

Background: iNOS/NO blocks TNFα-induced apoptosis by cGMP-dependent and cGMP-independent mechanisms in hepatocytes.
Result: TLR4-dependent iNOS expression in hepatocytes leads to NO/cGMP/PKG-dependent TACE/ADAM17 and iRhom2 phosphorylation and interaction, TACE/ADAM17 activation/surface translocation, and TNFR1 shedding.
Conclusion: iNOS expression leads to rapid TNFR1 shedding through NO/cGMP/PKG-dependent translocation and activation of TACE/ADAM17.
Significance: This novel mechanism for iNOS/NO/cGMP-induced TACE activation and translocation may limit TNFα-mediated signaling.
Keywords: Apoptosis, Nitric Oxide, Proteolytic Enzymes, Toll-like Receptors (TLR), Tumor Necrosis Factor (TNF), ADAM17, Hepatocytes, Liver, TNFα-converting Enzyme, Cyclic Nucleotides
Abstract
We and others have previously shown that the inducible nitric-oxide synthase (iNOS) and nitric oxide (NO) are hepatoprotective in a number of circumstances, including endotoxemia. In vitro, hepatocytes are protected from tumor necrosis factor (TNF) α-induced apoptosis via cGMP-dependent and cGMP-independent mechanisms. We have shown that the cGMP-dependent protective mechanisms involve the inhibition of death-inducing signaling complex formation. We show here that LPS-induced iNOS expression leads to rapid TNF receptor shedding from the surface of hepatocytes via NO/cGMP/protein kinase G-dependent activation and surface translocation of TNFα-converting enzyme (TACE/ADAM17). The activation of TACE is associated with the up-regulation of iRhom2 as well as the interaction and phosphorylation of TACE and iRhom2, which are also NO/cGMP/protein kinase G-dependent. These findings suggest that one mechanism of iNOS/NO-mediated protection of hepatocytes involves the rapid shedding of TNF receptor 1 to limit TNFα signaling.
Introduction
Tumor necrosis factor (TNF)2 α is a proinflammatory cytokine that plays important roles in the pathophysiology of inflammatory conditions, such as sepsis (1–3). Excessive TNFα production can also have detrimental effects, including the induction of apoptosis in parenchymal cells of organs, such as the liver (4, 5); this contributes to the pathogenesis and organ damage related to sepsis and inflammation. The effects of TNFα are mediated via two cell surface receptors: TNF receptor (TNFR) 1 and TNFR2 (6). TNFR1 is the main cell surface receptor for TNFα on hepatocytes (7–9), the major parenchymal cell type in the liver. Activation of the death domain in TNFR1 leads to apoptotic cell death (10–13), but other TNFR1 signaling pathways that activate mitogen-activated protein kinases, NFκB, or signal transduction and activators of transcription pathways can be protective (14–16).
Hepatocytes express large amounts of TNFR1 (17, 18), and activation of this receptor by TNFα during inflammation can lead to hepatocyte apoptosis (19). However, TNFR1 can also be cleaved from the cell surface through proteolytic ectodomain shedding driven by activation of TNFα-converting enzyme (TACE/ADAM17). Cleavage of TNFR1 by TACE leads to the release of soluble TNFR1 (sTNFR1), which not only reduces the number of receptors available on the outside of hepatocytes but also binds to circulating TNFα to prevent its inflammatory effects (20–23). The formation of sTNFR1 is a constitutive process (24–26) but is increased under inflammatory disease conditions or after stimulation with cytokines TNFα, interleukin (IL)-1β, and IL-10 as well as with lipopolysaccharide (LPS; endotoxin) (27–29).
The signaling pathways controlling TACE activation and extracellular receptor shedding are unclear, although previous studies have shown that TACE can mediate the shedding of multiple cell surface receptors, including TNFR1, on monocytes and macrophages (30–32). One study suggested that nitric oxide (NO) from an NO donor can S-nitrosylate TACE, resulting in increased cell surface receptor shedding in monocytes and macrophages (33). Additionally, stimulation of cells with NO alone was sufficient to cause significantly increased TNFR1 shedding, whereas TNFα stimulation alone results in only minor increases in sTNFR1 levels. These previous data suggested that NO mediates the activation of TACE, although the exact pathways involved were not identified. Recently, two studies showed that iRhom2 (also known as RHDBF2, RHBDL5, or RHBDL6), an intracellular protease that is localized to the endoplasmic reticulum, is critical to the maturation and trafficking of TACE to the cell surface for TNFα maturation (34, 35). In this study, we further elucidate the pathways involved in NO-mediated TACE activation and show that LPS stimulation up-regulates iNOS expression in mouse hepatocytes, leading to TACE and iRhom2 phosphorylation in a cyclic GMP (cGMP)/protein kinase G (PKG)-dependent manner. This is associated with TACE localization to the cell surface to cause TNFR1 shedding. Our data provide evidence of a novel mechanism for the protective effects of iNOS in preventing TNFα-mediated cell death and inflammation in the liver.
EXPERIMENTAL PROCEDURES
Reagents
Ultrapure LPS (E. coli 0111:B4/LPS-EB Ultrapure) was from InvivoGen (San Diego, CA). Williams' medium E, penicillin, streptomycin, l-glutamine, and HEPES were purchased from Invitrogen. Insulin (Humulin) was acquired from Eli Lilly, and fetal calf serum was from Hyclone Laboratories (Green Bay, WI). Mouse TNFα was obtained from R&D Systems (Minneapolis, MN). Unless otherwise indicated, all other chemicals and reagents were from Sigma-Aldrich.
Animals
Experimental protocols were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh. C57BL/6 (WT) mice were purchased from Charles River (Wilmington, MA) at 8–12 weeks of age. iNOS-deficient (−/−) mice were bred in our facility. These mice have been backcrossed at least six times and are a generous gift from Dr. Victor Laubach. TLR4−/− mice on a C57BL/6 background were generated in our laboratory and bred in our facility. Mice were injected intraperitoneally with saline (control) or LPS (5 mg/kg) and/or iNOS inhibitor 1400W (2.5 mg/kg). Blood and the liver were collected at time points up to 12 h after injection.
Hepatocyte Isolation and Cell Culture
Hepatocytes were isolated from mice by an in situ collagenase (type VI; Sigma) perfusion technique modified as described previously (36). Hepatocyte purity exceeded 99% by flow cytometric assay, and viability was typically over 85% by trypan blue exclusion. Hepatocytes (150,000 cells/ml) were plated on gelatin-coated culture plates or coverslips precoated with collagen I (BD Pharmingen) in Williams' medium E with 10% calf serum, 15 mm HEPES, 10−6 m insulin, 2 mm l-glutamine, 100 units/ml penicillin, and 100 units/ml streptomycin. Hepatocytes were allowed to attach to plates overnight, and prior to treatments, cell culture medium was changed to medium containing 5% calf serum.
Immunofluorescence
Hepatocytes plated on coverslips were treated as described and then fixed with 2% (w/v) paraformaldehyde for 15 min. Residual paraformaldehyde was removed in multiple PBS wash steps. Cells were then permeabilized with 0.1% Triton X-100, washed in PBS and PBB (0.5% bovine serum albumin (BSA) in PBS), and blocked with 2% BSA in PBS for 1 h with further blocking as needed overnight at 4 °C with whole mouse IgG (1:100 dilution). Non-permeabilized cells were used for counting cell surface TACE in five randomly selected fields per treatment group. Rabbit anti-TACE antibody (Abcam, Cambridge, MA) and mouse anti-iNOS antibody were added at a 1:1000 dilution for 10 h at 4 °C. Secondary antibody labeled with Cy3 or Cy5 at a 1:1000 dilution was added before visualization with an Olympus FluoView 500 confocal microscope.
Immunofluorescence Staining and Confocal Microscopy on Liver Sections
Livers were fixed in 2% paraformaldehyde, and the previously described standardized protocol for cryopreservation was performed (7). Livers were sectioned in a cryostat and stained as follows. 5-μm liver sections were incubated with 2% BSA in PBS for 1 h followed by five washes with PBS + 0.5% BSA. The samples were then incubated with rabbit anti-TACE (1-h incubation), anti-TNFR1 (overnight incubation), and anti-actin as described above. Samples were washed five times with PBS + 0.5% BSA followed by incubation in the appropriate Alexa Fluor 488 (1:500; Invitrogen) and Cy3 (1:1000; Jackson ImmunoResearch Laboratories) secondary antibodies diluted in PBS + 0.5% BSA. Samples were washed three times with PBS + 0.5% BSA followed by a single wash with PBS before 30-s incubation with Hoechst nuclear stain. The nuclear stain was removed, and samples were washed with PBS before placing a coverslip using Gelvatol (23 g of poly(vinyl alcohol 2000), 50 ml of glycerol, 0.1% sodium azide to 100 ml of PBS). Positively stained cells in six random fields were imaged on a FluoView 1000 confocal scanning microscope (Olympus). Imaging conditions were maintained at identical settings within each antibody labeling experiment with original gating performed using the negative control.
Preparation of Cell Lysates, Western Blotting, and Co-immunoprecipitation Analysis
Treated hepatocytes were washed twice in PBS and lysed with 1× cell lysis buffer (Cell Signaling Technology) containing 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerol phosphate, 1 mm Na3VO4, 1 μg/ml leupeptin, and 1 μg/ml phenylmethylsulfonyl fluoride (PMSF) on ice for 10 min. Liver samples were homogenized with a glass Dounce homogenizer in 1× radioimmune precipitation assay buffer (cell lysis buffer; Cell Signaling Technology) containing 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Nonidet P-40, 1% sodium deoxycholate, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3V04, 1 μg/ml leupeptin, and 1 mm PMSF in double distilled H2O. The protein content of cell lysates was determined by BCA protein assay (Pierce). For Western blotting, equal protein amounts were separated by SDS-PAGE and transferred onto a nitrocellulose membrane followed by immunostaining with optimized dilutions of primary antibodies rabbit anti-ADAM17 (Ab2051; for intracellular TACE), rabbit anti-ADAM17 (Ab39163; for activated TACE), rabbit anti-iNOS (Ab152323), rabbit anti-iRhom2 (sc-138584, Santa Cruz Biotechnology), anti-TNFR1 (Abcam), and rabbit anti-(active) caspase-3 (Ab2302) diluted in 0.5% BSA and 1× TBS-Tween 20. Horseradish peroxidase-conjugated secondary antibodies were then used in a standard enhanced chemiluminescence reaction according to the manufacturer's instructions (Pierce). For co-immunoprecipitation, whole cell lysates were incubated overnight with rabbit anti-TNFR1 antibody, and immune complexes were then precipitated with protein A/G-agarose beads for 6 h and then washed several times with immunoprecipitation buffer (20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Nonidet P-40, 1% sodium deoxycholate, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3V04, 1 μg/ml leupeptin, and 1 mm PMSF). Immunoprecipitated proteins were eluted with 2× SDS loading buffer (0.5 m Tris-HCl (pH 6.8), 2 ml of glycerol, 10% (w/v) SDS, 0.1% (w/v) bromphenol blue, and 5% β-mercaptoethanol in water) and then analyzed by Western blot as above.
Cross-linking Experiments
Cross-linking was carried out as described by the manufacturer (Pierce). Briefly, the cells were washed with PBS (137 mm NaCl, 0.67 mm KCl, 8 mm Na2HPO4, and 1.4 mm KH2PO4) three times and incubated in 2 mm dithiobis(sulfosuccinimidylpropionate) diluted in DMSO (Pierce) for 30 min at room temperature. The reaction was stopped with 20 mm Tris (pH 7.5) for 15 min at room temperature followed by washing three times with PBS before use in the following studies.
Measurement of sTNFR1
Soluble TNFR1 was measured in plasma and cell culture supernatants. For cell culture supernatant, primary hepatocytes were plated in 6-well or 12-well plates and treated either with vehicle (PBS), 10 nm solution of TACE inhibitor (TAPI-1®, EMD Chemical) for 24 h, or other exogenous reagents as indicated. Release of TNFR1 into cell culture supernatants over a 24-h period was assayed by using a Quantikine® mouse sTNFR1 ELISA with a sensitivity of 7.8 pg/ml (R&D Systems) according to the manufacturer's instructions.
Quantification of TACE Activity
A fluorometric TACE activity assay kit (SensoLyte 520®) was used to measure TACE activation (expressed as activity in relative fluorescence units) in membrane (20,000 × g) fractions. Total TACE activity in liver and primary hepatocytes was measured using whole cell lysates following the manufacturer's instructions. To obtain liver homogenate, liver tissues were disrupted and passed through a cell strainer (0.75-μm pore size) in PBS. Homogenates were then treated according to the manufacturer's instructions.
Knockdown of TACE/ADAM17 with siRNA
Primary hepatocytes were cultured in 6-well plates overnight and then transfected with siRNA (5 nm) for 6 h using Lipofectamine 2000 (Invitrogen). Cells were cultured in complete Williams' medium E for up to 24 h before treatment with LPS. ON-TARGETplus siRNA-Mouse ADAM17 ORF and the non-targeting control siRNA duplexes were from Dharmacon (Lafayette, CO).
Statistical Analysis
Data are presented as means ± S.E. of at least three separate experiments. Experimental results were analyzed for their significance using analysis of variance in Excel and SigmaStat (Systat Software, San Jose, CA). Significance was established at the 95% confidence level (p < 0.05).
RESULTS
LPS-mediated Up-regulation of Liver TNFR1 Shedding and Circulating sTNFR1 Levels Is iNOS-dependent
The liver is a major organ affected by inflammatory responses to infection, and liver cell apoptosis contributes to the pathogenesis of sepsis (37, 38). Additionally, we previously showed that liver and hepatocyte apoptosis can be initiated through TNFα-TNFR1 signaling (7), and iNOS protects against this apoptosis (19, 37). One potential mechanism of protection by iNOS and NO is through the shedding of TNFR1 from the cell surface. Therefore, we wanted to determine whether sTNFR1 levels were induced in mice during endotoxemia and whether this was iNOS-dependent. C57BL/6 mice were injected intraperitoneally with saline (control), LPS (5 mg/kg), or iNOS-specific inhibitor 1400W (2.5 mg/kg) 30 min prior to injection with LPS. After 6, 6, or 12 h, liver tissue was collected and immunoblotted for iNOS and TNFR1. As expected, iNOS was up-regulated in the liver as early as 3 h after LPS and remained up-regulated until 12 h (Fig. 1A). Of note, treatment with 1400W suppressed LPS-induced iNOS up-regulation at all time points measured, showing that iNOS-derived NO contributes to further iNOS up-regulation (Fig. 1A). Interestingly, TNFR1 levels in the liver were reduced after LPS stimulation in vivo at all time points measured compared with controls, and this decrease was prevented by blocking iNOS with 1400W (Fig. 1B). To determine whether this decrease in cellular TNFR1 was due to shedding of TNFR1, we also measured sTNFR1 in circulation. Levels of sTNFR1 were significantly increased in the serum of LPS-treated mice compared with controls, and this was attenuated when iNOS was inhibited with 1400W (Fig. 1C). To confirm these findings, we also visualized the TNFR1 level in liver tissues by immunohistochemistry. Lower TNFR1 fluorescence was noted in the livers of LPS-treated animals compared with controls (Fig. 1D). This is consistent with the Western blot findings showing that cellular TNFR1 is decreased after LPS stimulation and is dependent on iNOS. Fig. 1E shows that active caspase-3 levels in the liver are reduced in association with iNOS expression and TNFR1 shedding, confirming our previous work showing that iNOS is protective in hepatocytes (53). Together, these data suggest that TNFR1 is up-regulated by LPS and then cleaved to release soluble TNFR1 in an iNOS-dependent manner.
FIGURE 1.

iNOS up-regulation is necessary for LPS-mediated membrane TNFR1 shedding in mouse liver. A, liver proteins taken from saline-, 1400W (iNOS inhibitor)-, LPS (5 mg/kg)-, and 1400W + LPS-injected mice were separated by 12% SDS-PAGE for detection of iNOS after LPS injection at 3, 6, and 12 h. B, intracellular TNFR1 proteins were visualized by Western blot. Migration of molecular mass markers in kilodaltons (kDa) is indicated. C, quantification of the levels of soluble TNFR1 (pg/ml) shed from the membrane of liver cells of the same groups of animals analyzed in A and B after 12 h. Values are from at least three independent experiments (*, #, and ##, p < 0.01). D, liver cells displayed cytoplasmic immunohistochemical staining of TNFR1 shown in red (Cy3). Images represent three to six animals per group and five microscopic fields per animal. Scale bar, 20 μm. E, levels of active caspase-3 in the liver from WT mice injected with saline, 1400W, LPS, or 1400W combined with LPS. Inhibiting iNOS with 1400W induces cell death in the liver. Error bars indicate mean ± S.E.
To further examine the effect of LPS and iNOS on TNFR1 and sTNFR1, we next moved to an in vitro system of primary hepatocytes isolated from C57BL/6 (WT) and iNOS−/− mice. Hepatocytes were treated with 100 ng/ml LPS for time points up to 12 h, and the sTNFR1 levels in supernatants were measured. Soluble TNFR1 levels were increased in the supernatants of WT cells as early as 30 min after LPS stimulation, and this increased significantly by 2 h (Fig. 2A). Levels of sTNFR1 peaked in WT supernatants at 8 h and remained significantly elevated until 12 h (Fig. 2A). Levels of sTNFR1 in iNOS−/− supernatants increased slightly compared with controls after 2 h but remained at significantly lower levels compared with WT (Fig. 2A).
FIGURE 2.

iNOS is essential for TNFR1 shedding into cell culture medium. A, LPS induces TNFR1 shedding into the extracellular medium. ELISA was used to measure the amount of TNFR1 in medium, and raw optical density values were converted to soluble TNFR1 concentrations via a standard curve followed by normalization to PBS-treated hepatocytes. At 8 h, an 18-fold increase in optical density was measured for WT 100 ng/ml LPS-treated cells (*, p < 0.01; 0.5 versus 8 h by paired t test; n = 4–6 per time point; * represents mean ± S.E.). At 12 h, a 21-fold increase in optical density was measured for both WT and iNOS knock-out (KO) hepatocytes (*, p < 0.01; WT versus iNOS KO at 12 h by paired t test; n = 4–6 per time point; * represents mean ± S.E.). B, TNFR1 released into the medium from cells subjected to LPS at different concentrations (0.1–100 ng/ml) and saline (PBS) as a control. Culture medium was sampled, and raw optical density was normalized to the signal from untreated (medium only) cells. A 20-fold increase in soluble TNFR1 was noted after 12 h of 100 ng/ml LPS-treated cells compared with untreated cells (*, p < 0.01; n = 4–6 per condition; * and # represent mean ± S.E.), and a 13-fold increase was noted in soluble TNFR1 in WT hepatocytes versus iNOS KO hepatocytes (#, p < 0.01; n = 4–6 per condition). C, WT and iNOS KO hepatocytes were treated with different concentrations of LPS and analyzed by Western blot at 12 h after treatment. Error bars indicate mean ± S.E.
We next determined whether there was a concentration-dependent effect of LPS on sTNFR1 release and cellular TNFR1 levels. WT and iNOS−/− hepatocytes were treated with PBS (saline; control) or LPS at concentrations from 0.1 to 100 ng/ml for 12 h, and the levels of sTNFR1 in supernatants and TNFR1 in whole cell lysates were measured. LPS at a concentration as low as 0.1 ng/ml was sufficient to significantly increase sTNFR1 levels in WT supernatants compared with controls (Fig. 2B). Levels of sTNFR1 significantly increased in a dose-dependent manner in WT supernatants but once again were not increased significantly in the supernatants from iNOS−/− hepatocytes even at the highest concentration of 100 ng/ml (Fig. 2B). Similar to the results presented above, LPS treatment of WT hepatocytes decreased cell lysate levels of TNFR1 as measured by Western blot, and the decrease was most apparent at concentrations of 10 ng/ml and above (Fig. 2C). This TNFR1 decrease was not seen in hepatocyte cell lysates from iNOS−/− mice (Fig. 2C). Taken together, these data confirm our in vivo findings that LPS induces TNFR1 shedding from hepatocytes in an iNOS-dependent manner.
LPS-induced TACE Activity Is iNOS-dependent
Having determined that LPS-induced iNOS activity is required for LPS-induced TNFR1 shedding, we next wanted to determine the mechanism of iNOS-mediated TNFR1 cleavage. TACE/ADAM17 has previously been shown to be activated through S-nitrosylation induced by NO donors in monocytes and macrophages (33) to induce TNFR shedding. Therefore, we investigated the activation of TACE using a commercially available TACE activation assay in our in vitro model of LPS-stimulated primary hepatocytes. WT and iNOS−/− hepatocytes were pretreated with a TACE inhibitor (TAPI-1; 10 nm), siRNA specifically targeting TACE (or control/negative siRNA), 1400W (iNOS inhibitor; 2.5 μm), or saline control. Groups of hepatocytes were then further stimulated with LPS (100 ng/ml), and after 12 h, cell lysates and supernatants were collected for analysis of TACE activity and sTNFR1 levels, respectively. siRNA knockdown of TACE by at least 75% of control levels was confirmed by Western blot (Fig. 3A, inset). LPS treatment significantly increased activated TACE levels in WT hepatocytes but not iNOS−/− hepatocytes, and LPS-induced TACE activity was significantly inhibited with iNOS inhibition, TAPI-1 addition, and knockdown of TACE using siRNA (Fig. 3A). Soluble TNFR1 levels in WT supernatants paralleled TACE activation and were significantly increased after LPS treatment, and this increase was prevented by inhibition of iNOS (1400W) or TACE (TAPI-1 and TACE siRNA) (Fig. 3B). These data suggest that TNFR1 shedding in response to LPS is dependent not only on iNOS but also on TACE.
FIGURE 3.

TACE activity is affected in iNOS KO hepatocytes. A, primary hepatocytes from WT and iNOS KO mice were isolated and incubated with 100 ng/ml LPS or an equivalent dilution of saline (PBS). Cells were also incubated with LPS combined with a pharmacological inhibitor of TACE (TAPI-1; 10 nm). A separate group of cells was treated with TACE siRNA (5 nm) or negative (Neg.) siRNA (5 nm) to knock down intracellular TACE. Following 6 h of siRNA incubation, hepatocytes were treated with 100 ng/ml LPS. Hepatocytes were also incubated with iNOS inhibitor 1400W with or without LPS for a total of 12 h. Total TACE activity was measured in whole cell lysates. Values are relative fluorescence units at an excitation/emission of 490/520 nm normalized to blank (assay buffer). Error bars indicate mean ± S.E. Triplicate measurements for each treatment groups are shown and are from three independent experiments. A, inset, TACE knockdown reduced the amount of activated TACE. B, quantification of the level of soluble TNFR1 in medium after treatment as indicated in A. C, mice were injected with 5 mg/kg LPS or an equivalent volume of saline for 12 h. At the end of the injection, livers were homogenized, and supernatants were collected. The resulting lysates were used for measuring TACE activity according to the manufacturer's instructions. D, TACE activation in WT and iNOS KO hepatocytes harvested from C assayed by immunoblot using monoclonal anti-TACE directed against the cytoplasmic domain of TACE and monoclonal anti-TACE directed against a region of the activated TACE.
To confirm our findings, we also determined TACE activation in the livers of LPS-treated or control WT and iNOS−/− mice. Mice were treated intraperitoneally as described previously with either saline (control) or 5 mg/kg LPS, and TACE activation was determined in livers at 12 h. Similar to our in vitro results, we found that LPS significantly increased TACE activation in WT mice but not in iNOS−/− mice, and this increase in TACE activation was inhibited by iNOS inhibition with 1400W (Fig. 3C).
TACE has been shown to localize to the cell surface once activated to cleave receptors, such as TNFR1 (31, 40–43). Activated TACE is also glycosylated and can be differentially analyzed by Western blot using separate antibodies specific for total cellular TACE or activated TACE (43–47). We analyzed expression of total intracellular and activated TACE in liver cell lysates and membrane fractions from WT and iNOS−/− mice treated with and without LPS as described above. LPS strongly increased expression of total TACE in WT mouse liver at 12 h after LPS compared with controls (Fig. 3D). There was also an increase in intracellular TACE expression in iNOS−/− mouse liver after LPS treatment, although this was a lower level than in WT (Fig. 3D). However, activated TACE levels were significantly up-regulated in only WT mice after LPS but not in iNOS−/− mice (Fig. 3D). These results suggest that although TACE may be up-regulated to some degree in iNOS−/− liver during endotoxemia this TACE is not activated in the absence of functional iNOS protein.
To further investigate TACE activation and localization, we used fluorescence immunohistochemistry in both permeabilized and non-permeabilized liver sections to identify TACE within the cell and TACE located at the cell surface. Intracellular TACE levels were similar at base line between WT and iNOS−/− mouse liver (Fig. 4A). After LPS stimulation for 12 h, intracellular TACE levels increased in iNOS−/− liver but were decreased in WT liver (Fig. 4A). Cell surface TACE, however, was lower at base line in iNOS−/− liver compared with WT liver (Fig. 4B), and surface TACE levels did not increase in iNOS−/− liver after LPS stimulation (Fig. 4B). However, levels of cell surface TACE increased significantly in WT liver after LPS stimulation, suggesting that localization of TACE to the cell surface is dependent on iNOS. In vitro results were very similar to in vivo results (data not shown), suggesting iNOS-dependent TACE mobilization and activation after LPS stimulation.
FIGURE 4.

Translocation of TACE to the cell surface in mouse liver is dependent on iNOS. A, non-permeabilized liver sections harvested from WT and iNOS KO mice. Both groups of mice were injected with 5 mg/kg LPS or an equivalent dose of saline. B, permeabilized liver sections from WT and iNOS KO mice injected with LPS or an equivalent dose of saline. Sections were then stained for TACE using monoclonal anti-TACE. Liver cells displayed cytoplasmic immunohistochemical staining of TACE (Cy3). Images represent six to eight animals per group and five microscopic fields per animal. Scale bars, 20 μm.
TACE Activation Is cGMP-dependent
Having shown that TACE activation and localization to the cell surface are iNOS-dependent, we wanted to further understand how iNOS directs TACE to the cell surface. The second messenger cGMP is up-regulated in hepatocytes by NO/iNOS and has been implicated in the regulation of cellular apoptosis (48). Cyclic GMP is also able to activate protein kinases, such as PKG, which in turn can phosphorylate and activate target proteins (49, 50). Therefore, we investigated whether TACE activation was regulated by iNOS-derived cGMP.
Primary WT hepatocytes were left untreated or pretreated with 1400W (2.5 μm) or the soluble guanylyl cyclase inhibitor ODQ (10 μm) and then exposed to LPS (100 ng/ml) for up to 3 h. As shown previously, stimulation with LPS significantly increased sTNFR1 levels compared with untreated controls (Fig. 5A). Inhibition of either iNOS or cGMP significantly attenuated sTNFR1 release (Fig. 5A). We then asked whether stimulation of cells with exogenous cGMP would increase sTNFR1 levels. The cGMP analog 8-bromoguanosine 3′,5′-cyclic monophosphate (8-Br-cGMP) was added to cells at 800 μm for 30 or 60 min, and sTNFR1 levels were measured in supernatants. Stimulation with 8-Br-cGMP significantly increased sTNFR1 levels after just 30 min, and this increase was maintained until 1 h, although levels were roughly half those of LPS stimulation alone (Fig. 5B).
FIGURE 5.

TACE activation and translocation to the cell surface is dependent on cyclic GMP and NO, respectively. A, primary hepatocytes were incubated in 100 ng/ml LPS or pretreated with 1400W or ODQ (soluble guanylyl cyclase inhibitor; 10 μm) for at least 30 min before incubation with LPS. Cell medium was assayed for soluble TNFR1 after 3 h. The same medium was also assayed for cGMP (data not shown). B, levels of soluble TNFR1 in cell medium after 30 and 60 min of treatment with the cell-permeable cGMP analog 8-Br-cGMP (800 μm). Values are from at least three independent experiments (*, #, and ##, p < 0.05). To induce TACE activation, hepatocytes were incubated with the NO donor SNAP (80 μm) or with the cell-permeable cGMP analog 8-Br-cGMP (800 μm) for 30 min. To block TACE activation, cells were incubated with ODQ (10 μm) or with a PKG inhibitor (inhib.) (20 μm) before treatment with SNAP with 8-Br-cGMP (C and D, respectively). Lysates were separated by 4–20% gradient SDS-PAGE followed by immunoblotting with an anti-TACE (activated) antibody. E, hepatocytes were incubated in SNAP and cGMP for 30 min. Lysates were washed with PBS (supplemented with proteinase inhibitors) and subjected to subcellular centrifugation (20, 000 × g for 15 min at 4 °C) to isolate membrane fractions. Membrane fractions were reconstituted in TACE activity assay buffer (as supplied by the manufacturer) and analyzed (*, p < 0.05, SNAP versus untreated; and #, p < 0.05, cGMP versus untreated). To inhibit cGMP production, hepatocytes were incubated in ODQ (10 μm) for 30 min, and membrane fractions from the cells were analyzed for the level of TACE activity (##, p < 0.01, SNAP versus ODQ + SNAP). Values are from four independent experiments. F, the same experiment as in E was carried out with immunohistochemical analysis of the indicated factors. Scale bar, 20 μm. G, the number of active TACE (Cy3-positive puncta) on cellular membranes from E was counted. Error bars correspond to the mean ± S.E. Values are from five different fields per treatment and from three independent experiments (*, p < 0.01, SNAP versus untreated; #, p < 0.01, SNAP versus ODQ + SNAP). Error bars indicate mean ± S.E.
We then looked for activated TACE by Western blot in membrane fractions and lysates of WT hepatocytes that were stimulated for 30 min with a soluble guanylyl cyclase inhibitor (ODQ; 10 μm), a PKG inhibitor (20 μm) alone, or the NO donor SNAP (80 μm). As expected, SNAP alone induced expression of activated TACE (∼150 kDa), and this was inhibited by both ODQ and the PKG inhibitor (Fig. 5C). Similarly, TACE was activated by 8-Br-cGMP (800 μm) at 30 min (Fig. 5D), and this activation was inhibited in part by the PKG inhibitor. Taken together, these data suggest that blocking any part of the pathway from iNOS/NO to cGMP to PKG prevents TACE activation and sTNFR1 release from hepatocytes after LPS stimulation.
Because localization of TACE to the cell surface also indicates activation, we also determined the level of cell surface TACE in untreated hepatocytes or hepatocytes treated with SNAP, ODQ with SNAP, or with 8-Br-cGMP as above using immunofluorescence of non-permeabilized cells. There was a low level of cell surface TACE in untreated hepatocytes, and this was significantly increased by treatment with SNAP as expected (Fig. 5, E and F). Blocking cGMP with ODQ significantly reduced the level of cell surface TACE induced by SNAP (Fig. 5F). Similarly, stimulation of cells with 8-Br-cGMP increased the levels of cell surface TACE, although this increase was to a lower level than with SNAP (Fig. 5, F and G).
LPS-induced Increase in sTNFR1 Is TLR4-mediated
LPS is known to signal through the cell surface receptor Toll-like receptor (TLR) 4 complex. Therefore, we wanted to confirm that LPS-induced increases in sTNFR1 were mediated through TLR4. WT and TLR4−/− hepatocytes were treated with 100 ng/ml LPS for up to 6 h, and sTNFR1 levels were measured in supernatants. As expected, LPS stimulated a significant increase in sTNFR1 levels as early as 3 h, and this continued until the 6-h time point in WT cells; however, no increase was observed in sTNFR1 in supernatants from TLR4−/− cells (Fig. 6A). Cell lysates from LPS-treated WT and TLR4−/− hepatocytes were analyzed by Western blot for expression of iNOS, TNFR1, and activated TACE. Results show that in WT hepatocytes LPS (100 ng/ml) induced iNOS protein at 3 h (Fig. 6B). TACE was also activated after LPS stimulation, and this resulted in reduced cellular TNFR1 levels. Hepatocytes isolated from TLR4−/− mice did not express iNOS, and TACE was not activated in response to LPS.
FIGURE 6.
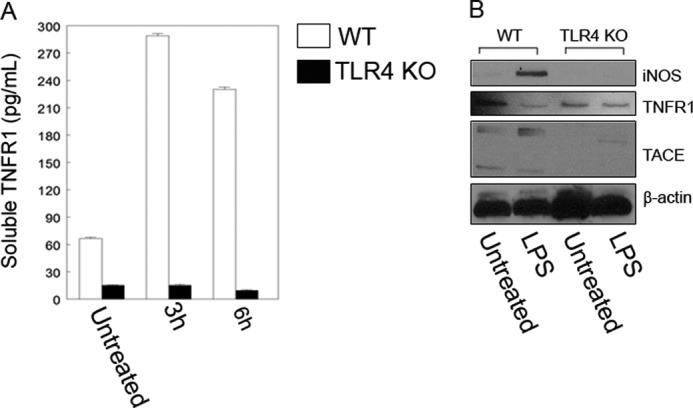
Multiple signaling pathways (TLR4, iRhom2, iNOS, and PKG) facilitate TACE activation in hepatocytes. A, TLR4 knock-out hepatocytes released lower levels of soluble TNFR1 into culture medium when incubated in 100 ng/ml LPS for up to 6 h. Error bars indicate the mean ± S.E. B, triplicate measurements for each treatment groups are shown and are from three independent experiments. Western blotting was performed to confirm iNOS and activated TACE expression in WT cells after the cells were incubated in 100 ng/ml LPS for 3 h. Activated TACE and membrane shedding of TNFR1 are dependent on TLR4 signaling as TLR4 knock-out abolished iNOS expression.
TACE Binds to iRhom2 in Hepatocytes in an iNOS/cGMP-dependent Manner
The involvement of iRhom2 is essential to TACE maturation and trafficking to the cell surface in hematopoietic cells (34, 35). To obtain evidence that iRhom2 is also involved in TACE maturation in hepatocytes, we first assessed iRhom2 expression. As shown in Fig. 7A, hepatocytes express low levels of iRhom2 at base line. LPS induced a marked increase in iRhom2 expression within 3 h that was partially prevented by iNOS inhibition with 1400W. The NO donor as well as 8-Br-cGMP also increased iRhom2 expression, whereas the PKG inhibitor blocked the cGMP-induced up-regulation of iRhom2 in hepatocytes. Immunoprecipitation experiments show that LPS treatment induced an association between TACE and iRhom2 that is iNOS-dependent (Fig. 7B).
FIGURE 7.

iRhom2 is expressed in hepatocytes after LPS treatment. A, representative Western blot of cells that were treated with LPS for up to 3 h or SNAP and cGMP for up to 1 h followed by Western blotting for iRhom2. Hepatocyte iRhom2 is expressed at low levels at base line, but iRhom2 protein levels are increased after LPS treatment. The increase in iRhom2 is likely to be dependent on the iNOS pathway as 1400W blocked some of the increase in iRhom2 expression. B, iNOS-derived NO was required for TACE and iRhom2 association. Cells were treated with LPS (3 h) followed by cross-linking (performed according to the manufacturer's instructions) and immunoprecipitation (IP) with TACE (activated) or with iRhom2 and immunoblotting (IB) for iRhom2 or TACE (activated), respectively. TACE and iRhom2 did not associate in iNOS KO primary hepatocytes (B). C, primary hepatocytes were treated with LPS (100 ng/ml for up to 3 h) followed by cross-linking and immunoprecipitation with TACE or iRhom2 and immunoblotting with phospho-Ser or with phospho-Thr. Phosphorylation of TACE was observed on both a serine and threonine after LPS stimulation, but phosphorylation of the endoplasmic reticulum-associated pseudoprotease iRhom2 was observed only on a serine after LPS stimulation. D, to provide evidence that the phosphorylation of TACE and iRhom2 could be PKG-dependent, cells were incubated with a PKG inhibitor (inhib.) (20 μm for at least 30 min) before LPS treatment. Phosphorylation of TACE and iRhom2 is likely to be dependent on the cGMP/PKG pathway as addition of the PKG inhibitor before LPS treatment blocked the phosphorylation of TACE and iRhom2. E, to further provide evidence that cGMP-activated PKG phosphorylated TACE and iRhom2, cells were treated as described above followed by cross-linking, immunoprecipitation, and immunoblotting.
iNOS/NO/cGMP/PKG Are Required for the Phosphorylation of TACE and iRhom2
It is known that phosphorylation of TACE occurs at a serine and a threonine (51–55). We assessed whether TACE and iRhom2 are phosphorylated in hepatocytes under our experimental conditions. As shown in Fig. 7C, iNOS-generated NO leads to the phosphorylation of TACE and iRhom2 in LPS-treated hepatocytes.
To test the hypothesis that PKG is involved in the phosphorylation of TACE and iRhom2, we performed TACE and iRhom2 immunoprecipitation following treatment with LPS (in the presence or absence of a PKG inhibitor) and cGMP (in the presence or absence of a PKG inhibitor). Because PKG is a serine/threonine kinase, we performed immunoprecipitation using antibodies to phosphoserine and phosphothreonine (phospho-Ser and phospho-Thr, respectively). As shown in Fig. 7, D and E, both LPS and cGMP exposure leads to the phosphorylation of TACE and iRhom2. The phosphorylation of TACE was observed on a serine and a threonine, whereas the phosphorylation of iRhom2 was observed only on a serine. The phosphorylation of both TACE and iRhom2 resulting from LPS or cGMP exposure was PKG-dependent.
Fig. 8 is our proposed model of TLR4/iNOS/NO/cGMP/PKG-induced TACE phosphorylation and translocation to the cell surface: (i) LPS induces iNOS expression through a TLR4-dependent mechanism. (ii) iNOS enzyme generates NO, leading to the production of cGMP by soluble guanylyl cyclase. (iii) cGMP activates PKG. (iv) PKG phosphorylates TACE; TACE can undergo a conformational change, which can serve as a substrate for iRhom2; and the phosphorylation of iRhom2 can facilitate the translocation to the cell surface to cleave membrane TNFR1.
FIGURE 8.
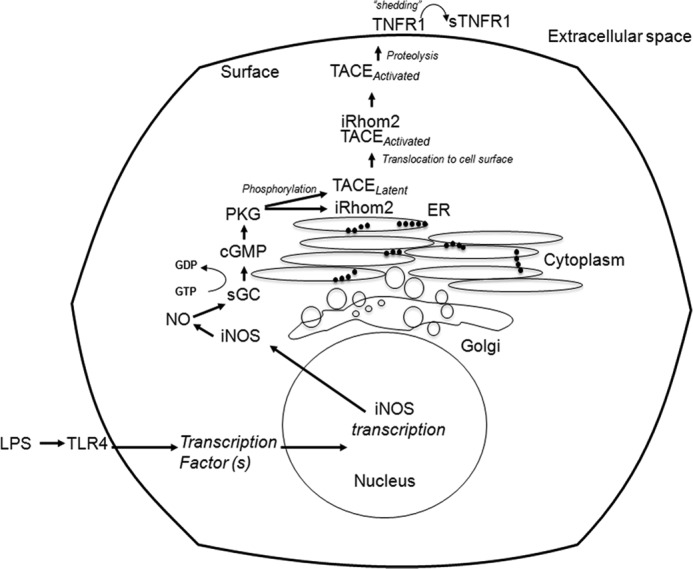
Schematic diagram of our proposed model of PKG-induced TACE phosphorylation and translocation to the cell surface. (i) LPS induces iNOS expression through a TLR4-dependent mechanism. (ii) iNOS enzyme generates NO, leading to the production of cGMP by soluble guanylyl cyclase (sGC). (iii) cGMP activates PKG. (iv) PKG phosphorylates TACE and iRhom2 (both are endoplasmic reticulum (ER)-associated enzymes), and phosphorylated TACE undergoes a conformational change, which can serve as a substrate for iRhom2. The phosphorylation of iRhom2 can facilitate the translocation of TACE to the cell surface to cleave membrane TNFR1.
DISCUSSION
Shedding of the TNFR1 is thought to limit the effects of TNFα by both binding soluble TNFα and limiting TNFα recognition by cells. Expression of iNOS can limit TNFα-mediated toxicity in hepatocytes; therefore, we carried out studies to examine the effects of iNOS and NO on TNFR1 shedding in LPS-treated hepatocytes. Our studies show that LPS induces TNFR1 shedding in a TLR4-, iNOS-, and NO-dependent manner. TNFR1 shedding was dependent on TACE activation and translocation to the cell surface, which in turn were dependent on cGMP and PKG. The activation and translocation of TACE was associated with a cGMP/PKG-dependent phosphorylation and interaction of both TACE and iRhom2. These observations identify a novel iNOS/NO/cGMP/PKG-dependent pathway in hepatocytes for the activation and translocation of TACE involving iRhom2.
The metalloproteinase enzyme TACE/ADAM17 is an α-secretase that is known to play an important role in the cleavage of several membrane receptors, such as those for TNFα, IL-6, and EGF (27). The mechanism by which TACE recognizes and cleaves specific membrane receptors is not fully understood but may be cell type- and model-dependent (27). It has been previously shown that TACE glycosylation and phosphorylation may be important in either the localization of TACE to the membrane (30) or its activation (51–54). In our model of endotoxemia, cGMP and PKG activation were important for localization of TACE to the cell membrane, leading to TNFR1 shedding. We provide evidence that phosphorylation of TACE and iRhom2 is driven through the kinase activity of PKG. The cytoplasmic tail of TACE contains serine, threonine, and tyrosine residues that represent potential phosphorylation sites, and phosphorylation of one or more of these sites has been observed to occur in response to a wide range of stimuli, including LPS (32). Other studies have shown that TACE can be phosphorylated by kinases, such as p38 MAPK. This kinase has been found to phosphorylate TACE in lymphocytes and monocytes (62). Phosphorylation of the cytoplasmic domain of TACE can occur not only on a threonine but also on a serine (55–58). We demonstrate that PKG is involved in the serine and threonine phosphorylation of TACE in response to NO and cGMP in hepatocytes.
Recently, iRhom2 was found to facilitate the translocation of TACE to the cell surface in monocytes for the maturation of TNFα (34, 35). To our knowledge, our study is the first to show that iRhom2 plays an important role in TNFR1 regulation and processing but more importantly that TACE and iRhom2 are targets for phosphorylation by PKG. We extend these previous observations to show that iRhom2 is expressed and rapidly up-regulated in hepatocytes exposed to LPS. We implicate iRhom2 in TNFR1 shedding by showing the interaction between TACE and iRhom2 in response to iNOS, NO, and cGMP/PKG. There is some evidence that reactive oxygen species may be required to activate TACE (59). It is thought that endothelial TACE is activated by mitochondrial reactive oxygen species in response to changes in calcium levels. In other models and in CHO or HL-60 cells, TACE activation has been shown to be ATP- (60), calcium-, or mitogen-activated protein kinase-dependent (61–63). Nitrosylation of TACE in macrophages has been shown to be involved in TACE activation for TNFα maturation (33). Altogether, these data suggest that activation of TACE may also vary in a cell type- and stimulus-dependent manner. Our study clearly implicates iNOS/NO signaling and cGMP/PKG activity in TACE activation and localization to the cell surface to cause TNFR1 shedding from hepatocytes, and we show that this pathway is active both in the liver in vivo in our model of endotoxemia and in LPS-stimulated hepatocytes. This mechanism of TACE activation could be important in the regulation of liver cell damage in the setting of sustained TNFα production. Combined with a previous report (33), this raises the possibility that NO can regulate TACE localization and activation by multiple mechanisms. Our results also raise the possibility that targeting iNOS, cGMP, PKG, or iRhom2 in the setting of acute inflammation could have the unwanted consequence of preventing TNFR1 shedding.
LPS (or endotoxin) is an important pathogen-associated molecular pattern that is found on the surface of Gram-negative bacteria. Therefore, LPS is an important mediator of inflammation during Gram-negative sepsis. Endotoxin can also be released into the circulation in situations where the gut barrier becomes compromised, such as during septic shock (64). The liver is therefore a frontline organ that recognizes and clears LPS from the circulation (65) and therefore is also an organ that can be easily damaged through excessive inflammation and cell death during sepsis. We have previously shown that TLR4 complex on hepatocytes is important for LPS uptake by these cells (66). We now extend these previous observations to show that hepatocyte TLR4 is required for LPS-induced iNOS up-regulation leading to TNFR1 shedding. We did not elucidate the postreceptor signaling pathways involved in iNOS up-regulation but do provide evidence that NO is part of a feed forward mechanism for iNOS expression in this pathway. Inhibition of iNOS activity markedly suppressed iNOS expression. The mechanisms involved in this NO-dependent iNOS up-regulation are unknown. Hepatocytes have cellular mechanisms to help protect them from excessive inflammation induced by LPS, including the up-regulation of iNOS (67, 68). LPS signaling through TLR4 is known to increase proinflammatory cytokine production, including TNFα, from inflammatory cells, such as macrophages. LPS/TLR4 signaling can also up-regulate iNOS production in hepatocytes, and we have shown in this study that TACE activation is TLR4-dependent. TLRs have been implicated in TACE activation previously (69), but our study now provides a definitive mechanism for TLR-mediated TACE activation to increase TNFR1 release and provide cellular protection.
The mechanisms by which iNOS/NO prevent cell death in hepatocytes include cGMP-dependent and -independent pathways. The cGMP-independent pathways involve nitrosylation and inhibition of caspase-3 and -8 (39, 70, 71), whereas the cGMP-dependent pathway leads to the down-regulation of the proapoptotic protein BNIP3 in hepatocytes (72). Cyclic nucleotides can also block death-inducing signaling complex formation in response to TNFα in hepatocytes (19, 73). Results from this study suggest that rapid TNFR1 shedding through iNOS/NO/cGMP/PKG-mediated TACE activation is one mechanism to limit cell death by blocking death-inducing signaling complex formation.
Acknowledgments
We thank Debra Williams, Danielle Reiser, Carol Meiers, and Amanda Chipman for expert technical advice and/or administrative assistance. We are grateful to Dr. Qian Sun for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R37-GM-044100 and R01-GM-050441.
- TNF
- tumor necrosis factor
- TACE
- TNFα-converting enzyme
- ADAM
- a disintegrin and metalloproteinase
- iNOS
- inducible nitric-oxide synthase
- iRhom2
- inactive Rhomboid 2
- TNFR
- TNF receptor
- PKG
- protein kinase G
- SNAP
- S-nitroso-N-acetyl-dl-penicillamine
- ODQ
- 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- 8-Br-cGMP
- 8-bromoguanosine 3′,5′-cyclic monophosphate
- TLR
- Toll-like receptor
- sTNFR1
- soluble TNFR1.
REFERENCES
- 1. Leon L. R., White A. A., Kluger M. J. (1998) Role of IL-6 and TNF in thermoregulation and survival during sepsis in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 275, R269–R277 [DOI] [PubMed] [Google Scholar]
- 2. Tracey K. J. (1991) Tumor necrosis factor (cachectin) in the biology of septic shock syndrome. Circ. Shock 35, 123–128 [PubMed] [Google Scholar]
- 3. Tracey K. J., Lowry S. F., Cerami A. (1988) The pathophysiologic role of cachectin/TNF in septic shock and cachexia. Ann. Inst. Pasteur Immunol. 139, 311–317 [DOI] [PubMed] [Google Scholar]
- 4. Leist M., Gantner F., Bohlinger I., Tiegs G., Germann P. G., Wendel A. (1995) Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am. J. Pathol. 146, 1220–1234 [PMC free article] [PubMed] [Google Scholar]
- 5. Rüdiger H. A., Clavien P. A. (2002) Tumor necrosis factor α, but not Fas, mediates hepatocellular apoptosis in the murine ischemic liver. Gastroenterology 122, 202–210 [DOI] [PubMed] [Google Scholar]
- 6. Smith C. A., Farrah T., Goodwin R. G. (1994) The TNF receptor superfamily of cellular and viral proteins: activation, costimulation, and death. Cell 76, 959–962 [DOI] [PubMed] [Google Scholar]
- 7. Eum H. A., Vallabhaneni R., Wang Y., Loughran P. A., Stolz D. B., Billiar T. R. (2011) Characterization of DISC formation and TNFR1 translocation to mitochondria in TNF-α-treated hepatocytes. Am. J. Pathol. 179, 1221–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eum H. A., Billiar T. R. (2011) TNF/TNF receptor 1-mediated apoptosis in hepatocytes. Adv. Exp. Med. Biol. 691, 617–624 [DOI] [PubMed] [Google Scholar]
- 9. Yamada Y., Webber E. M., Kirillova I., Peschon J. J., Fausto N. (1998) Analysis of liver regeneration in mice lacking type 1 or type 2 tumor necrosis factor receptor: requirement for type 1 but not type 2 receptor. Hepatology 28, 959–970 [DOI] [PubMed] [Google Scholar]
- 10. Cabal-Hierro L., Lazo P. S. (2012) Signal transduction by tumor necrosis factor receptors. Cell. Signal. 24, 1297–1305 [DOI] [PubMed] [Google Scholar]
- 11. Takada Y., Aggarwal B. B. (2003) Genetic deletion of the tumor necrosis factor receptor p60 or p80 sensitizes macrophages to lipopolysaccharide-induced nuclear factor-κB, mitogen-activated protein kinases, and apoptosis. J. Biol. Chem. 278, 23390–23397 [DOI] [PubMed] [Google Scholar]
- 12. Grell M., Wajant H., Zimmermann G., Scheurich P. (1998) The type 1 receptor (CD120a) is the high affinity receptor for soluble tumor necrosis factor. Proc. Natl. Acad. Sci. U.S.A. 95, 570–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bazzoni F., Beutler B. (1996) The tumor necrosis factor ligand and receptor families. N. Engl. J. Med. 334, 1717–1725 [DOI] [PubMed] [Google Scholar]
- 14. Hsu H., Xiong J., Goeddel D. V. (1995) The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell 81, 495–504 [DOI] [PubMed] [Google Scholar]
- 15. Hsu H., Shu H. B., Pan M. G., Goeddel D. V. (1996) TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 84, 299–308 [DOI] [PubMed] [Google Scholar]
- 16. Liu Z. G., Hsu H., Goeddel D. V., Karin M. (1996) Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell 87, 565–576 [DOI] [PubMed] [Google Scholar]
- 17. Costelli P., Aoki P., Zingaro B., Carbó N., Reffo P., Lopez-Soriano F. J., Bonelli G., Argilés J. M., Baccino F. M. (2003) Mice lacking TNFα receptors 1 and 2 are resistant to death and fulminant liver injury induced by agonistic anti-Fas antibody. Cell Death Differ. 10, 997–1004 [DOI] [PubMed] [Google Scholar]
- 18. Kim P. K., Wang Y., Gambotto A., Kim Y. M., Weller R., Zuckerbraun B. S., Hua Y., Watkins S. C., Billiar T. R. (2002) Hepatocyte Fas-associating death domain protein/mediator of receptor-induced toxicity (FADD/MORT1) levels increase in response to pro-apoptotic stimuli. J. Biol. Chem. 277, 38855–38862 [DOI] [PubMed] [Google Scholar]
- 19. Wang Y., Kim P. K., Peng X., Loughran P., Vodovotz Y., Zhang B., Billiar T. R. (2006) Cyclic AMP and cyclic GMP suppress TNFα-induced hepatocyte apoptosis by inhibiting FADD up-regulation via a protein kinase A-dependent pathway. Apoptosis 11, 441–451 [DOI] [PubMed] [Google Scholar]
- 20. Garton K. J., Gough P. J., Raines E. W. (2006) Emerging roles for ectodomain shedding in the regulation of inflammatory responses. J. Leukoc. Biol. 79, 1105–1116 [DOI] [PubMed] [Google Scholar]
- 21. Murthy A., Defamie V., Smookler D. S., Di Grappa M. A., Horiuchi K., Federici M., Sibilia M., Blobel C. P., Khokha R. (2010) Ectodomain shedding of EGFR ligands and TNFR1 dictates hepatocyte apoptosis during fulminant hepatitis in mice. J. Clin. Investig. 120, 2731–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fernandez-Botran R., Chilton P. M., Ma Y. (1996) Soluble cytokine receptors: their roles in immunoregulation, disease, and therapy. Adv. Immunol. 63, 269–336 [DOI] [PubMed] [Google Scholar]
- 23. Mohler K. M., Torrance D. S., Smith C. A., Goodwin R. G., Stremler K. E., Fung V. P., Madani H., Widmer M. B. (1993) Soluble tumor necrosis factor (TNF) receptors are effective therapeutic agents in lethal endotoxemia and function simultaneously as both TNF carriers and TNF antagonists. J. Immunol. 151, 1548–1561 [PubMed] [Google Scholar]
- 24. Hawari F. I., Rouhani F. N., Cui X., Yu Z. X., Buckley C., Kaler M., Levine S. J. (2004) Release of full-length 55-kDa TNF receptor 1 in exosome-like vesicles: a mechanism for generation of soluble cytokine receptors. Proc. Natl. Acad. Sci. U.S.A. 101, 1297–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Iglesias J., Marik P. E., Levine J. S. (2003) Elevated serum levels of the type I and type II receptors for tumor necrosis factor-α as predictive factors for ARF in patients with septic shock. Am. J. Kidney Dis. 41, 62–75 [DOI] [PubMed] [Google Scholar]
- 26. Islam A., Adamik B., Hawari F. I., Ma G., Rouhani F. N., Zhang J., Levine S. J. (2006) Extracellular TNFR1 release requires the calcium-dependent formation of a nucleobindin 2-ARTS-1 complex. J. Biol. Chem. 281, 6860–6873 [DOI] [PubMed] [Google Scholar]
- 27. Refsum S. E., Halliday M. I., Campbell G., McCaigue M., Rowlands B. J., Boston V. E. (1996) Modulation of TNFα and IL-6 in a peritonitis model using pentoxifylline. J. Pediatr. Surg. 31, 928–930 [DOI] [PubMed] [Google Scholar]
- 28. Xanthoulea S., Pasparakis M., Kousteni S., Brakebusch C., Wallach D., Bauer J., Lassmann H., Kollias G. (2004) Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J. Exp. Med. 200, 367–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Doedens J. R., Mahimkar R. M., Black R. A. (2003) TACE/ADAM-17 enzymatic activity is increased in response to cellular stimulation. Biochem. Biophys. Res. Commun. 308, 331–338 [DOI] [PubMed] [Google Scholar]
- 30. Reddy P., Slack J. L., Davis R., Cerretti D. P., Kozlosky C. J., Blanton R. A., Shows D., Peschon J. J., Black R. A. (2000) Functional analysis of the domain structure of tumor necrosis factor-α converting enzyme. J. Biol. Chem. 275, 14608–14614 [DOI] [PubMed] [Google Scholar]
- 31. Black R. A. (2002) Tumor necrosis factor-α converting enzyme. Int. J. Biochem. Cell Biol. 34, 1–5 [DOI] [PubMed] [Google Scholar]
- 32. Santos D. O., Lorré K., de Boer M., Van Heuverswyn H. (1999) Shedding of soluble receptor for tumor necrosis factor α induced by M. leprae or LPS from human mononuclear cells. Nihon Hansenbyo Gakkai Zasshi 68, 185–193 [DOI] [PubMed] [Google Scholar]
- 33. Zhang Z., Kolls J. K., Oliver P., Good D., Schwarzenberger P. O., Joshi M. S., Ponthier J. L., Lancaster J. R., Jr. (2000) Activation of tumor necrosis factor-α-converting enzyme-mediated ectodomain shedding by nitric oxide. J. Biol. Chem. 275, 15839–15844 [DOI] [PubMed] [Google Scholar]
- 34. McIlwain D. R., Lang P. A., Maretzky T., Hamada K., Ohishi K., Maney S. K., Berger T., Murthy A., Duncan G., Xu H. C., Lang K. S., Häussinger D., Wakeham A., Itie-Youten A., Khokha R., Ohashi P. S., Blobel C. P., Mak T. W. (2012) iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science 335, 229–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Adrain C., Zettl M., Christova Y., Taylor N., Freeman M. (2012) Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 335, 225–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Seglen P. O., Solheim A. E. (1977) Protein degradation in isolated rat hepatocytes. Acta Biol. Med. Ger. 36, 1789–1804 [PubMed] [Google Scholar]
- 37. Oberholzer A., Oberholzer C., Bahjat F. R., Edwards C. K., 3rd, Moldawer L. L. (2001) Genetic determinants of lipopolysaccharide and d-galactosamine-mediated hepatocellular apoptosis and lethality. J. Endotoxin. Res. 7, 375–380 [PubMed] [Google Scholar]
- 38. Hotchkiss R. S., Strasser A., McDunn J. E., Swanson P. E. (2009) Cell death. N. Engl. J. Med. 361, 1570–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim Y. M., Kim T. H., Chung H. T., Talanian R. V., Yin X. M., Billiar T. R. (2000) Nitric oxide prevents tumor necrosis factor α-induced rat hepatocyte apoptosis by the interruption of mitochondrial apoptotic signaling through S-nitrosylation of caspase-8. Hepatology 32, 770–778 [DOI] [PubMed] [Google Scholar]
- 40. Peiretti F., Canault M., Deprez-Beauclair P., Berthet V., Bonardo B., Juhan-Vague I., Nalbone G. (2003) Intracellular maturation and transport of tumor necrosis factor α converting enzyme. Exp. Cell Res. 285, 278–285 [DOI] [PubMed] [Google Scholar]
- 41. Hinkle C. L., Sunnarborg S. W., Loiselle D., Parker C. E., Stevenson M., Russell W. E., Lee D. C. (2004) Selective roles for tumor necrosis factor α-converting enzyme/ADAM17 in the shedding of the epidermal growth factor receptor ligand family: the juxtamembrane stalk determines cleavage efficiency. J. Biol. Chem. 279, 24179–24188 [DOI] [PubMed] [Google Scholar]
- 42. Srour N., Lebel A., McMahon S., Fournier I., Fugère M., Day R., Dubois C. M. (2003) TACE/ADAM-17 maturation and activation of sheddase activity require proprotein convertase activity. FEBS Lett. 554, 275–283 [DOI] [PubMed] [Google Scholar]
- 43. Endres K., Anders A., Kojro E., Gilbert S., Fahrenholz F., Postina R. (2003) Tumor necrosis factor-α converting enzyme is processed by proprotein-convertases to its mature form which is degraded upon phorbol ester stimulation. Eur. J. Biochem. 270, 2386–2393 [DOI] [PubMed] [Google Scholar]
- 44. Xu P., Derynck R. (2010) Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol. Cell 37, 551–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Podbilewicz B. (2004) Sweet control of cell migration, cytokinesis and organogenesis. Nat. Cell Biol. 6, 9–11 [DOI] [PubMed] [Google Scholar]
- 46. Nishiwaki K., Kubota Y., Chigira Y., Roy S. K., Suzuki M., Schvarzstein M., Jigami Y., Hisamoto N., Matsumoto K. (2004) An NDPase links ADAM protease glycosylation with organ morphogenesis in C. elegans. Nat. Cell Biol. 6, 31–37 [DOI] [PubMed] [Google Scholar]
- 47. Kubota Y., Sano M., Goda S., Suzuki N., Nishiwaki K. (2006) The conserved oligomeric Golgi complex acts in organ morphogenesis via glycosylation of an ADAM protease in C. elegans. Development 133, 263–273 [DOI] [PubMed] [Google Scholar]
- 48. Billiar T. R., Curran R. D., Harbrecht B. G., Stadler J., Williams D. L., Ochoa J. B., Di Silvio M., Simmons R. L., Murray S. A. (1992) Association between synthesis and release of cGMP and nitric oxide biosynthesis by hepatocytes. Am. J. Physiol. Cell Physiol. 262, C1077–C1082 [DOI] [PubMed] [Google Scholar]
- 49. Rangaswami H., Schwappacher R., Marathe N., Zhuang S., Casteel D. E., Haas B., Chen Y., Pfeifer A., Kato H., Shattil S., Boss G. R., Pilz R. B. (2010) Cyclic GMP and protein kinase G control a Src-containing mechanosome in osteoblasts. Sci. Signal. 3, ra91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Marathe N., Rangaswami H., Zhuang S., Boss G. R., Pilz R. B. (2012) Pro-survival effects of 17β-estradiol on osteocytes are mediated by nitric oxide/cGMP via differential actions of cGMP-dependent protein kinases I and II. J. Biol. Chem. 287, 978–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Black R. A., Rauch C. T., Kozlosky C. J., Peschon J. J., Slack J. L., Wolfson M. F., Castner B. J., Stocking K. L., Reddy P., Srinivasan S., Nelson N., Boiani N., Schooley K. A., Gerhart M., Davis R., Fitzner J. N., Johnson R. S., Paxton R. J., March C. J., Cerretti D. P. (1997) A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 385, 729–733 [DOI] [PubMed] [Google Scholar]
- 52. Doedens J. R., Black R. A. (2000) Stimulation-induced down-regulation of tumor necrosis factor-α converting enzyme. J. Biol. Chem. 275, 14598–14607 [DOI] [PubMed] [Google Scholar]
- 53. Primakoff P., Myles D. G. (2000) The ADAM gene family: surface proteins with adhesion and protease activity. Trends Genet. 16, 83–87 [DOI] [PubMed] [Google Scholar]
- 54. Wolfsberg T. G., Straight P. D., Gerena R. L., Huovila A. P., Primakoff P., Myles D. G., White J. M. (1995) ADAM, a widely distributed and developmentally regulated gene family encoding membrane proteins with a disintegrin and metalloprotease domain. Dev. Biol. 169, 378–383 [DOI] [PubMed] [Google Scholar]
- 55. Díaz-Rodríguez E., Montero J. C., Esparís-Ogando A., Yuste L., Pandiella A. (2002) Extracellular signal-regulated kinase phosphorylates tumor necrosis factor α-converting enzyme at threonine 735: a potential role in regulated shedding. Mol. Biol. Cell 13, 2031–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fan H., Turck C. W., Derynck R. (2003) Characterization of growth factor-induced serine phosphorylation of tumor necrosis factor-α converting enzyme and of an alternatively translated polypeptide. J. Biol. Chem. 278, 18617–18627 [DOI] [PubMed] [Google Scholar]
- 57. Lemjabbar-Alaoui H., Sidhu S. S., Mengistab A., Gallup M., Basbaum C. (2011) TACE/ADAM-17 phosphorylation by PKC-ϵ mediates premalignant changes in tobacco smoke-exposed lung cells. PLoS One 6, e17489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang Q., Thomas S. M., Lui V. W., Xi S., Siegfried J. M., Fan H., Smithgall T. E., Mills G. B., Grandis J. R. (2006) Phosphorylation of TNF-α converting enzyme by gastrin-releasing peptide induces amphiregulin release and EGF receptor activation. Proc. Natl. Acad. Sci. U.S.A. 103, 6901–6906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dada L. A., Sznajder J. I. (2011) Mitochondrial Ca2+ and ROS take center stage to orchestrate TNF-α-mediated inflammatory responses. J. Clin. Investig. 121, 1683–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Myers T. J., Brennaman L. H., Stevenson M., Higashiyama S., Russell W. E., Lee D. C., Sunnarborg S. W. (2009) Mitochondrial reactive oxygen species mediate GPCR-induced TACE/ADAM17-dependent transforming growth factor-α shedding. Mol. Biol. Cell 20, 5236–5249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rowlands D. J., Islam M. N., Das S. R., Huertas A., Quadri S. K., Horiuchi K., Inamdar N., Emin M. T., Lindert J., Ten V. S., Bhattacharya S., Bhattacharya J. (2011) Activation of TNFR1 ectodomain shedding by mitochondrial Ca2+ determines the severity of inflammation in mouse lung microvessels. J. Clin. Investig. 121, 1986–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Scott A. J., O'Dea K. P., O'Callaghan D., Williams L., Dokpesi J. O., Tatton L., Handy J. M., Hogg P. J., Takata M. (2011) Reactive oxygen species and p38 mitogen-activated protein kinase mediate tumor necrosis factor α-converting enzyme (TACE/ADAM-17) activation in primary human monocytes. J. Biol. Chem. 286, 35466–35476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Soond S. M., Everson B., Riches D. W., Murphy G. (2005) ERK-mediated phosphorylation of Thr735 in TNFα-converting enzyme and its potential role in TACE protein trafficking. J. Cell Sci. 118, 2371–2380 [DOI] [PubMed] [Google Scholar]
- 64. Swank G. M., Deitch E. A. (1996) Role of the gut in multiple organ failure: bacterial translocation and permeability changes. World J. Surg. 20, 411–417 [DOI] [PubMed] [Google Scholar]
- 65. Scott M. J., Liu S., Shapiro R. A., Vodovotz Y., Billiar T. R. (2009) Endotoxin uptake in mouse liver is blocked by endotoxin pretreatment through a suppressor of cytokine signaling-1-dependent mechanism. Hepatology 49, 1695–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Scott M. J., Billiar T. R. (2008) β2-Integrin-induced p38 MAPK activation is a key mediator in the CD14/TLR4/MD2-dependent uptake of lipopolysaccharide by hepatocytes. J. Biol. Chem. 283, 29433–29446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Billiar T. R., Curran R. D., Harbrecht B. G., Stuehr D. J., Demetris A. J., Simmons R. L. (1990) Modulation of nitrogen oxide synthesis in vivo: NG-monomethyl-l-arginine inhibits endotoxin-induced nitrate/nitrate biosynthesis while promoting hepatic damage. J. Leukoc. Biol. 48, 565–569 [DOI] [PubMed] [Google Scholar]
- 68. Tzeng E., Billiar T. R., Williams D. L., Li J., Lizonova A., Kovesdi I., Kim Y. M. (1998) Adenovirus-mediated inducible nitric oxide synthase gene transfer inhibits hepatocyte apoptosis. Surgery 124, 278–283 [PubMed] [Google Scholar]
- 69. Koff J. L., Shao M. X., Ueki I. F., Nadel J. A. (2008) Multiple TLRs activate EGFR via a signaling cascade to produce innate immune responses in airway epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L1068–L1075 [DOI] [PubMed] [Google Scholar]
- 70. Kim Y. M., Talanian R. V., Billiar T. R. (1997) Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J. Biol. Chem. 272, 31138–31148 [DOI] [PubMed] [Google Scholar]
- 71. Li J., Yang S., Billiar T.R. (2000) Cyclic nucleotides suppress tumor necrosis factor α-mediated apoptosis by inhibiting caspase activation and cytochrome c release in primary hepatocytes via a mechanism independent of Akt activation. J. Biol. Chem. 275, 13026–13034 [DOI] [PubMed] [Google Scholar]
- 72. Zamora R., Alarcon L., Vodovotz Y., Betten B., Kim P. K., Gibson K. F., Billiar T. R. (2001) Nitric oxide suppresses the expression of Bcl-2 binding protein BNIP3 in hepatocytes. J. Biol. Chem. 276, 46887–46895 [DOI] [PubMed] [Google Scholar]
- 73. Bhattacharjee R., Xiang W., Wang Y., Zhang X., Billiar T. R. (2012) cAMP prevents TNF-induced apoptosis through inhibiting DISC complex formation in rat hepatocytes. Biochem. Biophys. Res. Commun. 423, 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]