14:278–283. doi: 10.1016/j.drudis.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Coyne AG, Scott DE, Abell C. Drugging challenging targets using fragment-based approaches. Curr Opin Chem Biol. 2010;14:299–307. doi: 10.1016/j.cbpa.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 6.Jencks WP. On the attribution and additivity of binding energies. Proc Natl Acad Sci USA. 1981;78:4046–4050. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 8.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giamas G, Man YL, Hirner H, Bischof J, Kramer K, Khan K, et al. Kinases as targets in the treatment of solid tumors. Cell Signal. 2010;22:984–1002. doi: 10.1016/j.cellsig.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 10.Catapano LA, Manji HK. Kinases as drug targets in the treatment of bipolar disorder. Drug Discov Today. 2008;13:295–302. doi: 10.1016/j.drudis.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 11.Akritopoulou-Zanze I, Hajduk PJ. Kinase-targeted libraries: the design and synthesis of novel, potent, and selective kinase inhibitors. Drug Discov Today. 2009;14:291–297. doi: 10.1016/j.drudis.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Xiang Y, Hirth B, Asmussen G, Biemann H-P, Bishop KA, Good A, et al. The discovery of novel benzofuran-2-carboxylic acids as potent Pim-1 inhibitors. Bioorg Med Chem Lett. 2011;21:3050–3056. doi: 10.1016/j.bmcl.2011.03.030. [DOI] [PubMed] [Google Scholar]
- 13.Hughes SJ, Millan DS, Kilty IC, Lewthwaite RA, Mathias JP, O’Reilly MA, et al. Fragment based discovery of a novel and selective PI3 kinase inhibitor. Bioorg Med Chem Lett. 2011;21:6586–6590. doi: 10.1016/j.bmcl.2011.07.117. [DOI] [PubMed] [Google Scholar]
- 14.Erlanson DA, Arndt JW, Cancilla MT, Cao K, Elling RA, English N, et al. Discovery of a potent and highly selective PDK1 inhibitor via fragment-based drug discovery. Bioorg Med Chem Lett. 2011;21:3078–3083. doi: 10.1016/j.bmcl.2011.03.032. [DOI] [PubMed] [Google Scholar]
- 15.Frantz S. Drug discovery: playing dirty. Nature. 2005;437:942–943. doi: 10.1038/437942a. [DOI] [PubMed] [Google Scholar]
- 16.Ghoreschi K, Laurence A, O’Shea JJ. Selectivity and therapeutic inhibition of kinases: to be or not to be? Nat Immunol. 2009;10:356–360. doi: 10.1038/ni.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hesterkamp T, Whittaker M. Fragment-based activity space: smaller is better. Curr Opin Chem Biol. 2008;12:260–268. doi: 10.1016/j.cbpa.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Hubbard RE, Murray JB. Experiences in fragment-based lead discovery. Methods Enzymol. 2011;493:509–531. doi: 10.1016/B978-0-12-381274-2.00020-0. [DOI] [PubMed] [Google Scholar]
- 19.Hoffer L, Renaud J-P, Horvath D. Fragment-based drug design: computational and experimental state of the art. Comb Chem High Throughput Screen. 2011;14:500–520. doi: 10.2174/138620711795767884. [DOI] [PubMed] [Google Scholar]
- 20.Congreve M, Carr R, Murray C, Jhoti H. A “Rule of Three” for fragment-based lead. Drug Discov Today. 2003;8:876–877. doi: 10.1016/S1359-6446(03)02831-9. [DOI] [PubMed] [Google Scholar]
- 21.Larsson A, Jansson A, Aberg A, Nordlund P. Efficiency of hit generation and structural characterization in fragment-based ligand discovery. Curr Opin Chem Biol. 2011;15:482–488. doi: 10.1016/j.cbpa.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 22.Zopf D, Ohlson S. Weak-affinity chromatography. Nature. 1990;346:87–88. doi: 10.1038/346087a0. [DOI] [Google Scholar]
- 23.Bergström M, Liu S, Kiick KL, Ohlson S. Cholera toxin inhibitors studied with high-performance liquid affinity chromatography: a robust method to evaluate receptor–ligand interactions. Chem Biol Drug Des. 2009;73:132–141. doi: 10.1111/j.1747-0285.2008.00758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engström HA, Johansson R, Koch-Schmidt P, Gregorius K, Ohlson S, Bergström M. Evaluation of a glucose sensing antibody using weak affinity chromatography. Biomed Chromatogr. 2008;22:272–277. doi: 10.1002/bmc.924. [DOI] [PubMed] [Google Scholar]
- 25.Haginaka J. Protein-based chiral stationary phases for high-performance liquid chromatography enantioseparations. J Chromatogr A. 2001;906:253–273. doi: 10.1016/S0021-9673(00)00504-5. [DOI] [PubMed] [Google Scholar]
- 26.Duong-Thi M-D, Meiby E, Bergström M, Fex T, Isaksson R, Ohlson S. Weak affinity chromatography as a new approach for fragment screening in drug discovery. Anal Biochem. 2011;414:138–146. doi: 10.1016/j.ab.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 27.Dumitriu A, Pacheco CD, Wilk JB, Strathearn KE, Latourelle JC, Goldwurm S, et al. Cyclin-G-associated kinase modifies α-synuclein expression levels and toxicity in Parkinson’s disease: results from the GenePD Study. Hum Mol Genet. 2011;20:1478–1487. doi: 10.1093/hmg/ddr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rhodes SL, Sinsheimer JS, Bordelon Y, Bronstein JM, Ritz B. Replication of GWAS associations for GAK and MAPT in Parkinson’s disease. Ann Hum Genet. 2011;75:195–200. doi: 10.1111/j.1469-1809.2010.00616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Savitsky P, Bray J, Cooper CDO, Marsden BD, Mahajan P, Burgess-Brown NA, et al. High-throughput production of human proteins for crystallization: the SGC experience. J Struct Biol. 2010;172:3–13. doi: 10.1016/j.jsb.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strandh M, Andersson HS, Ohlson S. Weak affinity chromatography. Methods Mol Biol. 2000;147:7–23. doi: 10.1007/978-1-60327-261-2_2. [DOI] [PubMed] [Google Scholar]
- 31.Kasai K, Oda Y, Nishikata M, Ishii S. Frontal affinity-chromatography—theory for its application to studies on specific interactions of biomolecules. J Chromatogr. 1986;376:33–47. doi: 10.1016/S0378-4347(00)80822-1. [DOI] [PubMed] [Google Scholar]
- 32.Fedorov O, Marsden B, Pogacic V, Rellos P, Müller S, Bullock AN, et al. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc Natl Acad Sci USA. 2007;104:20523–20528. doi: 10.1073/pnas.0708800104. [DOI] [PMC free article] [PubMed] [Google Scholar]