

Abstract
Astrocytes contribute to many neuronal functions, including synaptogenesis, but their role in the development of synaptic plasticity remains unclear. Presynaptic muting of hippocampal glutamatergic terminals defends against excitotoxicity. Here we studied the role of astrocytes in the development of presynaptic muting at glutamatergic synapses in rat hippocampal neurons. We found that astrocytes were critical for the development of depolarization-dependent and Gi/o-dependent presynaptic muting. The ability of cAMP analogues to modulate presynaptic function was also impaired by astrocyte deficiency. Although astrocyte deprivation resulted in postsynaptic glutamate receptor deficits, this effect appeared independent of astrocytes' role in presynaptic muting. Muting was restored with chronic, but not acute, treatment with astrocyte-conditioned medium, indicating that a soluble factor is permissive for muting. Astrocyte-derived thrombospondins (TSPs) are likely responsible because TSP1 mimicked the effect of conditioned medium, and gabapentin, a high-affinity antagonist of TSP binding to the α2δ-1 calcium channel subunit, mimicked astrocyte deprivation. We found evidence that protein kinase A activity is abnormal in astrocyte-deprived neurons but restored by TSP1, so protein kinase A dysfunction may provide a mechanism by which muting is disrupted during astrocyte deficiency. In summary our results suggest an important role for astrocyte-derived TSPs, acting through α2δ-1, in maturation of a potentially important form of presynaptic plasticity.
Introduction
Correct development and plasticity of glutamate synapses is critical to brain function. Imbalances between excitatory and inhibitory neurotransmitter actions are postulated to underlie brain disturbances as diverse as schizophrenia (Lewis and Moghaddam, 2006), developmental disability (Rubenstein and Merzenich, 2003; Wetmore and Garner, 2010), and epilepsy (McCormick and Contreras, 2001). Diverse cues regulate synaptic development; neuron-to-neuron signaling is clearly important for synaptogenesis (Craig et al., 2006), but astrocytic factors are also important (Eroglu and Barres, 2010; Pfrieger, 2010). Still unclear is the breadth of astrocyte involvement in synapse development and the signaling systems affected. Astrocytic cues likely guide postsynaptic function, presynaptic function, synaptic plasticity, and combinations of these elements.
Our group and others have studied a form of adaptive presynaptic plasticity termed muting whereby the number of terminals releasing transmitter is reduced. Mute presynaptic terminals are found in a variety of preparations (Tong et al., 1996; Bolshakov et al., 1997; Kannenberg et al., 1999; Ma et al., 1999; Kim et al., 2003; Losonczy et al., 2004; Moulder et al., 2004; Cousin and Evans, 2011; Crawford and Mennerick, 2012). Reversible muting of hippocampal glutamatergic synapses occurs in cultured autaptic neurons, conventional mass cultures, and acute hippocampal slices (Moulder et al., 2004; Crawford et al., 2011). Muting is induced by electrical stimulation, depolarization challenges, prolonged Gi/o G-protein activation, and hypoxia (Moulder et al., 2004, 2006, 2008; Crawford et al., 2011; Hogins et al., 2011; Crawford and Mennerick, 2012). Muting may play a role in defending against excitotoxic glutamate release (Hogins et al., 2011), but it is also engaged by physiological changes in activity (Moulder et al., 2006). Despite the wide variety of preparations and conditions under which muting occurs (Crawford and Mennerick, 2012), factors controlling development of muting have not been explored.
Muting occurs in single-neuron networks in culture, but because these neurons contact astrocytes it is unclear whether muting is cell autonomous or whether astrocyte signals modulate or induce muting. Here we explored muting mechanisms in a reduced environment where astrocyte numbers could be controlled. We found that global, permissive astrocytic signaling rather than local, instructive astrocytic signaling is important for the development of muting. Astrocyte deprivation prevented muting in response to both depolarization and G-protein-coupled receptor stimulation. Chronic, but not acute, treatment with astrocyte-conditioned medium rescued muting. Thrombospondins (TSPs), astrocyte-derived glycoproteins previously implicated in glutamate synapse development (Asch et al., 1986; Christopherson et al., 2005; Eroglu et al., 2009; Xu et al., 2010), also rescued muting. Gabapentin, an antagonist of the TSP receptor α2δ-1 (Gee et al., 1996; Eroglu et al., 2009), mimicked the effects of astrocyte deprivation by preventing muting competence. Gabapentin additionally prevented adaptive network plasticity. Although the downstream pathways initiated by TSP binding to α2δ-1 remain unclear, astrocyte deprivation led to altered protein kinase A (PKA) activity, as suggested by abnormal responses to cAMP analogues and hyperphosphorylation of PKA substrates. TSP treatment corrected these abnormalities. Our results suggest that astrocyte-derived TSPs are important developmental modulators of presynaptic plasticity.
Materials and Methods
Primary hippocampal cultures.
All experiments were performed in accordance with National Institutes of Health guidelines and were approved by the Washington University Animal Studies Committee. For microcultures, culture dishes (35 mm) were first covered with a thin layer of 0.15% agarose and then stamped with 150- to 200-μm-diameter microdots of 0.5 mg/ml collagen using a polydimethylsiloxane microstamp as previously described (Moulder et al., 2007). Cortical astrocytes from postnatal d 4 male and female rat pups were seeded on these islands in Eagle's medium (Life Technologies) supplemented with 5% heat-inactivated horse serum, 5% fetal bovine serum, 17 mm d-glucose, 400 μm glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. Glial preplates were maintained in a humidified and atmospherically controlled incubator (5% CO2/95% air at 37°C) before neuronal plating. Astrocyte-poor cultures were prepared 7–10 d after glial plating by fixation with 4% paraformaldehyde/0.2% glutaraldehyde in PBS at room temperature (RT) for 5 min before at least 3 washes in PBS. Alternatively, 70% ethanol at −20°C was applied for 30 min before PBS rinses. Although most experiments used aldehyde fixation, both astrocyte fixation protocols produced the finding shown in Figure 1B. For mass cultures used in Figure 5 (see below), a confluent layer of polylysine/laminin was applied to the culture dish before neuronal plating.
Figure 1.

Astrocyte deprivation impairs presynaptic muting but not G-protein activation. A, Phase-contrast and fluorescence images of autaptic neurons on viable (control) or nonviable (4% paraformaldehyde/0.2% glutaraldehyde-fixed) glial “islands” acquired 30 min after 5 μg/ml calcein-AM treatment. Fewer islands contained live (calcein-positive) astrocytes in fixed cultures (control: 98.8 ± 0.9% live islands; fixed: 17.6 ± 2.8% live islands; n = 6 dishes; p = 7.9 × 10−11, Student's unpaired t test). Scale bar, 40 μm. B, Representative autaptic EPSCs from astrocyte-rich or astrocyte-poor microcultures recorded in normal saline up to 1 h after 4 h control treatment (30 mm NaCl) or after 4 h depolarization (30 mm KCl). C, Summary of peak EPSC amplitudes from DIV 10–14 astrocyte-rich (+) or astrocyte-poor (−) autaptic neurons after indicated treatment times with 30 mm KCl (black bars), 10 nm CCPA (A1 adenosine receptor agonist) plus 50 μm baclofen (GABAB receptor agonist) (C+B; yellow bars), or 50 μm Rp-cAMPS (Rp; cyan bars). EPSCs were normalized to the average EPSCs from control-treated sibling neurons for each recording day (n = 14–22 neurons; *p < 0.05 vs treatment controls, Student's unpaired t test). D, EPSCs from astrocyte-rich or astrocyte-poor cultures after acute perfusion with recording saline (control) or 20 μm baclofen. E, EPSCs from astrocyte-rich or astrocyte-poor cultures after acute perfusion with recording saline (control) or 10 nm CCPA. The same neurons were used for experiments in D and E. D, E, Summary data are given in the Results.
Figure 5.

Gabapentin prevents the development of muting competence. A, Representative DIV 10–12 autaptic EPSCs after 4 h control treatment (30 mm NaCl) or depolarized (30 mm KCl). Astrocyte-rich cultures were treated with 32 μm gabapentin at DIV 7 (1× GBP). B, Representative DIV 10–12 autaptic EPSCs after 4 h control or depolarized. Astrocyte-rich cultures were treated with 32 μm gabapentin at DIV 7 followed by half-media switches of fresh 32 μm gabapentin in astrocyte-conditioned medium every day (3–5× GBP). Summary data for A and B are given in the Results. C, Spontaneous activity in DIV 11–13 mass cultures after 4 h control treatment (30 mm NaCl) or depolarized (30 mm KCl). Cultures were treated at DIV 7 with or without 32 μm gabapentin followed by daily half media switches with astrocyte-conditioned medium (CM) or CM plus 32 μm gabapentin (3–5× GBP). EPSCs and IPSCs were measured simultaneously at −35 mV. D, Summary of total spontaneous synaptic activity (EPSC + IPSC points) in DIV 11–13 mass cultures, normalized to control (CM, NaCl) for each recording day (n = 23 neurons; *p < 0.05, Bonferroni corrected).
Hippocampal neurons were dispersed on fixed or live astrocyte beds for microcultures while astrocytes and neurons were coplated for mass cultures as previously described (Mennerick et al., 1995). Briefly, male and female postnatal d 0–3 Sprague Dawley rat hippocampi were dissected, incubated with 1 mg/ml papain, and mechanically dissociated. To encourage the formation of autaptic (solitary) neurons in microcultures, cells were plated at low density (∼100 cells/mm2). For mass cultures, cells were plated at high density (∼650 cells/mm2) to encourage neuron–astrocyte cocultures to form. After 3–4 days in vitro (DIV), 6.7 μm cytosine arabinoside was added to inhibit cell division. One half of the culture medium was exchanged with Neurobasal medium (Life Technologies) plus B27 supplement at DIV 4–5. Conditioned media were from astrocyte cultures or from neuron–astrocyte cocultures; similar results were obtained with both. Non-conditioned medium was fresh Neurobasal without supplements. Unless otherwise stated, all experiments were performed at 10–14 DIV, and controls were sibling cultures used the same day.
Electrophysiology.
Whole-cell voltage-clamp recordings were performed on autaptic neurons in micro-island culture unless otherwise stated. Data were collected using pClamp 9 software with a Multiclamp 700B or Axopatch 200B amplifier and Digidata1322A data acquisition board (Molecular Devices). Recording saline typically contained the following (in mm): 138 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 10 HEPES, and 25 μm d-aminophosphonovalerate (d-APV; Tocris Bioscience) at a pH of 7.25. For current-clamp experiments, recording saline contained 50 μm d-APV and 1 μm NBQX. For experiments requiring NMDA receptor activation, 10 μm glycine (Tocris Bioscience) was added to the saline solution and MgCl2 and D-APV were excluded. Pipette solution contained the following (in mm): 140 potassium gluconate, 4 NaCl, 0.5 CaCl2, 5 EGTA, and 10 HEPES at a pH of 7.25. For experiments in which both EPSCs and IPSCs were collected during the same recording session from autaptic neurons, potassium gluconate was replaced with 140 mm KCl. For network activity experiments that measured spontaneous EPSCs and IPSCs in mass cultures, 130 mm cesium methanesulfonate replaced potassium gluconate.
Recordings were performed at room temperature. Electrode pipettes were pulled from borosilicate glass (World Precision Instruments) and typically had 3–6 MΩ resistance, except for current-clamp experiments in which resistances were 4–7 MΩ. Access resistance was compensated 85–100% for autaptic EPSC and IPSC recordings, and membrane potential was typically held at −70 mV. For network activity experiments in mass cultures, neurons were voltage-clamped at −30 mV or −35 mV, midway between IPSC and EPSC reversal potentials, to measure spontaneous network activity due to excitation and inhibition simultaneously. Autaptic EPSCs and IPSCs were evoked by a 1.5 ms pulse depolarization to 0 mV. Paired-pulse responses were obtained by evoking two such depolarizing pulses 50 ms apart. Signals were sampled at 10 kHz and low-pass filtered at 4 kHz, except for miniature EPSCs collected at 5 kHz and filtered at 1 kHz and spontaneous EPSCs and IPSCs collected at 5 kHz and filtered at 2 kHz. Autaptic PSCs were recorded up to 1 h after switch to recording saline. When solutions were applied acutely to neurons, a multibarrel perfusion system was used with a common port placed within 0.5 mm of the neuron; solution exchange times were ∼100 ms. Wherever possible, acute effects of drugs were evaluated by interleaving control and drug treatments and averaging 2–5 responses per condition.
Glutamate receptor blockers (50 μm d-APV and 1 μm NBQX) were present during all depolarization challenges (30 mm KCl for depolarization or NaCl as a nondepolarized osmotic control) to prevent toxicity and NMDA receptor-dependent plasticity. Control treatments for all other experiments consisted of vehicle treatment unless otherwise specified.
Immunostaining and microscopy.
FM1-43 dye labeling was performed as previously described (Moulder et al., 2010). Briefly, microcultures were treated for 2 min with 10 μm fixable FM1-43 (FM1-43FX; Life Technologies) in recording saline supplemented with 45 mm KCl and 1 μm NBQX. Immediately following, the culture dishes were briefly washed (5 s) in saline supplemented with 500 μm Advasep-7 (CyDex Pharmaceuticals) and 1 μm NBQX. Cells rested during 10 min of washes with saline plus 1 μm NBQX and were then fixed for 10 min.
Fixation before immunostaining was 4% paraformaldehyde at RT, 4% paraformaldehyde/0.2% glutaraldehyde at RT, 4% paraformaldehyde/0.02% glutaraldehyde at RT, or 100% methanol at −20°C for 10 min. All culture dishes were washed with PBS at RT followed by 2–4% normal goat serum in PBS blocking solution plus 0% (for surface protein levels), 0.1%, or 0.04% Triton X-100. Primary antibodies were vesicular glutamate transporter 1 (vGluT-1; 1:2000; Millipore), GluA2/GluR2 (1:500, Millipore), microtubule-associated protein 2 (MAP2; 1:2000; Millipore), phospho-synapsin (1:400; Millipore), synapsin (1:2000; Millipore), phospho-cAMP response element binding protein (phospho-CREB; 1:400; Millipore), GABA (1:500), and phospho-dynamin 1 (1:100; Santa Cruz Biotechnology). Primary antibodies were applied for 2–3 h at RT before PBS wash, 30–40 min secondary antibody incubation with Alexa Fluor conjugates (Life Technologies), and glass coverslipping with Fluoromount G (Southern Biotechnology Associates).
FM1-43FX and immunostaining images were acquired on an inverted Eclipse TE2000-S microscope with a 60× objective (1.4 numerical aperture) using a C1 scanning confocal laser (488, 543, and/or 633 nm) and EZ-C1 software (Nikon). Alternating laser lines were used to obtain z-stack images while all gain and acquisition settings were held constant within a given experiment. Two-dimensional projected images were created and analyzed using MetaMorph 7 software (Universal Imaging).
Live cell images were obtained on an Eclipse TE2000-S inverted microscope (Nikon) with a 40× objective (0.6 numerical aperture; Nikon) and a metal halide lamp. Images were acquired with MetaMorph 7 software and a cooled 12-bit CCD camera (Photometrics).
Data analysis.
Data were analyzed and graphed using MetaMorph 7, Clampfit 9 (Molecular Devices), Mini Analysis 6 (Synaptosoft Inc.), Excel 2007 (Microsoft), and/or SigmaPlot 10 (Systat) software. Unless otherwise stated, data are displayed as mean ± SEM, and Student's unpaired t test was used to compare 2 groups while a Bonferroni correction was applied when experiments contained >2 groups. A corrected p value of < 0.05 was required to reach significance. The reported n refers to the sample size of each group within an experiment.
At least 3 PSC amplitudes were averaged for each autaptic neuron. For each recording day, PSC amplitudes from experimental treatments were normalized to the average amplitude in the control-treated sibling culture. For dual-component autaptic EPSCs, peak EPSC amplitude estimated the AMPA receptor component while the average current 80–100 ms following the peak estimated the NMDA receptor component (Hestrin et al., 1990). Miniature EPSCs were elicited by perfusion of 100 mm sucrose and analyzed using Mini Analysis software with manual confirmation. The first 57 miniature EPSC interevent interval and amplitude values were used for Kolmogorov–Smirnov tests while values for all miniature EPSCs from a given neuron were averaged for Mann–Whitney U tests.
At least 10 fields per culture dish were used to calculate the percentage of live (calcein-positive) islands. For the percentage of FM1-43-positive vGluT-1 synapses, regions of interest were manually drawn around 10 vGluT-1 puncta per field for 5 fields per culture dish. As previously described (Crawford et al., 2011), FM1-43 images were thresholded before analyzing vGluT-1-defined regions, and synapses were considered active if at least 10 pixels reached threshold. Similarly, 10 vGluT-1-defined regions were manually drawn per field before analyzing synapsin and phospho-synapsin images. The number and intensity of vGluT-1-positive synapses in autaptic neurons was quantified via automatic detection in thresholded images using MetaMorph 7 software. Most immunostaining experiments used glutamatergic autaptic neurons, identified by vGluT-1 immunoreactivity. Glutamatergic neurons on multicell islands were analyzed for nuclear phospho-CREB experiments, however, and antibody incompatibility precluded double labeling in phospho-dynamin 1 experiments. For display in figures, all images within a panel were pseudocolored and adjusted for brightness and contrast equivalently.
Materials.
Rp-cAMPS and 2-chloro-N6-cyclopentyladenosine (CCPA) were obtained from Tocris Bioscience, kainic acid was purchased from BioVectra, calcein-AM was purchased from AnaSpec, human recombinant thrombospondin 1 (TSP1) was obtained from Hematologic Technologies, and tumor necrosis factor α (TNFα) was obtained from EMD Millipore. All other materials were obtained from Sigma-Aldrich unless otherwise specified.
Results
Failure to mute in glutamatergic neurons from astrocyte-poor cultures
We manipulated glial signaling in rat hippocampal cultures by growing neurons on fixed or live astrocyte beds (Fig. 1A). This strategy, similar to prior studies using fixation to kill one cell type before coculturing with other cell types (Amur-Umarjee et al., 1993; Solà et al., 2002; Alexanian, 2005), permitted identical culture conditions for astrocyte-rich and astrocyte-poor cultures except for the number of viable astrocytes. It also allowed control neurons the full benefit of local and global astrocytic cues, unlike preparations where control neurons only receive soluble astrocytic signals (Kaech and Banker, 2006). Electrophysiological measures of input resistance (astrocyte-rich: 189.3 ± 39.3 MΩ; astrocyte-poor: 212.4 ± 76.3 MΩ; n = 11–12 neurons; p = 0.77, Student's unpaired t test), cell capacitance (astrocyte-rich: 73.3 ± 7.3 pF; astrocyte-poor: 94.0 ± 10.3 pF; n = 14–16 neurons; p = 0.11, Student's unpaired t test), and resting membrane potential (astrocyte-rich: −58.8 ± 1.7 mV; astrocyte-poor: −61.9 ± 2.6 mV; n = 10 neurons; p = 0.32, Student's unpaired t test) were similar in glutamatergic neurons from astrocyte-rich and astrocyte-poor cultures, suggesting no differences in membrane surface area or expression of background leak channels. We also detected no qualitative differences in morphological complexity between neurons grown in these two culture conditions, so gross differences in neuronal development were not apparent.
To test whether astrocytes are important for muting of glutamatergic synapses, we attempted to induce muting in neurons from astrocyte-rich and astrocyte-poor cultures. EPSCs from DIV 10–14 autaptic neurons in astrocyte-rich microcultures were persistently depressed with as little as 60 min depolarization, as assessed up to 1 h following the removal of depolarization (Fig. 1B,C), while 30 min of depolarization yielded insignificant muting (p = 0.45). We have shown previously that this depolarization-induced depression is selective for glutamate synapses and arises from muting in the absence of synapse loss or postsynaptic receptor changes (Moulder et al., 2004, 2006). By contrast, neurons in astrocyte-poor cultures exhibited no EPSC depression with up to 4 h depolarization, which produced strong muting in astrocyte-rich cultures (Fig. 1B,C). Neurons from astrocyte-poor cultures perfused for 10 s with 30 mm KCl in recording saline depolarized to the same steady-state membrane potential as those from astrocyte-rich cultures (astrocyte-rich: −17.3 ± 2.5 mV; astrocyte-poor: −17.4 ± 2.0 mV; n = 10 neurons; p = 0.97, Student's unpaired t test), suggesting that the induction protocol depolarizes neurons from both conditions similarly. Muting, therefore, is impaired in astrocyte-poor cultures.
To evaluate an alternative method of muting induction, we incubated cells in Gi/o-coupled receptor agonists CCPA (an A1 adenosine receptor agonist) and baclofen (a GABAB receptor agonist) or in PKA antagonist Rp-cAMPS. Both treatments induce presynaptic muting (Moulder et al., 2008; Crawford et al., 2011). These agents depressed EPSCs in astrocyte-rich cultures, as expected, but significantly increased EPSC amplitudes in astrocyte-poor cultures (Fig. 1C). This unexpected reversal of EPSC depression indicated an abnormality in muting induction. Meanwhile, canonical acute presynaptic depression, mediated by Gβγ subunit signaling (Brown and Sihra, 2008), did not differ in astrocyte-poor cultures; autaptic EPSCs were acutely depressed by both the GABAB receptor agonist baclofen (astrocyte-rich: 74.9 ± 4.5%; astrocyte-poor: 63.9 ± 5.5%; n = 11; p = 0.14, Student's paired t test; Fig. 1D) and the A1 adenosine receptor agonist CCPA (astrocyte-rich: 61.0 ± 6.2%; astrocyte-poor: 46.6 ± 4.6%; n = 11; p = 0.08, Student's paired t test; Fig. 1E) similarly in astrocyte-rich and astrocyte-poor cultures. The muting deficit in astrocyte-deprived neurons, therefore, is likely downstream of depolarization and Gαi/o subunit-mediated cAMP signaling.
Normal basal presynaptic function in astrocyte-poor cultures
The basal EPSC amplitude was smaller in astrocyte-deprived cultures than in astrocyte-rich cultures (astrocyte-rich: −18.3 ± 5.7 nA; astrocyte-poor: −5.0 ± 1.4 nA; n = 15–16; p = 0.037, Student's unpaired t test); however, IPSCs were similar (astrocyte-rich: −7.6 ± 2.5 nA; astrocyte-poor: −8.7 ± 1.9 nA; n = 14 neurons; p = 0.74, Student's unpaired t test), suggesting that the basal deficit in amplitude in astrocyte-poor cultures localized specifically to glutamatergic synapses. The decreased EPSC amplitude was not explained by differences in local astrocytic signaling because neurons in astrocyte-deprived cultures on islands with some of the few contaminating live astrocytes did not have larger EPSCs than neurons associated with nonviable islands, as assessed by a vital stain (viable islands: −2.0 ± 0.5 nA; nonviable islands: −4.8 ± 1.5 nA; n = 13–16 neurons; p = 0.13, Student's unpaired t test; Fig. 2A). A global deficit in astrocyte-poor cultures, therefore, likely explains the basal EPSC depression.
Figure 2.
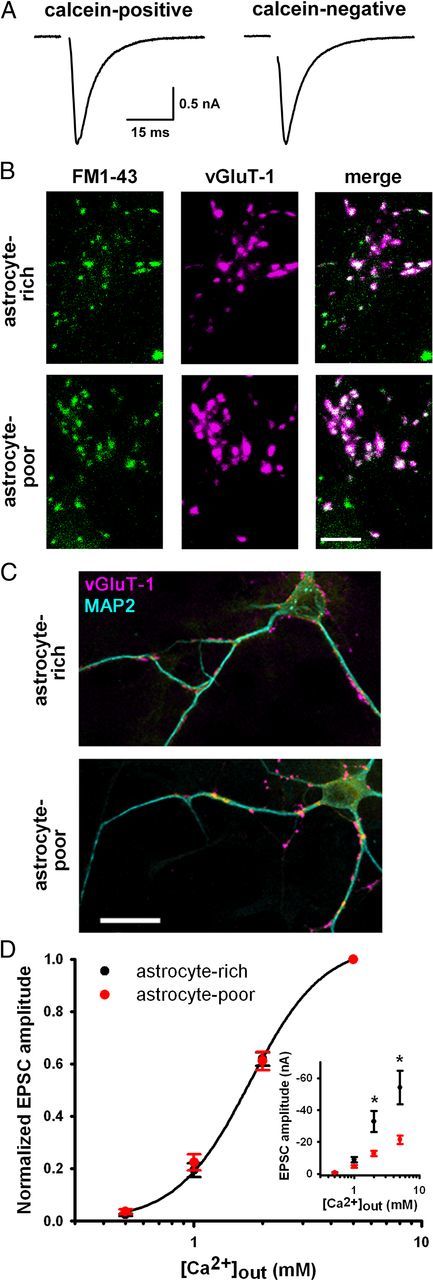
Astrocyte deprivation does not impair basal presynaptic function. A, EPSCs from autaptic neurons in astrocyte-poor cultures from islands with residual live astrocytes (calcein-positive) or no live astrocytes (calcein-negative). B, Images of 10 μm FM1-43FX labeling of presynaptic terminals (green) after 2 min 45 mm KCl and subsequent vGluT-1 immunostaining (magenta) in autaptic neurons from astrocyte-rich and astrocyte-poor cultures (30 min, −20°C, 70% EtOH-fixed astrocytes). Scale bar, 5 μm. C, vGluT-1 (magenta) and MAP2 (cyan) immunostaining in autaptic neurons showing similar density and intensity of presynaptic terminals. Scale bar, 20 μm. Summary results for A–C are given in the Results. D, Calcium concentration–response curves of autaptic EPSC amplitudes from astrocyte-rich or astrocyte-poor cultures normalized for each neuron to values obtained in 5 mm external calcium (n = 21–22). Data from astrocyte-rich cultures were fitted to the Hill equation (EC50 value: 1.8 mm; Hill coefficient: 2.7). Inset, Non-normalized values from the same dataset demonstrating basal depression of EPSCs in astrocyte-poor cultures at higher calcium concentrations. *p < 0.05, Student's unpaired t test, Bonferroni corrected.
Decreased functionally active presynaptic terminals did not explain the basal EPSC depression or the deficient stimulus-induced muting in astrocyte-poor cultures. The percentage of basally active terminals, as measured via stimulus-induced FM1-43FX dye labeling of vesicles at vGluT-1-immunoreactive synapses, was similar in astrocyte-rich and astrocyte-poor cultures (astrocyte-rich: 48.0 ± 4.1%; astrocyte-poor: 54.5 ± 7.0%; n = 8 dishes; p = 0.44, Student's unpaired t test; Fig. 2B). Furthermore, neither the number (astrocyte-rich: 188.3 ± 35.7 puncta; astrocyte-poor: 174.1 ± 21.8 puncta; n = 35 neurons; p = 0.74, Student's unpaired t test) nor the intensity [astrocyte-rich: 410 ± 58 × 1000 arbitrary units (AU); astrocyte-poor: 388 ± 65 × 1000 AU; n = 35 neurons; p = 0.80, Student's unpaired t test] of vGluT-1 puncta per cell differed, suggesting normal synapse and vesicle numbers (Fig. 2C). Paired-pulse EPSC depression was also normal, suggesting similar vesicle release probability (astrocyte-rich: 5.1 ± 9.2%; astrocyte-poor: 18.2 ± 8.6%; n = 15–16; p = 0.3, Student's unpaired t test), and normalized calcium concentration–response relationships were similar in astrocyte-rich and astrocyte-poor cultures (Fig. 2D). EPSCs remained depressed in astrocyte-poor cultures at the highest calcium concentration (5 mm; Fig. 2D), suggesting that deficits did not arise from altered coupling of calcium influx to vesicle fusion. No deficits in presynaptic function, therefore, were detected with reduced astrocytic signaling.
Basal postsynaptic deficits in astrocyte-poor cultures
A postsynaptic deficit, rather, appeared to explain the basal EPSC depression in astrocyte-poor cultures. AMPA receptor and NMDA receptor components of EPSCs were reduced in parallel (astrocyte-rich: 0.34 ± 0.06 NMDA/AMPA receptor EPSC ratio; astrocyte-poor: 0.47 ± 0.12 NMDA/AMPA receptor EPSC ratio; n = 14–15 neurons; p = 0.34, Student's unpaired t test; Fig. 3A), and this parallel depression was associated with decreased responsiveness to exogenously applied AMPA receptor agonist kainic acid (20 μm; astrocyte-rich: −12.2 ± 1.7 pA/pF; astrocyte-poor: −5.9 ± 1.0 pA/pF; n = 14–15 neurons; p = 0.004, Student's unpaired t test; Fig. 3B) and NMDA receptor agonist NMDA (10 μm; astrocyte-rich: −12.9 ± 1.7 pA/pF; astrocyte-poor: −7.3 ± 0.8 pA/pF; n = 14–15 neurons; p = 0.003, Student's unpaired t test; Fig. 3B). The preserved ratio of NMDA to AMPA receptor responses to exogenous agonists (astrocyte-rich: 1.26 ± 0.36; astrocyte-poor: 1.93 ± 0.41; n = 14–15 neurons; p = 0.16, Student's unpaired t test; Fig. 3B) further implies that NMDA receptors and AMPA receptors were reduced in parallel and that both synaptic and extrasynaptic receptor levels were decreased. Additionally, miniature EPSC (mEPSC) amplitude was reduced (astrocyte-rich: 19.7 ± 2.1 pA; astrocyte-poor: 13.4 ± 1.1 pA; n = 10 neurons; p < 0.01, Mann–Whitney U test; Fig. 3C), further suggesting that postsynaptic receptor levels were decreased in astrocyte-poor cultures. Frequency of mEPSCs also trended toward reduction (astrocyte-rich: 0.39 ± 0.22 s interevent interval; astrocyte-poor: 1.03 ± 0.28 s interevent interval; n = 10 neurons; p > 0.05, Mann–Whitney U test; Fig. 3C), but it was unclear whether this resulted from an undetected presynaptic change or whether the smallest mEPSCs fell below the detection threshold. Immunoreactivity for surface levels of the AMPA receptor subunit GluA2 was also reduced, with fewer vGluT-1-defined synapses colocalizing with GluA2 (astrocyte-rich: 66.7 ± 2.3%; astrocyte-poor: 59.0 ± 2.9%; n = 46–48 fields; p = 0.039, Student's unpaired t test). Together, these results suggest that neurons in astrocyte-poor cultures have fewer functional glutamate receptors.
Figure 3.

Glutamate receptor levels are decreased in astrocyte-poor cultures. A, Representative dual-component (NMDA and AMPA receptor-mediated) EPSCs elicited in autaptic neurons from astrocyte-rich or astrocyte-poor cultures. Note the change in vertical scale bar. B, Top, Currents from autaptic neurons in astrocyte-rich or astrocyte-poor cultures in response to acute kainic acid (KA) perfusion, a nondesensitizing AMPA receptor agonist. Bottom, Currents from autaptic neurons in astrocyte-rich or astrocyte-poor cultures in response to acute NMDA perfusion to selectively activate NMDA receptors. C, Top, representative miniature EPSCs from astrocyte-rich or astrocyte-poor cultures elicited by acute perfusion of 100 mm sucrose. Bottom, Cumulative probability plots of amplitude (p < 0.001 with D = 0.2175) and interevent interval (p < 0.001 with D = 0.3860) of miniature EPSCs (n = 10; Kolmogorov–Smirnov test). Additional summary data for A–C are given in the Results.
To ask whether the decreased basal EPSC amplitude contributed to the muting deficit in astrocyte-poor cultures, we attempted to induce muting in young (DIV 7–8), astrocyte-rich cultures. Despite smaller basal EPSC amplitudes similar to those found in DIV 10–14 astrocyte-poor cultures (−3.75 ± 0.95 nA; n = 17), these younger neurons in astrocyte-rich cultures expressed muting in response to a 4 h depolarization challenge (Fig. 4A,E). Conversely, older (DIV 21), astrocyte-poor cultures with larger basal EPSC amplitudes (10.27 ± 4.02 nA; n = 12) did not exhibit muting (Fig. 4E). Together with prior published studies suggesting that postsynaptic glutamate receptor function is not required for muting induction (Moulder et al., 2004, 2006), these data suggest that the postsynaptic changes caused by astrocyte deficiency did not explain the muting deficits. Because glial modulation of glutamate receptor levels and function has been studied previously (Beattie et al., 2002; Stellwagen and Malenka, 2006; Perea et al., 2009; Sullivan et al., 2011), we did not further characterize the mechanisms responsible for this postsynaptic deficit in astrocyte-poor cultures.
Figure 4.

Soluble, astrocyte-derived thrombospondin rescues presynaptic muting competence. A–D, Representative DIV 7–12 autaptic EPSCs after 4 h control treatment (30 mm NaCl) or depolarization (30 mm KCl). Cultures were astrocyte-rich and challenged at DIV 7 without additional treatments (A), astrocyte-poor, treated with astrocyte-conditioned medium (CM) at DIV 7, and challenged at DIV 10–12 (B), astrocyte-poor and challenged at DIV 10–12 in CM (C), or astrocyte-poor, treated with 5 μg/ml human thrombospondin 1 (TSP) at DIV 7, and challenged at DIV 10–12 (D). E, Summary of EPSC amplitudes after 4 h 30 mm KCl or 50 μm Rp-cAMPS (Rp) with indicated pretreatments and/or cotreatments (n = 12–20 neurons; *p < 0.05 vs treatment controls, Student's unpaired t test). EPSCs were normalized as in Figure 1C. +, Astrocyte-rich cultures; −, astrocyte-poor cultures; GBP, 32 μM gabapentin; TNFα, 1 ng/ml tumor necrosis factor α; FM, fresh, non-conditioned medium.
Thrombospondin as an astrocyte-derived permissive factor for muting
Astrocytes could directly instruct muting during depolarization (e.g., through gliotransmission), or they could permit development of muting competence (e.g., through global cues). In support of the latter, conditioned medium from mature, live-astrocyte cultures added to astrocyte-poor cultures at DIV 7 rescued muting, even when conditioned medium was replaced with fresh medium during the depolarization challenge (Fig. 4B,E). Conditioned medium was ineffective if applied only during the muting induction protocol (Fig. 4C,E). Thus, glial factors are not released instructively during muting induction and are, instead, released permissively during synapse development. Because older (DIV 21), astrocyte-poor cultures failed to express muting (Fig. 4E), it is unlikely that muting competence was delayed in the absence of astrocytes. Rather, it appears that astrocytes provide a fundamental developmental cue that permits muting competence.
We hypothesized that astrocytes release a permissive substance for muting development rather than remove a nonpermissive factor. Astrocyte-derived TSPs foster presynaptic maturation via binding to the neuronal α2δ-1 subunit of voltage-gated calcium channels or to neuroligin 1 (Eroglu et al., 2009; Xu et al., 2010). Synaptogenic effects of TSPs are mediated redundantly by the 5 TSP isoforms (Eroglu et al., 2009), and TSP1 is expressed strongly in hippocampus (Adams and Tucker, 2000). We, therefore, tested effects of adding human TSP1 (5 μg/ml) to astrocyte-poor cultures. Incubation in human TSP1 starting at DIV 7 rescued presynaptic muting competence in astrocyte-poor cultures (Fig. 4D,E). In contrast, TNFα (1 ng/ml), an astrocyte-derived factor involved in postsynaptic plasticity (Beattie et al., 2002; Stellwagen et al., 2005; Stellwagen and Malenka, 2006; Steinmetz and Turrigiano, 2010), did not rescue muting in astrocyte-poor cultures (Fig. 4E). These data suggest that TSP1 is capable of promoting muting competence but do not clarify whether TSPs are the endogenous astrocytic factors responsible.
Prevention of muting by gabapentin, a blocker of thrombospondin-α2δ-1 binding
To test for the receptor through which TSP1 promotes muting, we blocked TSP binding to the candidate receptor α2δ-1 using gabapentin, a clinically used drug for the treatment of human neuropathic pain and epilepsy. Gabapentin interferes with TSP binding to α2δ-1 and prevents TSP's synaptogenic effects in retinal ganglion neurons (Gee et al., 1996; Eroglu et al., 2009). In astrocyte-poor cultures, gabapentin (32 μm) abolished conditioned medium rescue of depolarization-induced muting (Fig. 4E), suggesting that TSPs are likely the astrocyte-derived regulators of muting competence. We also evaluated the effect of gabapentin in live-astrocyte cultures, where ongoing production of astrocytic TSPs was an additional consideration. In these cultures, a single treatment with gabapentin at DIV 7 was ineffective in preventing silencing, as assessed at DIV 10–12 (control: 100.0 ± 22.6%; depolarized 29.8 ± 7.9%; n = 12 neurons; p=0.008, Student's unpaired t test; Fig. 5A); however, 3–5 d maintenance of gabapentin by partial medium exchange in astrocyte-rich cultures eliminated depolarization-induced muting (control: 100.0 ± 20.0%; depolarized 103.6 ± 19.8%; n = 12 neurons; p = 0.90, Student's unpaired t test; Fig. 5B). These results suggest that gabapentin also blocks endogenously released factors important for muting when the ongoing production of the factors is taken into account.
To test whether using gabapentin to block endogenous TSP could have implications for neural network function, we measured spontaneous network activity after muting induction in control and gabapentin-treated cultures. These astrocyte-rich, conventional mass cultures exhibited decreased spontaneous activity after muting induction (Fig. 5C,D), consistent with an adaptive role for muting (Hogins et al., 2011; Crawford and Mennerick, 2012). In gabapentin-treated cultures, however, a nonsignificant increase in network activity was observed (p = 0.16, Bonferroni corrected), but network activity did not change after the muting induction protocol (Fig. 5C,D), suggesting that muting was not induced. Together, these results argue that TSP binding to α2δ-1 fosters development of presynaptic muting competence.
PKA-dependent phosphorylation normalization by thrombospondin in astrocyte-poor cultures
Although the signaling pathways downstream of α2δ-1 are unclear, we hypothesized that PKA signaling, which regulates muting (Moulder et al., 2008), is altered in astrocyte-deprived neurons. This hypothesis was based in part on the observations that both Gi/o-coupled receptor agonist-induced muting and Rp-cAMPS-induced muting were dysfunctional (Fig. 1C). To test whether PKA activity was normal in astrocyte-poor cultures, we measured phosphorylation levels of PKA substrates in neurons. We found that phosphorylation levels of the presynaptic protein synapsin at its PKA phosphorylation site were increased in vGluT-1-defined synapses in astrocyte-poor cultures (Fig. 6A,B). Immunoreactivity of total synapsin levels was unchanged, however (astrocyte-rich: 28.6 ± 1.8 × 1000 AU; astrocyte-poor: 30.2 ± 1.7 × 1000 AU; n = 35 neurons; p = 0.51, Student's unpaired t test; Fig. 6C). Phosphorylation of CREB, another PKA substrate, was also abnormally high in neuronal nuclei from GABA-negative neurons in astrocyte-deprived cultures, as measured via immunoreactivity (astrocyte-rich: 3.8 ± 0.3 × 1000 AU; astrocyte-poor 15.5 ± 1.2 × 1000 AU; n = 60–89 neurons; p = 3.7 × 10−13, Student's unpaired t test; Fig. 7A). In contrast, phosphorylation of dynamin 1, a PKC substrate, was unchanged in astrocyte-poor cultures (Fig. 7B), suggesting that hyperphosphorylation was restricted to PKA phosphorylation sites. TSP1 incubation in astrocyte-poor cultures reversed abnormal presynaptic phospho-synapsin (Fig. 6A,B), so TSP appears to restore normal PKA targeting and/or activity. Furthermore, TSP1 incubation reinstated normal Rp-cAMPS-induced persistent EPSC depression in astrocyte-poor cultures (Fig. 4E). Together, these results suggest that TSP promotes muting competence via normalization of signaling downstream of cAMP pathways.
Figure 6.

PKA-dependent hyperphosphorylation of synapsin is normalized by thrombospondin. A, Phospho-synapsin (PKA phosphorylation site) immunostaining in DIV 10–12 autaptic neurons from astrocyte-rich or astrocyte-poor cultures with or without 5 μg/ml TSP treatment at DIV 7. Red circles are representative regions of interest defined by vGluT-1 immunoreactivity (data not shown). Scale bar, 5 μm. B, Summary of background-subtracted phospho-synapsin at vGluT-1-positive synapses in DIV 10–12 autaptic neurons (n = 35 neurons; *p < 0.05, Bonferroni corrected). C, Synapsin immunostaining at vGluT-1-defined synapses (red circles are representative) in autaptic neurons from astrocyte-rich or astrocyte-poor cultures. Summary immunoreactivity values are given in the Results. Scale bar, 5 μm.
Figure 7.

A PKA target, but not a non-PKA target, is hyperphosphorylated in astrocyte-poor cultures. A, MAP2, GABA, and phospho-CREB (PKA phosphorylation site) immunoreactivity in neurons on multicell islands from astrocyte-rich or astrocyte-poor cultures. Summary pCREB immunoreactivity values are given in the Results. Scale bar, 20 μm. B, Left, Phospho-dynamin 1 (PKC phosphorylation site) immunoreactivity in autaptic neurons from astrocyte-rich and astrocyte-poor cultures. Scale bar, 20 μm. Right, Quantification of background-subtracted phospho-dynamin 1 intensity from a single primary and two secondary dendrites averaged per neuron. As a positive control, we treated for 30 min with phorbol ester (1 μm PDBu), which increased PKC-dependent phosphorylation of dynamin 1 (n = 30 neurons; *p < 0.05, Bonferroni corrected).
Discussion
Here we have demonstrated that astrocytes serve as permissive partners in the development of a form of persistent synaptic depression: presynaptic muting. In glutamatergic neurons from astrocyte-poor cultures, basal presynaptic function remained intact, suggesting that astrocytes modulate presynaptic plasticity competence differently than they regulate other types of presynaptic development. Astrocytic signaling was permissive, but not acutely instructive, for muting induction. Astrocyte-derived TSPs promoted the development of muting ability, as induced with varied stimuli, likely through α2δ-1 calcium channel subunits. Although the signaling pathways immediately downstream of α2δ-1 remain to be clarified, PKA appears to be an important downstream effector. PKA-dependent phosphorylation and synaptic behavior were abnormal in astrocyte-poor cultures but restored by TSP1 treatment, providing a potential mechanism by which TSP modulates muting competence.
TSPs were originally discovered in human blood platelets (Baenziger et al., 1971). In the brain, at least 4 TSPs are expressed in various regions and cell types (Iruela-Arispe et al., 1993; Adams and Tucker, 2000). TSPs are released from astrocytes and promote synaptogenesis (Asch et al., 1986; Christopherson et al., 2005; Xu et al., 2010), but because TSP-induced glutamate synapses in retinal ganglion cells are postsynaptically silent (Christopherson et al., 2005; Eroglu et al., 2009), TSPs may be involved mainly in presynaptic rather than postsynaptic differentiation. This is especially interesting because postsynaptic TSP actions produce these presynaptic effects (Eroglu et al., 2009; Xu et al., 2010). TSP increases the total number of synapses in retinal ganglion cells (Christopherson et al., 2005; Eroglu et al., 2009) but accelerates synaptogenesis without altering total synapse number in hippocampal neurons (Xu et al., 2010). Glial cells exhibit regional differences in synaptogenic effects (Steinmetz et al., 2006). This regional difference could explain why astrocyte deprivation failed to change total presynaptic terminal number in our study of hippocampal neurons and why TSP's effects on hippocampal synaptic plasticity have been previously overlooked. Although muting is found in a variety of preparations (Crawford and Mennerick, 2012), future work should clarify whether TSP's effects on muting are brain region-specific or whether they constitute a widespread mechanism. Because TSP expression increases after injury and is important for behavioral recovery after stroke, TSP may be important generally for adaptive neuronal responses (Lin et al., 2003; Liauw et al., 2008; but see Kim et al., 2012). Presynaptic muting, which reduced network activity during an excitatory insult during this study and protected neurons against hypoxia in a prior study (Hogins et al., 2011), could be one such TSP-mediated synaptic mechanism contributing to adaptive neural responses during brain insults.
TSP promoted muting competence through its interaction with the α2δ-1 calcium channel subunit. This subunit serves as the TSP receptor for synaptogenesis in retinal ganglion cells (Eroglu et al., 2009) and as the receptor for the antinociceptive and anticonvulsant drug gabapentin (Gee et al., 1996). Gabapentin's therapeutic effects may arise from alterations in calcium channel organization (Arikkath and Campbell, 2003; Field et al., 2006; Bauer et al., 2009; Bauer et al., 2010; Hoppa et al., 2012), among other neuronal changes (Freiman et al., 2001; Gu and Huang, 2001; Stefani et al., 2001; Surges et al., 2003). Gabapentin binding to α2δ-1 antagonizes TSP-induced synaptic development in retinal ganglion neurons (Eroglu et al., 2009), and we found that gabapentin prevented the development of muting competence in hippocampal neurons, implicating TSP binding to α2δ-1 in this effect. Interestingly, calcium channel function was not detectably altered in astrocyte-poor cultures in our study, as evidenced by the lack of change in vesicle release probability and calcium concentration–response curves. Consistent with prior literature (Eroglu et al., 2009; Hoppa et al., 2012), this suggests that thrombospondin's and gabapentin's effects do not arise via alterations in calcium channel function. These results emphasize that future work needs to clarify the mechanism by which α2δ-1 alters muting competence.
Muting incompetence caused by gabapentin could also have clinical implications. Previous studies have demonstrated a wide variety of effects of gabapentin on synaptic and behavioral plasticity including no effect (Cilio et al., 2001; Heidegger et al., 2010), reduced plasticity (Blake et al., 2007; Eroglu et al., 2009; Kurokawa et al., 2011), and enhanced learning (Buccafusco et al., 2010). In our study, gabapentin blocked adaptive network changes in response to a stimulus known to selectively induce muting. Although we saw a nonsignificant increase in total network activity after gabapentin treatment, this likely does not explain the lack of muting since muting is promoted by increased network activity (Moulder et al., 2004, 2006). The prevention of adaptive muting appears counterintuitive for gabapentin because it is a therapeutic agent. Our study and a previous study (Eroglu et al., 2009) suggest that synaptogenic periods might be particularly vulnerable to unintended effects of gabapentin. Therapeutic benefits have been hypothesized to arise from gabapentin's block of synaptogenesis (Eroglu et al., 2009; Lo et al., 2011) and reduction of calcium channel function or trafficking (Rogawski and Löscher, 2004). Because synapse formation was not hindered and calcium channels did not differ functionally in astrocyte-poor cultures compared with astrocyte-rich cultures, these effects of gabapentin likely did not contribute to muting prevention. Gabapentin's effects on muting may instead work through a different molecular pathway and become detrimental to the neural network only after a secondary excitatory insult. Future work should clarify the contexts under which multiple effects of gabapentin alter neuronal function.
Our work implicates PKA signaling in TSP's promotion of muting competence. Gi/o-linked receptor agonists and PKA inhibitors, which normally produce muting (Moulder et al., 2008; Crawford et al., 2011), persistently potentiated EPSCs in astrocyte-poor cultures. Additionally, PKA substrates in the presynaptic terminal and the nucleus were hyperphosphorylated in astrocyte-deprived cultures, indicating an abnormal increase in PKA site phosphorylation or a decrease in dephosphorylation. This surprising synaptic facilitation may suggest that multiple cAMP-dependent pathways regulate synaptic transmission. It is possible that PKA inhibition normally suppresses transmission by dominating or masking other potentiating effects that are then revealed in the context of astrocyte deprivation and hyperphosphorylated PKA substrates. Astrocyte-released TSPs, therefore, may restrain PKA activity, or phosphorylation levels of its substrates, during synaptic development so that PKA inhibition can produce a meaningful decrease in synaptic function upon exposure to the depolarization challenge. Other explanatory models are possible, and future work may distinguish them.
In addition to effects on synaptic plasticity, we also observed effects of astrocyte deprivation on basal postsynaptic function. Neuronal responses to AMPA and NMDA receptor agonists were depressed in parallel, supporting prior work suggesting that astrocytes are important for postsynaptic development (Beattie et al., 2002; Stellwagen and Malenka, 2006; Perea et al., 2009; Sullivan et al., 2011). Because neurons near live astrocytes in astrocyte-poor cultures produced EPSCs that were statistically indistinguishable from those on dead astrocytes, depressed basal EPSCs likely resulted from a deficit in global astrocytic signaling rather than a deficit in local signaling. It is unlikely that the basal EPSC deficit caused the deficit in muting because the failure to mute persisted in older, astrocyte-deprived cultures with larger basal EPSCs. Furthermore, muting persists in the presence of complete blockade of AMPA and NMDA receptor function (Moulder et al., 2004, 2006). Our results do not exclude a role for TSP and α2δ-1 in basal postsynaptic development, however, but TSP's role in muting appears independent of postsynaptic receptor function.
In summary, astrocyte-derived TSPs permit presynaptic muting at hippocampal glutamate terminals. TSPs likely act through α2δ-1 binding to normalize PKA signaling in developing synapses. These results reveal a novel mechanism by which glial-neuronal communication controls adaptive synaptic malleability.
Footnotes
This work was supported by U.S. National Institutes of Health Grants NS066611 (D.C.C.), MH078823 (S.M.), and NS52823 (S.M.). We thank Ann Benz for help with cultures and lab members for advice and support.
The authors declare no competing financial interests.
References
- Adams JC, Tucker RP. The thrombospondin type 1 repeat (TSR) superfamily: diverse proteins with related roles in neuronal development. Dev Dyn. 2000;218:280–299. doi: 10.1002/(SICI)1097-0177(200006)218:2<280::AID-DVDY4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Alexanian AR. Neural stem cells induce bone-marrow-derived mesenchymal stem cells to generate neural stem-like cells via juxtacrine and paracrine interactions. Exp Cell Res. 2005;310:383–391. doi: 10.1016/j.yexcr.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Amur-Umarjee S, Phan T, Campagnoni AT. Myelin basic protein mRNA translocation in oligodendrocytes is inhibited by astrocytes in vitro. J Neurosci Res. 1993;36:99–110. doi: 10.1002/jnr.490360111. [DOI] [PubMed] [Google Scholar]
- Arikkath J, Campbell KP. Auxiliary subunits: essential components of the voltage-gated calcium channel complex. Curr Opin Neurobiol. 2003;13:298–307. doi: 10.1016/s0959-4388(03)00066-7. [DOI] [PubMed] [Google Scholar]
- Asch AS, Leung LL, Shapiro J, Nachman RL. Human brain glial cells synthesize thrombospondin. Proc Natl Acad Sci U S A. 1986;83:2904–2908. doi: 10.1073/pnas.83.9.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baenziger NL, Brodie GN, Majerus PW. A thrombin-sensitive protein of human platelet membranes. Proc Natl Acad Sci U S A. 1971;68:240–243. doi: 10.1073/pnas.68.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CS, Nieto-Rostro M, Rahman W, Tran-Van-Minh A, Ferron L, Douglas L, Kadurin I, Sri Ranjan Y, Fernandez-Alacid L, Millar NS, Dickenson AH, Lujan R, Dolphin AC. The increased trafficking of the calcium channel subunit alpha2delta-1 to presynaptic terminals in neuropathic pain is inhibited by the alpha2delta ligand pregabalin. J Neurosci. 2009;29:4076–4088. doi: 10.1523/JNEUROSCI.0356-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CS, Tran-Van-Minh A, Kadurin I, Dolphin AC. A new look at calcium channel alpha2delta subunits. Curr Opin Neurobiol. 2010;20:563–571. doi: 10.1016/j.conb.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Blake MG, Boccia MM, Acosta GB, Höcht C, Baratti CM. Opposite effects of a single versus repeated doses of gabapentin on retention performance of an inhibitory avoidance response in mice. Neurobiol Learn Mem. 2007;87:192–200. doi: 10.1016/j.nlm.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Golan H, Kandel ER, Siegelbaum SA. Recruitment of new sites of synaptic transmission during the cAMP-dependent late phase of LTP at CA3-CA1 synapses in the hippocampus. Neuron. 1997;19:635–651. doi: 10.1016/s0896-6273(00)80377-3. [DOI] [PubMed] [Google Scholar]
- Brown DA, Sihra TS. Presynaptic signaling by heterotrimeric G-proteins. Handb Exp Pharmacol. 2008;184:207–260. doi: 10.1007/978-3-540-74805-2_8. [DOI] [PubMed] [Google Scholar]
- Buccafusco JJ, Terry AV, Jr, Vazdarjanova A, Snutch TP, Arneric SP. Treatments for neuropathic pain differentially affect delayed matching accuracy by macaques: effects of amitriptyline and gabapentin. Pain. 2010;148:446–453. doi: 10.1016/j.pain.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Cilio MR, Bolanos AR, Liu Z, Schmid R, Yang Y, Stafstrom CE, Mikati MA, Holmes GL. Anticonvulsant action and long-term effects of gabapentin in the immature brain. Neuropharmacology. 2001;40:139–147. doi: 10.1016/s0028-3908(00)00103-9. [DOI] [PubMed] [Google Scholar]
- Cousin MA, Evans GJ. Activation of silent and weak synapses by cAMP-dependent protein kinase in cultured cerebellar granule neurons. J Physiol. 2011;589:1943–1955. doi: 10.1113/jphysiol.2010.200477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AM, Graf ER, Linhoff MW. How to build a central synapse: clues from cell culture. Trends Neurosci. 2006;29:8–20. doi: 10.1016/j.tins.2005.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford DC, Mennerick S. Presynaptically silent synapses: dormancy and awakening of presynaptic vesicle release. Neuroscientist. 2012;18:216–223. doi: 10.1177/1073858411418525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford DC, Chang CY, Hyrc KL, Mennerick S. Calcium-independent inhibitory G-protein signaling induces persistent presynaptic muting of hippocampal synapses. J Neurosci. 2011;31:979–991. doi: 10.1523/JNEUROSCI.4960-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010;468:223–231. doi: 10.1038/nature09612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field MJ, Cox PJ, Stott E, Melrose H, Offord J, Su TZ, Bramwell S, Corradini L, England S, Winks J, Kinloch RA, Hendrich J, Dolphin AC, Webb T, Williams D. Identification of the alpha2-delta-1 subunit of voltage-dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. Proc Natl Acad Sci U S A. 2006;103:17537–17542. doi: 10.1073/pnas.0409066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiman TM, Kukolja J, Heinemeyer J, Eckhardt K, Aranda H, Rominger A, Dooley DJ, Zentner J, Feuerstein TJ. Modulation of K+-evoked [3H]-noradrenaline release from rat and human brain slices by gabapentin: involvement of KATP channels. Naunyn Schmiedebergs Arch Pharmacol. 2001;363:537–542. doi: 10.1007/s002100100408. [DOI] [PubMed] [Google Scholar]
- Gee NS, Brown JP, Dissanayake VU, Offord J, Thurlow R, Woodruff GN. The novel anticonvulsant drug, gabapentin (Neurontin), binds to the alpha2delta subunit of a calcium channel. J Biol Chem. 1996;271:5768–5776. doi: 10.1074/jbc.271.10.5768. [DOI] [PubMed] [Google Scholar]
- Gu Y, Huang LY. Gabapentin actions on N-methyl-d-aspartate receptor channels are protein kinase C-dependent. Pain. 2001;93:85–92. doi: 10.1016/S0304-3959(01)00297-4. [DOI] [PubMed] [Google Scholar]
- Heidegger T, Krakow K, Ziemann U. Effects of antiepileptic drugs on associative LTP-like plasticity in human motor cortex. Eur J Neurosci. 2010;32:1215–1222. doi: 10.1111/j.1460-9568.2010.07375.x. [DOI] [PubMed] [Google Scholar]
- Hestrin S, Nicoll RA, Perkel DJ, Sah P. Analysis of excitatory synaptic action in pyramidal cells using whole-cell recording from rat hippocampal slices. J Physiol. 1990;422:203–225. doi: 10.1113/jphysiol.1990.sp017980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogins J, Crawford DC, Jiang X, Mennerick S. Presynaptic silencing is an endogenous neuroprotectant during excitotoxic insults. Neurobiol Dis. 2011;43:516–525. doi: 10.1016/j.nbd.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA. alpha2delta expression sets presynaptic calcium channel abundance and release probability. Nature. 2012;486:122–125. doi: 10.1038/nature11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iruela-Arispe ML, Liska DJ, Sage EH, Bornstein P. Differential expression of thrombospondin 1, 2, and 3 during murine development. Dev Dyn. 1993;197:40–56. doi: 10.1002/aja.1001970105. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kannenberg K, Sieghart W, Reuter H. Clusters of GABAA receptors on cultured hippocampal cells correlate only partially with functional synapses. Eur J Neurosci. 1999;11:1256–1264. doi: 10.1046/j.1460-9568.1999.00533.x. [DOI] [PubMed] [Google Scholar]
- Kim DS, Li KW, Boroujerdi A, Peter Yu Y, Zhou CY, Deng P, Park J, Zhang X, Lee J, Corpe M, Sharp K, Steward O, Eroglu C, Barres B, Zaucke F, Xu ZC, Luo ZD. Thrombospondin-4 contributes to spinal sensitization and neuropathic pain states. J Neurosci. 2012;32:8977–8987. doi: 10.1523/JNEUROSCI.6494-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Udo H, Li HL, Youn TY, Chen M, Kandel ER, Bailey CH. Presynaptic activation of silent synapses and growth of new synapses contribute to intermediate and long-term facilitation in Aplysia. Neuron. 2003;40:151–165. doi: 10.1016/s0896-6273(03)00595-6. [DOI] [PubMed] [Google Scholar]
- Kurokawa K, Shibasaki M, Mizuno K, Ohkuma S. Gabapentin blocks methamphetamine-induced sensitization and conditioned place preference via inhibition of alpha(2)/delta-1 subunits of the voltage-gated calcium channels. Neuroscience. 2011;176:328–335. doi: 10.1016/j.neuroscience.2010.11.062. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Moghaddam B. Cognitive dysfunction in schizophrenia: convergence of gamma-aminobutyric acid and glutamate alterations. Arch Neurol. 2006;63:1372–1376. doi: 10.1001/archneur.63.10.1372. [DOI] [PubMed] [Google Scholar]
- Liauw J, Hoang S, Choi M, Eroglu C, Choi M, Sun GH, Percy M, Wildman-Tobriner B, Bliss T, Guzman RG, Barres BA, Steinberg GK. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cereb Blood Flow Metab. 2008;28:1722–1732. doi: 10.1038/jcbfm.2008.65. [DOI] [PubMed] [Google Scholar]
- Lin TN, Kim GM, Chen JJ, Cheung WM, He YY, Hsu CY. Differential regulation of thrombospondin-1 and thrombospondin-2 after focal cerebral ischemia/reperfusion. Stroke. 2003;34:177–186. doi: 10.1161/01.str.0000047100.84604.ba. [DOI] [PubMed] [Google Scholar]
- Lo FS, Zhao S, Erzurumlu RS. Astrocytes promote peripheral nerve injury-induced reactive synaptogenesis in the neonatal CNS. J Neurophysiol. 2011;106:2876–2887. doi: 10.1152/jn.00312.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losonczy A, Biró AA, Nusser Z. Persistently active cannabinoid receptors mute a subpopulation of hippocampal interneurons. Proc Natl Acad Sci U S A. 2004;101:1362–1367. doi: 10.1073/pnas.0304752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Zablow L, Kandel ER, Siegelbaum SA. Cyclic AMP induces functional presynaptic boutons in hippocampal CA3-CA1 neuronal cultures. Nat Neurosci. 1999;2:24–30. doi: 10.1038/4525. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Contreras D. On the cellular and network bases of epileptic seizures. Annu Rev Physiol. 2001;63:815–846. doi: 10.1146/annurev.physiol.63.1.815. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Que J, Benz A, Zorumski CF. Passive and synaptic properties of hippocampal neurons grown in microcultures and in mass cultures. J Neurophysiol. 1995;73:320–332. doi: 10.1152/jn.1995.73.1.320. [DOI] [PubMed] [Google Scholar]
- Moulder KL, Meeks JP, Shute AA, Hamilton CK, de Erausquin G, Mennerick S. Plastic elimination of functional glutamate release sites by depolarization. Neuron. 2004;42:423–435. doi: 10.1016/s0896-6273(04)00184-9. [DOI] [PubMed] [Google Scholar]
- Moulder KL, Jiang X, Taylor AA, Olney JW, Mennerick S. Physiological activity depresses synaptic function through an effect on vesicle priming. J Neurosci. 2006;26:6618–6626. doi: 10.1523/JNEUROSCI.5498-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Jiang X, Taylor AA, Shin W, Gillis KD, Mennerick S. Vesicle pool heterogeneity at hippocampal glutamate and GABA synapses. J Neurosci. 2007;27:9846–9854. doi: 10.1523/JNEUROSCI.2803-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Jiang X, Chang C, Taylor AA, Benz AM, Conti AC, Muglia LJ, Mennerick S. A specific role for Ca2+-dependent adenylyl cyclases in recovery from adaptive presynaptic silencing. J Neurosci. 2008;28:5159–5168. doi: 10.1523/JNEUROSCI.5317-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Jiang X, Taylor AA, Benz AM, Mennerick S. Presynaptically silent synapses studied with light microscopy. J Vis Exp. 2010;35:e1676. doi: 10.3791/1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 2009;32:421–431. doi: 10.1016/j.tins.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Pfrieger FW. Role of glial cells in the formation and maintenance of synapses. Brain Res Rev. 2010;63:39–46. doi: 10.1016/j.brainresrev.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Rogawski MA, Löscher W. The neurobiology of antiepileptic drugs for the treatment of nonepileptic conditions. Nat Med. 2004;10:685–692. doi: 10.1038/nm1074. [DOI] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solà C, Casal C, Tusell JM, Serratosa J. Astrocytes enhance lipopolysaccharide-induced nitric oxide production by microglial cells. Eur J Neurosci. 2002;16:1275–1283. doi: 10.1046/j.1460-9568.2002.02199.x. [DOI] [PubMed] [Google Scholar]
- Stefani A, Spadoni F, Giacomini P, Lavaroni F, Bernardi G. The effects of gabapentin on different ligand- and voltage-gated currents in isolated cortical neurons. Epilepsy Res. 2001;43:239–248. doi: 10.1016/s0920-1211(00)00201-1. [DOI] [PubMed] [Google Scholar]
- Steinmetz CC, Turrigiano GG. Tumor necrosis factor-alpha signaling maintains the ability of cortical synapses to express synaptic scaling. J Neurosci. 2010;30:14685–14690. doi: 10.1523/JNEUROSCI.2210-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz CC, Buard I, Claudepierre T, Nägler K, Pfrieger FW. Regional variations in the glial influence on synapse development in the mouse CNS. J Physiol. 2006;577:249–261. doi: 10.1113/jphysiol.2006.117358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan SJ, Esguerra M, Wickham RJ, Romero GE, Coyle JT, Miller RF. Serine racemase deletion abolishes light-evoked NMDA receptor currents in retinal ganglion cells. J Physiol. 2011;589:5997–6006. doi: 10.1113/jphysiol.2011.217059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surges R, Freiman TM, Feuerstein TJ. Gabapentin increases the hyperpolarization-activated cation current Ih in rat CA1 pyramidal cells. Epilepsia. 2003;44:150–156. doi: 10.1046/j.1528-1157.2003.36802.x. [DOI] [PubMed] [Google Scholar]
- Tong G, Malenka RC, Nicoll RA. Long-term potentiation in cultures of single hippocampal granule cells: a presynaptic form of plasticity. Neuron. 1996;16:1147–1157. doi: 10.1016/s0896-6273(00)80141-5. [DOI] [PubMed] [Google Scholar]
- Wetmore DZ, Garner CC. Emerging pharmacotherapies for neurodevelopmental disorders. J Dev Behav Pediatr. 2010;31:564–581. doi: 10.1097/DBP.0b013e3181ee3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Xiao N, Xia J. Thrombospondin 1 accelerates synaptogenesis in hippocampal neurons through neuroligin 1. Nat Neurosci. 2010;13:22–24. doi: 10.1038/nn.2459. [DOI] [PubMed] [Google Scholar]