

Background: CTRP12 is an insulin-sensitizing adipokine that exists in two isoforms.
Results: CTRP12 is a multimeric glycoprotein subjected to multiple posttranslational modifications. Cleavage generates an isoform with an altered oligomeric state and signaling specificity.
Conclusion: Differential proteolytic processing generates functionally distinct isoforms of CTRP12.
Significance: Cleavage-dependent modulation of oligomeric state represents a novel mechanism controlling signaling specificity and function of a hormone.
Keywords: Adipokines, Akt, Cell Signaling, MAP Kinases (MAPKs), Signaling
Abstract
Adipose tissue-derived adipokines are an important class of secreted metabolic regulators that mediate tissue cross-talk to control systemic energy balance. We recently described C1q/TNF-related protein-12 (CTRP12), a novel insulin-sensitizing adipokine that regulates glucose metabolism in liver and adipose tissue. However, the biochemical properties of CTRP12 and its naturally occurring cleaved isoform have not been characterized. Here, we show that CTRP12 is a secreted hormone subjected to multiple functionally relevant posttranslational modifications at highly conserved residues. For example, Asn39 is glycosylated, whereas Cys85 mediates the assembly of higher order oligomeric structure. Endopeptidase cleavage at Lys91 generates a cleaved globular gCTRP12 isoform, the expression of which is increased by insulin. PCSK3/furin was identified as the major proprotein convertase expressed by adipocytes that mediates the endogenous cleavage of CTRP12. Cleavage at Lys91 is context-dependent: mutation of the charged Arg93 to Ala on the P2′ position enhanced cleavage, and triple mutations (K90A/K91A/R93A) abolished cleavage. Importantly, the two isoforms of CTRP12 differ in oligomeric structures and are functionally distinct. The full-length protein forms trimers and larger complexes, and the cleaved isoform consisted of predominantly dimers. Whereas full-length fCTRP12 strongly activated Akt signaling in H4IIE hepatocytes and 3T3-L1 adipocytes, gCTRP12 preferentially activated MAP kinase (ERK1/2 and p38 MAPK) signaling. Further, only fCTRP12 improved insulin-stimulated glucose uptake in adipocytes. These results reveal a novel mechanism controlling signaling specificity and function of a hormone via cleavage-dependent alteration in oligomeric state.
Introduction
Adipose tissue-derived adipokines are an important class of metabolic regulators (1, 2). These secreted hormones and cytokines function in an autocrine, a paracrine, and/or an endocrine manner to modulate whole body insulin sensitivity and energy balance, either acting directly on metabolic tissues (e.g. skeletal muscle and liver) or indirectly through their regulatory roles on inflammatory processes (1, 2).
Our recent effort to discover novel metabolic regulators has led to the identification of a family of 15 secreted proteins designated as CTRP1–CTRP153 (3–7). CTRPs are part of the larger C1q family, all of which share a C-terminal globular domain homologous to the immune complement C1q (8). Recent studies suggest that CTRPs regulate glucose and fatty acid metabolism (3–7, 9–11), as well as playing diverse roles in the immune (12, 13), skeletal (14, 15), vascular (16, 17), and sensory systems (18–20).
CTRP12 is an insulin-sensitizing adipokine produced predominantly by adipose tissue in humans; in mice, it is more widely expressed (10). Whereas expression and circulating levels of CTRP12 are decreased in the obese state, antidiabetic drugs such as rosiglitazone enhance its expression in adipocytes. Recombinant CTRP12 administration acutely lowers blood glucose in wild-type mice, as well as in various mouse models (leptin-deficient ob/ob and diet-induced obese) of obesity and diabetes. Further, a moderate increase in circulating levels of CTRP12 via adenovirus-mediated overexpression is sufficient to improve insulin sensitivity in obese and insulin-resistant mice, in part by enhancing Akt signaling in liver and adipose tissue as well as by reducing the circulating levels of resistin known to induce hepatic insulin resistance. Independent of insulin, CTRP12 promotes glucose uptake in adipocytes and suppresses de novo glucose production in hepatocytes via the PI3 kinase-Akt signaling pathway (10).
Although the general metabolic function of CTRP12 has been established, important questions concerning its regulation remain unanswered. Recombinant and endogenous CTRP12 exist in two isoforms: the full-length protein and a proteolytically processed globular isoform. In the present study, we provide evidence that the two naturally occurring isoforms differ in higher order oligomeric structure due to differential processing by an endopeptidase. Proteolytic cleavage generates a functionally distinct isoform with altered signaling specificity, revealing a novel cleavage-dependent mechanism controlling CTRP12 function.
MATERIALS AND METHODS
Antibodies and Chemicals
Mouse monoclonal anti-FLAG M2 antibody was obtained from Sigma. Rabbit antibodies that recognize IRS1, phospho-AKT (Thr308), Akt, phospho-p44/42 MAPK (Thr202/Tyr204), phospho-p38 MAPK (Thr180/Tyr182), p44/42 MAPK, and p38 MAPK were obtained from Cell Signaling Technology. Anti-phospho-IRS1 (Tyr612) was obtained from BIOSOURCE. Anti-CTRP12-specific antibody was developed as described previously (10). Recombinant insulin was obtained from Sigma. PNGase F was obtained from New England Biolabs.
Site-directed Mutagenesis
High fidelity Pfu polymerase from Stratagene was used in site-directed mutagenesis of CTRP12 cDNA to generate N39A, C85A, and R93A single-substitution proteins or the (K90A/K91A/R93A) triple-substitution protein (designated as 3M) according to the manufacturer's protocol. Primers used in site-directed mutagenesis were as follows: N39A forward, 5′-CGTGTGGATTCCCCCGCTATTACCACGTCCAAC-3′ and reverse, 5′-GTTGGACGTGGTAATAGCGGGGGAATCCACACG-3′; C85A forward, 5′-GTCCTCTAGAAAACGGGCTCGTGGCCGGGACAAG-3′ and reverse, 5′-CTTGTCCCGGCCACGAGCCCGTTTTCTAGAGGAC-3′; R93A forward, 5′-CCGGGACAAGAAGTCGGCAGGCCTCTCAGGTCTC-3′ and reverse, 5′-GAGACCTGAGAGGCCTGCCGACTTCTTGTCCCGG-3′; triple mutant (3M) forward, 5′-CGGTGTCGTGGCCGGGACGCGGCGTCGGCAGGCCTCTCAGGTCTC-3′ and reverse, 5′-GAGACCTGAGAGGCCTGCCGACGCCGCGTCCCGGCCACGACACCG-3′. The R93A mutant was used as a PCR template to generate 3M. Successful mutagenesis was confirmed by DNA sequencing.
Protein Purification
Recombinant CTRP12 and its R93A mutant and triple mutant (3M) were produced and purified as described (10). In brief, GripTiteTM 293 cells (Invitrogen) were transfected with pcDNA3.1 encoding a C-terminal FLAG-tagged CTRP12 and its single or triple mutants using the calcium phosphate method. To generate gCTRP12, we cloned the entire globular isoform (corresponding to residues 92–308) downstream of the endogenous signal peptide (residues 1–21). Supernatants (serum-free Opti-MEM; Invitrogen) of cells were collected and subjected to affinity chromatography using anti-FLAG M2 Affinity Gel (Sigma) according to the manufacturer's protocol. Protein was eluted with 100 μg/ml FLAG peptide in PBS buffer. Protein was dialyzed against HEPES buffer (20 mm HEPES, 135 mm NaCl, pH 8.0) in a 10-kDa cut-off Slide-A-Lyzer dialysis cassette (Thermo Scientific), concentrated using a 10-kDa cut-off Amicon Ultra centrifugal filter (Millipore), aliquoted, and stored at −80 °C. Protein concentration was determined using the Coomassie Plus Protein Assay Reagent (Thermo Scientific).
Cell Culture
GripTiteTM 293 cells were cultured in DMEM (Invitrogen) containing 10% fetal bovine serum (FBS), 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. Mouse 3T3-L1 preadipocytes were cultured in high glucose DMEM containing 10% calf serum and antibiotics. 3T3-L1 adipocytes were differentiated as described previously (9). In brief, mouse 3T3-L1 preadipocytes were cultured until fully confluent. Differentiation was induced 2 days after cells reached confluence by culturing cells for 2 days in DMEM with 10% FBS supplemented with 1 μg/ml bovine insulin (Sigma), 0.25 μm dexamethasone (Sigma), and 0.5 mm 3-isobutyl-1-methylxanthine (Sigma), followed by DMEM/FBS containing only insulin (1 μg/ml) for an additional 2 days. Differentiated adipocytes were maintained thereafter in DMEM/FBS for another 4 days. Rat H4IIE hepatocytes were cultured in DMEM containing 10% FBS and antibiotics.
Western Blot Analysis
For Western blot analyses of signaling proteins, cells were serum-starved for 2 h and then treated as indicated in the figure legends. Cells were subsequently washed once with PBS and lysed in cell lysis/loading buffer (50 mm Tris, 2% SDS, 1% β-mercaptoethanol, 6% glycerol, 0.01% bromphenol blue). Cell lysates were then sonicated and incubated at 100 °C for 5 min. Proteins were separated in 4–12% Bis-Tris NuPAGE gel (Invitrogen), immunoblotted onto 0.2 μm Protran BA 83 nitrocellulose membrane (Whatman), blocked with 2% nonfat milk for 1 h, and probed with primary antibodies overnight. Immunoblots were washed three times (10 min each) in PBS containing 0.1% Tween 20 and incubated with horseradish peroxidase-conjugated secondary antibody (Amersham Biosciences) (1:5000) for 1 h. Blots were washed three times (10 min each) in PBS containing 0.1% Tween 20, developed in ECL reagent (Millipore) for 2–5 min and visualized with MultiImage III FluorChem Q (Alpha Innotech). Quantifications of signal intensity were performed using Alphaview Software (Alpha Innotech).
Lentiviral-mediated Knockdown of Furin in Adipocytes
Lentivirus particles containing an empty pLK0.1 vector or pLK0.1 encoding an shRNA against furin were obtained from Sigma. Differentiated 3T3-L1 adipocytes (day 8) were treated with the control or furin-shRNA lentivirus at a multiplicity of infection of 1:100. Infected adipocytes were subsequently selected by medium containing 1 μg/ml puromycin for 4 days. Adipocytes were then harvested for immunoblot analyses.
Glucose Uptake Assay in Adipocytes
Glucose uptake assay was performed as described previously (10). Briefly, differentiated 3T3-L1 adipocytes were serum-starved in low glucose DMEM for 2 h. Adipocytes were treated with vehicle or 10 μg/ml gCTRP12 or 10 μg/ml fCTRP12 for 30 min, with or without insulin (100 nm) for 15 min. Cells were then incubated with uptake medium containing 0.5 μCi/ml 2-deoxy-d-[1-14C]glucose in Krebs-Ringer-HEPES buffer (25 mm HEPES-NaOH, pH 7.4, 120 mm NaCl, 5 mm KCl, 1.2 mm MgSO4, 1.3 mm CaCl2,1.3 mm KH2PO4) supplemented with 0.5% BSA for 10 min. Uptake was stopped by aspirating the medium and washing extensively with ice-cold PBS buffer. Cells were lysed with 0.1% Triton X-100 in PBS buffer. An aliquot of cell lysate from each sample was obtained for protein content analysis using bicinchoninic acid assay kit (Pierce). Radioactivity of cell lysates was counted in Ecoscint® scintillation mixture (National Diagnostics) using a Beckman LS-6000 Liquid Scintillation Counter. Radioactivity in each sample was normalized against protein content.
Gel Filtration Chromatographic Analysis
Supernatants (500 μl) from transfected HEK 293T cells containing FLAG-tagged CTRP12 (WT), gCTRP12, R93A mutant, and triple mutant (3M) were sequentially loaded onto an AKTA FPLC and fractionated through a 10/30 GL Superdex 200 column (GE Healthcare) in PBS. The collected fractions (0.5 ml each) were subjected to Western blot analysis using the anti-FLAG antibody.
Quantitative Real-time PCR Analysis
For expression analyses of Pcsk genes, total RNAs were isolated from cells using TRIzol (Invitrogen) and reverse-transcribed using Superscript II RNase H-Reverse Transcriptase (Invitrogen). Quantitative real-time PCR analyses were performed on an Applied Biosystems 7500 Sequence Detection System. Samples were analyzed in 25-μl reactions according to the standard protocol provided in the SYBR Green PCR Master Mix (Applied Biosystems). All expression levels were normalized to corresponding 18 S rRNA levels. Primers used in real-time PCR included the following: PCSK1 forward, 5′-TCTGCCATCGCCGAAGAAC-3′ and reverse, 5′-CCCACGTCACACGATCATCAT-3′; PCSK2 forward, 5′-GTGTGATGGTTTTTGCGTCTG-3′ and reverse, 5′-GGGAGCTTTCGGACTCCAA-3′; PCSK3/furin forward, 5′-TCGGTGACTATTACCACTTCTGG-3′ and reverse, 5′-CTCCTGATACACGTCCCTCTT-3′; PCSK4 forward, 5′-CGCTACACACCCAACGATGAG-3′ and reverse, 5′-CCTCCAATTCTAGCATTGAAGGC-3′; PCSK5 forward, 5′-CCATGCCCAGTCAACCTACTT-3′ and reverse, 5′-CAGGCTCCTTCGATATTCATGTC-3′; PCSK6/PACE4 forward, 5′-CTTCCATCCAAGGCTCTCCTA-3′ and reverse, 5′-GGTCTGCATTGGGACCATCA-3′; PCSK7 forward, 5′-GTCCAGCCTACTGGGCATAG-3′ and reverse, 5′-TGAAGTGGATACTGCGCTTGG-3′; 18 S rRNA forward, 5′-GCAATTATTCCCCATGAACG-3′ and reverse, 5′-GGCCTCACTAAACCATCCAA-3′.
Statistical Analysis
Comparisons were performed using two-tailed Student's t tests. Values were considered to be significant at p < 0.05. All data are presented as means ± S.E.
RESULTS
Cleavage, Glycosylation, and Oligomerization of CTRP12
Mouse CTRP12 contains four recognizable domains, three potential N-linked glycosylation sites that conform to NX(S/T) motif (21), four Cys residues, and an endopeptidase cleavage motif, KKXR, located in the N terminus (Fig. 1A). These features suggest that the protein likely undergoes multiple types of posttranslational modification. Importantly, the four Cys residues, the cleavage motif, and one of the N-glycosylation sites (Asn39) are highly conserved in divergent vertebrate species (Fig. 1B).
FIGURE 1.

Domain structure and conserved features of CTRP12. A, four domains of CTRP12 protein: a signal peptide (SP); an N-terminal domain with the highly conserved N-glycosylation site (Asn39), Cys85, and polybasic cleavage motif KKXR; a collagen domain with six Gly-X-Y repeats; and a globular C1q/TNF-like domain with three conserved Cys residues (Cys162, Cys241, and Cys246) and two potential N-glycosylation sites (Asn287 and Asn297). Endopeptidase cleavage at Lys91 (indicated by arrow) generates the gCTRP12 isoform. B, sequence alignment of mouse, human, chicken, Xenopus, and zebra fish CTRP12. Highlighted are the conserved Asn39 shown to be glycosylated in this study, the cleavage motif KKXR, and the Cys85 shown in this study to be important for disulfide-linked multimerization of CTRP12.
Two isoforms of the protein, differing in size, were produced when FLAG epitope-tagged CTRP12 was expressed in mammalian HEK 293T cells (Fig. 2A), indicating proteolytic processing of the recombinant CTRP12. Cleavage is physiologically relevant: endogenous CTRP12 secreted from differentiated 3T3-L1 adipocytes and CTRP12 circulating in human and mouse serum also exist in two isoforms: full-length fCTRP12 (∼40 kDa) and a cleaved gCTRP12 (∼25 kDa) (10). In serum, gCTRP12 is the predominant isoform. Because both isoforms were found in cell pellet and supernatant of CTRP12-expressing HEK 293T cells, proteolytic processing most likely occurred inside the cell prior to protein secretion (Fig. 2A). N-terminal sequencing of purified mouse recombinant gCTRP12 (Fig. 2B) indicated that cleavage occurred at Lys-91, at the predicted endopeptidase cleavage site (K90K91 ↓ X92R93) (10). Cleavage appears to be regulated; only a fraction of fCTRP12 is processed into gCTRP12.
FIGURE 2.

CTRP12 is a secreted multimeric glycoprotein. A, immunoblot analysis of CTRP12 expressed in HEK 293T cells. Empty vector (pcDNA3.1) was used as negative control. P, pellet; S, supernatant. B, Coomassie Blue staining of purified recombinant CTRP12 on a SDS-PAGE gel. C, CTRP12 treated with (+) or without (−) PNGase F. D, immunoblot analysis of wild-type (WT) CTRP12 and its N39A mutant. E, immunoblot analysis of gCTRP12 treated with (+) or without (−) PNGase F (left) and the gCTRP12 mutants. F, immunoblot analysis of WT CTRP12 and its C85A mutant treated with (+) or without (−) β-mercaptoethanol (β-ME). Arrows indicate the monomeric, trimeric, and higher order molecular mass oligomeric form of CTRP12.
Broad banding of fCTRP12 on immunoblot indicated the presence of a carbohydrate moiety (Fig. 2A). Consistent with this, treatment of PNGase F reduced the apparent molecular mass of fCTRP12 on an immunoblot (Fig. 2C), confirming the presence of N-linked oligosaccharides. Of the three potential N-glycosylation sites (Asn39, Asn287, and Asn297), only Asn39 is conserved from zebra fish to humans (Fig. 1B), suggesting that Asn39 is likely the glycan attachment site. Indeed, mutation of Asn39 to Ala resulted in a similar mobility shift on immunoblot, comparable with PNGase F (Fig. 2D), confirming that N-terminal Asn39 is posttranslationally modified with oligosaccharides. Treatment of gCTRP12 with PNGase F did not result in mobility shift of the protein on immunoblotting (Fig. 2E, left), suggesting the absence of N-linked glycans. Consistent with this, gCTRP12 protein harboring an N287A single mutation or N287A/N297A double mutations also showed no mobility shift on immunoblotting compared with wild-type gCTRP12 (Fig. 2E, right), indicating that Asn287 and Asn297 were not glycosylated when expressed in HEK 293T cells.
Members of the CTRP family form trimers and higher order multimeric complexes, mediated by conserved N-terminal Cys residues (5). Denaturing but nonreducing SDS-PAGE/immunoblot analysis indicated that CTRP12 also forms disulfide-linked oligomers (Fig. 2F). Oligomeric complexes can be disrupted by the addition of reducing agent (β-mercaptoethanol). As with other CTRPs (5), the conserved N-terminal Cys85 of CTRP12 mediates the higher order structure formation; mutation of Cys85 to Ala abolished multimeric complex formation (Fig. 2F). Together, these data indicate that CTRP12 is a secreted oligomeric glycoprotein subjected to endoproteolytic processing.
Insulin Enhances Endogenous CTRP12 Cleavage
We tested whether proteolytic processing of endogenous CTRP12 is constitutive or potentially subjected to hormonal regulation. We have shown previously that insulin can increase the expression of CTRP12 in mouse 3T3-L1 adipocytes (10). We therefore tested whether insulin plays a role in regulating the cleavage of CTRP12. As shown in Fig. 3, treatment of 3T3-L1 adipocytes with 10 nm insulin increased the expression of both fCTRP12 and gCTRP12. However, the insulin-induced increase in gCTRP12 protein levels in adipocytes was greater and occurred earlier compared with fCTRP12, indicating that insulin enhanced the cleavage of fCTRP12. Modulation of CTRP12 processing by a metabolic hormone suggests that proteolytic cleavage of the protein may be functionally relevant.
FIGURE 3.

Insulin increases the expression and cleavage of CTRP12. A, immunoblot analysis of endogenous CTRP12 produced by 3T3-L1 adipocytes stimulated with vehicle control or insulin (10 nm for 6 or 12 h) using a CTRP12-specific antibody. The blot was stripped and reprobed with the anti-β-tubulin antibody as loading controls. B, quantifications of gCTRP12 and fCTRP12 expression levels based on immunoblot shown in A. *, p < 0.05; **, p < 0.01. Results shown are means ± S.E.
Mutation That Enhances or Suppresses Cleavage
Amino acids surrounding a proteolytic cleavage site influence the specificity of endopeptidase cleavage (22). Interestingly, when the charged Arg93 (at the P2′ position of CTRP12) was substituted with Ala, the R93A mutant exhibited enhanced cleavage compared with WT CTRP12 (Fig. 4A). In contrast, proteolytic cleavage was largely abolished as expected when the polybasic residues KKXR were converted to Ala, as in the triple mutant, K90A/K91A/R93A (3M) (Fig. 4B). These results suggest that the two mutants can be used to investigate the functional significance of proteolytic cleavage.
FIGURE 4.
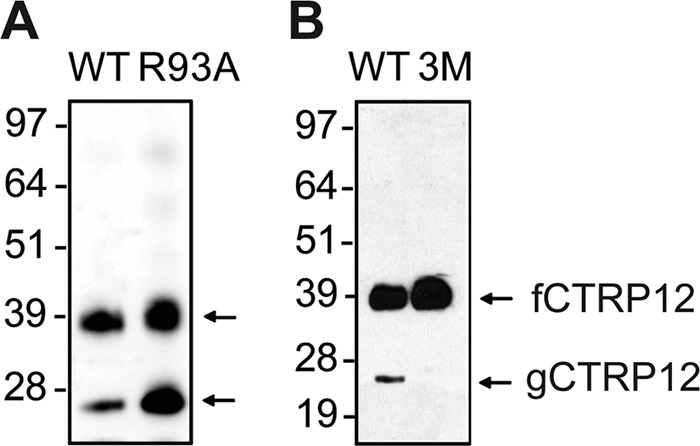
CTRP12 mutants with enhanced or suppressed cleavage. A, immunoblot analysis of secreted WT CTRP12 and its R93A mutant. B, immunoblot analysis of secreted WT CTRP12 and its triple mutant (K90A/K91A/R93A; designated as 3M). Media containing the recombinant proteins from transfected HEK 293T cells were collected 48 h after transfection and subjected to immunoblot analysis.
CTRP12 Is Cleaved by a Member of the Proprotein Convertases
Endoproteolytic cleavage of CTRP12 after a basic residue suggests the involvement of serine proteases of the proprotein convertase (PC) family. Nine members compose the PC family (designated as PCSK1–9) (23). Only the first seven members cleave C-terminal to a single (K/R) or pairs of basic residues ((K/R)(K/R)). Of these, PCSK3/furin shows the highest expression in adipose tissue and differentiated mouse 3T3-L1 adipocytes (Fig. 5, A and B). At the protein level, furin is expressed at higher levels in differentiated 3T3-L1 adipocytes and adipose tissue relative to undifferentiated 3T3-L1 preadipocytes (Fig. 5C). Because insulin increased the cleavage of CTRP12 (Fig. 3), we explored whether insulin also influences the expression of furin in adipocytes. Indeed, insulin treatment enhanced the expression of furin protein in adipocytes (Fig. 5D). Endogenous furin is also expressed robustly in HEK 293T cells (Fig. 5E), a cell type that supports the cleavage of CTRP12. Consistent with furin being a candidate convertase that can cleave CTRP12, co-expression of PCSK3/furin with WT CTRP12 in HEK 293T cells led to enhanced cleavage of the protein in a dose-dependent manner: increasing the expression of furin led to a proportional reduction in fCTRP12 and a simultaneous increase in cleaved gCTRP12 secreted into the conditioned medium (Fig. 5F). In contrast, co-expression of PCSK3/furin with the triple mutant (3M) largely abolished proteolytic cleavage of CTRP12, confirming that the cleavage site of furin is indeed at the polybasic KKSR motif (Fig. 5C). Minor residual cleavage of the 3M mutant may be the result of cleavage at nonpreferred basic residues (e.g. Arg74 and Arg75) adjacent to the Arg91 by furin or other members of the PC family expressed at low levels in HEK 293T cells. Consistent with this, residual gCTRP12 generated from the 3M mutant had a slightly larger apparent molecular mass on immunoblotting relative to gCTRP12 generated from the WT protein (Fig. 5F). This supports minor cleavage of the 3M mutant upstream of the preferred furin cleavage site at Lys91. Importantly, lentivirus-mediated knockdown of furin in adipocytes led to decreased cleavage of endogenous CTRP12 (Fig. 5G). Together, these results indicate that PCSK3/furin is likely the endopeptidase of the proprotein convertase family that generates the naturally occurring gCTRP12 isoform in adipocytes.
FIGURE 5.

PCSK3/furin cleaves fCTRP12 at the K90KSR motif. A and B, relative mRNA levels of different PC family members (PCSK1–7) in epididymal fat tissue (A) and in differentiated 3T3-L1 adipocytes (B). All quantitative real-time PCR values were normalized to 18 S rRNA. The expression of PCSK1 was set to 1, and expression levels of other PC family members were normalized against PCSK1. C, immunoblot analysis and quantification of furin expression in 3T3-L1 preadipocytes, mature 3T3-L1 adipocytes, and adipose tissue (mouse epididymal fat pad). Quantification of furin expression was normalized to β-tubulin. D, immunoblot blot analysis and quantification of furin expression in adipocytes treated with insulin (10 nm) for various time points. E, comparison of furin expression in HEK 293T cells and 3T3-L1 adipocytes. F, HEK 293T cells were co-transfected with wild-type (WT) CTRP12 or its 3M mutant construct and an increasing amount of plasmid DNA encoding PCSK3/furin (lanes 2–9). As a control, HEK 293T cells were also transfected with empty pCDNA3.1 plasmid (lane 1). Shown is the immunoblot analysis of secreted WT or 3M mutant protein found in the supernatant of transfected cells. NS, nonspecific band. G, knockdown of furin in adipocytes resulted in decreased endogenous CTRP12 cleavage. 3T3-L1 adipocytes were infected with lentiviruses encoding an empty vector or shRNA against furin. *, p < 0.05; **, p < 0.01. Error bars, S.E.
Signaling Specificities of fCTRP12 and gCTRP12 in Hepatocytes
The availability of recombinant wild-type, single, and triple mutant forms of CTRP12 enabled us to address the functional relevance and significance of fCTRP12 versus gCTRP12 isoforms. We have shown previously that recombinant CTRP12 induces the phosphorylation of IRS1, Akt, and p44/42 MAPK (also referred to as ERK1/2) in rat H4IIE hepatocytes (10). Compared with WT CTRP12, R93A mutant (with enhanced cleavage) had a reduced ability to induce IRS1 and Akt phosphorylation in H4IIE hepatocytes (Fig. 6, A and B). Conversely, the 3M mutant (uncleaved fCTRP12) retained its ability to stimulate IRS1 and Akt phosphorylation in a manner comparable with WT CTRP12 (Fig. 6, A and B). These results suggest that fCTRP12 preferentially activates Akt signaling.
FIGURE 6.

fCTRP12 and gCTRP12 preferentially activate Akt or MAPK signaling in H4IIE hepatocytes. A, immunoblot analysis of IRS1(Tyr612) and Akt (Thr308) phosphorylation in H4IIE hepatocytes stimulated with vehicle control or 10 μg/ml recombinant CTRP12 for 15 min. Blots were also probed for total IRS1 and Akt protein. B, quantification of phosphorylation as shown in A, normalized to total IRS1 and Akt protein, respectively. C, immunoblot analysis of p44/42 MAPK (Thr202/Tyr204) and p38 MAPK (Thr180/Tyr182) phosphorylation in H4IIE cells stimulated with vehicle control or 10 μg/ml CTRP12 for 15 min. D, quantification of phosphorylation as shown in C, normalized to their respective total protein. *, p < 0.05; **, p < 0.01; #, p < 0.001 (n = 3). Error bars, S.E.
However, opposite results were obtained when we examined the ability of WT, R93A, and 3M mutant to activate MAPKs in H4IIE hepatocytes. Both WT CTRP12 and R93A mutant were comparable in their ability to induce p44/42 (ERK1/2) and p38 MAPK phosphorylation (Fig. 6, C and D). The 3M mutant (uncleaved fCTRP12), however, only modestly stimulated p38 MAPK phosphorylation and had completely lost its ability to induce p44/42 MAPK phosphorylation (Fig. 6, C and D). Together, these results suggest that the two isoforms of CTRP12 are functionally distinct; whereas gCTRP12 preferentially activates p44/42 and p38 MAPK signaling, the full-length protein preferentially activates Akt signaling.
Signaling Specificities of fCTRP12 and gCTRP12 in Adipocytes
To address whether the preferential activation of different signal transduction pathways by the two isoforms of CTRP12 is restricted to H4IIE cells, we compared the ability of gCTRP12 and fCTRP12 to activate Akt and MAPK signaling in differentiated mouse 3T3-L1 adipocytes. The 3M mutant robustly induced Akt phosphorylation in a manner similar to the WT protein, whereas the R93A mutant only minimally enhanced Akt phosphorylation (Fig. 7, A and B). Conversely, the R93A mutant induced p44/42 MAPK phosphorylation in a manner comparable with the WT protein, whereas the 3M mutant failed to do so (Fig. 7, A and C). These results confirm and extend our findings that fCTRP12 and gCTRP12 isoforms are functionally distinct with regard to the signaling pathways they preferentially activate in different cell types.
FIGURE 7.

fCTRP12 and gCTRP12 preferentially activate Akt or MAPK signaling in 3T3-L1 adipocytes. A, immunoblot analysis of Akt (Thr308) and p44/42 MAPK (Thr202/Tyr204) phosphorylation in 3T3-L1 adipocytes stimulated with vehicle control or 10 μg/ml recombinant CTRP12 for 15 min. Blots were also probed for total IRS1 and Akt protein. B and C, quantification of Akt (B) and p44/42 MAPK (C) phosphorylation as shown in A, normalized to their respective total protein and then normalized to value obtained from cells treated with vehicle control. *, p < 0.05; **, p < 0.01; #, p < 0.001 (n = 3). Error bars, S.E.
To ensure that gCTRP12 is indeed the isoform that activates p44/42 MAPK signaling in 3T3-L1 adipocytes and H4IIE hepatocytes, we generated and tested the effect of recombinant gCTRP12 (corresponding to amino acid residues 92–308) on adipocytes and hepatocytes. Purified recombinant gCTRP12 migrated with the same apparent molecular mass on immunoblotting as the naturally cleaved gCTRP12 isoform generated from the full-length protein (Fig. 8A). Stimulation of 3T3-L1 adipocytes with recombinant gCTRP12 and fCTRP12 protein (uncleaved 3M mutant) indicated that fCTRP12 has a stronger ability to activate Akt signaling, whereas gCTRP12 is more potent at activating p44/42 MAPK signaling (Fig. 8B). Similar results were also obtained in H4IIE hepatocytes (Fig. 8C). These data confirm that gCTRP12 is indeed the isoform that activates p44/42 MAPK signaling. Further, our results also suggest that the C1q globular domain, and not the cleaved N-terminal portion (residue 22–91) derived from the full-length protein, is responsible for the activation of MAPK signaling.
FIGURE 8.

Activation of Akt and p44/42 MAPK signaling by recombinant gCTRP12 and fCTRP12. A, immunoblot analysis of recombinant WT CTRP12 and its gCTRP12 isoform. gCTRP12 protein consists of residue 1–21 (signal peptide) and 92–308. B, immunoblot analysis of Akt (Thr308) and p44/42 MAPK (Thr202/Tyr204) phosphorylation in 3T3-L1 adipocytes stimulated with fCTRP12 (the uncleaved 3M mutant) and gCTRP12. C, immunoblot analysis of Akt (Thr308) and p44/42 MAPK (Thr202/Tyr204) phosphorylation in H4IIE hepatocytes stimulated with fCTRP12 (uncleaved 3M mutant) and gCTRP12. Cells were stimulated with vehicle control or recombinant fCTRP12 (10 μg/ml) or gCTRP12 (10 μg/ml) for 15 min. D, 3T3-L1 adipocytes were treated with control vehicle buffer, 5 μg/ml gCTRP12, 5 μg/ml fCTRP12, with or without 100 nm insulin, and assayed for glucose uptake (n = 3). g12, gCTRP12; 3M, the uncleaved triple mutant. #, p < 0.001; *, p < 0.05 (n = 3). Error bars, S.E.
Next, we performed a glucose uptake assay in adipocytes to determine whether preferential activation of distinct signaling pathways by the two isoforms of CTRP12 has functional significance. As shown in Fig. 8D, both gCTRP12 and fCTRP12 (uncleaved 3M mutant) modestly increased glucose uptake in adipocytes without insulin. However, in the presence of insulin, only fCTRP12 further increased glucose uptake in adipocytes, in accordance with stronger activation of Akt phosphorylation by fCTRP12 (Fig. 8B). These results indicate that the two isoforms of CTRP12 have overlapping but different functions, with fCTRP12 better at improving insulin-stimulated glucose uptake in adipocytes.
The Two Isoforms of CTRP12 Differ in Oligomeric Structure
Because the conserved Cys85 that mediates disulfide-linked higher order structure formation is located N-terminal to Arg91, proteolytic cleavage is predicted to affect the oligomeric state of CTRP12. We performed gel filtration chromatographic analysis to test this possibility. For WT CTRP12, FPLC analysis revealed two oligomeric forms: the higher molecular mass (HMM, ∼120 kDa or larger) oligomer consisting of full-length protein and the lower molecular mass (LMM, ∼45 kDa) oligomer consisting of the cleaved gCTRP12 isoform (Fig. 9A). Based on the molecular mass makers and the peak elution fractions, the HMM oligomers consist of trimers (fractions 27 and 28) and larger complexes (fractions 18–26), and the LMM oligomers (fractions 30–32) are predominantly dimers. In accordance, recombinant gCTRP12 and the R93A mutant consist predominantly of the LMM oligomeric form (Fig. 9, B and D), whereas fCTRP12 (the uncleaved 3M mutant) consists predominantly of the HMM oligomeric form (Fig. 9C). These results suggest that the signaling specificity of full-length versus globular isoform of CTRP12 may, in part, be due to differences in their oligomeric structure (Fig. 10).
FIGURE 9.

fCTRP12 and gCTRP12 differ in oligomeric structure. Conditioned media from transfected HEK 293T cells containing wild-type (WT) CTRP12 (A), gCTRP12 (B), the 3M mutant (C), or the R93A mutant (D) protein were subjected to gel filtration chromatographic analysis. Arrows with the molecular mass markers of 669, 440, 158, 75, and 44 kDa correspond to the peak elution fraction of molecular standards thyroglobulin, ferritin, aldolase, conalbumin, and ovalbumin, respectively. WT CTRP12 consists of two distinct oligomeric forms: the higher molecular mass (HMM; ∼120 kDa or larger) oligomers composed of full-length protein, and the lower molecular mass (LMM; ∼45 kDa) oligomers consisted of the cleaved gCTRP12 isoform. Recombinant gCTRP12 and the R93A mutant consisted predominantly of the LMM oligomeric form (B and D), whereas the 3M mutant consisted predominantly of the HMM oligomeric form (C).
FIGURE 10.

Proposed model of cleavage-dependent regulation of oligomeric state and signaling specificity of CTRP12. In adipocytes, newly synthesized CTRP12 is differentially processed. Uncleaved CTRP12 is secreted as trimers and possibly larger complexes. Endoproteolytic cleavage of CTRP12 at Lys91 by an endopeptidase (i.e. PCSK3/furin) generates an isoform consisting predominantly of dimers. Importantly, differential processing resulted in two isoforms of CTRP12 with distinct function: fCTRP12 preferentially activated Akt; gCTRP12 preferentially activated MAPK signaling in hepatocytes and adipocytes. Unlike the gCTRP12, fCTRP12 can increase insulin-stimulated glucose uptake into adipocytes. These results revealed a novel mechanism controlling signaling and functional specificity of CTRP12 via cleavage-dependent alteration in oligomeric state.
DISCUSSION
In this study we provide a detailed biochemical characterization of CTRP12, an insulin-sensitizing adipokine that we and others have recently identified (10, 24). CTRP12 contains multiple types of posttranslational modifications likely relevant to its function. Its highly conserved Asn39 at the N terminus is glycosylated. Secreted proteins often contain N-linked oligosaccharides to facilitate proper folding in the endoplasmic reticulum, to enhance the half-life of the protein in circulation, and/or to provide an epitope recognized by receptors (25). Because proteolytic cleavage occurs after Asn39, the cleaved gCTRP12 isoform is predicted to be nonglycosylated. Indeed, PNGase F treatment of gCTRP12 did not result in mobility shift of the protein on immunoblot, indicating a lack of N-linked glycan. gCTRP12 with a single or double mutations of Asn187 and Asn197 also did not migrate differently on immunoblotting, further confirming the lack of N-glycosylation in the globular domain. In human and mouse serum, gCTRP12 is the predominant circulating isoform. Thus, it remains to be determined whether N-glycosylation affects the half-life of the protein or its target tissues in vivo.
Unique among all the CTRPs described to date is the proteolytic processing of CTRP12 that resulted in the generation of a cleaved gCTRP12 isoform that circulates in serum (10). Endoproteolytic processing occurs at Lys91 prior to protein secretion. Many secreted growth factors and hormones are processed by a family of nine highly conserved proprotein convertases (PCSK1–PCSK9) (23). The first seven members (PCSK1–7) cleave C-terminal to a single (K/R) or pairs of basic residues ((K/R)(K/R)), and PCSK8 and PCSK9 cleave C-terminal to nonbasic residues (23). PCSK5, 6, and 7 are widely expressed, but expression of other PCSKs is restricted to select tissues and cell types (23). We show that PCSK3/furin is likely the endogenous endopeptidase that cleaves CTRP12 to generate the gCTRP12 isoform in adipocytes. Human CTRP12 is also secreted as full-length and cleaved globular isoforms (10). In fact, Lys91 within the KKXR polybasic motif is conserved from Xenopus to humans (Fig. 1B). In teleost (e.g. zebra fish), the corresponding amino acid is Lys instead of Arg. Thus, endoproteolytic processing of CTRP12 is an evolutionarily conserved feature and likely reflects the distinct function of the two isoforms.
A shared biochemical feature of CTRPs is the formation of higher order oligomeric complexes. All CTRPs form trimers as their basic structural unit, mediated by the globular C1q domain (5). Several CTRPs are further assembled into disulfide-linked higher order structures greater than a trimer via the conserved N-terminal Cys residues (4–6). Similarly, secreted CTRP12 also forms disulfide-linked multimers, mediated by the conserved N-terminal Cys85. Because proteolytic cleavage occurs at Lys91 (C-terminal to Cys85), it is predicted that the gCTRP12 isoform that lacks Cys85 will differ from the uncleaved full-length protein in its oligomeric structure. Indeed, gel filtration chromatographic analysis revealed two distinct oligomeric forms of CTRP12 (HMM and LMM oligomers), which presumably correspond to the trimeric (and larger complexes) and dimeric form of CTRP12, respectively.
Differences in oligomeric state may account for the differential signal transduction pathway (Akt versus MAPK) activated by the two isoforms of CTRP12 in H4IIE hepatocytes and 3T3-L1 adipocytes. Although uncommon, such an oligomeric state-dependent mechanism controlling signaling specificity has been demonstrated previously for adiponectin, an important insulin-sensitizing adipokine. Adiponectin trimers were shown to preferentially activate AMP-activated protein kinase signaling in isolated rat muscle, whereas the octadecameric form (18-subunit) of adiponectin was shown to preferentially activate NF-κB signaling in myotubes (26, 27). Because the assembly of hexameric and octadecameric forms of adiponectin requires the conserved N-terminal Cys39, it has been suggested that the redox environment in the endoplasmic reticulum may dictate the proportion of hexameric/octadecameric versus trimeric form of adiponectin being secreted (28). In the case of CTRP12, our data suggest that the proportion of different oligomeric forms produced may be dependent on regulated cleavage by an endopeptidase such as PCSK3/furin.
Importantly, the two isoforms of CTRP12 differ in oligomeric state are functionally distinct. We demonstrate that fCTRP12 preferentially activated Akt, and, in contrast, gCTRP12 preferentially activated MAPK signaling in H4IIE hepatocytes and 3T3-L1 adipocytes. A functional assay revealed that fCTRP12, but not gCTRP12, increased insulin-simulated glucose uptake into adipocytes. These results illustrate that signaling and functional specificity of a hormone can be controlled by different oligomeric forms of a protein generated by endoproteolytic cleavage (Fig. 10). This differs from the many neuropeptides and hormones in which endopeptidase processing of the prohormones is a requisite step needed to generate mature and functioning hormones (23). Collectively, our biochemical characterizations established multiple posttranslational modifications of CTRP12 important for its biological function and revealed a novel mechanism controlling CTRP12 signaling and functional specificity via cleavage-dependent alteration in oligomeric state.
Acknowledgments
We thank Weijun Jin (SUNY Downstate Medical Center, Brooklyn, NY) for the FLAG epitope-tagged mouse furin-IRES-EGFP plasmid and Sharrol Bachas and Herschel Wade (Johns Hopkins University) for help with gel filtration chromatographic analysis.
This work was supported, in whole or in part, by National Institutes of Health Grant DK084171 (to G. W. W.). This work was also supported by American Heart Association Grant SDG2260721.
- CTRP
- C1q/TNF-related protein
- Bis-Tris
- N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid
- fCTRP
- full-length CTRP
- gCTRP
- globular CTRP
- HMM
- higher molecular mass
- LMM
- lower molecular mass
- PC
- prohormone convertase
- PCSK
- proprotein convertase subtilisin/kexin type
- PNGase F
- protein:N-glycosidase F.
REFERENCES
- 1. Deng Y., Scherer P. E. (2010) Adipokines as novel biomarkers and regulators of the metabolic syndrome. Ann. N.Y. Acad. Sci. 1212, E1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scherer P. E. (2006) Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes 55, 1537–1545 [DOI] [PubMed] [Google Scholar]
- 3. Wong G. W., Wang J., Hug C., Tsao T. S., Lodish H. F. (2004) A family of Acrp30/adiponectin structural and functional paralogs. Proc. Natl. Acad. Sci. U.S.A. 101, 10302–10307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wong G. W., Krawczyk S. A., Kitidis-Mitrokostas C., Ge G., Spooner E., Hug C., Gimeno R., Lodish H. F. (2009) Identification and characterization of CTRP9, a novel secreted glycoprotein, from adipose tissue that reduces serum glucose in mice and forms heterotrimers with adiponectin. FASEB J. 23, 241–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wong G. W., Krawczyk S. A., Kitidis-Mitrokostas C., Revett T., Gimeno R., Lodish H. F. (2008) Molecular, biochemical and functional characterizations of C1q/TNF family members: adipose-tissue-selective expression patterns, regulation by PPAR-γ agonist, cysteine-mediated oligomerizations, combinatorial associations, and metabolic functions. Biochem. J. 416, 161–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wei Z., Peterson J. M., Wong G. W. (2011) Metabolic regulation by C1q/TNF-related protein-13 (CTRP13): activation of AMP-activated protein kinase and suppression of fatty acid-induced JNK signaling. J. Biol. Chem. 286, 15652–15665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seldin M. M., Peterson J. M., Byerly M. S., Wei Z., Wong G. W. (2012) Myonectin (CTRP15), a novel myokine that links skeletal muscle to systemic lipid homeostasis. J. Biol. Chem. 287, 11968–11980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kishore U., Gaboriaud C., Waters P., Shrive A. K., Greenhough T. J., Reid K. B., Sim R. B., Arlaud G. J. (2004) C1q and tumor necrosis factor superfamily: modularity and versatility. Trends Immunol. 25, 551–561 [DOI] [PubMed] [Google Scholar]
- 9. Peterson J. M., Wei Z., Wong G. W. (2010) C1q/TNF-related protein-3 (CTRP3), a novel adipokine that regulates hepatic glucose output. J. Biol. Chem. 285, 39691–39701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wei Z., Peterson J. M., Lei X., Cebotaru L., Wolfgang M. J., Baldeviano G. C., Wong G. W. (2012) C1q/TNF-related protein-12 (CTRP12), a novel adipokine that improves insulin sensitivity and glycemic control in mouse models of obesity and diabetes. J. Biol. Chem. 287, 10301–10315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peterson J. M., Aja S., Wei Z., Wong G. W. (2012) CTRP1 protein enhances fatty acid oxidation via AMP-activated protein kinase (AMPK) activation and acetyl-CoA carboxylase (ACC) inhibition. J. Biol. Chem. 287, 1576–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kopp A., Bala M., Buechler C., Falk W., Gross P., Neumeier M., Schölmerich J., Schäffler A. (2010) C1q/TNF-related protein-3 represents a novel and endogenous lipopolysaccharide antagonist of the adipose tissue. Endocrinology 151, 5267–5278 [DOI] [PubMed] [Google Scholar]
- 13. Kopp A., Bala M., Weigert J., Büchler C., Neumeier M., Aslanidis C., Schölmerich J., Schäffler A. (2010) Effects of the new adiponectin paralogous protein CTRP-3 and of LPS on cytokine release from monocytes of patients with type 2 diabetes mellitus. Cytokine 49, 51–57 [DOI] [PubMed] [Google Scholar]
- 14. Maeda T., Abe M., Kurisu K., Jikko A., Furukawa S. (2001) Molecular cloning and characterization of a novel gene, CORS26, encoding a putative secretory protein and its possible involvement in skeletal development. J. Biol. Chem. 276, 3628–3634 [DOI] [PubMed] [Google Scholar]
- 15. Maeda T., Jikko A., Abe M., Yokohama-Tamaki T., Akiyama H., Furukawa S., Takigawa M., Wakisaka S. (2006) Cartducin, a paralog of Acrp30/adiponectin, is induced during chondrogenic differentiation and promotes proliferation of chondrogenic precursors and chondrocytes. J. Cell. Physiol. 206, 537–544 [DOI] [PubMed] [Google Scholar]
- 16. Lasser G., Guchhait P., Ellsworth J. L., Sheppard P., Lewis K., Bishop P., Cruz M. A., Lopez J. A., Fruebis J. (2006) C1qTNF-related protein-1 (CTRP-1): a vascular wall protein that inhibits collagen-induced platelet aggregation by blocking VWF binding to collagen. Blood 107, 423–430 [DOI] [PubMed] [Google Scholar]
- 17. Zheng Q., Yuan Y., Yi W., Lau W. B., Wang Y., Wang X., Sun Y., Lopez B. L., Christopher T. A., Peterson J. M., Wong G. W., Yu S., Yi D., Ma X. L. (2011) C1q/TNF-related proteins, a family of novel adipokines, induce vascular relaxation through the adiponectin receptor-1/AMPK/eNOS/nitric oxide signaling pathway. Arterioscler. Thromb. Vasc. Biol. 31, 2616–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hayward C., Shu X., Cideciyan A. V., Lennon A., Barran P., Zareparsi S., Sawyer L., Hendry G., Dhillon B., Milam A. H., Luthert P. J., Swaroop A., Hastie N. D., Jacobson S. G., Wright A. F. (2003) Mutation in a short-chain collagen gene, CTRP5, results in extracellular deposit formation in late-onset retinal degeneration: a genetic model for age-related macular degeneration. Hum. Mol. Genet. 12, 2657–2667 [DOI] [PubMed] [Google Scholar]
- 19. Ayyagari R., Mandal M. N., Karoukis A. J., Chen L., McLaren N. C., Lichter M., Wong D. T., Hitchcock P. F., Caruso R. C., Moroi S. E., Maumenee I. H., Sieving P. A. (2005) Late-onset macular degeneration and long anterior lens zonules result from a CTRP5 gene mutation. Invest. Ophthalmol. Vis. Sci. 46, 3363–3371 [DOI] [PubMed] [Google Scholar]
- 20. Chavali V. R., Khan N. W., Cukras C. A., Bartsch D. U., Jablonski M. M., Ayyagari R. (2011) A CTRP5 gene S163R mutation knock-in mouse model for late-onset retinal degeneration. Hum. Mol. Genet. 20, 2000–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yan A., Lennarz W. J. (2005) Unraveling the mechanism of protein N-glycosylation. J. Biol. Chem. 280, 3121–3124 [DOI] [PubMed] [Google Scholar]
- 22. Basak S., Chrétien M., Mbikay M., Basak A. (2004) In vitro elucidation of substrate specificity and bioassay of proprotein convertase 4 using intramolecularly quenched fluorogenic peptides. Biochem. J. 380, 505–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seidah N. G. (2011) The proprotein convertases, 20 years later. Methods Mol. Biol. 768, 23–57 [DOI] [PubMed] [Google Scholar]
- 24. Enomoto T., Ohashi K., Shibata R., Higuchi A., Maruyama S., Izumiya Y., Walsh K., Murohara T., Ouchi N. (2011) Adipolin/C1qdc2/CTRP12 protein functions as an adipokine that improves glucose metabolism. J. Biol. Chem. 286, 34552–34558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jaeken J., Matthijs G. (2007) Congenital disorders of glycosylation: a rapidly expanding disease family. Annu. Rev. Genomics Hum. Genet. 8, 261–278 [DOI] [PubMed] [Google Scholar]
- 26. Tsao T. S., Murrey H. E., Hug C., Lee D. H., Lodish H. F. (2002) Oligomerization state-dependent activation of NF-κB signaling pathway by adipocyte complement-related protein of 30 kDa (Acrp30). J. Biol. Chem. 277, 29359–29362 [DOI] [PubMed] [Google Scholar]
- 27. Tsao T. S., Tomas E., Murrey H. E., Hug C., Lee D. H., Ruderman N. B., Heuser J. E., Lodish H. F. (2003) Role of disulfide bonds in Acrp30/adiponectin structure and signaling specificity: different oligomers activate different signal transduction pathways. J. Biol. Chem. 278, 50810–50817 [DOI] [PubMed] [Google Scholar]
- 28. Briggs D. B., Giron R. M., Malinowski P. R., Nuñez M., Tsao T. S. (2011) Role of redox environment on the oligomerization of higher molecular weight adiponectin. BMC Biochem. 12, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]