

Introduction
In the 10 years since Daly reviewed1 the potential of adenosine receptors as drug targets, considerable advances have been made in the area of purinergic receptor related research such that there is little doubt remaining that adenosine, as well as adenosine 5′-triphosphate (ATP) and related nucleotides, functions as both neurohumoral agents and autacoids regulating the process of cell to cell communication.2
The techniques of molecular pharmacology have been extensively used to delineate purinergic receptor function, resulting in the identification of several receptor subclasses that subserve discrete physiological functions (Table I).2 And more recently, the two major classes of adenosine receptors, the A1 and A2, have been cloned,3,4 offering the potential to model the receptor–ligand interaction from the receptor side.5
Table I.
Purinoceptor Subtypes
| potencya | |||
|---|---|---|---|
| subtype | agonists | antagonists | location |
| P1 (Adenosine) Receptors | |||
| A1 | CPA > R-PIA > NECA ≫ MPEA > CGS 21680 | CPX > XAC > CPT > 8-PT | hippocampus, adipocytes, atrioventicular node |
| A1a | R-PIA > NECA > S-PIA ≫ MeAdo > CV 1808 ≫ CV 1674 | XAC = PD 113,297 > XCC | rat brainb |
| A1b | R-PIA > NECA ≫ S-PIA > CV 1808 ≫ MeAdo > CV 1674 | XAC ≥ XCC > PD 113,297 | guinea pig ileumb |
| A2a “high affinity” | APEC ≈ CGS21680 ≈ CGS 22492 ≈ NECA ≫ CPA ≈ CV1674c > N6-MeADO | CGS15943 > XAC ≈ PD115,199 > CPX > XCC | striatum, platelets, neutrophils, coronary vasculature, olfactory tubercule |
| A2b “low affinity” | NECA ≫ CGS21680 ≈ N6-MeADO > CV1674c | XCC ≈ XAC ≫ CPX, 5′MTA | brain |
| A3 | brain | ||
| P2 (ATP) Receptors | |||
| P2t | 2-MeSADP > ADP | ATP, AMP | platelet |
| P2u “nucleotide receptor” | UTP = ATP > ADP > 2-MeSATP | hepatocytes, bovine aorta smooth muscle, Ehrlich ascites tumor cells, HL–60 cells, rat renal mesangial cells, neutrophils, fibroblasts | |
| P2x | α,β-MeATP > ATP | suramin | bladder, vas deferens, ear artery |
| P2y | 2-MeSATP > ATP = ADP > UTP | reactive blue 2 | taenia coli, endothelium, turkey erythrocytes |
| P2z | ATP−4 > ATP | DIDSd | mast cells, lymphocytes |
| P3 Receptors | |||
| P3 | UTP, ATP, APPCP | 8-PST | prejunctional-rat caudal artery, vas deferens |
On the ligand front, structure–activity relationships (SAR) studies (Figure 1) for derivatives of adenosine (1), as agonists, and of theophylline (2), as antagonists, have revealed selective agents,6–8 and potent and selective A1- and A2-receptor agonists are now available. Newer antagonist ligands include a large number of 8-substituted xanthine derivatives, some of them over 10000-fold more potent than the parent compound 2, as well as numerous classes of non-xanthine heterocyclic compounds8 described in further detail below.
Figure 1.

The structures of adenosine (1) and theophylline (2), showing the effects of structural modifications at various sites on receptor binding.
The exceptional progress in the preclinical area, both chemical and biological, has not however been paralleled in the clinic. Very few adenosine agonists and antagonists have entered clinical trials and none of these, to the authors’ knowledge, have been successful.7 The only approved compound known to produce its therapeutic actions via a direct interaction with adenosine receptors is adenosine itself, used for the treatment of supraventricular tachycardia (SVT),9 a use designated by the U.S. Food and Drug Administration in their coveted 1A category, indicating a drug for major unmet medical need. Additional potential uses for adenosine include cardiac imaging,10 in cardioplegic solutions11 to delay the onset of ischemic contractions, and as a cardioprotectant in postischemic reperfusion.12 While caffeine and theophylline represent prototypic, albeit weak, adenosine antagonists, second generation forms of these compounds with improved antagonist activity for use as cardiotonics, cognition enhancers or antiasthmatics have not been forthcoming despite considerable chemical effort.8
The reasons for the limited progress in adenosine therapeutics are several-fold and include the ubiquity of action of adenosine (and ATP) on a variety of diverse tissue systems, a paucity of receptor selective ligands that are orally bioavailable and soluble, lack of knowledge of disease states involving a purinergic etiology, and probably most importantly, a failure to target adenosine agents in terms of unmet therapeutic need.13 Thus, agonists have been routinely targeted toward hypertension, an area where these agents have probable CNS and renal side effects and compare unfavorably with the many excellent and efficacious antihypertensive agents currently available in the clinic.13
In the present perspective, advances in knowledge related to adenosine function at the molecular level will be reviewed together with information on the structure–activity relationships for a number of pharmacophore series interacting with adenosine receptors. Therapeutic areas where improved adenosine ligands may represent potentially important therapeutic agents will also be indicated.
Adenosine Receptor Ligands
Development of Adenosine Agonists
Adenosine 1 has been extensively used as a probe for the study of adenosine systems in mammalian tissues since the initial reports on the cardiovascular actions of the purine some 60 years ago.14 The metabolic lability of adenosine precluded its use as an antihypertensive agent in the early 1930s,15,16 but more stable analogues have been synthesized (Table II), focusing primarily on modifications of the N6-, 2-, and 5′-positions.6 Among these are N6-cyclohexyladenosine (CHA, 6), (R)-N6-(2-phenyl-1-methylethyl)adenosine (R-PIA, 9), 2-chloroadenosine (2-CADO, 22) and N-ethyladenosine-5′-uronamide (NECA, 30). The availability of these agents provided the tools by which adenosine receptors have been classified into A1, and A2a, and A2b subtypes, on the basis of the pharmacology of radioligand bindingl7,18 (Table I). A1a, A1b,19 and A320 subtypes have also been proposed. Well-documented species differences in adenosine receptors21,22 have, however, provided a spurious basis for adenosine receptor classification,23 which can only serve to complicate nomenclature issues. Structure–efficacy relationships for adenosine receptor ligands have not been well defined, especially in regard to partial agonist activity.24,25
Table II.
Structures of Adenosine Agonists and Their Affinities at A1 and A2 Adenosine Receptorsa
 | |||||
|---|---|---|---|---|---|
| Ki | |||||
| compd | R2 | R1f | A1 | A2 | A2/A1 |
| 3 | 0.59 | 462 | 780 | ||
| 4 | Cl | 0.6 | 950 | 1500 | |
| 5 | NH2 | 8.3 | 6100 | 730 | |
| 6 | 1.3 | 514 | 400 | ||
| 7 | 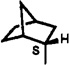 |
0.3 | 1390 | 4600 | |
| 8 | 7.0 | 4920 | 700 | ||
| 9 |  |
1.2 | 124 | 100 | |
| 10 |  |
0.94b | - | - | |
| 11 | 4.6 | 663 | 140 | ||
| 12 |  |
0.7b | - | - | |
| 13 | 4.1 | - | - | ||
| 14 |  |
2.0b | - | - | |
| 15 |  |
6.8 | 25 | 3.7 | |
| 16 |  |
142 | 4.4 | 0.31 | |
| 17 |  |
5.2 | 4.9 | 0.94 | |
| 18 | 0.85 | 210 | 250 | ||
| 19 |  |
0.22 | 8400 | 38000 | |
| 20 | 0.47 | 191 | 410 | ||
| 21 | 7.1 | - | - | ||
 | |||||
|---|---|---|---|---|---|
| Ki | |||||
| compd | R1 | A1 | A2 | A2/A1 | |
| R5 = CH2OH | |||||
| 22 | Cl | 9.3 | 63 | 6.8 | |
| 23 | 560 | 119 | 0.21 | ||
| 24 | 12000 | 22 | 0.0018 | ||
| 25 | 2700 | 13 | 0.0048 | ||
| 26 | 1500 | 22 | 0.014 | ||
| 27 | 48 | 11 | 0.22 | ||
| 28 | C≡C—(CH2)3CH3 | 147 | 4.1 | 0.028 | |
| 29 | C≡C—(CH2)5CH3 | 211 | 12 | 0.057 | |
| R5 = CONHCH2CH3 | |||||
| 30 | H | 6.3 | 10.3 | 1.6 | |
| 31 | 2600 | 15 | 0.0058 | ||
| 32 | 240 | 5.7 | 0.024 | ||
| 33 |  |
- | 1.0d | - | |
| 34 | 280 | 7.1e | 0.025 | ||
Unless noted, Ki values for binding experiments (using [3H]CHA or [3H]PIA at A1 or [3H]NECA at A2, in rat brain unless indicated) are given in nM; data if from refs 6, 18, 29, 30, 33, 34, 43, and 167.
Kd value for radioligand binding to rat brain membranes.
FITC = fluorescein isothiocyanate, which forms a thiourea linkage.
Versus [3H]PIA.
Versus [125I]PAPA-APEC in bovine striatum.
R1 = H, unless noted.
N6-substituted analogues of adenosine have generally proven to be A1-receptor selective, with N6-cyclopentyladenosine (CPA, 3) and CHA (6) being 400–800-fold selective.6 N6-bicycloalkyladenosines are even more A1 selective with N6-endo-norborn-2-yladenosine (S-ENBA, 7) being 4700-fold selective for the A1 receptor.6 Combined substitutions at the N6- and 2-positions have yielded 2-chloro-CPA (CCPA, 4) which is 1500-fold A1 selective.26,164 A computer-generated model of the N6 region of the A1 receptor (Figure 2) has been shown to accurately predict the affinities of a number of N6-substituted adenosines.27
Figure 2.

Computer-generated models of the N6 regions of adenosine A1 and A2 receptors. For the A1 model, areas are indicated where hydrophobic substituents may lead to enhanced affinity. These are designated S1, S1A, S2, S3, S3A, A(ryl), B(ulk), and C(ycloalkyl). In both models, shaded areas indicate the receptor boundaries (adapted from ref 27 and 31).
The 5′-substituted adenosine analogue, NECA (30), has been extensively used to define tissue responses mediated by A2 receptor activation.2,7 This analogue is however nonselective in its interactions with adenosine receptors, being approximately equipotent (Ki ≈ 10 nM) at both A1 and A2 receptors.18 Ascribing effects elicited by NECA to A2 receptor-mediated processes can only be validated if such effects are not seen with equivalent doses/concentrations of A1 selective ligands such as CPA 3 or CHA 6. In the seminal A2-receptor binding assay developed by Bruns and co-workers,18 the A1 component of the binding profile of NECA was eliminated by the use of 50 nM CPA.
Other ribose modifications, especially at the 2′ and 3′ positions are generally not well tolerated a t the binding site: 2′ substitution abolishes affinity altogether, and an unsubstituted 3′-hydroxyl group is required for high efficacy.6,28
A2-selective adenosine agonists have been developed more recently. Although most N6-substituted adenosine derivatives are A1 selective,29 a series of N6-(2,2-diphenylethyl)-substituted adenosine analogues includes potent and A2-selective compounds like CI 936 (15) and DPMA (16, N6-[2-(3,5-dimethoxyphenyl)-2-(2-methylphenyl)ethyl]adenosine), the racemate of which is 30-fold selective for the A2 receptor.30 The design of this series was aided by a computer-generated model (Figure 2) of the N6 region of the A2 receptor.31
The 2-(arylamino)adenosine analogue, CV 1808 (23)32 (Table II) while only moderately potent (Ki ~ 100 nM) at A2 receptors has a modest 5-fold selectivity versus the A1 receptor.18 Evaluation of 2-position modifications of 30 led to the identification of CGS 21680 (31)33 which is 140-fold selective for the A2 receptor, with a Ki value of 21 nM. CGS 22492 2-[(cyclohexylethyl)amino]adenosine (24) and the 2-cydohexenyl analogue, CGS 22989 (25), two monosubstituted adenosine derivatives related to CV 1808, are 530- and 210-fold selective, respectively, for the A2 receptor with Ki values in the range of 13–22 nM.34 CGS 21680 has been derivatized as iodo-PAPA-APEC (33, Table II) and used as a probe to explore the A2a receptor (see below).35 The availability of [3H]CGS 2168036 was instrumental to the identification of the cloned A2 receptor.4
Further structural modifications at the 2-position led to development of the 2-alkoxyadenosines37,38 and the 2-alkynyladenosines.39 2-(2-Cyclohexylethoxy)adenosine (26) (CHEA; Table II) has an EC50 value of 1 nM at the A2 receptor in heart modulating coronary vasodilation, resulting in an 8700-fold selectivity for the A2 receptor37 in this functional model of adenosine receptor selectivity. The 2-aralkoxyadenosine derivative 2-[2-(4-methylphenyl)ethoxy]adenosine (27) (MPEA) is 44000-fold selective for the coronary A2 receptor with an EC50 value of 190 pM.38 Evaluation of these 2-alkoxy compounds in binding assays in rat brain tissue39 indicated that while both are active at the A2 receptor (MPEA, Ki = 11 nM; CHEA, Ki = 22 nM), MPEA is only 5-fold A2 selective while CHEA is 73-fold selective, a marked contrast to the 8700- and 44000-fold selectivity for the A2 receptor seen in the guinea pig tissue models.37–39 These results do not appear to be species dependent since affinity in guinea pig tissues is very comparable with that seen in rat brain binding assays.
The 2-alkynyl derivatives, 2-hexynyladenosine (2-HNA, 28) and 2-octynyladenosine (2-ONA; YT-146, 29), are potent A2 agonists (Ki values 4 and 12 nM) with 36- and 17-fold selectivity, respectively, for the A2 receptor in binding assays.40 In the spontaneously hypertensive rat (SHR), 2-HNA and 2-ONA are 390- and 260-fold selective for the A2 receptor, mediating blood pressure lowering. Like 2-HNA and 2-ONA, the differences in selectivity between binding and functional test procedures for CGS 21680 are more modest41 than those reported for the alkoxyadenosines.37,38 Furthermore, the A1-selective agonist, CPA is considerably less selective in these functional assays than has been reported by numerous laboratories using binding assays. There is thus a considerable need for caution in comparing in vitro binding assay activity with classical functional paradigms. The latter determine efficacy as well as activity and depend in large part on the choice of tissue used, an issue that has caused concern within the context of receptor classification42 and requires further study in regard to the delineation of adenosine receptor function. This issue is typified by recent biochemical data on 5′-methylthioadenosine (MTA),43 a potent A1-receptor agonist (EC50 = 90 nM), with weak partial agonist activity at the PC12 A2a receptor (EC50 = 8.9 µM) and antagonist activity at the human VC13 cell A2b, receptor (Ki = 8.2 µM).
Few purine ring modifications can be tolerated, but 1-deazaadenosines retain high affinity. Thus, 1-deaza-2-chloro-N6-cyclopentyladenoeine is a potent and A1-selective agonist.44
Development of Adenosine Antagonists
Structures reported as adenosine receptor antagonists are shown in Table III. The prototypic adenosine receptor antagonists were the xanthines, theophylline (2), and caffeine (35).45 Since then a multitude of xanthines has been synthesized and studied as antagonists at A1 and A2 receptors.8 Numerous structurally diverse non-xanthine antagonists (Figure 3) have also been identified during the last decade, many of which have only poor to moderate affinity and are not well defined in terms of SAR. Three classes of related heterocycles comprise more active entities and are termed the “tricyclic” non-xanthine antagonists. These include the triazoloquinazolines,46 the triazoloquinoxalines,47,48 and the imidazoquinolines.49 Other potent and A1-selective antagonists have been derived from adenine and include the 2-phenyl-7-deazaadenines such as ADPEP (60) (A1, 4.7 nM; A2, 3710 nM)50 and the N6-substituted 9-methyladenines,51 including N-0861 53 [(±)-N6-endo-norbornyl-9-methyladenine (A1, 10 nM; A2, 6100 nM in bovine brain)].52 The structures and binding activity of some relevant compounds are shown in Figure 2 and Table IV.
Table III.
Classes of Adenosine Receptor Antagonists
| chemical class | example |
|---|---|
| adenines | N-0861 (53),52 N6-butyl-8-phenyladenine61 |
| adenosines, ribose-modified | 5′-deoxy-5′-methylthioadenosine152 |
| barbiturates | DMBB153 |
| benzimidazoles | 1-methyl-2-phenylimidazole154 |
| benzo[1,2-c:5,4-c′]dipyrazoles | 1,7-dihydro-3,5,8-trimethylbenzo[1,2-c:5,4-c′]dipyrazole155 |
| benzo[b]furanes | 5-(3-hydroxypropyl)-7-methoxy-2-(3′-methoxy-4′-hydroxyphenyl)-3-benzo[b]-furancarbaldehyde53 |
| benzo[g]pteridine-2,4-diones | alloxazine57 |
| β-carbolines | β-carboline-3-ethylcarboxylate154 |
| 7-deazadenines | (see pyrrolo[2,3-d]pyrimidines) |
| dibenz[b,f]azepines | carbamazepine156 |
| imidazo[1,2-a]pyrazines | SC-12154 |
| imidazo[4,5-b]pyridines | sulmazole154 |
| imidazo[4,5-c]quinolines | CPPIQA (55)49 |
| imidazo[4,5-e][1,4]diazepine-5,8-diones | 4,7-dipropyl-1-benzyl-4,5-tetrahydro-6H-imidazo[4,5-e][1,4]diazepine-5,8-dione157 |
| imidazo[4,5-f]quinazoline-7,9-diones | prox-benzotheophylline59 |
| imidazo[4,5-g]quinazoline-6,8-diones | lin-benzotheophylline59 |
| imidazolidines | DPI154 |
| pteridine-2,4-diones | lumazine57 |
| pyrazolo[3,4-b]pyridines | cartazolate, ethazolate, tracazolate158 |
| pyrazolo[3,4-d]pyrimidines | DJB-KK,159 APPP (57)61 |
| pyrazolo[4,3-d]pyrimidines | 5-(2-amino-4-chlorophenyl)-1,3-dimethylpyrazolo[4,3-d]pyrimidin-7-one160 |
| pyrazolo[4,3-c]quinolines | CGS 821669 |
| pyrimidines | amiloride161 |
| pyrimido[4,5-b](tetrahydro)indoles | 4-amino-9-phenyl-9H-pyrimido[4,5-b]indole50 |
| pyrrolo[2,3-d]pyrimidines(7-deazaadenines) | ADPEP (55)50 |
| quinazolines | ADQZ61 |
| thiazolo[2,3-b]quinazolines | HTQZ (58)61 |
| thiazolo[4,5-d]pyrimidine-5,7-diones | 4,6-dimethyl-2-phenyl-4,5,6,7-tetrahydrothiazolo[4,5-d]pyrimidine-5,7-dione154 |
| thiazolo[5,4-d]pyrimidine-5,7-diones | DJB-W159 |
| [1,2,4]triazolo[4,3-b]pyridazines | CL 218872154 |
| [1,2,4]triazolo[1,5-c]quinazolines | CGS 15943 (51)46 |
| [1,2,4]triazolo[4,3-a]quinoxalines | CP 66713 (53), CP 68247 (54)48 |
| xanthines | caffeine (33), theophylline (2), CPX (39),58 PACPX (48),18 XAC (50)62 |
| xanthines, benzo-separated | (see imidazo[4,5]quinazolinediones) |
| xanthines, mesoionic | anhydro-6,8-di-n-propyl-5-hydroxy-7-oxothiazolo[3,2-a]pyrimidinium hydroxide154 |
| xanthine-7-ribosides | 1,3-dibutylxanthine-7-riboside162 |
Figure 3.

The structures of non-xanthine adenosine antagonists. (See text and Table III for description.)
Table IV.
Affinities of Some Relevant Adenosine Antagonists
 | |||||||
|---|---|---|---|---|---|---|---|
| compd | R1 | R3 | R8 | R7h | A1, nMa | A2, nMb | A2/A1 |
| Xanthines | |||||||
| 2 theophylline | Me | Me | H | 8500 | 25000 | 3.018 | |
| 35 caffeine | Me | Me | H | Me | 29000 | 48000 | 1.718 |
| 36 DMPX | CH≡CCH2 | Me | H | Me | 22000 | 9600 | 0.4460 |
| 37 CHCi | Me | Me | cyclohexyl | Me | 2.0c | 0.19d | 0.095155 |
| 38 CPT | Me | Me | cyclopentyl | 11 | 1400 | 13018 | |
| 39 CPX | Pr | Pr | cyclopentyl | 0.46 | 340 | 74056 | |
| 40 KFM19 | Pr | Pr | 10.5 | 1510 | 14467 | ||
| 41 | Pr | Pr |  |
8c | 20d | 2.566 | |
| 42 KF15372 | Pr | Pr |  |
3e | 430 | 14065 | |
| 43 KW-3902 | Pr | Pr | 1.3 | 380 | 29068 | ||
| 44 I-BW-A844U | Pr |  |
cyclopentyl | 0.23 | 2000 | 870055 | |
| 45 | Pr | Pr |  |
6.9 | 160 | 2370 | |
| 46 8-PT | Me | Me | phenyl | 86 | 850 | 9.918 | |
| 47 8-PST | Me | Me | p-(SO3H)phenyl | 2600 | 1500 | 5.818,64 | |
| 48 PACPX | Pr | Pr |  |
2.5 | 92 | 3718 | |
| 49 XCC | Pr | Pr | 58 | 2200 | 5762 | ||
| 50 XAC | Pr | Pr | 1.2 | 60 | 5062 | ||
| 51 I-PAPA-XAC | Pr | Pr |  |
0.1f | - | ref 108 | |
| 52 m-DITC-XAC | Pr | Pr |  |
2.4 | 343 | 144113 | |
| 53 N-0861 | 10g | 6100g | 61052 | ||||
| 54 CGS 15943 | 21 | 3.3 | 0.1646 | ||||
| 55 CP 66713 | 270 | 21 | 0.07848 | ||||
| 56 CP 68247 | 28 | >100000 | >300048 | ||||
| 57 CPPIQA | 10 | 450 | 4545 | ||||
| 58 APPP | 23 | 35 | 1.561 | ||||
| 59 HTQZ | 3100 | 120 | 0.0461 | ||||
| 60 ADPEP | 4.7 | 3700 | 79050 | ||||
Ki or IC50 values in nM, displacement of [3H]PIA or [3H]CHA in rat brain cortical membranes, unless indicated otherwise.
Displacement of [3H]NECA in rat brain striatal membranes (in the presence of 50 nM CPA), unless indicated otherwise.
Ki for antagonism of adenylate cyclase inhibition in rat adipocytes.
Ki for antagonism of adenylate cyclase activation in human platelets.
Ki or IC50 value for displacement of [3H]CHA in guinea pig forebrain membranes.
Kd in bovine brain cortical membranes.
In bovine brain.
R7 = H, unless noted.
Not selective in adenylate cyclase assays.
In general, for high affinity at adenosine receptors the following criteria have to be fulfilled: adenosine receptor antagonists are (i) flat, (ii) aromatic or π-electron rich, (iii) nitrogen-containing heterocycles, often 6:5-fused. Hydrophobic substituents may greatly enhance affinity, whereas hydrophilic substituents are usually not tolerated, which renders many of the high-affinity antagonists quite insoluble in water. One notable exception to the general pattern is the naturally occurring benzo[b]furan (61) (containing an O rather than an N 6:5-fused heterocycle), which has an affinity of 17 nM in bovine A1 binding53 and may provide an important new lead to further non-xanthine, non-nitrogen-containing adenosine receptor antagonists.
The SAR for xanthines at adenosine receptors is summarized in Figure 1b. Theophylline (1,3-dimethylxanthine) has only moderate affinity and is essentially nonselective (A1, 8.5 µM; A2, 25 µM).18 Increasing chain length at positions 1 and 3 increases affinity. 1,3-Dipropyl substitution is optimal, resulting in a 19-fold increase in affinity at A1 receptors.54 In the 3-position, the A1 receptor has considerably more bulk tolerance than the A2 receptor. For example, BW-A844U (l-propyl-3-(4-amino-3-iodophenethyl)-8-cyclopentylxanthine, 44) is 8700-fold A1 selective, whereas the parent compound, CPX (1,3-dipropyl-8-cyclopentylxanthine, 39) is equipotent yet only 740-fold selective.55,56 Substitutions at the 7-position are usually not favorable57,58 while 9-substitutions are detrimental to affinity.57,59 Although certain alkyl modifications at the 1-, 3-, and 7-positions (e.g. DMPX, 36) may favor A2 affinity to some extent,60 no xanthine (or non-xanthine) antagonists with appreciable potency and selectivity at A2 receptors have yet been reported. Thus the lack of potent, selective A2-receptor antagonists remains a major obstacle in the characterization of the function of adenosine A2 receptors.
Phenyl or cycloalkyl substitutions in the 8-position may yield highly potent and, in many cases, also highly A1-selective antagonists including PACPX (48),8,18 CPX (39),61 and XAC (50).62,63 These compounds have proven valuable as radioligands and/or pharmacological tools, and 8-(p-sulfophenyl)theophylline (8-PST, 47)64 is a useful peripheral acting antagonist. Some interesting newer 8-substituted xanthines include 8-(dicyclopropylmethyl)-1,3-dipropylxanthine (KF15372, 42) which is even more potent and A1 selective than CPX in guinea pig forebrain.65 8-[trans-4-(Acetamidomethyl)cyclohexyl]-l,3-dipropylxanthine (41) has surprisingly high potency at A2 receptors, unlike other cycloalkylxanthines (Ki for antagonism of adenylate cyclase activity in rat adipocytes (A1, 8 nM) or human platelets (A2, 20 nM)).66 KFM 19 (40, (±)-8-(3-oxocyclopentyl)-l,3-dipropylxanthine) is a potent A1-selective compound with sufficient aqueous solubility to display good bioavailability. It is currently under development as a potential cognition enhancer.67 KW 3902 (43, 8-noradamant-3-yl-1,3-dipropylxanthine, A1 = 1.3 nM, A2 = 380 nM) has potent diuretic and renal protective activity.68
One of the first non-xanthine adenosine receptor antagonists identified was CGS 8216,69 a pyrazoloquinoline whose predominant activity was at the benzodiazepine receptor. Subsequent SAR work on this novel heterocycle led to the identification of the triazoloquinazoline, CGS 15943 (54), a potent adenosine receptor antagonist with 7-fold selectivity and an IC50 of 3 nM at the A2 receptor.46 CGS 15943 while under development as a potential antiasthmatic was found to be a potent skin irritant and was discontinued.
Another series of tricyclic antagonists, the triazoloquinoxalines,48 which for a time were in clinical trials as antidepressants, were later found to be adenosine antagonists, including, in dependence of the ring substitutions, both A2-receptor-selective adenosine antagonists, such as CP 66713 (55) and some highly A1-receptor-selective antagonists, such as CP 68247 (56).48
A third series of tricyclic non-xanthine antagonists was developed on the basis of a computer model (Figure 4) for the steric, electrostatic, and hydrophobic features of A1 receptor antagonists.49 This model, which assumes that xanthines bind to the receptor backward, i.e. the purine ring of xanthines overlaps the purine ring of adenosine but is upside down, identifies regions with a distinct pattern of negative and positive electrostatic potential in antagonists, as well as regions where hydrophobic substituents may greatly enhance affinity. Furthermore, it is hypothesized that a nitrogen atom at the position corresponding to N7 in adenosine may act as a hydrogen-bond acceptor. A series of imidazoquinolines, designed and synthesized to test this model, resulted in some potent adenosine antagonists, including CPPIQA (57) (A1, 10 nM; A2, 450 nM).49 A second computer-generated model of the antagonist binding site of the adenosine receptor assumes that N6 substituents of agonists and C8 substituents of xanthine antagonists bind to the same region of the receptor.70 This model has correctly predicted the receptors’ preference for the R-isomer of 8-(2-phenyl-1-methylethyl)xanthines, e.g. 45, analogous to its preference for R-PIA. Further studies will be required to reconcile these two seemingly incompatible models.
Figure 4.

Computer-generated model of the antagonist binding site of the adenosine A1 receptor. Indicated is the molecular electrostatic potential (at the +5 and −5 kcal/mol level) in the plane of the 6:5-fused heterocycle that is common to xanthines and many non-xanthine adenosine antagonists. R (corresponds to the N6 position of adenosine) and dotted lines indicate regions where hydrophobic substitution may enhance affinity. N7 is thought to act as a hydrogen bond acceptor (adapted from ref 49).
Similarities in the shape and charge distribution of the tricyclic antagonists suggest a common binding mode, although the SAR’s differ to some extent. In each case, the basic ring structure is nonselective or slightly A2 selective, but this may be altered by various substitutions. Whereas in the triazoloquinoxaline series substitution at the exocyclic amino group may impart very high A1 affinity and selectivity (e.g. CP-68,247 56),48 analogous substitution in the triazoloquinazolines often diminishes rather than enhances A1 affinity.47 On the other hand, the 2-furyl group is essential for the high affinity of CGS 15943,46 but analogous substitution in the triazoloquinoxaline series is not feasible because of the presence of a tertiary nitrogen atom in the corresponding position. Interestingly, in the imidazoquinoline series substituents at both positions can enhance A1 affinity considerably, although the effects are not necessarily additive.49
This lack of additivity has been shown to occur in a number of cases in both adenosine agonists and antagonists. Explanations for this phenomenon include (i) induction of a conformational change in the receptor by one substituent, thereby altering the binding environment for the second substituent, (ii) dissimilar or even multiple binding modes for similar compounds, (iii) direct interactions between nearby sites, and (iv) a “loose fit” concept, which assumes that the heterocyclic antagonist pharmacophore is amply accommodated by the receptor; high affinity would then be achieved by a substituent that anchors the heterocycle to the receptor, at the same time hampering the optimal orientation for a substituent at another site.49 The latter explanation agrees well with the seemingly endless array of structural variations of heterocycles that the receptor accepts as antagonists.
Solubility has been a major issue with both xanthine and non-xanthine heterocycle antagonists of the adenosine receptor and has led to anomalous biological results as in the case of CP-66,713 (55).48 While 8-phenyl substitution in the xanthine pharmacophore increases receptor blocking activity, it also markedly decreases solubility. While 8-phenyltheophylline (46) is 100-fold more active at the A1 receptor than theophylline (2), it is some 6000-fold less soluble.71 Addition of charged side chains to the 8-phenyl substituent, as in the case of 8-PST (47),64 XCC (49), and XAC (50),62,63 or the substitution of a cyclopentyl for the phenyl group can improve solubility, as for cyclopentyltheophylline (CPT, 38).61 Bruns, in developing a ratio concept relating solubility to receptor affinity,71 has proposed that the greater the ratio, the more optimal the compound.
Indirect Modulation of Adenosine Function
In addition to the design of ligands that directly interact with adenosine receptors, the actions of adenosine may also be potentiated via inhibition of uptake,72–74 by allosteric modulation of receptor function,75,76 or by compounds that act to enhance the free levels of adenosine.77 A potential permissive role wherein A2-receptor activation can influence A1-mediated responses has also been postulated.78,79 The precise mechanism for this effect is unknown as there appears to be no clear SAR for the observed effects.79 It is noteworthy, however, in regard to the CNS effects of adenosine agonists, that agents that increase CAMP also have the potential to increase blood–brain barrier permeability.80 Thus “classical” A2-receptor agonists have the potential to increase the activity of A1 ligands by increasing their access to the brain.
Dipyridamole (63) (Figure 5), mioflazine, and its analogue, R 75231 (62), are adenosine transport inhibitors that have clinical utility as coronary vasodilators and hypnotic agents.81,82 PD 81,723 (64) and related 3-benzoylthiophenes are selective enhancers of the binding of adenosine to A1 receptors.75,76 They also potentiate the inhibitory effects of the purine in adenylate cyclase76 and electrophysiological paradigms.83 By analogy with the benzodiazepines at the benzodiazepine–GABA-A receptor complex84 and various modulators of the N-methyl-d-aspartate receptor complex,85 it has been postulated that an adenosine binding enhancer would have therapeutic potential with fewer side effects than administered agonists, in that it amplifies the action(s) of endogenous, situationally generated adenosine.77 AICA riboside (acadesine, 65) is the prototypic adenosine “site and event specific” potentiator which is in phase III clinical trials for cardiac ischemia86 with additional indications in type II diabetes. An orally active analogue, GP-1-468-3, is also under development.87 Adenosine deaminase inhibitors like deoxycoformycin (66)88 may also have therapeutic potential in a manner similar to AICA riboside although the in vivo efficacy of such agents requires considerable improvement.89
Figure 5.

Agents for indirect modulation of adenosine function through transport (62 and 63) or metabolic processes (65 and 66), or at an allosteric site on the A1 receptor (64). See text for description.
The anti-inflammatory actions of the anticancer agent, methotrexate, have been tentatively related to its ability to elevate endogenous extracellular adenosine levels,90 resulting in a putative reduction in neutrophil free radical formation presumably due to A2-receptor activation.91 The molecular target for the actions of methotrexate is thought to be via the AICA riboside formed due to methotrexate inhibition of AICA riboside transformylase.90
ATP Receptor Ligands
Progress in the related area of purine nucleotide neurotransmission, specifically P2-receptor targets, has been hampered by the lack of selective antagonists, a lack of availability of those agonists generally accepted as efficacious, and the lack of general binding assays. ATP receptors can be classified into four major subclasses (Table II) termed P2x, P2y, P2t, and P2z92. The P2t receptor is actually an ADP rather than ATP receptor. Furthermore, a UTP (uridine triphosphate) receptor, distinct from the adenine nucleotide receptors already described, has been termed P2u or nucleotide receptor.93,94 A P3 receptor has also been postulated.95,96 Preliminary evidence suggests the existence of a “dipurinergic” cell surface recognition site for the “alarmone”, Ap4A (Figure 6), a dinucleotide tetraphosphate.97
Figure 6.

Structure of Ap4A.
The concept that ATP, an intimate component of cellular energy as well as the other energy-rich nucleotides, could function as a neuromodulatory substance was not widely accepted until very recently. ATP is the principal agent thought to be responsible for the phenomenon of “nonadrenergic, noncholinergic” (NANC) neurotransmission.98
Current concensus would support a role for ATP as a neuroeffector agent although the physiological and pathophysiological function has yet to be determined. Roles as an anticancer agent99 and in the treatment of shock100 have been suggested.
Second Messenger Systems
The effects of adenosine on cell function were initially described in terms of agonist actions on cAMP production, A1-receptor activation leading to adenylate cyclase inhibition and A2-receptor activation leading to adenylate cyclase stimulation. Subsequently, multiple second messenger systems for adenosine have been identified including stimulation of phosphatidylinositol (PI) turnover, potassium and calcium channel activation, and cyclic GMP formation.101,102 The latter effect may occur via modulation of nitric oxide formation103 although this is somewhat controversial.104 All of these second messenger systems have the potential to elicit multiple effects within the cell leading to complex effects on cell responsiveness. Adenosine acts both pre- and postsynaptically to alter cell excitability and to suppress the release of a diverse number of neuromodulators, such as excitatory amino acids, acetylcholine, dopamine, and norepinephrine. Thus, adenosine acts as paracrine effector agent2 with the ability to antagonize the effects of many stimulatory neurotransmitters by inhibiting their release. The presynaptic actions of adenosine appear to predominate and may be more reflective of the actions observed with exogenously introduced adenosine agonists.
Receptor Structural Models
Chemical Approaches
Adenosine receptors have been affinity labeled using agonist and antagonist probes, often containing high specific radioactivity, carrier-free iodine-125 to facilitate identification of the labeled receptor. Two approaches have been used (i) indirect photoaffinity crosslinking, in which a radiolabeled receptor ligand containing a chemically reactive group, i.e. aryl amine, is first bound to the receptor and then exposed to a bifunctional reagent, such as SANPAH (N-succinimidyl 6-(4′-azido-2′-nitrophenylamino)hexanoate) designed both to acylate amines and generate a reactive nitrene; and (ii) direct photoaffinity labeling, where the ligand is preactivated for photolysis, leading to reaction with the receptor, that affords a higher percentage of available receptor being labeled.
The A1 receptor was labeled by photoaffinity crosslinking using the ligand [125I]APNEA (14, Table II) in combination with the cross-linker SANPAH105 and by converting the amine of [125I]APNEA, in advance, to a photolabile azido group, yielding [125I]AZPNEA. In both cases, a protein of molecular weight 36000 was labeled. The azide derived from N6-(4-amino-3-iodobenzyl)adenosine (12, [125I]ABA)106 and 2-azido-N6-[2-(p-hydroxyphenyl)-1-methylethyl]adenosine ([125I]AHPIA)107 (similar to 10, except that R1 = N3) have also been used to photoaffinity label the A1 receptor with similar results. Photoaffinity labeling the A1 receptor with an antagonist ligand, (see also ref 55) [125I]PAPA-XAC (51, Table IV), gave a molecular weight of 38000.108
Photoaffinity labeling of bovine brain A1 receptors, using azido-derivatized agonists (AZPNEA) and antagonists (preformed PAPA-XAC-SANPAH and azido-PAPA-XAC) in parallel, followed by partial peptide mapping identified identical peptide fragments when proteolysis was performed following photolabeling and denaturation.109 When ligands were first bound to the receptor in membranes, followed by limited proteolysis and irradiation, distinct and different peptide fragments were obtained providing evidence for different conformational states for agonist-occupied A1 receptors compared to the antagonist-occupied A1 receptors. These differences probably relate to the ability of an agonist to initiate a transmembrane signal, whereas an antagonist binds to the receptor without producing an effect.
The A2 receptor in bovine striatum was affinity labeled using the agonist [125I]PAPA-APEC (33) and found to be a single glycoprotein of molecular weight 45000.35 The A2 receptor in human striatum,110 rat PC12 cells,111 and frog erythrocytes111 have molecular weights in the 44000–47000 range, while that in the DDT1MF-2 (Syrian hamster) cell line112 has a molecular weight 42000. Furthermore human and rabbit striatal A2 receptors were found to undergo proteolytic cleavage,35,110 resulting in fragments of MW 37000 and 38000, respectively.
Results similar to those obtained with photoaffinity labels are found with chemical cross-linking agents. These studies use ADAC (18), APEC (32), and XAC (50), coupled to bifunctional alkylating and acylating cross-linking reagents, such as m- or p-phenylene diisothiocyanate (DITC) to provide a chemically reactive isothiocyanate group (NCS) on the ligand. For example, the A1-receptor protein is specifically labeled by DITC-XAC with MW 38000.113 In preliminary studies, the m- and p-DITC (34) conjugates of APEC appear to inhibit A2-adenosine receptors irreversibly.165
Purification to apparent homogeneity of cortical A1 receptors from rat114 and bovine115 brain have been achieved through affinity chromatography with agarose-coupled XAC.
Molecular Biology Approaches
Molecular modeling approaches concerning the mode of interaction of ligands with the adenosine receptor have of necessity focused on the ligand SAR (Figures 2 and 4), in the absence of knowledge regarding receptor structure. However, the recent cloning and sequencing of canine,3 rat,116,117 and bovine (G. Stiles, personal communication) A1 receptors and canine3,4 A2 receptors has now yielded valuable information on some of the structural features of the receptors. A schematical model is shown in Figure 7.
Figure 7.

A proposed model (121) for adenosine receptors deduced from the primary sequences, showing features common to both A1 and A2 receptors, including the seven transmembrane helices typical of G-protein-coupled receptors (I–VII), three extracellular loops (EI–III), three cytoplasmic loops (CI–III), two histidinyl residues (H) (possibly involved in ligand binding) a sodium binding site (Na), and sites for phosphorylation of serine and threonine residues (P) and glycosylation. Phosphorylation sites on the C-terminus apply to the A2-sequence only. A site for acylation (Ac) applies to A1 receptors only. The C-terminal sequences (beyond H-7) are approximately 34 and 119 residues in length for canine A1 and A2 receptors, respectively.
The adenosine receptor sequences conform with the seven transmembrane domain topology commonly predicted for G-protein-coupled receptors, with the amino terminus on the extracellular and the carboxy terminus on the cytoplasmic side of the membrane. The seven membrane spanning regions (designated H-I to -VII) likely consist of right-handed α-helices that are interconnected by three extracellular loops (E-I to -III) and three cytoplasmic loops (C-I to -III). Although adenosine receptors are known to be glycosylated,118–120 there are no potential glycosylation sites present near the amino terminus, in contrast to other G-protein-coupled receptors. Putative glycosylation sites have been identified on E-II: Asn159 for canine A1 receptors and Asn145 and Asn154 for canine A2 receptors.121 Cytoplasmic domains contain multiple serine and threonine residues that are potential substrates for phosphorylation by protein kinase A, protein kinase C, caseine kinase 2, and β-adrenoceptor kinase,121, which may be relevant to receptor desensitization mechanisms. Cytoplasmic domains may also be involved in G-protein interactions. The A1 receptor contains a potential site for palmitoylation in the carboxy terminus (canine A1 Cys309) which is of potential interest given the presence of a similar palmitoyl group in the β2 adrenergic receptor122 and the visual pigment rhodopsin123 is required for interaction with G-proteins. Both A1 and A2 receptors, which are known to be regulated by Na+,124 contain putative sites for interaction with Na+ at the cytoplasmic site of H-II (canine A1, Asp55; canine A2, Asp52). Two different histidine residues have been implicated in ligand binding to both A1125 and A2165 receptors, and a putative mode of interaction of histidine residues with adenosine receptor agonists and antagonists has been proposed.126 Likely candidates for these interactions are two conserved histidine residues in H-VI and H-VII (canine A1 His251 and His278).121 Site-directed mutagenesis and chemical modification studies together with recombinant DNA techniques5 will aid in understanding the nature of the ligand binding site(s), the potential role of glycosylation, phosphorylation, acylation, palmitoylation, and regulation by Na+ in the physiological aspects of receptor function, as well as in establishing which domains are important in interacting with G-proteins.
Therapeutic Targets and Future Aspects
Considerable effort has been expended in defining more precisely a physiological role for adenosine receptor related processes in the cardiovascular system.13,16 A broader based albeit somewhat more circumstantial effort has focused on the role(s) of the purine nucleoside in the central nervous system.127 In the latter system, such studies have frequently suffered from a highly reductionistic approach, wherein the effects of adenosine agonists and/or antagonists have been studied with only a single endpoint, biochemical or behavioral, and often only a single compound. In light of the largely theoretical basis for the etiology of many of the drug classes used to treat CNS disorders128 and the multiple actions of exogenously applied adenosine agonists or antagonists, the results obtained in such limited studies have tended to confuse rather than delineate the role of adenosine (and ATP) in the pathophysiology of both psychiatric and neurological disorders.
Adenosine has been implicated in the mechanisms of drugs effective in schizophrenia, depression, epilepsy, cognition, and anxiety129 reflecting both pre- and postsynaptic effects on neuronal function, the former via inhibition of transmitter release.2 Excitatory neurotransmission process are more sensitive to the inhibitory effects of the purine than inhibitory ones.166 Purinergic mechanisms may also be involved in processes related to pain, ischemia, and regulation of cerebral blood flow and substance abuse.129,130 CI 936 (15), a potent A2 receptor agonist, was profiled preclinically as an antipsychotic agent producing its actions via modulation of striatal dopamine systems.131 The compound entered clinical trials but was withdrawn.7 It is unknown whether clinical efficacy was observed.
Adenosine is an effective antiepileptic agent.132 Its endogenous production as the result of the ischemic episodes accompanying epileptic fits has led to the proposal that the purine functions as an endogenous anticonvulsant agent.133 Adenosine agonists prolong survival and improve cellular morphology, in particular in the hippocampus, in animal models of cerebral ischemia134 and represent one of the numerous experimental approaches to stroke therapy.135
Antagonists are central stimulants as evidenced by caffeine being the most widely used nonprescription/nonillicit drug currently in use.8 Attempts to develop xanthine antagonists as cognition enhancers or for use in senility have not been successful due to side effect liability as proconvulsants, cardiotonics, or diuretics. The CPX analogue KFM 19 (40), an A1-receptor-selective antagonist, is apparently devoid of these side effects67 and is being developed as a cognition enhancer with potential in Alzheimer’s disease. CPX can activate chloride flux in cell culture, suggesting a potential use in the treatment of cystic fibrosis.168
Selective A1 antagonists have protective effects in various models of renal failure. KW-3902 (43) is an effective diuretic at doses as low as 10 µg/kg.65,68 Targeting of adenosine antagonists to the kidney using a prodrug approach offers a potential approach to avoiding side effects.136 Similarly systems for the delivery of adenosine analogues to the brain are under development.137
Various xanthines have been shown at high concentration (1 mM) to increase nerve growth factor (NGF) production in cell culture system.138 Interestingly, Hoechst’s pentoxyfylline at similar concentrations is being used opportunistically in the treatment of AIDS-related infections because of its ability to modulate TNF (tumor necrosis factor) formation.139 Pentoxyfylline has also been reported to inhibit lymphocyte interleukin-2 receptor expression.140 The effects of this xanthine on superoxide anion production appear to occur independently of adenosine receptor antagonism.169
Adenosine is implicated in events related to both inflammation and the immune response.141 It is known to modulate neutrophil function via A2-receptor activation91 and also affect the interactions between B and T cells.141 The arthritic process may also involve a purinergic component142 although it is unclear which receptor subtype is involved. Adenosine levels in synovial fluid are reduced in rheumatoid arthritis.143
The purine also has effects on the pituitary–adrenocortical axis,144,145 increasing the release of a number of hormones, including dopamine, as part of a stress-related response.
Data on the potential therapeutic applications of compounds interacting with the various classes of P2 receptor is yet in its early stages due to a paucity of suitable ligands and their limited availability. The effectiveness of antagonists at these receptors is also somewhat controversial as is the selectivity of the limited number of agonists. Nonetheless, knowledge related to the extracellular actions of ATP and other nucleotides is increasing. ATP can function as a growth factor,146 acting to modulate the cytotoxic actions of TNF, an effect that appears to involve a permissive role related to P1-receptor activation. Most recently, aerosolized ATP or UTP, in conjunction with the sodium channel blocker, amiloride, has been reported to stimulate chloride secretion in cystic fibrosis patients. The effect probably involves interactions with nucleotide receptors.163,168 Clinical efficacy remains to be shown.
Recent studies147 with CGS 21680 (31) and CGS 22989 (25) have shown that the hypotensive actions of these A2 receptor agonists can be attenuated following a 2-week chronic administration regimen using osmotic pumps, an effect paralleled by a 25–32% decrease in brain A2-receptor number. Whether this response can be generalized to all adenosine agonists and is reflective of the situation that might occur with a repeated PO administration as opposed to steady-state administration remains to be determined.
In reviewing the various effects of adenosine in order to prioritize realistic options related to therapeutic targeting, it is important to recognize the limitations of purinergic therapy due to the ubiquitous nature of adenosine (and ATP) effects. Adenosine antagonists effective as cognition enhancers have potential use in the elderly population, a group that would not be especially tolerant of the cardiac stimulant or renal properties of such compounds. Similarly, the use of adenosine agonists as hypotensive agents may be anticipated to cause direct effects on the renin angiotensin system as well as elicit sedation via central mechanisms. The acceptance of such side effects in adenosine receptor ligands is dependent on the level of improvement ascribable to the use of a purinergic agent and the degree of unmet medical need. Thus agents that act to mimic (agonists) or potentiate (AICA riboside, uptake inhibitors) adenosine are targeted toward acute, life threatening situations such as SVT, reperfusion injury, stroke, and, perhaps, epilepsy. The use of such agents for the potential treatment of hypertension in light of the large number of highly efficacious and essentially side effect free agents currently available, consequently invites ridicule in the absence of any additional beneficial actions.13 The development of adenosine antagonists as selective cognition enhancers may be feasible should agents such as KFM 19 (40) prove to be selective for the CNS.
Yet the potential involvement of adenosine agonists and antagonists in inflammation, immunoregulation, and neuroendocrine function, areas in which pathophysiological mechanisms and mediators are just beginning to be understood and for which medications (like theophylline and β-receptor agonists in asthma) leave much to be desired, represents an important new arena for the study of purinergic mechanisms. The potential role of adenosine, and its related nucleotides, as paracrine homeostatic modulatory entities or as autocoids,148 may therefore also be reflected in the nature of the systems in which these agents act both physiologically and pathophysiologically, systems where malfunction is discrete and as global as the availability of the paracrine effector itself. An additional layer of complexity may also be reflected in the nature of the purinergic cascade149 where ATP, en route to adenosine via successive dephosphorylation steps produces products with discrete receptor selectivity and functionality.
While consensus related to the potential existence of A1a, A1b, and A3 receptors has yet to be obtained, it appears probable, baaed on experience from other transmitter systems that receptor subtypes represent the key to developing selective drugs and to understanding basic receptor functionality. Whether present data supporting the existence of adenosine receptor subtypes relates to the uniqueness of the experimental situations in which they were defined42 or is reflective of more generalized nuances of adenosine-elicited responses requires a more systematic evaluation of the effects of a number of agonists and antagonists in related systems and/or classification of receptors through cloning. As in the serotonin receptor field in the late 1970s, to which the adenosine area may loosely be compared, the major breakthroughs both from the functional and clinical viewpoint will be the selective ligands for the receptor subclasses. It is highly noteworthy that it took a decade and a considerable aggregate effort both within the pharmaceutical industry and in academia targeted toward A2-receptor-selective agonists to develop CGS 21680 (31), CGS 22492 (24), CHEA (26), and MPEA (27), from CV1808. The momentum that has been attained will hopefully result in a better understanding of the role of adenosine as the “signal of life”148 in tissue function and result in new classes of therapeutic agents, that acting via purinergic receptors, will effectively treat the disease challenges of the 21st century.
Acknowledgment
The authors would like to thank John Daly, Ray Olsson, and John Francis for preprints of their work.
Footnotes
References
- 1.Daly JW. Adenosine receptors: targets for future drugs. J. Med. Chem. 1982;25:197–207. doi: 10.1021/jm00345a001. [DOI] [PubMed] [Google Scholar]
- 2.Williams M. Adenosine: the prototypic neuromodulator. Neurochem. Intern. 1989;14:249–264. doi: 10.1016/0197-0186(89)90051-x. [DOI] [PubMed] [Google Scholar]
- 3.Libert F, Schiffmann S, Lefort A, Parmentier M, Gerard C, Dumont JE, Vanderhaegen JJ, Vassart G. The orphan receptor cDNA RDC7 encodes an A1 adenosine receptor. EMBO J. 1991;10:1677–1682. doi: 10.1002/j.1460-2075.1991.tb07691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maenhaut C, van Sande SJ, Libert F, Abramowicz M, Parmentier M, Vanderhaegen JJ, Dumont JE, Vassart G, Schiffmann S. RDC8 codes for an adenosine A2 receptor with physiological constitutive activity. Biochem. Biophys. Res. Commun. 1990;173:1169–1178. doi: 10.1016/s0006-291x(05)80909-x. [DOI] [PubMed] [Google Scholar]
- 5.Hollenberg MD. Receptor triggering and receptor regulation: Structure–activity relationship from the receptor’s point of view. J. Med. Chem. 1990;33:1275–1281. doi: 10.1021/jm00167a001. [DOI] [PubMed] [Google Scholar]
- 6.Trivedi BK, Bridges AJ, Bruns RF. Structure–activity relationships of adenosine A1 and A2 receptors. In: Williams M, editor. Adenosine and Adenosine Receptors. Humana: Clifton, NJ; 1990. pp. 57–103. [Google Scholar]
- 7.Jacobson KA, Trivedi BK, Churchill PC, Williams M. Novel therapeutics acting via purine receptors. Biochem. Pharmacol. 1991;41:1399–1410. doi: 10.1016/0006-2952(91)90555-j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williams M. Adenosine antagonists as therapeutic agents. Med. Res. Rev. 1989;9:219–243. doi: 10.1002/med.2610090205. [DOI] [PubMed] [Google Scholar]
- 9.Pantely GA, Bristow JD. Adenosine. Renewed interest in an old drug. Circulation. 1990;82:1854–1856. doi: 10.1161/01.cir.82.5.1854. [DOI] [PubMed] [Google Scholar]
- 10.Verani MS, Mahmanian JJ, Hixson JB, Boyce TM, Staudacher RA. Diagnosis of coronary artery disease by controlled coronary vasodilation with adenosine and thallium-201 in patients unable to exercise. Circulation. 1990;82:80–87. doi: 10.1161/01.cir.82.1.80. [DOI] [PubMed] [Google Scholar]
- 11.Ladey RD, Rhee JW, Van Wylen DGL, Mentzer RM., Jr Adenosine A1 receptor mediated protection of the globally ischemic isolated rat heart. J. Mol. Cell. Cardiol. 1990;22:39–47. doi: 10.1016/0022-2828(90)90970-d. [DOI] [PubMed] [Google Scholar]
- 12.Pitarys CJ, II, Virmani R, Vildibill HD, Jackson EK, Forman MB. Reduction of myocardial reperfusion injury by intravenous adenosine administered during the early reperfusion period. Circulation. 1991;83:237–247. doi: 10.1161/01.cir.83.1.237. [DOI] [PubMed] [Google Scholar]
- 13.Williams M. Purinergic pharmaceuticals for the 1990s. Nucleosides Nucleotides. 1991;10:1087–1089. [Google Scholar]
- 14.Drury AN, Szent-Györgyi A. The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J. Physiol. (Lond) 1929;68:213–237. doi: 10.1113/jphysiol.1929.sp002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Honey RM, Ritchie WT, Thompson WAR. The action of adenosine upon the human heart. Quart. J. Med. 1930;23:485–490. [Google Scholar]
- 16.Olsson RA, Pearson JD. Cardiovascular purinoceptors. Physiol. Rev. 1990;70:761–845. doi: 10.1152/physrev.1990.70.3.761. [DOI] [PubMed] [Google Scholar]
- 17.Hamprecht B, Van Calker D. Trends Phurmacol. Sci. 1985;6:153–154. [Google Scholar]
- 18.Brune RF, Lu GH, Pugsley TA. Characterization of the A2 adenosine receptor labeled by [3H]NECA in rat striatal membranes. Mol. Pharmacol. 1986;29:331–346. [PubMed] [Google Scholar]
- 19.Gustaffson LE, Wiklund CU, Wiklund NP, Stelius L. Subclassification of neuronal adenosine receptors. In: Jacobson KA, Daly JW, Manganiello V, editors. Purines in Cellular Signaling. Targets for New Drugs. Springer Verlag: New York; 1990. pp. 200–205. [Google Scholar]
- 20.Ribeiro JA, Sebastião AM. Adenosine receptors and calcium: basis for proposing a third (A3) adenosine receptor. Prog. Neurobiol. 1986;26:179–209. doi: 10.1016/0301-0082(86)90015-8. [DOI] [PubMed] [Google Scholar]
- 21.Ferkany JW, Valentine HL, Stone GA, Williams M. Adenosine A1 receptors in mammalian brain: species differences in their interactions with agonists and antagonists. Drug Dev. Res. 1986;9:85–93. [Google Scholar]
- 22.Stone GA, Jarvis MF, Sills MA, Weeks B, Snowhill EW, Williams M. Species differences in high-affinity adenosine A2 binding sites in striatal membranes from mammalian brain. Drug Dev. Res. 1988;15:31–46. [Google Scholar]
- 23.Weiner HL, Craddock-Royal B, Maaynai S. Tentative subclassification of the adenosine A1 (AD A1) receptor in mammalian hippocampus. Abstr. Soc. Neurosci. 1990;16:33.8. [Google Scholar]
- 24.Bazil CW, Minneman KP. An investigation of the low intrinsic activity of adenosine and its analogs at low affinity (A2) adenosine receptors in rat cerebral cortex. J. Neurochem. 1986;47:547–553. doi: 10.1111/j.1471-4159.1986.tb04534.x. [DOI] [PubMed] [Google Scholar]
- 25.Lohse M, Klotz KN, Schwabe U. Mechanism of A2 adenosine receptor activation. I. Blockade of A2 adenosine receptors by photoaffinity labeling. Mol. Pharmacol. 1991;39:517–523. [PubMed] [Google Scholar]
- 26.Lohse MJ, Klotz KN, Schwabe U, Cristalli G, Vittori S, Grifantini M. 2-Chloro-N6-cyclopentyladenosine: a highly selective agonist at A1 adenosine receptors. Naunyn Schmiedebergs’ Arch. Pharmacol. 1988;337:687–689. doi: 10.1007/BF00175797. [DOI] [PubMed] [Google Scholar]
- 27.van Galen PJM, Leusen FJJ, IJzerman AP, Soudijn W. Mapping the N6-region of the adenosine A1 receptor with computer graphics. Eur. J. Pharmaco1.-Mol. Pharmacol. Sect. 1989;172:19–27. doi: 10.1016/0922-4106(89)90041-2. [DOI] [PubMed] [Google Scholar]
- 28.Taylor MD, Moos WH, Hamilton HW, Szotek DS, Patt WC, Badger EW, Bristol JA, Bruns RF, Heffner TG, Mertz TE. Ribose-modified adenosine analogues as adenosine receptor agonists. J. Med. Chem. 1986;29:346–353. doi: 10.1021/jm00153a008. [DOI] [PubMed] [Google Scholar]
- 29.Daly JW, Padgett W, Thompson RD, Kusachi S, Bugni WJ, Olsson RA. Structure-activity relationships for N6-substituted adenosines at a brain A1-adenosine receptor with a comparison to an A2-adenosine receptor regulating coronary blood flow. Biochem. Pharmacol. 1986;35:2467–2481. doi: 10.1016/0006-2952(86)90042-0. [DOI] [PubMed] [Google Scholar]
- 30.Trivedi BK. Structure-activity relationships for adenosine agonists. In: Jacobson KA, Daly JW, Manganiello V, editors. Purines in Cellular Signaling. Targets for New Drugs. Springer Verlag: New York; 1990. pp. 136–145. [Google Scholar]
- 31.Ortwine DF, Bridges AJ, Humblet C, Trivedi BK. Adenosine agonists. Characterization of the N6-region of the adenosine A2 receptor via molecular modeling techniques. In: Jacobson KA, Daly JW, Manganiello V, editors. Purines in Cellular Signaling. Targets for New Drugs. Springer Verlag: New York; 1990. pp. 152–157. [Google Scholar]
- 32.Kawazoe K, Matsumato M, Tanabe S, Fujiwara M, Yanagimoto M, Hirata M, Kakiuchi K. Coronary and cardiohemodynamic effects of 2-phenyl-aminoadenosine (CV1808) in anaesthetized dogs and cats. Arzneim. Forsch. 1980;30:1083–1087. [PubMed] [Google Scholar]
- 33.Hutchison AJ, Williams M, de Jesus R, Yokoyama R, Oei HH, Ghai GR, Webb RL, Zoganas HC, Stone GA, Jarvis MF. 2-(Arylalkylamino)adenosin-5’-uronamides: a new class of highly selective adenosine A2 receptor ligands. J. Med. Chem. 1990;33:1919–1924. doi: 10.1021/jm00169a015. [DOI] [PubMed] [Google Scholar]
- 34.Francis JE, Webb RL, Ghai GR, Hutchiaon AJ, Moskal MA, de Jesus R, Yokoyama R, Rovinaki SL, Contrado N, Dotson R, Barclay B, Stone GA, Jarvis MF. Highly selective adenosine A2 receptor agonists in a series of N-alkylated 2-aminoadenosines. J. Med. Chem. 1991;34:2570–2579. doi: 10.1021/jm00112a035. [DOI] [PubMed] [Google Scholar]
- 35.Barrington WW, Jacobson KA, Hutchison AJ, Williams M, Stiles GL. Identification of the A2 adenosine receptor binding subunit by photoaffinity crosslinking. Proc. Natl. Acad. Sci. U.S.A. 1989;86:6572–6576. doi: 10.1073/pnas.86.17.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jarvis MF, Schulz R, Hutchison AJ, Do UH, Sills MA, Williams M. [3H] CGS 21680, a selective A2 adenosine receptor agonist ligand directly labels A2-receptors in rat brain. J. Pharmacol. Exp. Ther. 1989;251:888–893. [PubMed] [Google Scholar]
- 37.Ueeda M, Thompson RD, Arroyo LH, Olsson RA. 2-Alkoryadenosines: potent and selective agonists at the coronary artery A2 adenosine receptor. J. Med. Chem. 1991;34:1334–1339. doi: 10.1021/jm00108a014. [DOI] [PubMed] [Google Scholar]
- 38.Ueeda M, Thompson RD, Arroyo LH, Olsson RA. 2-Aralkoxyadenosines: potent and selective agonists at the coronary artery A2 adenosine receptor. J. Med. Chem. 1991;34:1340–1344. doi: 10.1021/jm00108a015. [DOI] [PubMed] [Google Scholar]
- 39.Ueeda M, Thompson RD, Padgett WL, Secunda S, Daly JW, Olsson RA. Cardiovascular actions of adenosines, but not adenosine receptors, differ in rat and guinea pig. Life Sci. 1991;49:1351–1358. doi: 10.1016/0024-3205(91)90199-l. [DOI] [PubMed] [Google Scholar]
- 40.Abiru T, Yamaguchi T, Watanabe Y, Kogi K, Aihara K, Matsuda A. The antihypertensive effect of 2-alkynyl-adenosines and their selective affinity for adenosine A2-receptors. Eur. J. Pharmacol. 1991;196:69–76. doi: 10.1016/0014-2999(91)90410-r. [DOI] [PubMed] [Google Scholar]
- 41.Hutchison AJ, Webb RL, Oei HH, Ghai GR, Zimmerman MB, Williams M. CGS 21680C, an A2 selective adenosine agonist with selective hypotensive activity. J. Pharmacol. Exp. Ther. 1989;251:47–55. [PubMed] [Google Scholar]
- 42.Black JW. Should we be concerned about the state of hormone receptor classification? In: Black JW, Jenkinson DH, Gerskowitch VP, editors. Perspectives on Receptor Classification. New York: Liss; 1987. pp. 11–15. [Google Scholar]
- 43.Daly JW, Padgett WL. Agonist activity of 2- and 5′-substituted adenosine analogs and their N6-cycloalkyl derivatives at A1- and A2-adenosine receptors coupled to adenylate cyclase. Biochem. Pharmacol. 1992 doi: 10.1016/0006-2952(92)90616-q. in press. [DOI] [PubMed] [Google Scholar]
- 44.Cristalli G, Franchetti P, Grifantini M, Vittori S, Klotz KN, Lohse MJ. Adenosine receptor agonists: synthesis and biological evaluation of 1-deaza analogues of adenosine derivatives. J. Med. Chem. 1988;31:1179–1183. doi: 10.1021/jm00401a018. [DOI] [PubMed] [Google Scholar]
- 45.Sattin A, Rall TW. The effect of adenosine and adenine nucleotides on the cyclic adenosine 3’, 5’-phosphate content of guinea pig cerebral slices. Mol. Pharmacol. 1970;6:13–23. [PubMed] [Google Scholar]
- 46.Francis JE, Cash WD, Psychoyos S, Ghai G, Wenk P, Friedmann RC, Atkins C, Warren V, Furness P, Hyun JL, et al. Structure-activity profile of a series of novel triazoloquinazoline adenosine antagonists. J. Med. Chem. 1988;31:1014–1020. doi: 10.1021/jm00400a022. [DOI] [PubMed] [Google Scholar]
- 47.Trivedi BK, Brune RF. [1,2,4]-Triazolo[4,3-a]-quinoxalin-4-amines: a new class of A1 receptor selective adenosine antagonists. J. Med. Chem. 1988;31:1011–1014. doi: 10.1021/jm00400a021. [DOI] [PubMed] [Google Scholar]
- 48.Sarges R, Howard HR, Browne RG, Lebel LA, Seymour PA, Koe BK. 4-Amino[1,2,4]triazolo[4,3-a]-quinoxalines. A novel class of potent adenosine receptor antagonists and potential rapid-onset antidepressants. J. Med. Chem. 1990;33:2240–2254. doi: 10.1021/jm00170a031. [DOI] [PubMed] [Google Scholar]
- 49.van Galen PJM, Nissen P, van Wijngaarden I, IJzerman AP, Soudijn W. 1H-imidazo[4,5-c]quinolin-4-amines: novel non-xanthine adenosine antagonists. J. Med. Chem. 1991;34:1202–1206. doi: 10.1021/jm00107a046. [DOI] [PubMed] [Google Scholar]
- 50.Müller CE, Hide I, Daly JW, Rothenhäusler K, Eger K. 7-Deaza-2-phenyladenines: structure-activity relationships of potent A1 selective adenosine receptor antagonists. J. Med. Chem. 1990;33:2822–2828. doi: 10.1021/jm00172a023. [DOI] [PubMed] [Google Scholar]
- 51.Ukena D, Padgett WL, Hong O, Daly JW, Daly DT, Olsson RA. N6-substituted 9-methyladenines: a new class of adenosine receptor antagonists. FEBS Lett. 1987;215:203–208. doi: 10.1016/0014-5793(87)80146-1. [DOI] [PubMed] [Google Scholar]
- 52.May JM, Martin PL, Miller JR. N-0861: A selective A1-adenosine receptor antagonist. FASEB J. 1991;5:1572. [Google Scholar]
- 53.Yang Z, Hon PM, Chui KY, Xu ZL, Chang HM, Lee CM, Cui YX, Wong H, Poon CD, Fung BM. Naturally occurring benzofuran: isolation, structure elucidation and total synthesis of 5-(3-Hydroxypropyl)-7-methoxy-2-(3′-methoxy-4′-hydroxyphenyl)-3-Benzo[b] furancarbaldehyde, a novel adenosine-A1 receptor ligand isolated from Salvia Militorrhiza bunge (Danshen) Tetrahedron Lett. 1991;32:2061–2064. [Google Scholar]
- 54.Daly JW. Adenosine agonists and antagonists. In: Jacobson KA, Daly JW, Manganiello V, editors. Purines in Cellular Signaling. Targets for New Drugs. New York: Springer Verlag; 1990. pp. 3–12. [Google Scholar]
- 55.Patel A, Craig RH, Daluge SM, Linden J. 125I-BW-A844U, an antagonist radioligand with high affinity and selectivity for adenosine A1 receptors, and 125I-azido-BW-A844U, a photoaffinity label. Mol. Pharmacol. 1988;33:585–591. [PubMed] [Google Scholar]
- 56.Bruns RF, Fergus JH, Badger EW, Bristol JA, Santay LA, Hartman JD, Hays SJ, Huang CC. Binding of the A1-selective adenosine antagonist 8-cyclopentyl-1,3-dipropylxanthine to rat brain membranes. Naunyn-Schmiedberg’ss Arch. Pharmacol. 1987;335:59–63. doi: 10.1007/BF00165037. [DOI] [PubMed] [Google Scholar]
- 57.Bruns RF. Adenosine antagonism by purines, pteridines and benzopteridines in human fibroblasts. Biochem. Pharmacol. 1981;30:325–333. doi: 10.1016/0006-2952(81)90062-9. [DOI] [PubMed] [Google Scholar]
- 58.Schwabe U, Ukena D, Lohse MJ. Xanthine derivatives as antagonists at A1 and A2 adenosine receptors. Naunyn Schmiedebergs’ Arch. Pharmacol. 1985;330:212–221. doi: 10.1007/BF00572436. [DOI] [PubMed] [Google Scholar]
- 59.Schneller SW, Ibay AC, Christ WJ, Bruns RF. Linear and proximal benzo-separated alkylated xanthines as adenosine-receptor antagonists. J. Med. Chem. 1989;32:2247–2254. doi: 10.1021/jm00130a004. [DOI] [PubMed] [Google Scholar]
- 60.Daly JW, Padgett WL, Shamim MT. Analogues of caffeine and theophylline: effect of structural alterations on affinity at adenosine receptors. J. Med. Chem. 1986;29:1305–1308. doi: 10.1021/jm00157a035. [DOI] [PubMed] [Google Scholar]
- 61.Bruns RF, Davis RE, Ninteman FW, Poschel BPH, Wiley JN, Heffner TG. Adenosine antagonists as pharmacological tools. In: Paton DM, editor. Adenosine and Adenine Nucleotides. Physiology and Pharmacology. London: Taylor & Francis; 1958. pp. 39–49. [Google Scholar]
- 62.Jacobson KA, Kirk KL, Padgett WL, Daly JW. Functionalized congeners of 1,3-dialkylxanthines: preparation of analogues with high affinity for adenosine receptors. J. Med. Chem. 1986;28:1334–1340. doi: 10.1021/jm00147a038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jacobson KA, Kirk KL, Padgett WL, Daly JW. A functionalized congener approach to adenosine receptor antagonists: amino acid conjugates of 1,3-dipropylxanthine. Mol. Pharmacol. 1986;29:26–133. [PMC free article] [PubMed] [Google Scholar]
- 64.Daly JW, Padgett W, Shamim MT, Butts LP, Waters J. 1,3-Dialkyl-8-(p-sulfophenyl)-xanthines: potent water-soluble antagonists for A1- and A2-adenosine receptors. J. Med. Chem. 1985;28:487–492. doi: 10.1021/jm00382a018. [DOI] [PubMed] [Google Scholar]
- 65.Shimada J, Suzuki F, Nonaka H, Karasawa A, Mizumoto H, Ohno T, Kubo K, Ishii A. 8-(Dicyclopropylmethyl)-1,3-dipropylxanthine: a potent and selective adenosine A1 antagonist with renal protective and diuretic activities. J. Med. Chem. 1991;34:466–469. doi: 10.1021/jm00105a072. [DOI] [PubMed] [Google Scholar]
- 66.Katsushima T, Nieves L, Wells JN. Structure–activity relationships of 8-cycloalkyl-1,3-dipropylxanthines as antagonists of adenosine receptors. J. Med. Chem. 1990;33:1906–1910. doi: 10.1021/jm00169a012. [DOI] [PubMed] [Google Scholar]
- 67.Schingnitz G, Küfner-Mühl U, Ensinger H, Lehr E, Kuhn FJ. Selective A1-antagonists for treatment of cognitive deficits. Nucleosides Nucleotides. 1991;10:1067–1076. [Google Scholar]
- 68.Suzuki F, Shimada J, Mizumoto H, Nonaka H, Ishii A, Karasawa A, Kubo K. KW-3902: A potent and selective adenosine A1 antagonist with renal protective and diuretic activity. Book of Abstracts; National Meeting of the American Chemical Society; Aug 25–30; New York City, NY. Washington, DC: American Chemical Society; 1991. Abstract MEDI112. [Google Scholar]
- 69.Czernik AJ, Petrack B, Kalinsky HJ, Psychoyos S, Cash WD, Tsai C, Rinehart RK, Granat FR, Lovell RA, Brundish DE, Wade R. CGS 8216 receptor binding characteristics of a potent benzodiazepine antagonist. Life Sci. 1982;30:363–372. doi: 10.1016/0024-3205(82)90573-2. [DOI] [PubMed] [Google Scholar]
- 70.Peet NP, Lentz NL, Meng EC, Dudley MW, Ogden AM, Demeter DA, Weintraub HJ, Bey P. A novel synthesis of xanthines: support for a new binding mode for xanthines with respect to adenosine at adenosine receptors. J. Med. Chem. 1990;33:3127–3130. doi: 10.1021/jm00174a004. [DOI] [PubMed] [Google Scholar]
- 71.Bruns RF, Fergus JH. Solubilities of adenosine antagonists determined by radioreceptor assay. J. Pharm. Pharmacol. 1989;41:590–594. doi: 10.1111/j.2042-7158.1989.tb06537.x. [DOI] [PubMed] [Google Scholar]
- 72.Plagemann PGW, Wohlhüter RM. Permeation of nucleosides, nucleic acid bases and nucleotides in animal cells. Curr. Top. Membr. Trans. 1980;14:225–330. [Google Scholar]
- 73.Jarvis SM. Kinetic and molecular properties of nucleoside transporters in animal cells. In: Gerlach E, Becker BF, editors. Topics and Perspectives in Adenosine Research. Berlin: Springer Verlag; 1987. pp. 102–117. [Google Scholar]
- 74.Wiley JS, Brocklebank AM, Snook MB, Jamieson GP, Sawyer WH, Craik JD, Cass CE, Robins MJ, McAdam DP, Paterson AR. A new fluorescent probe for the equilibrative inhibitor-sensitive nucleoside transporter. 5’-S-(2-aminoethyl)-N6-(4-nitrobenzyl)-5’-thioadenosine (SAENTA)-chi 2-fluorescein. Biochem. J. 1991;273:667–672. doi: 10.1042/bj2730667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bruns RF, Fergus JH. Allosteric enhancement of adenosine A1 receptor binding and function by 2-amino-3-benzoylthiophenes. Mol. Pharmacol. 1990;38:939–949. [PubMed] [Google Scholar]
- 76.Bruns RF, Fergus JH, Coughenour LL, Courtland GG, Pugsley TA, Dodd JH, Tinney FJ. Structure–activity relationships for enhancement of adenosine A1 receptor binding by 2-amino-3-benzoylthiophenes. Mol. Pharmacol. 1990;38:950–958. [PubMed] [Google Scholar]
- 77.Gruber HA, Hoffer ME, McAllister DR, Laikind PK, Lane TA, Schmid-Schoenbein GW, Engler RL. Increased adenosine concentration in blood from ischemic myocardium by AICA riboside. Effects on flow, granulocytes, and injury. Circulation. 1989;80:1400–1411. doi: 10.1161/01.cir.80.5.1400. [DOI] [PubMed] [Google Scholar]
- 78.Barraco RA, El-Ridi MR, Parizon M. The adenosine analog 5’-N-ethylcarboxamido adenosine, exerts mixed agonist actions of cardiorespiratory parameters in the intact but not decerebrate rat following microinjections into the nucleus tractus solitarius. Brain Res. 1990;530:54–72. doi: 10.1016/0006-8993(90)90657-w. [DOI] [PubMed] [Google Scholar]
- 79.Nikodijevič O, Sarges R, Daly JW, Jacobson KA. Behavioral effects of A1- and A2-selective adenosine agonists and antagonists; evidence for synergism and antagonism. J. Pharmacol. Exp. Ther. 1991;259:286–294. [PMC free article] [PubMed] [Google Scholar]
- 80.Rubin L, Porter S, Horner H, Yednock T. Blood–brain barrier model. Patent Pub. 1991 W091/05038. [Google Scholar]
- 81.IJzerman AP, Thedinga KH, Custers AFCM, Hoos B, Van Belle H. Inhibition of nucleoeide transport by a new series of compounds related to lidoflazine and mioflazine. Eur. J. Pharmacol. 1989;172:273–281. doi: 10.1016/0922-4106(89)90057-6. [DOI] [PubMed] [Google Scholar]
- 82.Pirovano IM, Van Belle H, IJzerman AP. Inhibition of nucleoside uptake in human erythrocytes by a new series of compounds related to lidoflazine and mioflazine. Eur. J. Pharmacol. 1990;189:419–422. doi: 10.1016/0922-4106(90)90040-5. [DOI] [PubMed] [Google Scholar]
- 83.Janusz CA, Bruns RF, Berman RF. Functional activity of the adenosine binding enhancer, PD 81,723, in the in vitro hippocampal slice. Brain Res. 1992 doi: 10.1016/0006-8993(91)90794-v. in press. [DOI] [PubMed] [Google Scholar]
- 84.Izquierdo I, Medina JH. GABAA receptor modulation of memory: the role of endogenous benzodiazepines. Trends Pharmacol. Sci. 1991;12:260–265. doi: 10.1016/0165-6147(91)90567-c. [DOI] [PubMed] [Google Scholar]
- 85.Young AB, Fagg GE. Excitatory amino acid receptors in the brain: membrane binding and receptor autoradiographic approaches. Trends Pharmacol. Sci. 1990;11:126–133. doi: 10.1016/0165-6147(90)90199-i. [DOI] [PubMed] [Google Scholar]
- 86.Gensia’s ischemia therapy, Arasine, in Phase III. Scrip. 1991;1630:23. [Google Scholar]
- 87.Marion Merrell Dow to evaluate Gensia ARA compound. Scrip. 1991;1643:12. [Google Scholar]
- 88.Agarwal RP. Adenosine deaminase. Measurement of activity and use of inhibitors. In: Paton DM, editor. Methods In Pharmacology. Vol. 6. Methods Used in Adenosine Research. New York: Plenum; 1985. pp. 109–125. [Google Scholar]
- 89.Phillis JW, Walter GA, Simpson RE. Brain adenosine and transmitter amino acid release from the ischemic rat cerebral cortex: effects of the adenosine deaminase inhibitor deoxycoformycin. J. Neurochem. 1991;56:644–650. doi: 10.1111/j.1471-4159.1991.tb08198.x. [DOI] [PubMed] [Google Scholar]
- 90.Cronstein BN, Eberle MA, Gruber HE, Levin RI. Methotrexate inhibits neutrophil function by stimulating adenosine release from connective tissue cells. Proc. Natl. Acad. Sci. U.S.A. 1991;88:2441–2445. doi: 10.1073/pnas.88.6.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cronstein BN, Daguma L, Nichols D, Hutchison AJ, Williams M. The adenosine/neutrophil paradox resolved human neutrophils possess both A1 and A2 receptors that promote chemotaxis and inhibit O2− generation, respectively. J. Clin. Invest. 1990;85:1150–1157. doi: 10.1172/JCI114547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gordon J. Extracellular ATP: effects, sources, and fate. Biochem. J. 1986;233:309–319. doi: 10.1042/bj2330309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.O’Connor SE, Dainty IA, Leff P. Further subclassification of ATP receptors based on agonist studies. Trends Pharmacol. Sci. 1991;12:137–141. doi: 10.1016/0165-6147(91)90530-6. [DOI] [PubMed] [Google Scholar]
- 94.Dubyak GR. Signal Transduction by P2-Purinergic Receptors for Extracellular ATP. Am. J. Respir. Cell. Mol. Biol. 1991;4:295–300. doi: 10.1165/ajrcmb/4.4.295. [DOI] [PubMed] [Google Scholar]
- 95.Forsyth KM, Bjur RA, Westfall DP. Nucleotide modulation of norepinephrine release from sympathetic nerves in the rat vas deferens. J. Pharmacol. Exp. Ther. 1991;256:821–826. [PubMed] [Google Scholar]
- 96.Westfall DP, Shinozuka K, Forsyth KM, Bjur RA. Modulation of norepinephrine release by ATP and adenosine. In: Jacobson KA, Daly JW, Manganiello V, editors. Purines in Cellular Signaling. Targets for New Drugs. New York: Springer Verlag; 1990. pp. 260–265. [Google Scholar]
- 97.Hilderman RH, Martin M, Zimmerman JK, Pivorun EB. Identification of a unique membrane receptor for adenosine 5′, 5″′-P1,P4-tetraphosphate. J. Biol. Chem. 1991;266:6915–6918. [PubMed] [Google Scholar]
- 98.Burnstock G. Purinergic mechanisms. Ann. N.Y. Acad. Sci. 1990;603:1–17. doi: 10.1111/j.1749-6632.1990.tb37657.x. [DOI] [PubMed] [Google Scholar]
- 99.Rapaport E. Mechanisms of anticancer activities of adenine nucleotides in tumor-bearing hosts. Ann. N.Y. Acad. Sci. 1990;603:142–150. doi: 10.1111/j.1749-6632.1990.tb37668.x. [DOI] [PubMed] [Google Scholar]
- 100.Chaudry IH. Use of ATP following shock and ischemia. Ann. N.Y. Acad. Sci. 1990;603:130–141. doi: 10.1111/j.1749-6632.1990.tb37667.x. [DOI] [PubMed] [Google Scholar]
- 101.Fredholm BB, Dunwiddie TV. How does adenosine inhibit transmitter release? Trends Pharmacol. Sci. 1988;9:130–134. doi: 10.1016/0165-6147(88)90194-0. [DOI] [PubMed] [Google Scholar]
- 102.Cooper DMF, Caldwell KK. Signal transduction mechanisms for adenosine. In: Williams M, editor. Adenosine and Adenosine Receptors. Clifton, NJ: Humana; 1990. pp. 105–141. [Google Scholar]
- 103.Burnstock G. Dual control of local blood flow by purines. Ann. N.Y. Acad. Sci. 1990;603:31–45. doi: 10.1111/j.1749-6632.1990.tb37659.x. [DOI] [PubMed] [Google Scholar]
- 104.Linden J, Prater MR, Sullivan GW, Johns RA, Patel A. Contamination of adenosine deaminase by superoxide dismutase. Stabilization of endothelium-derived relaxing factor. Biochem. Pharmacol. 1991;41:273–279. doi: 10.1016/0006-2952(91)90486-o. [DOI] [PubMed] [Google Scholar]
- 105.Stiles GA, Daly DT, Olsson RA. The A1 adenosine receptor. Identification of the binding subunit by photoaffinity crosslinking. J. Biol. Chem. 1985;260:10806–10811. [PubMed] [Google Scholar]
- 106.Choca JI, Kwatra MM, Hosey MM, Green RD. Specific photoaffinity labelling of inhibitory adenosine receptors. Biochem. Biophys. Res. Commun. 1985;131:115–121. doi: 10.1016/0006-291x(85)91778-4. [DOI] [PubMed] [Google Scholar]
- 107.Lohse MJ, Klotz KN, Schwabe U. Agonist photoaffinity labeling of A1 adenosine receptors: persistent activation reveals spare receptors. Mol. Pharmacol. 1986;30:403–409. [PubMed] [Google Scholar]
- 108.Stiles GL, Jacobson KA. A new high affinity, iodinated adenosine receptor antagonist as a radioligand/photoaffinity crosslinking probe. Mol. Pharmacol. 1987;32:184–188. [PMC free article] [PubMed] [Google Scholar]
- 109.Barrington WW, Jacobson KA, Stiles GL. Demonstration of distinct agonist and antagonist conformations of the A1 adenosine receptor. J. Biol. Chem. 1989;264:3157–3164. [PMC free article] [PubMed] [Google Scholar]
- 110.Ji XD, Stiles GL, van Galen PJM, Jacobson KA. Characterization of human striatal A2-adenosine receptors using radioligand binding and photoaffinity labeling. J. Recept. Res. 1992 doi: 10.3109/10799899209074789. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nanoff C, Jacobson KA, Stiles GL. The A2 Adenosine receptor—guanine nucleotide modulation of agonist binding is enhanced by proteolysis. Mol. Pharmacol. 1991;39:130–135. [PMC free article] [PubMed] [Google Scholar]
- 112.Ramkumar V, Barrington WW, Jacobson KA, Stiles GL. Demonstration of both A1 and A2 adenosine receptors in DDT1 MF-2 smooth muscle cells. Mol. Pharmacol. 1990;37:149–156. [PMC free article] [PubMed] [Google Scholar]
- 113.Stiles GL, Jacobson KA. High affinity acylating antagonists for the A1 adenosine receptor: identification of binding subunit. Mol. Pharmacol. 1988;34:724–728. [PMC free article] [PubMed] [Google Scholar]
- 114.Nakata H. Purification of A1 adenosine receptor from rat brain membranes. J. Biol. Chem. 1989;264:16545–16551. [PubMed] [Google Scholar]
- 115.Olah ME, Jacobson KA, Stiles GL. Purification and characterization of bovine cerebral cortex A1 adenosine receptor. Arch. Biochem. Biophys. 1990;243:440–446. doi: 10.1016/0003-9861(90)90665-l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mahan LC, McVittie LD, Smyk-Randall EM, Nakata H, Monsma FJ, Gerfen CR, Sibley DR. Cloning and expression of an A1 adenosine recepter from rat brain. Mol. Pharmacol. 1991;40:1–7. [PubMed] [Google Scholar]
- 117.Reppert SM, Weaver DR, Stehle JH, Rivkees SA. Molecular cloning of a rat A1-adenosine receptor that is widely expressed in brain and spinal cord. Mol. Endocrinol. 1991;5:1037–1048. doi: 10.1210/mend-5-8-1037. [DOI] [PubMed] [Google Scholar]
- 118.Klotz KN, Lohse MJ. The glycoprotein nature of A1 adenosine receptors. Biochem. Biophys. Res. Commun. 1986;140:406–413. doi: 10.1016/0006-291x(86)91105-8. [DOI] [PubMed] [Google Scholar]
- 119.Stiles GL. Photoaffinity cross-linked A1 adenosine receptor-binding subunits. Homologous glycoprotein expression by different tissues. J. Biol. Chem. 1986;261:10839–10843. [PubMed] [Google Scholar]
- 120.Barrington WW, Jacobson KA, Stiles GL. Glycoprotein nature of the A2-adenosine receptor binding subunit. Mol. Pharmacol. 1990;38:177–183. [PMC free article] [PubMed] [Google Scholar]
- 121.van Galen PJM, Stiles GL, Michaels GS, Jacobson KA. Adenosine A1 and A2 receptors: structure function relationships. Med. Res. Rev. 1992 doi: 10.1002/med.2610120502. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.O’Dowd BF, Hnatowich M, Caron MG, Lefkowtiz RJ, Bouvier M. Palmitoylation of the human β-adrenergic receptor. Mutation of Cys341 in the carboxyl tail leads to an uncoupled nonpalmitoylated form of the receptor. J. Biol. Chem. 1989;264:7564–7569. [PubMed] [Google Scholar]
- 123.Ovchinnikov YA, Abdulaev NG, Bogachuk AS. Two adjacent cysteine residues in the C-terminal cytoplasmic fragment of bovine rhodopsin are palmitylated. FEBS Lett. 1988;230:1–5. doi: 10.1016/0014-5793(88)80628-8. [DOI] [PubMed] [Google Scholar]
- 124.Stiles GL. A1 adenosine receptor-G protein coupling in bovine brain membranes: effects of guanine nucleotides, salt, and solubilization. J. Neurochem. 1988;51:1592–1598. doi: 10.1111/j.1471-4159.1988.tb01129.x. [DOI] [PubMed] [Google Scholar]
- 125.Klotz KN, Lobe MJ, Schwabe U. Chemical modification of A1 adenosine receptors in rat brain membranes. Evidence for histidine in different domains of the ligand binding site. J. Biol. Chem. 1988;263:17522–17526. [PubMed] [Google Scholar]
- 126.van de Wenden EM, van Galen PJM, IJzerman AP, Soudijn W. A model for the hydrogen bonding interactions between adenosine receptor ligands and histidyl residues in the adenosine A1 receptor binding site, based on AM1 calculations. J. Comput. Chem. (Theochem) 1991;231:175–184. [Google Scholar]
- 127.Jarvis MF, Williams M. Adenosine in central nervous system function. In: Williams M, editor. Adenosine and Adenosine Receptors. Clifton, NJ: Humana Press; 1990. pp. 423–474. [Google Scholar]
- 128.Williams M. Challenges in the search for CNS therapeutics in the 1990s. Curr. Opinion Therap. Patent. 1991;1:693–723. [Google Scholar]
- 129.Williams M. Purine nucleosides and nucleotides as central nervous system modulators. Ann. N.Y. Acad. Sci. 1990;603:93–107. doi: 10.1111/j.1749-6632.1990.tb37664.x. [DOI] [PubMed] [Google Scholar]
- 130.Diamond I, Gordon A. Use of adenosine agonists and antagonists in the treatment of alcohol abuse. 0 431 758 A2. Eur. Patent Application. 1991
- 131.Heffner TG, Wiley JN, Williams AE, Bruns RF, Coughenour LJ, Downs DA. Comparison of the behavioral effects of adenosine agonists and dopamine antagonists in mice. Psychopharmacology. 1989;98:31–37. doi: 10.1007/BF00442002. [DOI] [PubMed] [Google Scholar]
- 132.Dunwiddie TV, Worth T. Sedative and anticonvulsant actions of adenosine analogs in mouse and rat. J. Pharmacol. Exp. Ther. 1982;220:70–76. [PubMed] [Google Scholar]
- 133.Dragunow M, Goddard GV, Laverty R. Is adenosine an endogenous anticonvulsant? Epilepsia. 1985;26:480–487. doi: 10.1111/j.1528-1157.1985.tb05684.x. [DOI] [PubMed] [Google Scholar]
- 134.Evans MC, Swan JH, Meldrum BS. An adenosine analogue, 2-chloroadenosine protects against long term development of ischemic cell loss in the rat hippocampus. Neurosci. Lett. 1987;83:287–292. doi: 10.1016/0304-3940(87)90101-7. [DOI] [PubMed] [Google Scholar]
- 135.Jacobsen EJ, McCall JM. Acute ischemic and traumatic injury to the CNS. Ann. Rep. Med. Chem. 1990;25:31–40. [Google Scholar]
- 136.Barone S, Churchill PC, Jacobson KA. Adenosine receptor prodrugs. Towards kidney selective dialkylxanthines. J. Pharmacol. Exp. Ther. 1989;250:79–85. [PMC free article] [PubMed] [Google Scholar]
- 137.Anderson W, Pop E, Lee S-K, Bhagrath M, Bodor N, Brewster M. Brain-targeting chemical delivery systems for adenosine depresses locomotor behavior in rats. Med. Chem. Res. 1991;1:74–79. [Google Scholar]
- 138.Shinoda I, Furukawa Y, Furukawa S. Stimulation of nerve growth factor synthesis/secretion by propentofylline in cultured mouse astroglial cells. Biochem. Pharmacol. 1990;39:1813–1816. doi: 10.1016/0006-2952(90)90130-d. [DOI] [PubMed] [Google Scholar]
- 139.Scrip, Pentoxyfylline for AIDS? Scripta. 1991;1610:27. [Google Scholar]
- 140.Rao KMK, Currie MS, McCachren SS, Cohen HJ. Pentoxifylline and other methylxanthines inhibit interleukin-2 receptor expression in human lymphocytes. Cell. Immunol. 1991;135:314–325. doi: 10.1016/0008-8749(91)90276-h. [DOI] [PubMed] [Google Scholar]
- 141.Polmar SH, Fernadez-Mejia C, Birch RE. Adenosine receptors: immunologic aspects. In: Cooper DMF, Londos C, editors. Adenosine Receptors. New York: Liss; 1988. pp. 97–112. [Google Scholar]
- 142.Green PG, Basbaum AI, Helms C, Levine JD. Purinergic regulation of bradykinin-induced plasma extravasation and adjuvant-induced arthritis in the rat. Proc. Natl. Acad. Sci. U.S.A. 1991;88:4162–4165. doi: 10.1073/pnas.88.10.4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Herbert KE, Bhusate LL, Scott DL, Perett D. Purine metabolism in arthritis: 1. Synovial fluid adenosine concentrations are low in rheumatoid arthritis. Intern. J. Purine Pyrimidine Res. 1991;2:31–34. [Google Scholar]
- 144.Scaccianoce S, Navarra D, Di Sciullo A, Angelucci L, Endroczi E. Adenosine and pituitary-adrenocortical axis activity in the rat. Neuroendocrinology. 1989;50:464–468. doi: 10.1159/000125264. [DOI] [PubMed] [Google Scholar]
- 145.Schettini G, Landolfi E, Meucci O, Floria T, Grimaldi M, Ventra C, Marino A. Adenosine and its analogue (−)-N6-phenylisopropyladenosine modulate anterior pituitary adenylate cyclase activity and prolactin secretion in the rat. J. Mol. Endocrinol. 1990;5:69–76. doi: 10.1677/jme.0.0050069. [DOI] [PubMed] [Google Scholar]
- 146.Kinzer D, Lehmann V. Extracellular ATP and adenosine modulate tumor necrosis factor-induced lysis of L929 cells in the presence of actinomycin D. J. Immunol. 1991;146:2708–2711. [PubMed] [Google Scholar]
- 147.Sills MA, Webb RL, Hummel H, Francis JE, Stone GA. Chronic administration of the adenosine A2-selective agonists, CGS 21680C and CGS 22989, produces receptor down-regulation and tolerance to their hypotensive effects. Pharmacologist. 1991;33:176. [Google Scholar]
- 148.Engler RL. Adenosine: the signal of life? Circulation. 1991;84:951–954. doi: 10.1161/01.cir.84.2.951. [DOI] [PubMed] [Google Scholar]
- 149.Williams M. Purinergic receptors as drug targets. Drug News Persp. 1990;4:5–12. [Google Scholar]
- 150.McMillian MK, Soltoff SP, Cantley LC, Talamo BR. Extracellular ATP increases free cytosolic calcium in rat parotid acinar cells. Biochem. J. 1988;255:291–300. [PMC free article] [PubMed] [Google Scholar]
- 151.Soltoff SP, McMillian MK, Talamao BR. Coomassie brilliant blue G is a more potent antagonist of P2 purinergic responses than reactive blue 2 (Cibacron Blue 3GA) in rate parotid acinar cells. Biochem. Biophys. Res. Commun. 1989;165:1279–1285. doi: 10.1016/0006-291x(89)92741-1. [DOI] [PubMed] [Google Scholar]
- 152.Bruns RF. Adenosine receptor activation in human fibroblasts: nucleoside agonists and antagonists. Can. J. Physiol. Pharmacol. 1980;58:673–691. doi: 10.1139/y80-110. [DOI] [PubMed] [Google Scholar]
- 153.Lohse MJ, Böser S, Klotz KN, Schwabe U. Affinities of barbiturates for the GABA-receptor complex and A1 adenosine receptors: a possible explanation of their excitatory effects. Naunyn Schmiedeberg’s Arch. Pharmacol. 1987;336:211–217. doi: 10.1007/BF00165807. [DOI] [PubMed] [Google Scholar]
- 154.Daly JW, Hong O, Padgett WL, Shamim MT, Jacobson KA, Ukena D. Non-xanthine heterocycles: activity as antagonists of A1- and A2-adenosine receptors. Biochem. Pharmacol. 1988;37:655–664. doi: 10.1016/0006-2952(88)90139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Peet NP, Dickerson GA, Abdallah AH, Daly JW, Ukena D. Benzo[1,2-c:5,4-c′]dipyrazoles: non-xanthine adenosine antagonists. J. Med. Chem. 1988;31:2034–2039. doi: 10.1021/jm00118a032. [DOI] [PubMed] [Google Scholar]
- 156.Skerrit JH, Davies LP, Johnston GAR. A purinergic component in the anticonvulsant action of carbamazepine? Eur. J. Pharmacol. 1982;82:195–197. doi: 10.1016/0014-2999(82)90512-x. [DOI] [PubMed] [Google Scholar]
- 157.Daly JW, Hide I, Bridson PK. Imidazodiazepinediones: a new class of adenosine receptor antagonists. J. Med. Chem. 1990;33:2818–2821. doi: 10.1021/jm00172a022. [DOI] [PubMed] [Google Scholar]
- 158.Psychoyos S, Ford CJ, Phillip MA. Inhibition by etazolate (SQ 20009) and cartazolate (SQ 65396) of adenosine-stimulated [3H]cAMP formation in 2-[3H]adenine-prelabeled vesicles prepared from guinea pig cerebral cortex. Biochem. Pharmacol. 1982;31:1441–1442. doi: 10.1016/0006-2952(82)90041-7. [DOI] [PubMed] [Google Scholar]
- 159.Davies LP, Chow SC, Skerrit JH, Brown DJ, Johnston GAR. Pyrazolo[3,4-d]pyrimidines as adenosine antagonists. Life Sci. 1984;34:2117–2128. doi: 10.1016/0024-3205(84)90310-2. [DOI] [PubMed] [Google Scholar]
- 160.Hamilton HW, Ortwine DF, Worth DF, Bristol JA. Synthesis and structure–activity relationships of pyrazolo-[4,3-d]pyrimidin-7-ones as adenosine receptor antagonists. J. Med. Chem. 1987;30:91–96. doi: 10.1021/jm00384a016. [DOI] [PubMed] [Google Scholar]
- 161.Garritsen A, IJzerman AP, Beukers MW, Cragoe EJ, Soudijn W. Interaction of amiloride and its analogues with adenosine A1 receptors in calf brain. Biochem. Pharmacol. 1990;40:827–834. doi: 10.1016/0006-2952(90)90323-d. [DOI] [PubMed] [Google Scholar]
- 162.van Galen PJM, IJzerman AP, Soudijn W. Xanthine-7-ribosides as adenosine receptor antagonists. Nucleoside Nucleotide. 1990;9:276–291. [Google Scholar]
- 163.Knowles M, Lane LC, Boucher RC. Activation by extracellular nucleotides of chloride secretion in the airway epithelia of patients with cystic fibrosis. New Eng. J. Med. 1991;325:533–538. doi: 10.1056/NEJM199108223250802. [DOI] [PubMed] [Google Scholar]
- 164.Thompson RD, Secunda S, Daly JW, Olsson RA. Activity of N6-substituted 2-chloroadenosines at A1 and A2-adenosine receptors. J. Med. Chem. 1991;34:3388–3390. doi: 10.1021/jm00116a007. [DOI] [PubMed] [Google Scholar]
- 165.Jacobson KA, Stiles GL, Ji XD. J. Biol. Chem. Chemical modification and irreversible inhibition of striated A2a-adenosine receptors. Mol. Pharmacol. 1992 submitted. [PMC free article] [PubMed] [Google Scholar]
- 166.Yoon K-W, Rothman SM. Adenosine inhibits excitatory but not inhibitory synaptic transmission in the hippocampus. J. Neurosci. 1991;11:1375–1380. doi: 10.1523/JNEUROSCI.11-05-01375.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jacobson KA. Adenosine (P1) and ATP (P2) receptors. Comp. Med. Chem. 1990;3:601–642. [Google Scholar]
- 168.Eidelman O, Guay-Broder C, van Galen PJM, Jacobson KA, Fox C, Turner RJ, Cabantchile ZI, Pollard HB. A1-Adenosine antagonist drugs activate chloride efflux from cystic fibrosis cells. Proc. Natl. Acad. Sci. U.S.A. 1992 doi: 10.1073/pnas.89.12.5562. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Thiel M, Bardenheuer H, Pöch G, Madel C, Peter K. Pentoxitylline does not act via adenosine receptors in the inhibition of the superoxide anion production of human polymorphonuclear leukocytes. Biochem. Biophys. Res. Commun. 1991;180:53–58. doi: 10.1016/s0006-291x(05)81253-7. [DOI] [PubMed] [Google Scholar]
- 170.Sykes R. Comments. F.D.C. Reports. 1991;53(50):20. [Google Scholar]
- 171.Honore E, Fournier F, Collin T, Nargeot T, Guilbault P. Functional expression of Pzy purinoceptor in Xenopus oocyte injected with brain mRNA. Pflugen Arch. 1991;418:447–452. doi: 10.1007/BF00497772. [DOI] [PubMed] [Google Scholar]