

Background: Glutathionyl-hydroquinone reductases (GS-HQRs) are a newly recognized and widely distributed class of GSTs using GSH to reduce GS-hydroquinones to hydroquinone.
Results: Three Tyr and a Cys within the active site contribute to catalytic activity.
Conclusion: The residues constituting the active site and dimer interface are conserved among GS-HQRs.
Significance: This is a thorough characterization of GS-HQRs, providing the structural link to enzymatic activity.
Keywords: Biomineralization, Crystallography, Enzyme Mechanisms, Glutathione, Quinones, Reductase, Deglutathionylation, Glutathione S-Transferase
Abstract
Glutathionyl-hydroquinone reductases (GS- HQRs) are a newly identified group of glutathione transferases, and they are widely distributed in bacteria, halobacteria, fungi, and plants. GS-HQRs catalyze glutathione (GSH)-dependent reduction of glutathionyl-hydroquinones (GS-hydroquinones) to hydroquinones. GS-hydroquinones can be spontaneously formed from benzoquinones reacting with reduced GSH via Michael addition, and GS-HQRs convert the conjugates to hydroquinones. In this report we have determined the structures of two bacterial GS-HQRs, PcpF of Sphingobium chlorophenolicum and YqjG of Escherichia coli. The two structures and the previously reported structure of a fungal GS-HQR shared many features and displayed complete conservation for all the critical residues. Furthermore, we obtained the binary complex structures with GS-menadione, which in its reduced form, GS-menadiol, is a substrate. The structure revealed a large H-site that could accommodate various substituted hydroquinones and a hydrogen network of three Tyr residues that could provide the proton for reductive deglutathionylation. Mutation of the Tyr residues and the position of two GSH molecules confirmed the proposed mechanism of GS-HQRs. The conservation of GS-HQRs across bacteria, halobacteria, fungi, and plants potentiates the physiological role of these enzymes in quinone metabolism.
Introduction
The glutathione transferase (GST) superfamily has been mainly considered to be detoxification enzymes that transfer GSH to the electrophilic centers of various hydrophobic substrates. Despite their marginal level of sequential similarity, most of the cytosolic GSTs share a high degree of structural similarity consisting of a two-domain subunit. A thioredoxin-like N-terminal domain is mainly responsible for reduced glutathione (GSH) binding (the G-site), and a C-terminal helix-rich domain is involved in the binding of the second (hydrophobic) substrate (the H-site) (1). Sequence similarity, including the active site residues, has been the primary basis for the classification (1). The Alpha, Mu, and Pi classes of GSTs have a catalytic Tyr residue that stabilizes a thiolate ion of bound GSH (GS−), facilitating conjugation to the second substrate. In the Theta, Zeta, Kappa, Phi, and Tau classes, a Ser residue is adopted for the same catalytic function (2). In the Beta, Omega, Lambda, and dehydroascorbate reductase classes, a Cys residue plays a catalytic role (3).
Recently, a new class of GST, glutathionyl-hydroquinone reductases (GS-HQRs),3 have been found in bacteria, fungi, and plants (4, 5, 7, 8). Among cytosolic GSTs, the closest relatives for the GS-HQRs are the Omega-class GSTs; however, these GSTs display less than 20% sequence identity (4). Both classes have little typical GST activities but instead catalyze a GSH-dependent thioltransferase reaction, dehydroascorbate reduction, and dimethylarsinate reduction (4–7). Furthermore, GS-HQRs catalyze GSH-dependent reduction of GS-hydroquinones to hydroquinones at the expense of GSH (4, 5, 7). PcpF of Sphingobium chlorophenolicum, EcYqjG of Escherichia coli, CnYqjG of Cupriavidus necator, and ECM4 of Saccharomyces cerevisiae are representatives of GS-HQRs from bacteria and yeast. They all catalyze GSH-dependent reduction of GS-trichloro-p-hydroquinone (GS-TriCH) to TriCH (4) as well as the reduction of various other tested GS-hydroquinones, including GS-p-hydroquinone (GS-HQ), GS-methyl-p-hydroquinone, GS-hydroxyl-p-hydroquinone, and GS-menadiol to the corresponding hydroquinones (7). A GS-HQR from white rot fungus Phanerochaete chrysosporium (PcGHR1) also reduces GS-HQ to HQ at the expense of GSH (5). This unique activity of the widely distributed GS-HQRs is believed to offer a new route for reduction of benzoquinones that spontaneously react with GSH to produce GS-hydroquinones via Michael addition (7). GSH does not spontaneously reduce GS-hydroquinones to hydroquinones, but GS-HQRs irreversibly catalyze this reaction (8). Due to the irreversible nature, GS-hydroquinones are not detectable at the end of enzymatic reaction (7).
So far all the identified GS-HQRs contain the catalytic Cys residue at a position similar to the Omega-class GSTs despite distinct differences in their activities (4, 8). The structure of PcGHR1 has been determined with a GSH in the G-site (5). Overall it has the typical GST-fold; however, because of its unique structural properties including a dimerization mode, a new classification of Xi GST has been proposed (5). Additionally, this binary structure of PcGHR1 with GSH could not pinpoint the location and nature of H-site (5). Consequently, the explanation of the unique reduction mechanism of GS-HQRs has to rely on several lines of indirect evidence (9).
In this work we report the structures of two bacterial GS-HQRs, PcpF and EcYqjG. Their three-dimensional structure and dimer interaction are essentially the same as that of PcGHR1, establishing the structural signature of this newly discovered class of GST. Furthermore, we have obtained the complex structures of EcYqjG with GSH and GS-menadione, which in its reduced form, GS-menadiol, is a substrate. The structure revealed a large H-site that can accommodate various substituted hydroquinones and a hydrogen network that can provide the proton for reductive deglutathionylation.
EXPERIMENTAL PROCEDURES
Chemicals
Chemicals were obtained from Sigma or Fisher. Crystallization screens were obtained from Hampton Research. 3-Glutathionyl-2-methyl-1,4-naphthoquinone (GS-menadione) was synthesized as previously described (10). GS-HQ was synthesized by mixing equal volumes of a 101 mm solution of reduced glutathione (GSH) in distilled water and a 100 mm solution of p-benzoquinone in ethanol. GS-hydroquinone was spontaneously formed within 5 min and final concentrations of GS-hydroquinone were determined by absorbance at 309 nm.
Cloning and Enzyme Purification
The wild-type (WT) E. coli YqjG plasmid has been described previously (4). C63A, Y195F, Y253F, and Y296F mutants were generated with the QuikChange Lightening site-directed mutagenesis kit (Agilent Technologies) and sequenced. Confirmed mutant plasmids were electroporated into E. coli BL21 (DE3). All clones were grown on Luria-Bertani (LB) plates or in LB broth supplemented with kanamycin (30 mg/ml) at 37 °C.
Expression clones of PcpF and EcYqjG were grown in 1.5 liters of LB broth at 37 °C to an A600 nm = 0.6 and induced with 0.5 mm isopropyl-β-d-thiogalactopyranoside for 12 h at 25 °C. Cells were then harvested via centrifugation at 4200 × g for 10 min and resuspended in 20 mm Tris buffer, pH 7.5. Cells were lysed by sonication, and lysates were cleared by centrifugation at 27000 × g for 30 min. Each lysate was applied to DEAE-650 M (TOSOH Biosciences) and eluted at 200 mm NaCl. These fractions were concentrated, and buffer was exchanged into phenyl-Sepharose buffer (25% saturated ammonium sulfate, 20 mm Tris, pH 7.5) and run over phenyl-Sepharose (GE Healthcare Fast Flow 6) with elution below 10% saturation. Fractions were pooled, concentrated, and exchanged into 5 mm sodium phosphate buffer (pH 6.8). The protein was then further purified over hydroxyapatite (Bio-Rad) and eluted in the flow-through. These fractions were pooled, concentrated and exchanged into 20 mm Tris, pH 8.5, for the final column: MonoQ10/100GL (GE Healthcare). The pure wild-type and mutant enzymes were eluted at around 300 mm NaCl and were concentrated to between 20 and 40 mg/ml. All proteins were purified to apparent homogeneity as judged by SDS-PAGE analysis (supplemental Data 1). Normally ∼30 mg of pure proteins were obtained from 1.5 liters of cultures. The purified proteins were immediately used for crystallization at 4 °C. For activity analysis, the proteins were stored at −80 °C. PcpF and EcYqjG were stable at −80 °C with no apparent loss of activity for several weeks (4).
Data Collection and Structure Determination
Both PcpF and EcYqjG crystals were grown at 4 °C by the vapor diffusion method. 3-μl drops consisting of a 1:1 ratio of either PcpF or EcYqjG protein (15 mg/ml in 20 mm Tris, pH 8.5) and crystallization solution (1.6 m ammonium sulfate, 0.1 m MES monohydrate, pH 6.5, 10% v/v 1,4-dioxane) were equilibrated against 0.5 ml of the same solution. Although crystals of EcYqjG typically appeared within 5 days, it took more than a month for a crystal of PcpF to grow. Therefore, reduced glutathione (GSH) complex crystals were obtained by the addition of 100 mm GSH to the drop containing YqjG crystals 30 min before harvest. The same procedure was used for the GS-menadione complex crystals of EcYqjG with the exception that soaking time was 1 h. Before x-ray data collection, crystals were transferred into cryoprotectant (1.6 m ammonium sulfate, 0.1 m MES monohydrate, pH 6.5, 10% v/v 1,4-dioxane, 15% v/v glycerol) and flash-frozen in liquid nitrogen. The space group of PcpF was P213 with eight molecules in the asymmetric unit. The space group of EcYqjG crystals was P3121 and contained two molecules in the asymmetric unit. Diffraction data were collected up to 2.0 Å of resolution for the complex crystals. All diffraction data were collected at the Berkeley Advanced Light Source (ALS, beam line 8.2.1) and were processed and scaled with the HKL2000 package (11) and CrystalClear 1.3.6 (Rigaku/MSC). The statistics for the diffraction data are listed in Table 1. Initial phasing of apo-form YqjG diffraction data were done by the program AMoRe (12) through the coordinates of the glutathione transferase from P. chrysosporium (PcGSTO1, PDB code 3PPU) (5). Iterative model building and refinement took place using the programs COOT (13) and PHENIX (14). Initial phasing for PcpF was conducted using PHENIX (14) with YqjG as a starting model. All coordinates have been deposited in the Protein Data Bank: 4G0L (YqjG, GSH complex), 4G0K (YqjG, GS-menadione complex), 4G0I (apo YqjG), and 4FQU (Apo PcpF).
TABLE 1.
Crystallographic data for the EcYqjg apo and binary complexes and PcpF apo
| Wild-type EcYqjG apo | Wild-type EcYqjG GSH, 30-min soaked | Wild-type EcYqjG GS-menadione, 1-h soaked | Wild-type PcpF apo | |
|---|---|---|---|---|
| Data | ||||
| Wavelength (Å) | 1.03 | 1.03 | 1.03 | 1.03 |
| Resolution (Å) | 48.8-2.00 | 49.9-2.59 | 46.0-2.49 | 49.6-2.80 |
| Space group | P3121 | P3121 | P3121 | P213 |
| Cell dimensions (Å) | a = 149.147 | a = 148.791 | a = 148.410 | a = 242.845 |
| b = 149.147 | b = 148.791 | b = 148.410 | b = 242.845 | |
| c = 105.340 | c = 108.321 | c = 105.171 | c = 242.845 | |
| α = 90.00 | α = 90.00 | α = 90.00 | α = 90.00 | |
| β = 90.00 | β = 90.00 | β = 90.00 | β = 90.00 | |
| γ = 120.00 | γ = 120.00 | γ = 120.00 | γ = 90.00 | |
| Asymmetric unit | 2 | 2 | 2 | 8 |
| Total observations | 1,852,174 | 880,223 | 869,554 | 978,432 |
| Unique reflections | 84,714 | 41,935 | 42,491 | 243,097 |
| Completeness (%) | 87.4 | 95.9 | 91.3 | 91.3 |
| Rsyma | 0.089 | 0.086 | 0.120 | 0.084 |
| Refinement | ||||
| Resolution (Å) | 48.8-2.05 | 49.9-2.61 | 48.6-2.55 | 49.6-3.0 |
| Number of reflections | 74,305 | 40,245 | 39,684 | 162,7249 |
| Rcrystb | 0.19 | 0.18 | 0.19 | 0.20 |
| Rfreec | 0.23 | 0.22 | 0.23 | 0.25 |
| r.m.s.d. bonds (Å) | 0.017 | 0.004 | 0.026 | 0.017 |
| r.m.s.d. angles (°) | 1.795 | 0.905 | 2.426 | 1.860 |
| Number of atoms | ||||
| Protein and ligand | 5,410 | 5,199 | 5,220 | 19,674 |
| Water | 324 | 131 | 135 | 55 |
a Rsym = ΣIh − 〈Ih〉|/ΣIh, where <Ih > is the average intensity over symmetry equivalent reflections.
b Rcryst = Σ|Fobs − Fcalc|/ΣFobs, where summation is over the data used for refinement.
c Rfree was calculated as for Rcryst using 5% of the data that was excluded from refinement.
Multi-angle Light Scattering and Isothermal Titration Calorimetry (ITC)
Static light scattering was performed as previously described (4); however, the isocratic elution buffer for EcYqjG was 20 mm Tris, pH 7.5.
Isothermal titration calorimetric reactions were carried out in a VP-ITC instrument (MicroCal, Northhampton, MA). The protein was prepared for ITC by dialyzing into titration buffer (20 mm Tris, pH 7.5) for 24 h at 4 °C. The concentration of protein in the calorimetric reaction cell was diluted to 100 μm. All titrations were performed at 25 °C with a stirring speed of 300 rpm and 29 injections (10 μl each). Ligands were diluted into the same titration buffer and injected into the cell-containing protein solution, and the heats of binding were recorded. Ligands were also titrated against buffer to account for the heats of dilution. Ligand concentrations were adjusted to obtain significant heats of binding, and the time intervals between injections were also adjusted to ensure proper baseline equilibration. All samples were degassed before titration.
Enzyme Assays of Mutant EcYqjG
Mutant enzymes were purified with the same protocol as wild-type EcYqjG. Each kinetic reaction was performed with 1.5 μm enzyme and 6 mm GSH in 50 mm Tris, pH 7.5. The substrate (GS-HQ) concentration was varied from less than 0.5 Km to 4 times Km of wild-type EcYqjG. Initial rates were obtained from monitoring the decrease in absorbance at 309 nm for the first 15 s of each reaction. The experiments were repeated four times, and data were fitted using Origin 5.0.
pKa Titration Experiments
Titration reactions were performed according to the ITC method (15) with the following exceptions. Titration buffer was a mixture of 50 mm Tris, 50 mm potassium phosphate, and 5% glycerol with the following pH values: 6.2, 6.5, 6.7, 7.0, 7.2, 7.5, 7.7, or 8.2. Recombinant PcpF C248A was purified via nickel-nitrilotriacetic acid and dialyzed into each buffer before titration. The experiments were repeated three times, and the curve-fitting was performed in Origin 5.0.
Circular Dichroism (CD)
CD spectra for each wild-type mutant EcYqjG protein was measured using an AVIV 202SF spectropolarimeter (AVIV Biomedical, Inc.) at 25 °C at a concentration of 5 μm in phosphate buffer saline and recorded from 200 to 300 nm.
Computational Molecular Docking
The ligand molecule GSH was retrieved from the RCSB PDB (ID: GSH). The ligand dihedrals, bond angles, and distances were treated as flexible in the docking process, whereas the YqjG and bound GSH molecules were treated as a rigid receptor. These structures were converted into PDBQT format by Autodock Tools for usage in Autodock v 4.2 (16). As the first GSH binding site in EcYqjG had already been established crystallographically, the corresponding position was used to define the search space for the second GSH moiety. The atomic affinity grid was calculated using the Autogrid module. All search parameters were kept as default. A total of 100 docking runs were completed for GSH using Lamarckian Genetic Algorithm output, resulting in the 100 best conformations. The predicted binding orientations were finally subjected to cluster analysis with a 3 Å root mean square deviation (r.m.s.d.) cut-off, whereby the conformations within each cluster were ranked hierarchically in the order of increasing ΔGb, the free energy of binding.
RESULTS
Recombinant EcYqjG was purified, and its crystal structures of apo-form, GSH, and GS-menadione complexes, were determined at 2.7 Å resolution (Table 1). The apo-form of EcYqjG was crystallized in the trigonal space group, P3121, with two molecules per asymmetric unit assembled by a non-crystallographic 2-fold axis, and the structure was determined by the molecular replacement method using the coordinates of a GST from P. chrysosporium (PDB code 3PPU) (5). In turn, the structures of the binary complexes of EcYqjG and apo-form PcpF were determined using the coordinates of the deduced apo-form EcYqjG structure, and crystallographic data were reported in Table 1. Due to an observed intricate molecular interaction among eight molecules in the asymmetric unit of the PcpF crystal lattice, a complete tracing for all the residues was possible, which in turn helped a backbone tracing for the flexible coils located at the N terminus of EcYqjG. Due to the lengthy crystallization time (∼1 month) and slightly inferior resolution of PcpF, the binary complex structures with GSH were determined for only EcYqjG. Our attempted soaking experiments for both apo-crystals of PcpF and EcYqjG in GS-hydroquinone solution were not successful, resulting in severe and instant crystal damage that might be due to enzyme activity on that compound. However, soaking the apo-form EcYqjG crystal with GS-menadione, which is an analog of GS-menadiol, was successful. In the asymmetric unit of the EcYqjG crystal lattice, two molecules were assembled by a non-crystallographic 2-fold symmetry operation. The dimer status of EcYqjG was verified in solution by a multi-angle laser light scattering experiment (Fig. 1). Previously we determined the dimer state of PcpF (4). The observed dimers of EcYqjG and PcpF were superimposable, and the interface had an extensive network of symmetrically oriented intersubunit hydrogen bonds (Fig. 2A).
FIGURE 1.

Dimeric EcYqjG in solution. Elution profile for EcYqjG was monitored with multiangle laser light scattering (2 mg ml−1) shown as molecular mass (kDa) versus elution volume (ml). The solid line represents changes in absorption at 280 nm. The thick black cluster in the middle of the peaks indicates the calculated molecular mass. A.U., absorbance units.
FIGURE 2.

Ribbon diagram representing the crystal structures of EcYqjG and PcpF. A, backbone structures of PcpF (blue) and EcYqjG (green) are superimposed; the r.m.s.d. value of their Cα was 0.83 Å. The N and C termini were labeled as N and C, respectively; non-crystallographic 2-fold axis was indicated. B, shown is ribbon diagram of EcYqjG with the N-terminal extension unique to GS-HQRs colored in yellow, the N-terminal thioredoxin-like domain colored in red, and the C-terminal helix-rich domain in green. The bound glutathione (GSH) at the active site was shown in CPK model. All secondary structural elements were numbered following the convention together with GS-HQR unique βa, βb. and βc. These figures were generated using Open-Source PyMOLTM (v1.4).
Global Structure
The crystal structures of EcYqjG and PcpF showed an identical backbone topology, being superimposable with r.m.s.d. of 0.83 Å (Fig. 2A). Individual subunits of both enzymes had an N-terminal thioredoxin-like domain and a C-terminal α-helix rich domain. The observed classical N-terminal thioredoxin-like fold, which covered the residues 41–155 and 51–166 of PcpF and EcYqjG, respectively, consisted of a central four-stranded β-sheet flanked by two (α1 and α3) helices on one side and one helix (α2) on the other side of the sheet (Fig. 2B). The twisted central β-sheet constituting the core of EcYqjG or PcpF was arranged in the order of β2-β1-β3-β4, and most of its surface residues were hydrophobic. In addition, the N-terminal, ∼40 residues in PcpF and ∼50 residues in EcYqjG showed a flexible but unique fold. Particularly, two areas of PcpF, 3LLI5 and 8VWR10, which correspond to 3QLI5 and 8VWH10 in EcYqjG, formed a small three-stranded β-sheet together with 127YTG129 and 116YSG118 in PcpF and EcYqjG, respectively.
The Cα carbons among the apo-form and two complexes of EcYqjG were superimposable, with r.m.s.d. values of 0.31 and 0.26 Å between the apo-form and GS-menadione complex and the apo-form and GSH-complex, respectively, without including the N-terminal flexible region. However, there was an observed interdomain motion as discussed later. In addition, side chains of a few residues showed different conformations to accommodate the binding of GSH or GS-menadione.
GSH Binding Pocket
The Fo − Fc maps of both GSH and GS-menadione-soaked crystal data of EcYqjG clearly showed the corresponding electron density for the bound molecules located on one edge of the twisted β-sheet within the thioredoxin-like domain, which is a typical topological switch point observed in many redox-active enzymes containing thioredoxin-like folds (Fig. 3C). That is, the binding pocket for the GS moiety was established by four loops connecting α1 and β1, β2 and α2, α2 and β3, and β4 and α3 without any significantly participating residues from the C-terminal helix-rich domain. In both complex structures of EcYqjG, the GSH and GS-moieties in GS-menadione showed a similar conformation, and thus their positions were superimposable with each other. The apo-forms of both EcYqjG and PcpF had a few water molecules and sulfate ions at equivalent positions for the polar groups of the GS-moiety (Fig. 3A). The backbone of Val-133 and the side chains of Trp-96, Cys-63, Trp-65, Glu-148, and Ser-149 were in direct interaction with either GSH or the GS moiety of GS-menadione (Fig. 3, B and C). In detail, the carboxyl oxygen of the glutamyl residue in both GS-menadione and GSH were hydrogen-bonded to Og of Ser-149 and Oe of Glu-148. As mentioned above, the two short, but unique N-terminal β-strands (βA and βB) and the following coils up to Arg-31 established another layer for one side of the pocket together with another short β-strand (βC, Ser-128–Gly130). However, the carboxyl group of glycine in both GSH and GS-menadione did not show any interaction with neighboring residues. On the contrary, the carbonyl oxygen and amide nitrogen of its cysteine residue were within a hydrogen bond distance from the backbone nitrogen and oxygen of Val-133 (Ile-122 in PcpF), respectively. Considering the adjacent short βC, 127YTG129 (116YSG118 in PcpF), the cysteinyl conformation of attached ligands almost resembled a small β-sheet. In addition, the same carbonyl oxygen of GSH and GS-menadione established a hydrogen bond with the indole Ne atom of Trp-96. Both electron density and angular position of the sulfur atom in the GSH complex did not indicate any bond formation with Cys-63, and the distance between two sulfur atoms was 3.7 Å.
FIGURE 3.

G-site and H-site of EcYqjG and observed interaction with GSH and GS-menadione. A, shown is the active site of the apo-form EcYqjG. Water molecules and SO42− ion were represented by red spheres and a yellow-red tetrahedron, respectively. The tyrosine hydrogen-bond network was depicted with black dashed lines. Secondary structural elements and amino acid residues were numbered following the convention. N and C terminus were labeled with an N and C, respectively. B, the G-site of glutathione-complexed EcYqjG is shown. Glutathione (GSH) was depicted in orange with electron density surrounding it (blue mesh). C, shown is glutathionyl-menadione bound in the active site of GS-menadione complexed EcYqjG. D, the position of the second GSH molecule produced by Autodocking was shown (green) with respect to the crystallographic first GSH molecule (orange). The figures were generated by PyMOL v1.4 (Schrödinger Co). The experimental electron density maps were contoured at 1.0 δ level.
Superimposing these three EcYqjG structures, apo-form, GSH-complex, and menadione-complex, indicated that the distance between the N-terminal thioredoxin-like domain and the C-terminal helix-rich domain was shortened upon ligand binding. In particular, the α4 and α5 helices in the C-terminal domain approached toward the G-site upon ligand binding. Consequently, the sulfhydryl group of Cys-63 that was located 6.5 Å away from the phenolic hydroxyl group of Tyr-195 in apo-form approached up to 3.5 Å to the same group in GSH complex structure. It is also noticeable that the indole ring of Trp-65 in apo-form occupied a part of the GS binding pocket and yielded the position to the incoming ligand acting like a swinging gate.
H-site
The Fo − Fc maps generated with the data collected from the EcYqjG crystal soaked with GS-menadione showed the corresponding electron density for the menadione (2-methyl-1,4-napthoquinone) group connected to the GS group, which was somewhat diffused (Fig. 3C). The residues, such as Tyr-187, Asn-192, Tyr-195, Phe-199, and His-300, which all belong to C-terminal domain, established a good-sized pocket for the bulky moiety attached to the cysteinyl group of GSH. Both aromatic rings of Tyr-195 and Phe-199 in this pocket were in a parallel position with the menadione ring, although their inter-ring distances were longer than that for a classical stacking interaction. Overall, in contrast to the highly coordinated interaction for the GS moiety, the menadione moiety did not display any polar interaction, and one side of it was largely exposed to the solvent being surrounded with polar residues.
ITC
To confirm the differential substrate binding affinities between EcYqjG and PcpF, thermodynamic characterizations were done with ITC. As shown in Fig. 4, a significant amount of heat was released when these enzymes associated with GS-menadione, indicating that the binding interactions with that substrate analog had significant enthalpic contributions, −25.4 and −20.9 kcal/mol for EcYqjg and PcpF, respectively. In addition, ITC data revealed a slightly unfavorable entropic contribution, −47.7 and −45.5 cal/mol/degree for GS-menadione to EcYqjG and PcpF respectively, possibly indicating that the active site of the enzymes were slightly stabilized upon binding to the GS-menadione and only a few solvent molecules were freed from the pocket. These were consistent with structural data, as indicated by the significant reduction of the B values for the four loops constituting the binding pocket upon formation of the GS-menadione complexes. The calculated Kd values for GS-menadione from ITC data analysis were 4.3 and 1.6 μm for EcYqjG and PcpF, respectively. The thermodynamic profile for the interaction between GSH and EcYqjG was similar to GS-menadione, having an enthalpic contribution of −27.1 kcal/mol and an entropic contribution of −69.4 cal/mol/degree, yielding Kdvalues of 6.5 μm (supplemental Data 2). The enthalpic contributions of both GSH and GS-menadione are quite comparable, suggesting that the GSH-moiety is primarily responsible for binding. The entropic contributions were slightly different possibly due to the freeing of a small fraction of solvent from the H-site by the menadione moiety.
FIGURE 4.

Measurement of GS-menadione binding through ITC experiments. The trend of heat released by serial injections of GS-menadione into EcYqjG (square) or PcpF (circle) was monitored. GS-menadione showed the typical heat-releasing pattern. Solid lines represent the least square fits of the data.
Enzyme Kinetics of Site-directed Mutants
To confirm the catalytic roles of the residues suggested by the crystal structures, the corresponding mutants were generated and characterized. The similar shapes and intensities for the far UV-CD spectra indicate that the wild type and four mutants, Cys-63, Tyr-195, Tyr-253, and Tyr-296, contain a similar amount of secondary structures (supplemental Data 3). To confirm that the participating catalytic residues in EcYqjG were Cys-63, Tyr-195, Tyr-253, and Tyr-296, steady-state kinetics experiments were performed by holding the concentration of mutant EcYqjG enzymes and GSH constant at 1.5 μm and 6 mm, respectively, whereas GS-hydroquinone concentrations were varied from 0 to 1400 μm (Fig. 5). The calculated Km value for GS-hydroquinone of WT EcYqjG was 341 (±18.9) μm. The Vmax value of WT EcYqjG was 10.8 (±0.249) μmol min−1 mg−1. The mutants showed negligible activity, and thus meaningful kinetic parameters for them could not be determined. The specific activity of WT EcYqjG was 8.65 (±0.590) μmol min−1 mg−1 at saturating concentrations of GSH and GS-HQ, whereas the specific activities of C63A, Y195F, Y253F, and Y296F EcYqjG were 0.00 (±0.011), 0.189 (±0.063), 0.157 (± 0.053), and 0.397 (±0.031) μmol min−1 mg−1, respectively.
FIGURE 5.

Michaelis-Menten curve for wild type and mutants of EcYqjG. Plot of reaction velocity (y axis) with differing GS-HQ concentrations (x axis). Km (341 ± 18.9 μm) is indicated on the wild-type curve (dashed line). Kinetic curves for wild type (■), Y195F (▴), Y253F (▾), and Y296F (●) are displayed. Y195F and Y253F have approximately equivalent reaction rates, which lead overlap of the curves.
pKa Titration of Cys-63
The thiolate state of the active site Cys-63 was confirmed via the ITC method. Initial rates of reactions between iodoacetamide (100 mm) and PcpF C248A mutant (50 μm) at varying pH were dependent on the ionization state of the Cys-63, which is the only remaining Cys residue. The rise in reaction rate, which is directly proportional to the heat produced (dQ/dt), indicated that the Cys-63 was in thiol/thiolate equilibrium at the inflection point, pH 7.3 (Fig. 6). Thus, the Cys-63 is in the thiolate form at physiological pH ∼ 7.6.
FIGURE 6.

Thiolate pKa titration curve. Shown is a titration curve of the heats of reaction (dQ/dt), which is directly proportional to the initial rates of reaction, of 100 mm iodoacetamide with 50 μm PcpF C248A mutant versus the pH. Due to the dependence of reaction rate on the ionization state of the thiolate, inflection of the curve indicates the pKa of the catalytic thiolate, which is 7.3. The black line represents the least squares fit Boltzmann sigmoidal curve in Origin 5.0.
Molecular Docking for the Secondary GSH
The molecular docking experiment was conducted with the intention of identifying the binding position and conformation of the second GSH molecule or GS moiety of oxidized glutathione disulfide, GSSG. The results suggested two predominant locations for the second GSH molecule in the GSH binary complex of EcYqjG. Although both sites allowed a reasonable steric fit for a GSH molecule, only one of these sites agreed with the catalytic reaction, which was located directly above the first GSH in the G-site within disulfide bonding distance of their sulfur atoms. Additionally, the side chain of Arg-130 was located within a salt-bridge-forming distance from the docked GSH (Fig. 3D). The polarity of the docked GSH molecule at this location was parallel to the bound GSH in the G-site. The other location involved a small pocket largely created by the loop involving side chains of residues from 22 to 28. However, the resulting distance between the two sulfur atoms was ∼13 Å, suggesting its incompatibility for GSSG formation.
DISCUSSION
Structural Uniqueness of GS-HQRs
To establish a proper classification for GS-HQRs, and to identify a structural norm among them, a search of the PDB database was carried out through a DALI (17) and BLAST (18). The result showed that the most similar structure for both PcpF and EcYqjG was PcGHR1 (PDB code 3PPU), with a high Z-score of 21.9, and putative GST from Corynebacterium glutamicum (CgGST, PDB code 3M1G), with a Z-score of 18.9 (Fig. 7). After PcGHR1 and CgGST, Z-score values significantly dropped for 2,4-d-inducible GST from Glycine max (3FHS), a Phi GST, with a Z-score of 5.4 followed by a GST-like protein from Idiomarina loihiensis (4DEJ) with a Z-score of 4.2. In addition, a search for similar amino acid sequences in the PDB using BLAST (18) revealed that PcGHR1 again showed the highest score (322 bits) with 56% identity among matched amino acids to EcYqjG followed by the putative CgGST (214 bits, 39% identity). Although there were several other GST proteins such as human GST Omega 1 (hGSTO1, PDB code 1EEM) with 77 bits, 22% identity, their levels of sequence identity were low, and most of the BLAST alignments were mainly located in the C-terminal domains (175–246, 217–275). Although PcpF, EcYqjG, and PcGHR1 shared the basic GST-fold with other GSTs and GST-like proteins, the primary and tertiary structural comparison revealed several features unique to GS-HQRs that justified their designation to a unique branch in the GST superfamily together with CnYqjG, ScECM4, and AtECM4. Despite its structural similarity, CgGST showed substantial heterogeneity in critical residues, and the observed intersubunit interaction in CgCST dimer is totally different, confirming its remoteness from GS-HQRs (Fig. 7).
FIGURE 7.

Multiple sequence alignment of EcYqjG, PcpF, and homologues. The secondary structures of EcYqjG (and PcpF) are displayed above the aligned residues. Binding pocket and catalytic residues were indicated in yellow and red, respectively. Residues involved in forming the dimer interface are highlighted in blue. ScPcpF:PcpF was from S. chlorophenolicum; EcYqjG, YqjG was from E. coli; CnYqjG, YqjG was from C. necator; AtECM4, extracellular mutant 4 (ECM4) was from Arabidopsis thaliana; ScECM4, ECM4 was from S. cerevisiae; PcGHR1, glutathione-hydroquinone reductase from P. chrysosporium; CgGST, putative glutathione S-transferase (GST) was from C. glutamicum; hGSTO2-2, Omega class GST2 was from Homo sapiens; hGSTO1-1, Omega class GST1 was from Homo sapiens. Multiple sequence alignment was performed with ClustalW using a BLOSUM weighting matrix.
The substrate binding pockets of the three GS-HQRs were much deeper than those observed in other GSTs. Despite their heterogeneity in length, the backbone and side chains of the long and unique N-terminal extension of GS-HQRs were engaged in forming another layer for the secluded crevice. The residues establishing this deeper H-site, Tyr-187, Asn-192, Tyr-195, Phe-199, His-300, were conserved among GS-HQRs (Fig. 7). The residues constituting the G-site, which were in direct contact with bound GSH, Trp-96, Cys-63, Trp-65, Glu-148, and Ser-149, were also conserved among GS-HQRs. In particular, Tyr-195, Tyr-253, and Tyr-296 were clustered near the redox active sites forming a unique network among their hydroxyl groups. This interaction was completely absent in other classes of GSTs, but these three tyrosine residues were completely conserved among GS-HQRs (Fig. 7). In addition, EcYqjG, PcpF and PcGHR1 share a new type of dimer interface exclusively via their C-terminal helix-rich domains, different from other GSTs (5). In detail, there were hydrogen bond interactions between Gln-203, Tyr-206, Thr-255, and Arg-293 from one subunit and Ile-303, Leu-311, Asp-260, and Ser-310 from the other subunit. Considering all those interactions were observed near the noncrystallographic 2-fold axis, very strong intermolecular interactions existed between subunits. These participating residues in this dimer formation were conserved, and thus the oligomeric nature of GS-HQRs also revealed this uniqueness that separates them from other GSTs. Additionally, the above-mentioned Tyr network was located near the dimer interface; thus, the observed dimerization could be essential for its function.
Active Site and Catalytic Mechanism
The structure of a GS-HQR with a GS-adduct provides essential information for assessing a plausible mechanism by which GS-HQRs perform their unique catalysis (Fig. 8). At each of the two redox-active sites in the complex dimer of EcYqjG, either bound GSH or GS-menadione was observed. The G-site had significant interaction with the GS moiety, but the H-site did not display any apparent interaction with the menadione moiety of the bound GS-menadione. Thus, the enzyme bound GS-menadione mainly through its interaction with the GS moiety, as displayed by the similar order of affinity toward GS-menadione and GSH (Fig. 4, supplemental Data 2). Upon binding of either GSH or GS-menadione, the distance between the thioredoxin-like domain and the helix-rich domain was significantly shortened. Consequently, the hydroxyl oxygen of Tyr-195 moved to an approximate hydrogen-bond distance of 3.5 Å to the sulfur of the GS-menadione. Because the complex structure was achieved through soaking an apo-form crystal, which may have restricted the interdomain motion, the possibility of further approaching between domains in solution could not be eliminated. The PcpF Cys-53 and EcYqG Cys-63 residues are essential for catalytic activity, and the corresponding Cys is conserved among GSTs of the Omega class, Lambda class, dehydroascorbate reductases, and GS-HQRs (9). This Cys residue is located at the N-terminal end of α1 helix immediately followed by another highly conserved Pro residue as in PcpF and EcYqjG (Fig. 7). The proximity of the redox-active Cys residue at the positive end of the helix dipole has been proposed as a stabilizing mechanism for the Cys thiolate ion (19). This hypothesis predicts the pKa of the Cys residue in the enzyme to be significantly lower than that of the Cys in GSH. The experimental pKa of Cys-53 was 7.3 (Fig. 6), and the reported pKa of GSH is 8.3 (15). Therefore, Cys-53 would exist predominately as a thiolate at the physiological pH of 7.6. The pKa values may vary slightly among GS-HQRs, but the catalytic Cys should predominately be present as a thiolate ion in the enzymes at physiological pH. In general, the hydroquinone moieties attached to GS are electron-withdrawing groups, making the sulfur atom electron-deficient. Consequently, the Cys thiolate can effectively attack the corresponding electron-deficient sulfur atom of GS-hydroquinone molecules to form a mixed disulfide bond of Cys-S-SG and release hydroquinone (Fig. 8).
FIGURE 8.

Proposed reaction mechanism for GS-HQR. Proposed reaction mechanism in the active site of GS-HQRs. Residues are numbered according to the active site of EcYqjG. A, redox-active Cys attacks the sulfhydryl of GS-hydroquinone; for example, 2-glutathionyl-3,4,6-trichloro-p-hydroquinone, coupled with Tyr-195 general acid catalysis, forms a covalent glutathionyl-EcYqjG and yields a protonated hydroquinone. B, Tyr-195 acts as a general base producing thiolate from the incoming GSH. C, shown is the thiol exchange reaction in which GSSG, the oxidized glutathione, is produced through the transition state and leaves the active site. D, the original thiolate form of GS-HQR is thus regenerated. The arrows indicate the movement of two electrons. The figure was drawn with ChemDraw 12.0.
As shown in our docking experiment, EcYqjG and other GS-HQRs may accommodate a second GSH through an extended H-site that is highly electropositive (Fig. 9). Therefore, the mixed disulfide bond can be reduced by an incoming GSH to exchange the disulfide bond resulting in GS-SG formation and regeneration of the active enzyme (Fig. 8). The guanidinium side chain of Arg-130, which is conserved among GS-HQRs, was located at the proper distance from the carboxyl group of the second GSH glycine for a salt bridge. The temperature factors of the corresponding Arg-130 were high in apo-form and GSH, suggesting its possible locking role for the second GSH, contrary to the proposed locking role for the first GSH (5). The positions of the crystallographic first GSH and docked second GSH were superimposable to both GS-moieties of the G-SS-G molecule in the complex crystal structure of the Sigma class GST from Fasciola hepatica (PDB code 2WDU). However, in contrast to that Sigma GST whose active site is formed by the dimer interface, the second GSH molecule in EcYqjG was located in the deep H-site surrounded by the residues from one subunit, Arg-130, Asn-192, His-295, and His-300, which were all conserved among the GS-HQRs (Fig. 7).
FIGURE 9.
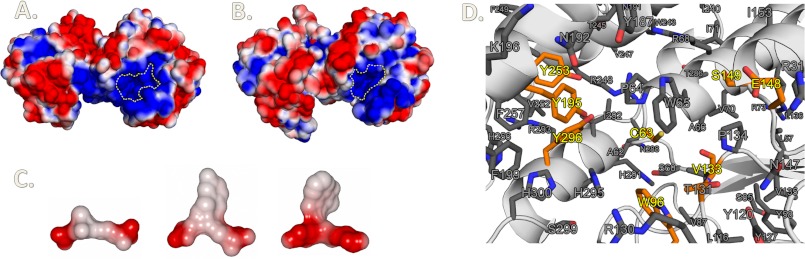
Electrostatic potential surfaces of EcYgjG, PcpF, and substrates along with the conserved residues in the active site of GS-HQRs. EcYqjG (A) and PcpF (B) dimers arranged in a non-crystallographic C2 symmetry. The molecular surface was shown with the corresponding electrostatic potential from −40 to +40 mV. red represents negative, blue represents positive, and white is uncharged or hydrophobic. The active sites were outlined with a dashed yellow line. C, electrostatic potential surfaces, also −40 to 40 mV, of the substrates are displayed: glutathione (left), GS-hydroquinone (middle), and GS-menadione (right). D, shown is a closer look at the highly conserved active sites of EcYqjG and GS-HQRs. Side chains of residues that were conserved across all GS-HQRs are shown. The residues for catalysis and direct binding of the substrate are colored orange. These figures were generated using Open-Source ePyMOLTM (v1.4).
Considering the observed distance between the hydroxyl group of Tyr-195 and the sulfur atom in the bound GSH, it is likely that Tyr-195 serves as a general acid/base catalyst for transfer reactions. Additionally, both hydroxyl groups of neighboring Tyr-253 and Tyr-296, of which mutations substantially reduced the enzyme activity (Fig. 5), were also within hydrogen-bonding distance of Tyr-195, probably increasing its activity (Fig. 8). The normal pKa of tyrosine is 10.2, which would make it difficult to ionize in neutral solutions. In the active sites of GS-HQRs, however, the pKa of tyrosine might be lowered by the intimate interaction of the hydroxyl groups of the neighboring two tyrosine residues, allowing it to function as an acid/base. In addition, the hydroxyl group of one of these tyrosine residues, Tyr-253, was within hydrogen-bonding distance from a tightly bound water molecule that was further connected to the bulk solvent through intricate hydrogen bond network, probably forming an effective proton shuttling channel. The pKa of a typical thiol group is ∼8.3 (20), and the Tyr-195 in the observed proton network can deprotonate the thiol group of incoming GSH-forming thiolate anion, allowing the thiol-disulfide exchange reaction between the mixed disulfide bond and GSH (Fig. 8). We have shown that GSH removal can also be established by other thiol-containing compounds, such as l-cysteine, DTT, and 2-mercaptoethanol (4) or even by the reducing agent NaBH4 (7). This reaction scheme is in agreement with our previous kinetic analysis that PcpF catalyzes GSH-dependent reduction of GS-TriCH via a ping-pong mechanism (4). In addition, PcGHR1 forms mixed disulfide bond with GSH after GS-hydroquinone reduction in the absence of small thiols (5).
Biological Relevance
We have previously shown that PcpF, EcYqjG, CnYqjG, and ScECM4 can deglutathionylate several GS-HQs with different substitutions, including a chlorine (TriCH), a methyl group (GS-MHQ), a hydroxyl group (GS-HHQ), and an aromatic ring (GS-menadiol) (7). Accordingly, the EcYqjG structure of GS-menadione complex displayed the relatively large size of the H-site and the lack of any specific interaction with the residues constituting the H-site, thus enabling the accommodation of these compounds. Given the diverse origins of the four enzymes, from E. coli, C. necator, S. chlorophenolicum, and S. cerevisiae, which share identical residues at all the critical positions (Fig. 7), we predict that other GS-HQRs such as the one from Arabidopsis should be able to use a variety of GS-hydroquinones as substrates. This prediction is in agreement with the report that a PcGHR1 reduces GS-hydroquinone to hydroquinone (5).
PcpF plays a maintenance role in pentachlorophenol degradation by S. chlorophenolicum (8). TriCH and DiCH are normal metabolic intermediates of pentachlorophenol degradation, whereas GS-TriCH is produced by an oxidatively damaged enzyme. PcpF channels the conjugates back to the degradation pathway. It is plausible that certain GS adducts can be more effectively reduced by selected members of the GS-HQRs, which is an important issue in terms of bioremediation. For example, PcpF is able to deglutathionylate S-(phenacyl) glutathione, but EcYqjG, CnYqjG, and ECM4 PcGHR1 do not use it as a substrate (5). In addition, the specific enzyme activity varied among these enzymes (7). Therefore, in lieu of bioremediation applications, in-depth investigation of GS-HQRs could provide a new approach for treating environmental pollutants that produce various quinone intermediates during biodegradation.
Despite their common presence in bacteria, fungi, and plants, the physiological roles of GS-HQRs have not been demonstrated. However, it is plausible that GS-HQR ability to reduce a variety of GS-HQs places this new class of GSTs in a central role for cellular redox cycling. Hydroquinones are metabolites of benzene and phenol derivatives (21) and tend to be converted into benzoquinones upon autoxidation (7, 22). Considering the spontaneous reaction between benzoquinones and GSH resulting in GS-hydroquinones, accumulation of GS-hydroquinones can be predicted to occur during the metabolism of various benzilic and phenolic compounds. For example, GS-menadione is spontaneously formed from menadione (vitamin K3) and GSH in perfused rat liver (23). GS-hydroquinones are more easily autoxidized than the corresponding hydroquinones, leading to elevated reactive oxygen species that are detrimental to the cell (24). GS-HQRs in bacteria, fungi, and plants convert GS-hydroquinones to hydroquinones, which can re-enter the metabolic pathway, probably minimizing their toxic effects to the cell and maximizing the efficiency of their metabolism. GS-HQRs have been found only in either single cell organisms or organisms that have no renal and central detoxifying systems. Therefore, it is tempting to speculate that GS-HQRs are a critical enzyme protecting cells from quinones and reactive oxygen species. Additionally, GS-HQRs can participate in quinone reduction through spontaneous reaction of quinones with GSH and then enzymatic reduction to hydroquinones (7). Furthermore, GS-HQRs in different organisms may have been adapted to deal with specific GS-hydroquinones, such as PcpF, which has a Km value of 4.4 μm for GS-TriCH, preventing accumulation of the conjugate during pentachlorophenol degradation in S. chlorophenolicum (4).
Summary
For the first time our results of EcYqjG and PcpF have provided a more comprehensive picture for the unique catalytic mechanism of the GS-HQRs than previously known. The GSH moiety of the substrate binds to the GS-HQR enzyme through several hydrogen bonds in a deep pocket that could allow various GS-HQs to remain bound adjacent to the redox active site. The sulfur atom of the GS moiety is within a hydrogen bond distance from a tyrosine cluster, which potentially catalyzes the acid/base reaction. The large H-site exposed to the solvent can accommodate various hydroquinones and the second GSH. Our structural and biochemical data for EcYqjG suggested that the deglutathionylation activity of GS-HQRs is through the binding of a loosely associated (i.e. diffusible) quinol moiety and a tightly bound GS moiety followed by the reduction by Cys-63 together with Tyr-195 and its two associated Tyr residues, which provide the proton for reduction. Due to the loose binding nature of the HQ-site, the GS-HQRs cannot hold a diffused-in electrophilic compound long enough to transfer GSH to it and, thus, lack transferase activity. Instead GS-HQR can adopt a broad substrate range of GS-hydroquinones. All the residues constituting both the G-site, the H-site, and the dimer interface were completely conserved among compared homologues. Having only a well defined pocket for GSH, GS-HQR substrate specificity is mainly defined by the presence of a glutathionyl moiety.
The conservation of GS-HQRs across all three domains of life potentiates the presence of these reductases in any prospective metabolic pathway. Thus, having a solid knowledge of the structure and mechanism of these GS-HQRs will be beneficial not only for the optimization of the PCP degradation pathway but also for the optimization of other pathways containing quinones.
This work was supported by National Science Foundation Grants MCB 1021148 and DBI 0959778 and by the M. J. Murdock Charitable Trust.

This article contains supplemental Data 1–3.
The atomic coordinates and structure factors (codes 4G0, 4G0K, 4G0I, and 4FQU) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- GS-HQR
- glutathionyl (GS)-hydroquinone reductases
- TriCH
- trichloro-p-hydroquinone
- GS-HQ
- GS-p-hydroquinone
- GS-menadione
- 3-glutathionyl-2-methyl-1,4-naphthoquinone
- ITC
- isothermal titration calorimetry
- r.m.s.d.
- root mean squared deviation.
REFERENCES
- 1. Sheehan D., Meade G., Foley V. M., Dowd C. (2001) Structure, function, and evolution of glutathione transferases. Implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 360, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li J., Xia Z., Ding J. (2005) Thioredoxin-like domain of human κ class glutathione transferase reveals sequence homology and structure similarity to the theta class enzyme. Protein Sci. 14, 2361–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dixon D. P., Davis B. G., Edwards R. (2002) Functional divergence in the glutathione transferase superfamily in plants. Identification of two classes with putative functions in redox homeostasis in Arabidopsis thaliana. J. Biol. Chem. 277, 30859–30869 [DOI] [PubMed] [Google Scholar]
- 4. Xun L., Belchik S. M., Xun R., Huang Y., Zhou H., Sanchez E., Kang C., Board P. G. (2010) S-Glutathionyl-(chloro)hydroquinone reductases. A novel class of glutathione transferases. Biochem. J. 428, 419–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meux E., Prosper P., Ngadin A., Didierjean C., Morel M., Dumarçay S., Lamant T., Jacquot J. P., Favier F., Gelhaye E. (2011) Glutathione transferases of Phanerochaete chrysosporium. S-Glutathionyl-p-hydroquinone reductase belongs to a new structural class. J. Biol. Chem. 286, 9162–9173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Whitbread A. K., Masoumi A., Tetlow N., Schmuck E., Coggan M., Board P. G. (2005) Characterization of the omega class of glutathione transferases. Methods Enzymol. 401, 78–99 [DOI] [PubMed] [Google Scholar]
- 7. Lam L. K., Zhang Z., Board P. G., Xun L. (2012) Reduction of benzoquinones to hydroquinones via spontaneous reaction with glutathione and enzymatic reaction by S-glutathionyl-hydroquinone reductases. Biochemistry 51, 5014–5021 [DOI] [PubMed] [Google Scholar]
- 8. Huang Y., Xun R., Chen G., Xun L. (2008) Maintenance role of glutahionyl-hydroquinone lyase (PcpF) in pentachlorophenol degradation by Sphingobium chlorophenolicum ATCC 39723. J. Bacteriol. 190, 7595–7600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Belchik S. M., Xun L. (2011) S-Glutathionyl-(chloro)hydroquinone reductases. A new class of glutathione transferases functioning as oxidoreductases. Drug Metab. Rev. 43, 307–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nickerson W. J., Falcone G., Strauss G. (1963) STUDIES ON QUINONE-THIOETHERS. I. MECHANISM OF FORMATION AND PROPERTIES OF THIODIONE. Biochemistry 2, 537–543 [DOI] [PubMed] [Google Scholar]
- 11. Otwinowski Z., Borek D., Majewski W., Minor W. (2003) Multiparametric scaling of diffraction intensities. Acta Crystallogr A. 59, 228–234 [DOI] [PubMed] [Google Scholar]
- 12. Navaza J. (2001) Implementation of molecular replacement in AMoRe. Acta Crystallogr D Biol Crystallogr. 57, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 13. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tajc S. G., Tolbert B. S., Basavappa R., Miller B. L. (2004) Direct determination of thiol pKa by isothermal titration microcalorimetry. J. Am. Chem. Soc. 126, 10508–10509 [DOI] [PubMed] [Google Scholar]
- 16. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J. (2009) AutoDock4 and AutoDockTools4. Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Holm L., Sander C. (1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233, 123–138 [DOI] [PubMed] [Google Scholar]
- 18. Altschul S. F., Madden T. L., Schäffer A. A, Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Gapped BLAST and PSI-BLAST. A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Board P. G., Coggan M., Chelvanayagam G., Easteal S., Jermiin L. S., Schulte G. K., Danley D. E., Hoth L. R., Griffor M. C., Kamath A. V., Rosner M. H., Chrunyk B. A., Perregaux D. E., Gabel C. A., Geoghegan K. F., Pandit J. (2000) Identification, characterization, and crystal structure of the Omega class glutathione transferases. J. Biol. Chem. 275, 24798–24806 [DOI] [PubMed] [Google Scholar]
- 20. Gilbert H. (1995) Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 251, 8–28 [DOI] [PubMed] [Google Scholar]
- 21. Nerland D. E., Pierce W. M., Jr. (1990) Identification of N-acetyl-S-(2,5-dihydroxyphenyl)-l-cysteine as a urinary metabolite of benzene, phenol, and hydroquinone. Drug Metab. Dispos. 18, 958–961 [PubMed] [Google Scholar]
- 22. Hill B. A., Kleiner H. E., Ryan E. A., Dulik D. M., Monks T. J., Lau S. S. (1993) Identification of multi-S-substituted conjugates of hydroquinone by HPLC-coulometric electrode array analysis and mass spectroscopy. Chem. Res. Toxicol. 6, 459–469 [DOI] [PubMed] [Google Scholar]
- 23. Wefers H., Sies H. (1983) Hepatic low-level chemiluminescence during redox cycling of menadione and the menadione-glutathione conjugate. Relation to glutathione and NAD(P)H:quinone reductase (DT-diaphorase) activity. Arch. Biochem. Biophys. 224, 568–578 [DOI] [PubMed] [Google Scholar]
- 24. Brunmark A., Cadenas E. (1988) Reductive addition of glutathione to p-benzoquinone, 2-hydroxy-benzoquinone, and p-benzoquinone epoxides. Effect of the hydroxy- and glutathionyl-substituents on p-benzohydroquinone autoxidation. Chem. Biol. Interact. 68, 273–298 [DOI] [PubMed] [Google Scholar]