

Abstract
The initial rate of Ca2+ movement across the inner-envelope membrane of pea (Pisum sativum L.) chloroplasts was directly measured by stopped-flow spectrofluorometry using membrane vesicles loaded with the Ca2+-sensitive fluorophore fura-2. Calibration of fura-2 fluorescence was achieved by combining a ratiometric method with Ca2+-selective minielectrodes to determine pCa values. The initial rate of Ca2+ influx in predominantly right-side-out inner-envelope membrane vesicles was greater than that in largely inside-out vesicles. Ca2+ movement was stimulated by an inwardly directed electrochemical proton gradient across the membrane vesicles, an effect that was diminished by the addition of valinomycin in the presence of K+. In addition, Ca2+ was shown to move across the membrane vesicles in the presence of a K+ diffusion potential gradient. The potential-stimulated rate of Ca2+ transport was slightly inhibited by diltiazem and greatly inhibited by ruthenium red. Other pharmacological agents such as LaCl3, verapamil, and nifedipine had little or no effect. These results indicate that Ca2+ transport across the chloroplast inner envelope can occur by a potential-stimulated uniport mechanism.
Ca2+ in plant cells has many key physiological functions; for example, as an intracellular second messenger it is especially important for the maintenance of cellular homeostasis and signal transduction pathways (Evans et al., 1991; Pineros and Tester, 1997). Therefore, the [Ca2+]cyt must be strictly regulated. The sequestration of Ca2+ into endomembrane compartments has been documented in detail for the ER and the vacuole (Evans et al., 1991). The chloroplast may also serve as a potential Ca2+ sink (Brand and Becker, 1984; Evans et al., 1991).
In addition to the potential role of chloroplasts in maintaining low resting [Ca2+]cyt, it has been proposed that the free [Ca2+] in the stroma regulates several key enzymes involved in photosynthetic CO2 assimilation, including Fru-1,6-bisphosphatase and sedoheptulose-1,7-bisphosphatase (Kreimer et al., 1988) and NAD+ kinase (Brand and Becker, 1984). Ca2+ is also essential for O2 evolution by PSII (Grove and Brudvig, 1998). Although the role of Ca2+ in the stroma and thylakoids has been studied in detail (Brand and Becker, 1984), there have been relatively few investigations on Ca2+ uptake by chloroplasts.
One study examining Ca2+ movement into intact wheat chloroplasts (Muto et al., 1982) indicated that the Ca2+ uptake occurs via an H+/Ca2+-antiport mechanism, and that the Km was only slightly higher than [Ca2+]cyt. Kreimer et al. (1985a, 1985b), who measured Ca2+ fluxes across the envelope of intact chloroplasts isolated from spinach, reported that Ca2+ transport into illuminated chloroplasts could occur via electrogenic uniport and that this was linked to photosynthetic electron transport.
The majority of the work done in this area has been carried out using intact chloroplasts; however, that system has some disadvantages. For example, it is difficult to monitor the nearly instantaneous influx or efflux of ions, and therefore it is difficult to resolve the initial rate kinetics of transport processes. In addition, the pH and ionic composition of the stroma are not easy to control. Another drawback to using intact chloroplasts is that it is difficult to examine the directionality of transport processes, because it is difficult to preload the chloroplasts with Ca2+ to measure efflux.
An alternative approach, which we used in this study, involves the use of membrane vesicles prepared from inner-envelope membranes isolated from intact chloroplasts. Vesicles have been shown to be competent for studying ion and metabolite movement across membranes (Sze, 1985). There are several advantages to using membrane vesicles over intact organelles. For example, vesicles can be loaded with fluorescent probes, allowing for the continuous fluorometric measurement of substrate and ion transport. When used along with stopped-flow spectrofluorometry, these processes can be monitored almost instantaneously with measurement times of less than 2 ms, which allows for the determination of initial rate kinetics of transport. This method has been used to measure the symport movement of protons with glycolate and d-glycerate (Young and McCarty, 1993) and the rapid proton-linked diffusion of nitrite as nitrous acid (Shingles et al., 1996) across the chloroplast inner-envelope membrane. Another advantage of this method is that the pH and ionic content of the intravesicular and external media can be easily manipulated, a procedure commonly used to study transport mechanisms. Finally, because inner-envelope vesicles of either right-side-out or inside-out orientation can be prepared (Shingles and McCarty, 1995), the directionality of transport processes can be characterized in detail.
Studies using liposomes loaded with the ratioable Ca2+ fluorescent probe fura-2 have been performed to measure Ca2+ transport catalyzed by reconstituted annexin ion channels (Berendes et al., 1993) and ionophores such as ionomycin and A23187 (Blau and Weissmann, 1988; Fasolato and Pozzan, 1989). In addition, fura-2 has been used to monitor voltage-dependent Ca2+ movement across the erythrocyte plasma membrane (Soldati et al., 1997). Preliminary experiments using chloroplast inner-envelope vesicles loaded with Ca2+-sensitive fluorophores indicated that these probes would be sensitive enough to detect small changes in intravesicular free [Ca2+], and therefore could be used to monitor Ca2+ movement across membranes (Cleveland and McCarty, 1995). In the present study this experimental procedure was used to directly calculate the initial rates of Ca2+ movement and to characterize further the properties of Ca2+ transport across the chloroplast inner envelope.
MATERIALS AND METHODS
Reagents
Fura-2 was purchased from Molecular Probes (Eugene, OR). All other reagents were of the highest grade commercially available. Stock solutions of buffer components were passed through a column containing Chelex-100 to reduce Ca2+ content before buffer preparation.
Plant Material
Pea (Pisum sativum L. cv Laxton's Progress No. 9) plants were grown from seed for 16 to 18 d in vermiculite in a controlled-environment growth cabinet (Revco, Asheville, NC) set for 16-h day (24°C)/8-h night (20°C) periods. Spinach was obtained at local markets.
Membrane Isolation
Intact chloroplasts were isolated according to the method of Joy and Mills (1987). Inner envelopes were prepared as described by Keegstra and Yousif (1986). Frozen, intact chloroplasts, equivalent to between 80 and 120 mg of chlorophyll, were thawed at 4°C, refrozen at −20°C, and thawed again at 4°C. Chloroplast rupture was facilitated by gentle homogenization using a pestle tissue grinder. The homogenate was centrifuged at 3,150g for 15 min. The resulting supernatants were collected and centrifuged at 27,000g for 90 min. Pellets were resuspended in 0.2 m Suc and placed on top of a 0.45/0.80/1.0 m Suc step gradient and centrifuged at 105,000g for 18 h. Inner-envelope membrane vesicles were recovered from the 0.80/1.0 m Suc interface. All of the operations described above were performed at 4°C. Inner envelopes were stored under liquid nitrogen.
Vesicle Preparations
Suspensions of purified inner envelopes or asolectin (20 mg) were diluted 4-fold in the appropriate resuspension buffer. The membranes were pelleted by centrifugation at 144,000g for 1 h at 4°C and then resuspended in the same buffer before vesicle-preparation protocols.
Membrane vesicles were also prepared using a hand-held, small-volume extrusion apparatus (Shingles and McCarty, 1995). Trace amounts of Ca2+ in the filters and apparatus were removed by passing through the apparatus a total of 5 mL of resuspension buffer containing 10 mm K-Hepes, pH 8.0, 100 mm KCl, 100 mm Suc, and 100 μm EGTA. A total of 1.0 to 2.0 mL of a membrane suspension containing 1.2 mm fura-2 and about 1 mg of inner-envelope protein or 15 to 20 mg of asolectin was then passed 9 to 11 times through the extrusion apparatus with a polycarbonate filter (100-nm pore size) in place.
Inner-envelope vesicles, predominantly of the inside-out orientation, were prepared by the freeze/thaw method described by Young and McCarty (1993) in a resuspension buffer containing 10 mm K-Hepes, pH 8.0, 50 mm KCl, 50 mm choline chloride, 100 mm Suc, 100 μm EGTA, and 1.2 mm fura-2.
All vesicle preparations were passed through a 1.6- × 10-cm Sephadex G-50 column equilibrated with the appropriate resuspension buffer to remove most of the external fura-2. To eliminate trace amounts of external probe, the eluted vesicles were diluted 4-fold in resuspension buffer and centrifuged at 144,000g for 20 min at 4°C. The pellet comprising fura-2-loaded membrane vesicles was resuspended in the same buffer and allowed to equilibrate for 1 to 2 h at 4°C.
Ca2+ Minielectrodes
The Ca2+ electrode was constructed as described by Baudet et al. (1994). A 3-cm length of 1.67-mm (o.d.) polyethylene tubing was dipped into a mixture of polyvinyl chloride and potassium tetrakis chlorophenyl borate dissolved in tetrahydrofuran and the Ca2+ ionophore N,N,N′,N′-tetracyclohexyl-3-oxapentanediamide dissolved in 2-nitrophenyl octyl ether and allowed to dry overnight. Dried electrodes were filled with 28.5 mm nitrilotriacetic acid, pH 8.0, and 1.13 mm CaCl2 to give approximately 1 μm free Ca2+ and an ionic strength of about 0.10 m. The electrodes were allowed to equilibrate in this buffer for at least 3 d before use.
Fluorescence Measurements
Fura-2 fluorescence emission was monitored at 512 nm with excitation at 340 nm (Fs) or 359 nm (Fis) using a modified spectrofluorometer (model SLM-SPF-500C, Olis, Bogart, GA) and a stopped-flow apparatus (Olis). All slits were set at 10 nm with a cutoff filter (LP47, Oriel, Stamford, CT) placed over the entrance to the emission monochromator. Chamber A contained 2.0 mL of vesicle suspension, and chamber B contained 2.0 mL of buffer of predetermined pH and composition so that the intravesicular osmotic and ionic strengths were closely balanced with those of the external medium. Chamber B also contained CaCl2 when used. Mixing was achieved by a nitrogen-driven piston at 80 p.s.i. All measurements were taken at 25°C.
Internal Buffering Capacity Measurements
To determine the βin of inner-envelope vesicles known amounts of CaCl2 were added to the vesicles in the stopped-flow apparatus and the resulting changes in external pCa were monitored over time. The βin relates the total number of moles of Ca2+ that cross the vesicle membrane in a given number of vesicles and the intravesicular pCa change that results under experimental conditions; accordingly, it is expressed in nanomoles of Ca2+ per pCa unit per milligram of inner-envelope protein.
In this assay fura-2 was present as an indicator of the external pCa. Spinach chloroplast inner-envelope vesicles were prepared via the freeze/thaw procedure described previously with the following exceptions. The resuspension buffer consisted of 10 mm K-Hepes, pH 8.0, 100 mm KCl, 100 mm Suc, and 100 μm EGTA. The intravesicular fura-2 was replaced by 1.2 mm 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid to avoid interference with the fluorescence of the external fura-2. Finally, the EGTA concentration in the elution buffer used to equilibrate the Sephadex G-50 column and to dilute the eluted vesicles before centrifugation was reduced to 20 μm. However, the concentrations of KCl, Suc, and K-Hepes, pH 8.0, in the elution buffer were identical to those in the resuspension buffer.
Vesicle suspensions were diluted with an equal volume of elution buffer supplemented with approximately 10 μm fura-2. The resulting mixture was loaded into chamber A of the stopped-flow apparatus. Chamber B contained buffer containing 100 mm choline chloride plus 32 μm CaCl2. The fluorescence of fura-2 was monitored for 2 min. The decrease in pCa immediately after mixing was used to calculate the βout. This decrease was followed by a gradual increase in external pCa for the next 2 min as the external and internal [Ca2+] reached equilibrium. The final overall extent of change in pCa allowed for the calculation of the combination of βin and βout. From these data, βin was calculated.
Assays
The modified TCA-Lowry procedure of Bensadoun and Weinstein (1976) was used to determine the amount of protein in the inner-envelope membrane vesicle preparations.
RESULTS
Calibration of Vesicles Loaded with Fura-2
The minielectrode used in this study was calibrated in a buffer containing 12 mm EGTA, 15 mm nitrilotriacetic acid, and 10 mm CaCl2. At the start of the calibration process, the pH of the buffer was 4.0. Under these conditions, almost all of the Ca2+ was unchelated. The concentration of free Ca2+ in the buffer was changed by varying the pH of the buffer from 4.0 to 9.0. This procedure allowed for the calibration of the minielectrode at millimolar and submicromolar [Ca2+]. The electrode gave a Nernstian response at both of these [Ca2+] ranges (Fig. 1A). At each pH the pCa was calculated using the MaxChelator program (Bers et al., 1994).
Figure 1.

Ca2+ minielectrode calibration. The pH of the calibration buffer containing 15 mm nitrilotriacetic acid, 12 mm EGTA, and 10 mm CaCl2 was varied from 4.0 to 9.0, thereby decreasing the free [Ca2+] (expressed as pCa) that was calculated using computer software. At each pH the reading from the electrode was recorded (A). Afterward, the excitation spectra of a suspension of inner-envelope vesicles loaded with 1.2 mm fura-2 were recorded in the presence of 1 μm ionomycin between 300 and 400 nm, with emission monitored at 512 nm after adding small amounts of CaCl2 (B). The pCa of the vesicle suspension was recorded using the minielectrode after each addition. The Fs (340 nm) and Fis (359 nm) data from each of the spectra were used to produce the pCa versus Fs/Fis calibration curve (C).
When small amounts of CaCl2 were added to membrane vesicles loaded with fura-2 in the presence of 1 μm ionomycin, thereby sequentially decreasing the intravesicular pCa (i.e. increasing the internal Ca2+ concentration), significant changes in the excitation spectrum were observed (Fig. 1B). Although ionomycin catalyzes Ca2+/H+ exchange, the changes in the spectrum were not attributable to pH effects, because fura-2 is insensitive to pH. The fluorescence change at 512 nm brought on by CaCl2 addition was greatest at an excitation wavelength of 340 nm, so this wavelength was used as the Fs. However, with excitation at 359 nm, the fluorescence was relatively insensitive to the change in [Ca2+] inside the vesicles, so this isoexcitation point was taken as the Fis. After each addition of CaCl2, the fluorescence values at Fs and Fis were determined from excitation spectra and the final pCa values were measured with the Ca2+ minielectrode. The plot of Fs/Fis versus pCa produced the curve shown in Figure 1C.
According to Grynkiewicz et al. (1985), the fluorescence ratio of fura-2 (R or Fs/Fis) can be related to [Ca2+] by the following equation:
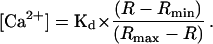 |
1 |
The plot shown in Figure 1C yielded a curve that fit the rearranged form of Equation 1:
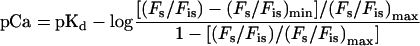 |
2 |
where (Fs/Fis)max is the fluorescence ratio at a pCa value at which essentially all of the fura-2 is bound to Ca2+, and (Fs/Fis)min is the ratio observed when all of the fura-2 is in the unchelated form. From the plot of this particular curve, the pKd was determined to be 7.0 and the (Fs/Fis)min and (Fs/Fis)max were equal to 0.66 and 1.4, respectively. These values were observed to vary only slightly among vesicle preparations; however, this calibration process was performed for each set of experiments. In each preparation the relationship between Ca2+ chelation by fura-2 and pCa was approximately linear between pCa 6.5 and 7.5.
Determination of the Internal Buffering Capacity of Inner-Envelope Vesicles
The internal buffering capacity of the inner-envelope vesicles must be known to directly calculate the actual rate of Ca2+ influx. The stopped-flow method used by Young and McCarty (1993) was used to determine buffering capacity by mixing a known amount of CaCl2 with a vesicle suspension. Fura-2 was present in the weakly buffered external medium as an indicator of the external pCa. The observed decrease in pCa immediately after mixing was used to calculate βout. This decrease was followed by a slow increase in pCa (the rebound phase) over the next 2 min as the extravesicular and intravesicular pCa reached equilibrium. The final change in pCa allows for the calculation of the internal and external buffering capacities.
In our experiments an average βin value of 7.0 ± 1.0 μm pCa unit−1 was obtained. Because the concentration of inner-envelope protein in the stopped-flow apparatus was 0.07 mg mL−1, the internal buffering capacity was calculated to be 100 ± 14 nmol Ca2+ pCa unit−1 mg−1 protein. The calculation of βin allows for the direct calculation of the initial rate of Ca2+ movement as the product of the initial rate in pCa units per second and the internal buffering capacity.
Ca2+ Movement across Vesicle Membranes
To investigate the diffusive component of Ca2+ influx, asolectin vesicles loaded with fura-2 were mixed with CaCl2 and the fluorescence emission after excitation at 340 nm (Fs) and 359 nm (Fis) was monitored over time. Asolectin is a protein-free lipid mixture, so Ca2+ would traverse these membranes via passive diffusion. When these vesicles were mixed with approximately 0.35 μm free Ca2+, no decrease in internal pCa was observed for the first 30 s (Fig. 2A). Even when the free [Ca2+] was increased to 10 to 25 μm, no change in fura-2 fluorescence was observed (data not shown), suggesting that the diffusive movement of Ca2+ across nonproteinaceous membranes was quite low. However, when 0.2 μm ionomycin was added to the asolectin vesicles and subsequently mixed with a buffer containing free Ca2+ at a concentration of 0.35 μm, a rapid Ca2+ influx at a rate of 1.0 pCa unit s−1 was observed (Fig. 2A), confirming that fura-2 was indeed present inside these vesicles and was still sensitive to changes in intravesicular pCa.
Figure 2.

Ca2+ influx in asolectin and chloroplast inner-envelope vesicles. A, Asolectin vesicles at pH 8.0 were mixed with pH 8.0 buffer containing CaCl2 such that the free external [Ca2+] after mixing was 0.35 μm. When used, the asolectin vesicles were preincubated with 0.2 μm ionomycin for 15 min. B, Inner-envelope vesicles (50 μg of protein) prepared by extrusion were mixed under the same conditions as in A with 1.0 μm free Ca2+. C, Inner-envelope vesicles (50 μg of protein) prepared by the freeze/thaw method were mixed with 1.0 μm free Ca2+. Data were fit to the single exponential equation using the graphing program Kaleidagraph (Synergy Software, Reading, PA).
Membranes that were extruded were shown to be predominantly right-side-out, whereas membranes subjected to a freeze/thaw treatment were largely inside-out in orientation (Shingles and McCarty, 1995). The addition of 1.0 μm free Ca2+ to inner-envelope membrane vesicles prepared by extrusion resulted in a decrease in intravesicular pCa during the first 30 s (Fig. 2B). The initial rate of Ca2+ transport under these conditions was equal to 0.06 pCa unit s−1. However, when inner-envelope membrane vesicles prepared by the freeze/thaw method were used (Fig. 2C), the observed initial rate of Ca2+ influx was approximately 0.03 pCa unit s−1. When vesicles of either orientation were mixed with Ca2+ in the presence of 0.2 μm ionomycin, the added Ca2+ equilibrated within the 1st s (data not shown).
Effect of a Proton Gradient on Ca2+ Influx into Inner-Envelope Vesicles
To investigate the possibility that a proton gradient may stimulate Ca2+ uptake, Ca2+ influx into inner-envelope vesicles prepared by extrusion was monitored in the stopped-flow apparatus under several conditions. In these assays the free external [Ca2+] was equal to 0.4 μm. When the external and internal pH were equivalent, the initial rate of Ca2+ transport was determined to be 1.1 pCa units mg−1 protein s−1 (Fig. 3). However, when the vesicles in pH 8.0 buffer (containing 100 mm KCl) were mixed with the same buffer, giving a final external pH of 7.0 in the presence of Ca2+, the initial rate increased to approximately 2.3 pCa units mg−1 protein s−1 (Fig. 3). This observation may be consistent with a Ca2+/H+-symport mechanism for Ca2+ movement across inner-envelope vesicles, or it may reflect the dependence of Ca2+ uptake on pH. Because the proton gradient has a ΔΨ component, the results are also consistent with the potential-stimulated uniport mechanism described by Kreimer et al. (1985b). A pH difference of 1.0 unit corresponds to a ΔΨ of −59 mV (lumen negative) at 25°C. In the presence of 2 nm valinomycin, which dissipates the potential gradient while essentially leaving the magnitude of the pH gradient unaffected, the stimulation of Ca2+ movement by the pH increase disappeared (Fig. 3). Finally, when the vesicles were mixed with pH 8.0 buffer containing 100 mm choline chloride in the presence of valinomycin, resulting in the formation of a ΔΨ of about −16 mV, the initial rate of Ca2+ uptake was equivalent to that produced in the pH-increase experiment (Fig. 3). Therefore, it appears that membrane potential, rather than external pH or the pH gradient, is the cause of the stimulation of Ca2+ uptake by a pH increase.
Figure 3.

pH and potential gradient effects on Ca2+ transport across inner-envelope vesicles. Extruded inner-envelope vesicles (15 μg of protein) at pH 8.0 were mixed with various buffers containing Ca2+ such that after mixing the free external [Ca2+] was approximately 0.4 μm. When the external pH was 7.0 in the absence of valinomycin, the ΔΨ was calculated to be −59 mV using the Nernst equation. When used, 2 nm valinomycin was preincubated with vesicles for 30 min on ice. A membrane potential of about −16 mV was imposed across the vesicle membranes by mixing vesicles in 100 mm KCl, 10 mm K-Hepes, 100 mm Suc, and 100 μm EGTA with an equal volume of the same buffer in which the 100 mm KCl was replaced with 100 mm choline chloride. The instantaneous equilibration of K+ in the presence of valinomycin resulted in a negatively charged vesicle interior.
The Effect of the Membrane Potential on Ca2+ Influx
It is interesting to note that a ΔΨ of −59 mV resulted in the same Ca2+ uptake rate as a ΔΨ of −16 mV (Fig. 3). To further investigate the effect of ΔΨ on Ca2+ uptake, Ca2+ influx was measured as the magnitude of ΔΨ was varied and the external free [Ca2+] was kept constant at 0.65 μm. As illustrated in Figure 4, a significant increase in the initial rate was observed as the magnitude of the membrane potential was increased from 0 to −15.8 mV (lumen negative). Furthermore, the stimulation of Ca2+ uptake by ΔΨ appears to be nearly saturated at the highest ΔΨ used.
Figure 4.

Potential gradient effect on Ca2+ transport across inner-envelope vesicles. Ca2+ influx into inner-envelope vesicles prepared by extrusion was monitored under varying magnitudes of ΔΨ generated by adjusting the KCl and choline chloride solutions used in Figure 3. The intravesicular [K+] was maintained at 110 mm. The ΔΨ was varied by changing the external [K+]. Inner-envelope vesicles (15 μg of protein) were incubated with 2 nm valinomycin for 30 min before each experiment.
Ca2+ Concentration Dependence of Initial Rates of Ca2+ Movement
To determine the relationship between the initial rate of Ca2+ influx and external free [Ca2+], inner-envelope vesicles were mixed with buffers so that the ΔΨ was maintained at approximately −16 mV while free [Ca2+] was varied in the submicromolar range. As seen in Figure 5, the observed increase in initial rate with respect to increasing free [Ca2+] was linear over the range studied. Also, because the initial internal pCa in the majority of the vesicle preparations ranged from 7.2 to 7.3 units, it follows that the initial rate of Ca2+ influx would be minimal when the external [Ca2+] was equal to 0.06 μm (Fig. 5), which corresponds to an external pCa of 7.22 units. Both Muto et al. (1982) and Kreimer et al. (1985b) used Ca2+ concentrations in their studies that exceeded the estimated physiological concentration of 0.05 to 0.4 μm in the plant cell cytosol (Evans et al., 1991). In fact, the Km for Ca2+ uptake by isolated chloroplasts was determined to be 188 μm (Kreimer et al., 1985b), which indicates that Ca2+ transport would not be saturable at low physiological levels, as seen in Figure 5. A limitation of these experiments is that Ca2+ concentrations higher than about 1 μm cannot be measured reliably, because fura-2 fluorescence becomes saturated when the intravesicular [Ca2+] exceeds this value.
Figure 5.

Effect of [Ca2+] on transport across inner-envelope vesicles. Extruded inner-envelope vesicles (15 μg of protein) were mixed with buffer containing various concentrations of free Ca2+, and Ca2+ influx was monitored. In each experiment a membrane potential of approximately −16 mV was imposed across the vesicle membranes, as described in Figure 3.
Effect of Inhibitors on Ca2+ Influx
In a previous study (Kreimer et al., 1985a, 1985b) Ca2+ influx into intact chloroplasts was shown to be inhibited by micromolar amounts of ruthenium red. Similar results were observed in this study. When 0.4 μm Ca2+ was present in the external medium and ΔΨ was −16 mV (lumen negative), the initial rate of Ca2+ influx was equal to 2.8 pCa units mg−1 protein s−1. In the presence of 10 μm ruthenium red, the observed Ca2+-transport activity was 0.1 pCa unit mg−1 protein s−1, an inhibition of 96% (Table I).
Table I.
Effect of inhibitors on Ca2+ influx into pea chloroplast inner-envelope membranes prepared by extrusion
| Inhibitor | pCa
|
|
|---|---|---|
| Initial rate | Inhibition | |
| units mg−1 protein s−1 | % | |
| No membrane potential | ||
| Control | 1.05 | 0 |
| + Diltiazem | 0.16 | 85 |
| + Ruthenium red | 0.10 | 90 |
| Membrane potential of −16 mV | ||
| Control | 2.80 | 0 |
| + LaCl3 | 2.93 | 0 |
| + Diltiazem | 2.13 | 25 |
| + Ruthenium red | 0.13 | 96 |
Ca2+ movement was monitored for 30 s in the absence and presence of known Ca2+-channel blockers at a concentration of 10 μm. In each experiment a membrane potential of −16 mV (when used) was imposed across the vesicle membranes and the [Ca2+] in the external medium was equal to 0.4 μm. The amount of inner-envelope protein in each experiment was approximately 15 μg.
Other pharmacological agents known to block Ca2+-channel activity were also tested for their effects on Ca2+ movement. Diltiazem inhibited the non-potential-dependent Ca2+ influx by approximately 85%, but only inhibited potential-stimulated Ca2+ influx by 25% (Table I). LaCl3 and two other known Ca2+-channel blockers, nifedipine and verapamil, had little inhibitory effect on Ca2+ transport. Other divalent cations that might compete for Ca2+ uptake were not tested because of their known effects on fura-2 fluorescence. However, an independent study on divalent cation uptake could be performed using fura-2-loaded membrane vesicles.
DISCUSSION
In this study we have demonstrated that a sensitive Ca2+ probe, fura-2, can be loaded into chloroplast inner-envelope vesicles and used to monitor Ca2+ transport across this membrane. This system has several advantages, including control of the buffer components on both sides of the membrane, the analysis of Ca2+ movement under essentially zero trans conditions, and sensitivity to Ca2+ at submicromolar levels. Combined with stopped-flow spectrofluorometry, the movement of Ca2+ can be followed with a resolution time of 2 ms (Fig. 2). In addition, previous methods used to produce membrane vesicles of right-side-out or inside-out orientation (Shingles and McCarty, 1995) allow for the evaluation of the sidedness of Ca2+ movement.
Previous studies using intact chloroplasts have demonstrated light-stimulated Ca2+ uptake (Muto et al., 1982; Kreimer et al., 1985b). The mechanism by which Ca2+ influx occurs in chloroplasts is in dispute. Muto et al. (1982) claimed the presence of a Ca2+/H+ antiporter, whereas Kreimer et al. (1985b) concluded that this process was mediated by an electrogenic uniport-carrier system. In our inner-envelope vesicle preparations we were not able to measure any Ca2+-linked, proton transport using membrane vesicles loaded with the pH-sensitive fluorophore pyranine (data not shown). This would seem to indicate that Ca2+ does not move across these membrane vesicles by a Ca2+/H+-antiport mechanism. Huang et al. (1993) reported that an antibody raised against a portion of a putative Ca2+-ATPase recognizes a 95-kD polypeptide in chloroplast inner-envelope preparations, suggesting that Ca2+ pumping might also be a mechanism for moving Ca2+ across this membrane. However, they were not able to show any Ca2+-dependent ATPase activity or ATP-dependent Ca2+ uptake in their inner-envelope preparations from pea chloroplasts. Similarly, we were not able to measure either of these activities using fura-2-loaded vesicles or a 45Ca assay in the presence or absence of added calmodulin (data not shown).
Because the rate at which Ca2+ diffuses across nonproteinaceous membranes is very low (Fig. 2A), the movement of Ca2+ across the chloroplast inner-envelope membrane observed in this study indicates the presence of a Ca2+ uniporter in our vesicle preparations. The influx of Ca2+ into predominantly right-side-out inner-envelope vesicles was stimulated by a negative ΔΨ and was very sensitive to micromolar amounts of ruthenium red (Table I). These results are in agreement with those of previous studies using intact chloroplasts (Kreimer et al., 1985a, 1985b). Furthermore, a high magnitude of ΔΨ may not be necessary to fully stimulate Ca2+-transport activity (Fig. 4). The observation that Ca2+ influx does not increase at voltages more negative than −11.8 mV indicates that uptake is not dependent on an electrophoretic driving force, but, rather, may occur through a voltage-dependent channel. In addition, the fact that Ca2+ influx does increase with Ca2+ concentration at a constant ΔΨ of −16 mV (Fig. 5) indicates that this channel would be Ca2+ specific.
The initial rate of Ca2+ influx at a free Ca2+ concentration of 0.2 to 0.4 μm was determined to range between 1.0 and 3.0 pCa units mg−1 protein s−1 (Fig. 5). Under these conditions the βin of the vesicles was equal to approximately 100 nmol Ca2+ pCa unit−1 mg−1 inner-envelope protein. Assuming that there is 0.1 mg of inner-envelope protein per mg of chlorophyll (Young and McCarty, 1993), the initial rate was calculated to range from 0.54 to 1.62 μmol Ca2+ h−1 mg−1 chlorophyll at 25°C. These rates are 10-fold lower than the rates calculated by Kreimer et al. (1985b); however, we used a [Ca2+] of up to 100 times lower than that used in the previous study.
Antagonists to Ca2+ channels have been frequently used to characterize transport processes. Various drugs such as verapamil, nifedipine, and diltiazem have been frequently used as inhibitors of voltage-dependent Ca2+ channels (Triggle, 1990; Takahashi et al., 1997). In this study Ca2+ influx under nonpotential conditions was greatly inhibited by diltiazem and ruthenium red (Table I). The weak inhibition by diltiazem under potential gradient conditions suggests that there may be more than one pathway for Ca2+ uptake across chloroplast inner envelopes. Furthermore, LaCl3 can be used to inhibit a ruthenium-red-insensitive Ca2+/H+ exchanger in liver and kidney mitochondria (Saris and Allshire, 1989). In chloroplast inner-envelope membranes LaCl3 had no effect, indicating that Ca2+ does not move by a Ca2+/H+ antiporter. In contrast, ruthenium red, which inhibits the plant mitochondrial Ca2+ uniporter (Wilson and Graesser, 1976), almost completely inhibited Ca2+ movement across chloroplast inner-envelope vesicles (Table I).
Chloroplasts have been reported to contain from 4 to 23 mm total Ca2+ (Portis and Heldt, 1976). The concentration of free Ca2+ in the stroma of chloroplasts kept in the dark was determined to be 2.4 to 6.3 μm (Kreimer et al., 1988), which indicates that most of the Ca2+ is sequestered at binding sites within the chloroplast. Johnson et al. (1995), using a transgenic tobacco line in which expressed apoaequorin was targeted to the chloroplast stroma, estimated that under light conditions, chloroplasts maintain basal free [Ca2+] in the low nanomolar range (150 nm), indicating that even more Ca2+-binding sites, such as those on PSII (Grove and Brudvig, 1998), become available during illumination. However, when plants are transferred from light to dark conditions, a transient increase in free stromal [Ca2+], which peaks at 5 to 10 μm, takes place (Johnson et al., 1995). The transient nature of the stromal [Ca2+] increase as plants are switched from light to dark suggests that there may be a mechanism for Ca2+ efflux across the inner envelope, and also indicates that Ca2+ efflux may be slower than influx. Under certain conditions, Kreimer et al. (1985a, 1985b) observed an efflux rather than a net influx of Ca2+ into intact chloroplasts; however, they were unable to conclude whether efflux occurred via a reversal of uniport.
Inner-envelope membranes prepared by the freeze/thaw method produce vesicles of predominantly inside-out orientation (Shingles and McCarty, 1995). When vesicles in pH 8.0 buffer prepared by the freeze/thaw method were mixed with 1 μm free Ca2+, a significant Ca2+ influx was observed (Fig. 2C). This movement was also inhibited by ruthenium red but not as strongly as in predominantly right-side-out vesicles (data not shown). Therefore, it is possible that the Ca2+ uniporter may be reversed and may catalyze the net efflux of Ca2+ after chloroplasts are transferred from light to dark conditions. However, because the initial rates measured with predominantly inside-out vesicles were one-third of those measured with predominantly right-side-out vesicles, there is a directional preference to Ca2+ transport across the chloroplast inner envelope favoring Ca2+ uptake.
In this study the initial rate of Ca2+ uniport activity across the pea chloroplast inner envelope under physiological free [Ca2+] was measured to be 0.5 to 1.6 μmol h−1 mg−1 chlorophyll and was demonstrated to be stimulated by a negative membrane potential. It has been demonstrated that an inwardly directed pH gradient of 0.25 to 0.5 unit can be maintained across the chloroplast envelope by the H+-ATPase present in the inner-envelope membrane (Shingles and McCarty, 1994). This H+ gradient also represents a ΔΨ of 15 to 30 mV, enough to couple this ATPase to the potential-stimulated Ca2+ uniporter. This mechanism could result in the efficient uptake of Ca2+ by the chloroplasts as well as the activation of photosynthetic CO2 assimilation.
ACKNOWLEDGMENT
We thank Leonard Beaton for cultivating the pea plants and preparing the chloroplasts.
Abbreviations:
- βin
internal Ca2+-buffering capacity
- βout
external Ca2+-buffering capacity
- [Ca2+]cyt
free [Ca2+] in the cytosol
- ΔΨ
membrane potential difference
- Fs
fluorescence at a Ca-sensitive wavelength
- Fis
fluorescence at a Ca-insensitive wavelength
Footnotes
This work was supported by the U.S. Department of Energy (grant no. DE-FG02-92ER 200 280).
LITERATURE CITED
- Baudet S, Hove-Madsen L, Bers DM. How to make and use Ca2+-specific mini- and microelectrodes. Methods Cell Biol. 1994;40:93–113. [PubMed] [Google Scholar]
- Bensadoun A, Weinstein D. Assay of proteins in the presence of interfering materials. Anal Biochem. 1976;70:241–250. doi: 10.1016/s0003-2697(76)80064-4. [DOI] [PubMed] [Google Scholar]
- Berendes R, Burger A, Voges D, Demange P, Huber R. Calcium influx through annexin V ion channels into large unilamellar vesicles measured with fura-2. FEBS Lett. 1993;317:131–134. doi: 10.1016/0014-5793(93)81507-v. [DOI] [PubMed] [Google Scholar]
- Bers DM, Patton CW, Nuccitelli R. A practical guide to the preparation of Ca2+ buffers. Methods Cell Biol. 1994;40:3–29. doi: 10.1016/s0091-679x(08)61108-5. [DOI] [PubMed] [Google Scholar]
- Blau L, Weissmann G. Transmembrane Ca2+ movements mediated by ionomycin and phosphatidate in liposomes with fura 2 entrapped. Biochemistry. 1988;27:5661–5666. doi: 10.1021/bi00415a040. [DOI] [PubMed] [Google Scholar]
- Brand JJ, Becker DW. Evidence for direct roles of Ca2+ in photosynthesis. J Bioenerg Biomembr. 1984;16:239–249. doi: 10.1007/BF00744278. [DOI] [PubMed] [Google Scholar]
- Cleveland MJ, McCarty RE. Calcium influx in chloroplast inner envelope vesicles loaded with the fluorescent probe mag-indo-1 (abstract no. 391) Plant Physiol. 1995;108:S-84. [Google Scholar]
- Evans DE, Briars SA, Williams LE. Active Ca2+ transport by plant cell membranes. J Exp Bot. 1991;42:285–303. [Google Scholar]
- Fasolato C, Pozzan T. Effect of membrane potential on divalent cation transport catalyzed by the “electroneutral” ionophores A23187 and ionomycin. J Biol Chem. 1989;264:19630–19636. [PubMed] [Google Scholar]
- Grove GN, Brudvig GW. Calcium binding studies on photosystem II using a Ca2+-selective electrode. Biochemistry. 1998;37:1532–1539. doi: 10.1021/bi971356z. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Huang L, Berkelman T, Franklin AE, Hoffman NE. Characterization of a gene encoding a Ca2+-ATPase-like protein in the plastid envelope. Proc Natl Acad Sci USA. 1993;90:10066–10070. doi: 10.1073/pnas.90.21.10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CH, Knight MR, Kondo T, Masson P, Sedbrook J, Haley A, Trewavas A. Circadian oscillations of cytosolic and chloroplastic free Ca2+ in plants. Science. 1995;269:1863–1865. doi: 10.1126/science.7569925. [DOI] [PubMed] [Google Scholar]
- Joy KW, Mills WR. Purification of chloroplasts using silica solutions. Methods Enzymol. 1987;148:179–188. [Google Scholar]
- Keegstra K, Yousif AE. Isolation and characterization of chloroplast envelope membranes. Methods Enzymol. 1986;118:316–325. [Google Scholar]
- Kreimer G, Melkonian M, Holtum JAM, Latzko E. Characterization of Ca2+ fluxes across the envelope of intact spinach chloroplasts. Planta. 1985a;166:515–523. doi: 10.1007/BF00391276. [DOI] [PubMed] [Google Scholar]
- Kreimer G, Melkonian M, Holtum JAM, Latzko E. Stromal free Ca2+ concentration and light-mediated activation of chloroplast fructose-1,6-bisphosphatase. Plant Physiol. 1988;86:423–428. doi: 10.1104/pp.86.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreimer G, Melkonian M, Latzko E. An electrogenic uniport mediates light-dependent Ca2+ influx into intact spinach chloroplasts. FEBS Lett. 1985b;180:253–258. [Google Scholar]
- Muto S, Izawa S, Miyachi S. Light-induced Ca2+ uptake by intact chloroplasts. FEBS Lett. 1982;139:250–254. [Google Scholar]
- Pineros M, Tester M. Calcium channels in higher plant cells: selectivity, regulation and pharmacology. J Exp Bot. 1997;48:551–577. doi: 10.1093/jxb/48.Special_Issue.551. [DOI] [PubMed] [Google Scholar]
- Portis AR, Jr, Heldt HW. Light-dependent changes of the Mg2+ concentration in the stroma in relation to the Mg2+ dependency of CO2 fixation in intact chloroplasts. Biochim Biophys Acta. 1976;449:434–446. doi: 10.1016/0005-2728(76)90154-7. [DOI] [PubMed] [Google Scholar]
- Saris NL, Allshire A. Calcium ion transport in mitochondria. Methods Enzymol. 1989;174:68–85. doi: 10.1016/0076-6879(89)74011-8. [DOI] [PubMed] [Google Scholar]
- Shingles R, McCarty RE. Direct measurement of ATP-dependent proton concentration changes and characterization of a K+-stimulated ATPase in pea chloroplast inner envelope vesicles. Plant Physiol. 1994;106:731–737. doi: 10.1104/pp.106.2.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shingles R, McCarty RE. Production of membrane vesicles by extrusion: size distribution, enzyme activity, and orientation of plasma membrane and chloroplast inner-envelope membrane vesicles. Anal Biochem. 1995;229:92–98. doi: 10.1006/abio.1995.1383. [DOI] [PubMed] [Google Scholar]
- Shingles R, Roh MH, McCarty RE. Nitrite transport in chloroplast inner envelope vesicles. I. Direct measurement of proton-linked transport. Plant Physiol. 1996;112:1375–1381. doi: 10.1104/pp.112.3.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldati L, Spaventa R, Vezzoli G, Zerbi S, Adamo D, Caumo A, Rivera R, Bianchi G. Characterization of voltage-dependent Ca2+ influx in human erythrocytes by fura-2. Biochem Biophys Res Commun. 1997;236:549–554. doi: 10.1006/bbrc.1997.7002. [DOI] [PubMed] [Google Scholar]
- Sze H. H+-translocating ATPases: advances using membrane vesicles. Annu Rev Plant Physiol. 1985;36:175–208. [Google Scholar]
- Takahashi K, Isobe M, Knight MR, Trewavas AJ, Muto S. Hypoosmotic shock induces increases in cytosolic Ca2+ in tobacco suspension-cultured cells. Plant Physiol. 1997;113:587–594. doi: 10.1104/pp.113.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triggle DJ. Calcium, Ca2+ channels, and Ca2+ channel antagonists. Can J Physiol Pharmacol. 1990;68:1474–1481. doi: 10.1139/y90-224. [DOI] [PubMed] [Google Scholar]
- Wilson RH, Graesser RJ. Ion transport in plant mitochondria. In: Pirson A, Zimmermann MH, editors. Encyclopedia of Plant Physiology, Ed 1, Vol 3. Berlin: Springer-Verlag; 1976. pp. 377–397. [Google Scholar]
- Young XK, McCarty RE. Assay of proton-coupled glycolate and d-glycerate transport into chloroplast inner envelope membrane vesicles by stopped-flow fluorescence. Plant Physiol. 1993;101:793–799. doi: 10.1104/pp.101.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]