

Background: The flagellar motor generates bidirectional rotation using the proteins FliG, FliM, and CheY and membrane-bound stator complexes.
Results: The structures of portions of FliG and FliM in complex allow for insights into switching and structure.
Conclusion: FliGMC and FliMM in a 1:1 complex may form the C-ring of the motor.
Significance: Understanding motor structure is key to understanding its mechanism.
Keywords: Bacteria, Chemotaxis, Molecular Motors, Protein Complexes, Protein Structure
Abstract
The flagellar motor is one type of propulsion device of motile bacteria. The cytoplasmic ring (C-ring) of the motor interacts with the stator to generate torque in clockwise and counterclockwise directions. The C-ring is composed of three proteins, FliM, FliN, and FliG. Together they form the “switch complex” and regulate switching and torque generation. Here we report the crystal structure of the middle domain of FliM in complex with the middle and C-terminal domains of FliG that shows the interaction surface and orientations of the proteins. In the complex, FliG assumes a compact conformation in which the middle and C-terminal domains (FliGMC) collapse and stack together similarly to the recently published structure of a mutant of FliGMC with a clockwise rotational bias. This intramolecular stacking of the domains is distinct from the intermolecular stacking seen in other structures of FliG. We fit the complex structure into the three-dimensional reconstructions of the motor and propose that the cytoplasmic ring is assembled from 34 FliG and FliM molecules in a 1:1 fashion.
Introduction
The flagellar motor is a rotary nano-turbine that drives motile bacteria. The motor generates torque through the free energy of an electrochemical gradient of extracellular cations flowing through the stator in the membrane to the cytoplasmic rotor components (1, 2). The stator proteins MotA and MotB form a membrane channel (MotAB) that generates torque at the interaction site between MotAB and the rotor protein FliG (3, 4). The rotation direction results from modulation of the FliG/MotAB interaction that is regulated by the “switch complex” composed of FliM, FliG, and FliN. Multiple copies of each protein component form the cytoplasmic ring (C-ring)3 of the motor (5–8). The structure of the C-ring has been visualized by cryo-EM tomography, and models of how the motor proteins fit into the C-ring have been published by several laboratories (24). X-ray crystallography has supplied the structure of parts of every protein that comprise the C-ring, yet an absolute arrangement has not emerged. The structure of the middle domain of FliM (FliMM) has been solved alone (Protein Data Bank (PDB) 2HP7) and in complex (PDB 3SOH) with the middle domain of FliG (FliGM). In each structure, alone or in complex, FliMM has assumed identical conformations, suggesting that the structure of FliM in the C-ring has been observed in the crystal structures.
The three domains of FliG, N-terminal, middle, and C-terminal, have also been crystallized, but in each case, the domains assume different orientations with respect to one another. The structures show the domains of FliG in two conformations either held apart by extended linker helices as seen in the 1LKV and 3HJL structures or collapsed atop each other as seen in the 3AJC structure. An interesting observation is that in all these structures, the middle and C-terminal domains of FliG form a pseudo-armadillo repeat motif (ARM) stack. The apparent ARM stack can form both intermolecularly (PDB 1LKV and 3HJL) or intramolecularly (PDB 3AJC), but the orientation and interactions of the two domains in all these structures are similar.
Here we report the 1.9 Å resolution crystal structure of the middle domain of FliM (FliMM) in complex with the middle (FliGM) and C-terminal (FliGC) domains of FliG (FliGMC) from Thermotoga maritima (see Fig. 1A). FliGMC assumes a compact conformation similar to the recently published FliGMC (PDB 3AJC) structure from the Namba laboratory (9). Our structure also contains an apparent intramolecular ARM stack as well as the same interface between FliGM and FliMM as seen in the 3SOH structure. Our structure was able to be modeled into the three-dimensional cryo-tomography reconstructions of the motor provided by DeRosier laboratory, allowing for a proposal of the three-dimensional arrangement of the motor structure to be formed.
FIGURE 1.

The FliGMC-FliMM complex. A, the FliGMC-FliMM complex. B, expanded view of the ARM stacking region. A green dashed line is shown for the residues that could not be modeled in the structure. C, expanded view of the FliGMC and FliMM binding interface. Hydrogen bonding residues and resulting hydrogen bonds are shown in magenta. The black residues are the buried hydrophobic residues at the binding interface. D, the FliGMC structure sequence. Helices are underlined, and the disorder residues are marked with a strikethrough.
EXPERIMENTAL PROCEDURES
FliG (residues 115–327) and FliM (residues 46–230) were cloned into the pJY5 vector. Proteins were expressed using BL-21 Rosetta cells. The cell pellets were suspended in 25 mm Tris, pH 7.5, 400 mm NaCl, 10 mm imidazole, 1 mm EDTA and lysed with a French pressure cell. The lysate was heat shocked at 80 °C for 15 min followed by centrifugation at 13 rpm in a Beckman JA-10 rotor. The clarified lysate was passed through a nickel affinity column and eluted with 200 mm imidazole. The eluant was cleaved with tobacco etch virus protease in 50 mm Tris, pH 8.0. Following cleavage, the proteins were passed for a second time through a nickel affinity column. The flow-through was collected and exchanged into 10 mm Tris, pH 7.9, 10 mm NaCl. The proteins were mixed in a 1:1 ratio of 75 μm each and crystallized in 50 mm calcium acetate, 0.1 m sodium cacodylate, pH 6.0, and 25% v/v 2-methyl-2,4-pentanediol at 20 °C in a hanging drop plate. The crystals were frozen in liquid nitrogen after being dipped into cryo-buffer containing 0.1 m Tris, pH 8.8, 8% PEG 8000, 35% glycerol. The data sets were collected at beamline 5.0.2 at the Advanced Light Source in Berkeley, CA. Images were processed and scaled using HKL2000 (10). Because the data were highly anisotropic, ellipsoidal truncation and anisotropic scaling were performed using the UCLA Molecular Biology Institute diffraction anisotropy server (11). The structure was solved using T. maritima FliGMC (PDB 1LKV) and T. maritima FliM (PDB 2HP7) coordinates as a template for molecular replacement, refined using PHENIX (12). The structure was manually built in COOT (13).
RESULTS AND DISCUSSION
The FliGMC-FliMM complex (Fig. 1A) was generated by mixing truncated forms of FliG (residues 115–327) and FliM (residues 46–230) in an equimolar ratio. Optimized crystals displayed diffraction with Bragg spacing of 1.9 Å. The structure was solved by molecular replacement using the previously solved structures of FliG (PDB 1LKV) and FliM (PDB 2HP7) as search models (7, 14). The complex was refined to a resolution of 1.9 Å with an Rfree of 24.7% and an Rwork of 21.7%. Complete refinement statistics are shown in Table 1. The overall architecture of the complex is cylindrical with a vertical height of ∼100 Å and a diameter of ∼25 Å, with a contact surface area between FliM and FliG of 703 Å2 (15, 16). Four of the C-terminal residues of FliM and seven residues of a loop in FliG were not observed in the electron density and were presumed disordered. The unstructured region of FliG does not contain conserved residues, suggesting that this is a variable region among species. Furthermore, the missing residues precede the conserved Gly-Gly hinge located between the domains of FliG and may be unstructured due to the inherent flexibility in this region of the protein (17).
TABLE 1.
Data collection and refinement statistics (molecular replacement)
| Crystal 1 | |
|---|---|
| Data collection | |
| Space group | C121 |
| Cell dimensions | |
| a, b, c (Å) | 125.96, 45.72, 98.84 |
| α, β, γ (°) | 90.0, 100.85, 90.0 |
| Resolution (Å) | 50–1.93 (2.0–1.93)a |
| Rsym or Rmerge | 0.076 (0.44) |
| I/σI | 17.3 (1.65) |
| Completeness (%) | 99.5 (99.8) |
| Redundancy | 5.3 (4.6) |
| Refinement | |
| Resolution (Å) | 50–1.93 |
| No. of reflections | 38,740 |
| Rwork/Rfree | 21.7/24.7 |
| No. of atoms | 3249 |
| Protein | 3140 |
| Ligand/ion | |
| Water | 109 |
| B-factors | |
| Protein | 32.27 |
| Ligand/ion | |
| Water | 29.0 |
| r.m.s deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.0 |
a Highest resolution shell is shown in parentheses.
Upon binding to FliGMC, FliMM does not undergo any significant conformational changes, with an r.m.s deviation of 0.6 Å as compared with the FliMM 2.0 Å resolution structure (PDB 2HP7) or the 3.5 Å resolution structure of FliMM bound to FliGM both previously solved by the Blair and Crane laboratories (7, 18). FliMM consists of three β-strands (β1–β3) and three α-helices (α1–α3) that form a pseudo-symmetric α/β/α three-layered sandwich. The highly conserved GGXG sequence motif, located in the loop between helices α3–α1′, is involved in FliG binding and confirmed by both the 3SOH and this structure (6, 19). In the 2HP7 structure, the two glycine residues of the GGXG motif are resolved, but the following four residues (PGEN) are disordered (7). Upon binding to FliGM, the loop in FliMM surrounding the GGXG motif becomes ordered and stabilized and is observable in both the 3SOH and this structure. The FliGM/FliMM interaction surface is positioned directly above the secondary binding site of CheY located on FliMM that was identified by NMR experiments (20). When the response regulator CheY is phosphorylated (CheY-P), it is able to bind to FliM to signal the switch in rotation direction. The proximity of the CheY-P binding site to the FliGM/FliMM interface could explain how the switch signal is propagated from FliM through FliG toward the MotAB complex.
FliGMC in complex with FliMM is predominantly α-helical consisting of 14 helices arranged in an extended structure (Fig. 1A). A right-handed super-helix is generated from the stacking of pseudo-armadillo repeats from helices 1–4 (ARMM) and helices 6–8 (ARMC) (Fig. 1B), mimicking the stacking observed in eukaryotic ARM motifs (21, 22). The ARMM/ARMC stacking results in the burial of 14 hydrophobic residues forming a contact area of 745 Å2 (15, 16). HelixMC lies below ARMM and interacts with helices 1–2 of FliGMC as well as parts of FliMM. The FliMM loop between helices α1′ and α3 and residues within the helices themselves interact with ARMM and HelixMC of FliG, forming a network of bonding interactions. Fig. 1C shows the packing of hydrophobic residues and hydrogen bonding between the two proteins. The C-terminal domain (HelicesC1–6) is located above the intramolecular ARM stack with the charged HelixC5 positioned on top of the domain, free to interact with MotAB (3). The C-terminal domain of FliG is principally connected to the ARM stack through the loop between helices 8 and C1, suggesting that changes in the position of FliGC relative to the ARM stack may be a possible component of motor switching.
In two of the four FliG structures (PDB 1LKV and 3HJL), the domains are held separate by extended linker helices (Fig. 2, C and D) (14, 23). Brown et al. (14) suggested that HelixMC is stabilized through crystal contacts and postulated that this region would be flexible in vivo if the crystal packing represented nonphysiological interactions. Furthermore, Van Way et al. (17) used bioinformatic analysis in conjunction with mutational experiments to show that the conserved Gly-Gly pair, at the end of HelixMC, acts as a flexible hinge between the two domains of FliGMC. The 3AJC structure (Fig. 2B) shows HelixMC in a new position as compared with the 1LKV and 3HJL structures. The new position of HelixMC was likely a product of the crystal packing facilitated by domain mobility through the Gly-Gly pair (9). In the FliGMC-FliMM complex, HelixMC is packed against FliMM beneath ARMM as opposed to being pointed away from the ARM stack as in the 3AJC structure. The multiple positions of HelixMC observed in the structures suggest a degree of freedom that is likely limited when FliGM binds to FliMM, allowing HelixMC to pack against FliMM. In the 3AJC structure, the position of HelixMC contacts the neighboring molecule, and movement of HelixMC in response to CheY-P has been suggested to play a role in switching (9, 24). HelixMC in our complex structure is not in contact with neighboring molecules as it is in the 1LKV and 3AJC structures. This suggests that it may play a role in switching but not through the communication of multiple FliG monomers.
FIGURE 2.
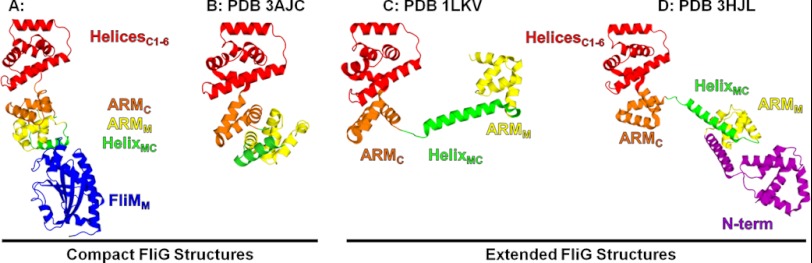
Comparison of the FliG structures. Four of the published FliG structures show that FliG can assume either a compact or an extended structure. Overall the individual domains retain their fold with their orientation varying in each structure. A, FliGMC-FliMM complex. B, FliGMC (3AJC) (9). C, FliGMC (1LKV) (14). D, FliG full-length (3HJL) (23).
The majority of the C-ring models argue that the ARM stacking observed in the FliG structures is a central structural feature of the C-ring. Alignments of the individual ARM domains of FliGMC-FliMM complex versus the ARM domains of 1LKV, 3HJL, and 3AJC show r.m.s deviation values <1.0 Å. Thus when the ARM domains stack, the ARM domain structures do not change significantly whether the domains come from the same molecule or from adjacent molecules. The crystal packing of FliG structures 1LKV and 3HJL show ARM stacking between ARMC and ARMM of symmetry-related copies of FliG (Fig. 3, C and D) (14, 23). This results in an ARMC-ARMM+1 stack. In the FliGMC-FliMM complex, the ARM stacking is observed within a single FliG protomer (Fig. 3A). The intramolecular ARM stacking is also assumed in the 3AJC structure (Fig. 3B), although the residues connecting the domains are not observed (9). We propose that ARM stacking is driven by the requirement to bury the hydrophobic residues that comprise the ARM domains, and the intramolecular stack is driven by the binding to FliM as seen in the complex. Determining whether the ARM stacking is intra- or intermolecular in the C-ring requires further experimentation, but in the co-crystal, the presence of FliM apparently drove the ARM domains to stack within a single FliGMC monomer, suggesting that the in vivo interaction could be intramolecular.
FIGURE 3.
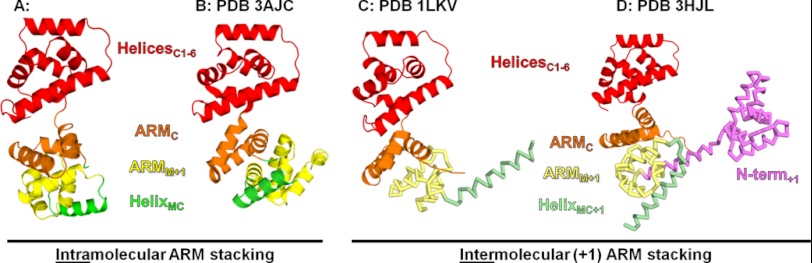
Comparison of the ARM stacking observed in structures and crystal packing. The (+1) designates that these domains are from a neighboring molecule in the crystal packing. For clarity, the domains from neighboring molecules are shown in ribbon form and pale colors. A, ARM stacking observed in the FliGMC-FliMM complex structure. B, ARM stacking observed in the 3AJC crystal (9). C, ARM stacking observed in the 1LKV crystal (14). D, ARM stacking observed in the 3HJL crystal (23).
The intramolecular ARM stacking we observed in the crystal may represent a preassembly conformation of FliG. In complex with FliM, FliG in the intramolecular stacking configuration may be a precursor building block of the C-ring, and as they are assembled into the motor, FliG may undergo a transformation to the intermolecular interaction. The in vivo stacking may be represented by either the stacking we observed in the co-complex structure or the intermolecular stacking observed in the other FliG structures. If the intermolecular stacking proposal were correct, the rearrangement from cytoplasmic intramolecular stacking to the motor assembled intermolecular stacking would require the 745 Å2 of contact surface between the ARM domains to be broken and reassembled during the transition. This transition would have to overcome the energetics of exposing the 14 buried hydrophobic residues to reassemble the ARM stacks in the motor in the exact way they were previously, but now in the intermolecular stacked arrangement. Further experimentation on intact isolated motors will be necessary to unambiguously determine the arrangement of FliG inside the C-ring.
FliG and FliM have been extensively studied by mutational analysis and biophysical techniques to determine the interaction site or sites (17, 19, 20, 25, 26). FliGM contains a conserved EHPQ motif that when mutated disrupts the binding to FliMM. These same residues were shown to be in contact in the FliGM-FliMM co-crystal (PDB 3SOH) and in the complex presented here. The interaction surface between FliMM and FliGM has also been analyzed by NMR and chemical cross-linking. In NMR experiments, the resonances assigned to the EHP(Q/R) motif broaden when labeled FliGM is titrated with FliMM, suggesting that the surfaces seen in the crystal interact in solution as well (20). Cross-linking experiments were designed from the 3SOH structure and showed that FliGM is able to cross-link to FliMM in vivo, confirming that these faces are in contact in the assembled motor.
Blair and co-workers (7, 18) have established that mutations in the conserved hydrophobic patch in ARMC also affect binding to FliM and have performed chemical cross-linking of intact cells to determine whether FliGC is in contact with FliMM in vivo (6, 19). Their cross-linking results show that the hydrophobic patch of FliGC binds to the GGXG surface on FliMM as FliGM. In previous work (20), we used NMR to monitor the interactions of FliGC and FliMM. This showed chemical shifts in the resonances corresponding to the HelicesC1–6 region of FliGC and not in the ARMC region, suggesting that the cross-linking results may not be present in solution. Composition gradient multiangle light scattering was also performed to measure the affinity of FliGC and FliMM, resulting in a Kd of 580 μm. (data not shown). This low affinity suggests that the interaction would probably not survive EM extraction. A model for the C-ring presented by Blair and co-workers (7, 18) imagines that a portion of the FliG molecules in the C-ring interacts with two FliMM domains, one via the FliGM domain and the other via the FliGC domain. The remaining FliG molecules interact with FliMM via the FliGC domain only. DeRosier and co-workers (27) suggest that this unusual arrangement allows for matching the 26–34-fold stoichiometry difference inferred from averaging of the EM reconstructions of the flagellar motor. The yields of the cross-links they observe between these domains are relatively low, suggesting that these interactions may reflect the behavior of a subpopulation of both FliM and FliG.
The FliGMC-FliMM complex was modeled into the high-resolution maps of the motor derived from three-dimensional electron micrograph reconstructions of a clockwise locked motor from Salmonella typhimurium (27, 28). The C-ring from the three-dimensional reconstructions consists of two parts, an inner lobe with 26-fold symmetry and an outer lobe with 34-fold symmetry. The inner lobe of the C-ring disappears in three-dimensional reconstructions of a motor deleted for FliGN (29). This suggests that FliGN is only present in the inner lobe and that the outer lobe contains FliGMC, FliMM, and FliN that is mirrored by our construction of the C-ring.
The outer lobe is divided into three parts consisting of an upper, middle, and lower section. The upper section faces the membrane and is bridged to the middle section by a thin strip of density. Below the middle section is a ring that is believed to contain the FliN tetramer and the C terminus of FliM. The FliGMC-FliMM complex structure was docked into the density by the program VEDA with a correlation coefficient of 66.6 and an R-factor of 58.0. The alignment positioned the ridge of charged residues in FliGC toward the membrane in position to interact with the MotAB membrane channel (Fig. 4A). FliGM aligns nicely into density between the upper and middle sections, interacting with both FliMM and FliG. The orientation of FliMM in the middle lobe places the FliMM-FliMM subunit interfaces in close proximity, as identified previously by disulfide cross-linking experiments (7). Once we were satisfied with the position of our complex, the FliN tetramer was docked into the lower section of density. An expanded view of the docking is shown in Fig. 4C. The orientation of FliN to FliM was estimated from cross-linking experimental data and then refined by the VEDA fitting macro. Our x-ray structure and EM model fitting places the FliGC domain, FliM, and FliN subunits similarly to the model proposed by Lee et al. (15) without invoking the crystal packing ARMC-ARMM+1 notion they proposed. Furthermore, we observed a partial melting of HelixMC, suggesting that HelixNM might be destabilized as well in the complex. This has major implications with regard to some of the rigid body movements proposed for these proteins during the flagellar motions (9, 18, 23).
FIGURE 4.

Modeling the FliGMC-FliMM complex crystal structure and FliN tetramer into three-dimensional reconstructions of the motor. Maps of the flagellar motor were provided by the DeRosier laboratory. VEDA was used to position the structures into the density; a total of 34 copies of the FliGMC-FliMM complex were positioned in the density as well as 34 copies of the FliN tetramer (30). A, side view of the assembled C-ring. B, bottom up view of the C-ring. C, expanded view of a portion of the C-ring illustrating the fit of the proteins inside of the density.
The symmetry of the C-ring observed in the three-dimensional reconstructions shows a shift from 34-fold to 26-fold when going from the outer to inner rings, respectively (Fig. 4B). The modeling of the C-ring presented here shows FliG and FliM in a 1:1 stoichiometry, which implies that the mismatch in the symmetry is managed in the connection from the outer to inner rings. We postulate that only 26 out of the 34 of the FliGN domains are able to bind to the 26 FliF copies located in the inner ring. The eight unbound FliGN domains would most likely be located unbound inside the inner space of the C-ring and would not participate in torque transfer from the outer to inner rings. This would allow for the motor to reconcile the symmetry mismatch in the simplest method possible. The symmetry mismatch may be a dynamic part of motor assembly. The three-dimensional reconstructions showed that the C-ring varies between 31-, 33-, and 34-fold symmetries, and the free FliGN domains may explain the ability of the C-ring to assume different copy numbers by displacing FliG-FliM complexes when necessary.
Acknowledgments
We thank Dr. David DeRosier for providing the three-dimensional maps of the flagellar motor. We also thank Dr. Gael Goret for the review of the density fitting.
The atomic coordinates and structure factors (code 4FHR) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- C-ring
- cytoplasmic ring
- ARM
- armadillo repeat motif
- r.m.s.
- root mean square.
REFERENCES
- 1. Blair D. F. (2003) Flagellar movement driven by proton translocation. FEBS Lett. 545, 86–95 [DOI] [PubMed] [Google Scholar]
- 2. Berg H. C. (2003) The rotary motor of bacterial flagella. Annu. Rev. Biochem. 72, 19–54 [DOI] [PubMed] [Google Scholar]
- 3. Lloyd S. A., Blair D. F. (1997) Charged residues of the rotor protein FliG essential for torque generation in the flagellar motor of Escherichia coli. J. Mol. Biol. 266, 733–744 [DOI] [PubMed] [Google Scholar]
- 4. Yakushi T., Yang J., Fukuoka H., Homma M., Blair D. F. (2006) Roles of charged residues of rotor and stator in flagellar rotation: comparative study using H+-driven and Na+-driven motors in Escherichia coli. J. Bacteriol. 188, 1466–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Irikura V. M., Kihara M., Yamaguchi S., Sockett H., Macnab R. (1993) Salmonella typhimurium fliG and fliN mutations causing defects in assembly, rotation, and switching of the flagellar motor. J. Bacteriol. 175, 802–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marykwas D. L., Berg H. C. (1996) A mutational analysis of the interaction between FliG and FliM, two components of the flagellar motor of Escherichia coli. J. Bacteriol. 178, 1289–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Park S. Y., Lowder B., Bilwes A. M., Blair D. F., Crane B. R. (2006) Structure of FliM provides insight into assembly of the switch complex in the bacterial flagella motor. Proc. Natl. Acad. Sci. U.S.A. 103, 11886–11891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tang H., Braun T. F., Blair D. F. (1996) Motility protein complexes in the bacterial flagellar motor. J. Mol. Biol. 261, 209–221 [DOI] [PubMed] [Google Scholar]
- 9. Minamino T., Imada K., Kinoshita M., Nakamura S., Morimoto Y. V., Namba K. (2011) Structural insight into the rotational switching mechanism of the bacterial flagellar motor. PLoS Biol. 9, e1000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Method Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 11. Strong M., Sawaya M. R., Wang S., Phillips M., Cascio D., Eisenberg D. (2006) Toward the structural genomics of complexes: crystal structure of a PE-PPE protein complex from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 103, 8060–8065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 14. Brown P. N., Hill C. P., Blair D. F. (2002) Crystal structure of the middle and C-terminal domains of the flagellar rotor protein FliG. EMBO J. 21, 3225–3234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee B., Richards F. M. (1971) The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 55, 379–400 [DOI] [PubMed] [Google Scholar]
- 16. Saff E., Kuijlaars A. B. (1997) Distributing many points on a sphere. Mathematical Intelligencer 19, 5–11 [Google Scholar]
- 17. Van Way S. M., Millas S. G., Lee A. H., Manson M. D. (2004) Rusty, jammed, and well-oiled hinges: mutations affecting the interdomain region of FliG, a rotor element of the Escherichia coli flagellar motor. J. Bacteriol. 186, 3173–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paul K., Gonzalez-Bonet G., Bilwes A. M., Crane B. R., Blair D. (2011) Architecture of the flagellar rotor. EMBO J. 30, 2962–2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Passmore S. E., Meas R., Marykwas D. L. (2008) Analysis of the FliM/FliG motor protein interaction by two-hybrid mutation suppression analysis. Microbiology 154, 714–724 [DOI] [PubMed] [Google Scholar]
- 20. Dyer C. M., Vartanian A. S., Zhou H., Dahlquist F. W. (2009) A molecular mechanism of bacterial flagellar motor switching. J. Mol. Biol. 388, 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Andrade M. A., Petosa C., O'Donoghue S. I., Müller C. W., Bork P. (2001) Comparison of ARM and HEAT protein repeats. J. Mol. Biol. 309, 1–18 [DOI] [PubMed] [Google Scholar]
- 22. Parmeggiani F., Pellarin R., Larsen A. P., Varadamsetty G., Stumpp M. T., Zerbe O., Caflisch A., Plückthun A. (2008) Designed armadillo repeat proteins as general peptide-binding scaffolds: consensus design and computational optimization of the hydrophobic core. J. Mol. Biol. 376, 1282–1304 [DOI] [PubMed] [Google Scholar]
- 23. Lee L. K., Ginsburg M. A., Crovace C., Donohoe M., Stock D. (2010) Structure of the torque ring of the flagellar motor and the molecular basis for rotational switching. Nature 466, 996–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stock D., Namba K., Lee L. K. (2012) Nanorotors and self-assembling macromolecular machines: the torque ring of the bacterial flagellar motor. Curr. Opin. Biotechnol. 23, 545–554 [DOI] [PubMed] [Google Scholar]
- 25. Brown P. N., Terrazas M., Paul K., Blair D. F. (2007) Mutational analysis of the flagellar protein FliG: sites of interaction with FliM and implications for organization of the switch complex. J. Bacteriol. 189, 305–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kihara M., Miller G. U., Macnab R. M. (2000) Deletion analysis of the flagellar switch protein FliG of Salmonella. J. Bacteriol. 182, 3022–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thomas D. R., Francis N. R., Xu C., DeRosier D. J. (2006) The three-dimensional structure of the flagellar rotor from a clockwise-locked mutant of Salmonella enterica serovar Typhimurium. J. Bacteriol. 188, 7039–7048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Computut. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 29. Thomas D., Morgan D. G., DeRosier D. J. (2001) Structures of bacterial flagellar motors from two FliF-FliG gene fusion mutants. J. Bacteriol. 183, 6404–6412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brown P. N., Mathews M. A., Joss L. A., Hill C. P., Blair D. F. (2005) Crystal structure of the flagellar rotor protein FliN from Thermotoga maritima. J. Bacteriol. 187, 2890–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]