

Background: The mechanism of motor neuron degeneration in amyotrophic lateral sclerosis (ALS) is poorly understood.
Results: The peroxisome proliferator-activated receptor γ (PPARγ) can be activated by lipid peroxidation metabolites in ALS motor neurons, and this can prompt the expression of antioxidant enzymes.
Conclusion: PPARγ can exert a direct protective effect in ALS motor neurons.
Significance: PPARγ transcriptional co-activators may represent therapeutic targets in ALS.
Keywords: Amyotropic Lateral Sclerosis (Lou Gehrig's Disease), Lipid Peroxidation, Neurodegeneration, Oxidative Stress, Peroxisome Proliferator-activated Receptor (PPAR)
Abstract
Recent evidence highlights the peroxisome proliferator-activated receptors (PPARs) as critical neuroprotective factors in several neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS). To gain new mechanistic insights into the role of these receptors in the context of ALS, here we investigated how PPAR transcriptional activity varies in hSOD1G93A ALS transgenic mice. We demonstrate that PPARγ-driven transcription selectively increases in the spinal cord of symptomatic hSOD1G93A mice. This phenomenon correlates with the up-regulation of target genes, such as lipoprotein lipase and glutathione S-transferase α-2, which are implicated in scavenging lipid peroxidation by-products. Such events are associated with enhanced PPARγ immunoreactivity within motor neuronal nuclei. This observation, and the fact that PPARγ displays increased responsiveness in cultured hSOD1G93A motor neurons, points to a role for this receptor in neutralizing deleterious lipoperoxidation derivatives within the motor cells. Consistently, in both motor neuron-like cultures and animal models, we report that PPARγ is activated by lipid peroxidation end products, such as 4-hydroxynonenal, whose levels are elevated in the cerebrospinal fluid and spinal cord from ALS patients. We propose that the accumulation of critical concentrations of lipid peroxidation adducts during ALS progression leads to the activation of PPARγ in motor neurons. This in turn triggers self-protective mechanisms that involve the up-regulation of lipid detoxification enzymes, such as lipoprotein lipase and glutathione S-transferase α-2. Our findings indicate that anticipating natural protective reactions by pharmacologically modulating PPARγ transcriptional activity may attenuate neurodegeneration by limiting the damage induced by lipid peroxidation derivatives.
Introduction
Amyotrophic lateral sclerosis (ALS) is the most common motor neuron disorder of the adult life. In 90–95% of clinical cases the disease occurs with no apparent genetic linkage, whereas in the remaining 5–10% of cases ALS is inherited, usually in an autosomal dominant manner (familial ALS, fALS). Both forms of the disease cause degeneration of motor neurons, leading to paralysis and death within 3–5 years from the appearance of the first symptoms. The discovery that mutations in the gene coding for the enzyme Cu,Zn-superoxide dismutase (SOD1)3 are the most common cause of fALS (20%) prompted the generation of transgenic animal models, which allowed investigations into disease pathogenesis (Refs. 1–4; for review see Ref. 5). Currently, there is no pharmacological intervention or cure to halt the progression of this disorder. The sole FDA approved drug for the treatment of ALS, i.e. riluzole, is effective in extending the patient lifespan only by 3–6 months (6). Thus, the identification of novel pharmacological targets and strategies for therapeutic intervention in ALS is highly desirable. A class of signaling molecules, whose relevance for neurodegenerative diseases has become lately evident, is represented by the peroxisome proliferator-activated receptors (PPARs), a family of nuclear receptors that includes three members, i.e. PPARα, -β/δ, and -γ (7, 8).
In the central nervous system (CNS), PPARs are expressed by neurons as well as glial cells (9), and can be activated by natural lipids and/or their metabolites, including oxidized fatty acids (10). Growing evidence indicates that activation of PPARs, particularly PPARγ, effectively attenuates neurodegeneration and disease progression in animal models of several neurodegenerative disorders (8). However, the specific mechanisms by which these receptors positively affect the course of these diseases have not been elucidated. The beneficial effects of the PPARγ agonist pioglitazone, described in early pharmacological studies on ALS transgenic mice, were mainly ascribed to the receptor anti-inflammatory activity (11, 12). PPARs can in fact control several aspects of the inflammatory response by repressing the expression of inflammatory genes through interaction with other transcription factors, such as the nuclear factor-κB and the activator protein 1 (7). However, PPARs can modulate gene expression also directly by binding, as heterodimers with the retinoid X receptors, to specific DNA response elements (PPREs) in the promoter or enhancer regions of target genes. By this means, they exert several homeostatic functions that are crucial for the activity and survival of neural cells (7). Among the functions fulfilled by the PPARs, there is, for example, the capacity to control the expression of enzymes that are critically involved in detoxifying neurons from a wide variety of stress stimuli (13). These PPAR-dependent self-protective mechanisms appear to be relevant for survival of ALS motor neurons. Indeed, expression of PGC-1α, a transcriptional co-activator of PPARγ, was recently described to prevent mutant SOD1-induced neuronal cell death in vitro (14). Furthermore, both ubiquitous and neuron-specific expression of PGC-1α was reported to reduce motor neuron degeneration and alleviate the disease outcome in transgenic mice carrying the G93A mutant form of human SOD1 (hSOD1G93A) (15, 16). Altogether, these observations strongly suggest that fine-tuning PPAR transcriptional activity within the CNS may represent an attractive approach to limit the progression of ALS.
To gain further insights into PPAR functions during the progression of the disease, here we bred mice genetically engineered to highlight PPAR transcriptional activity (PPRE-Luc mice; Ref. 17) with the hSOD1G93AALS mouse line (2). We demonstrate that transcriptional activity of PPARs significantly increases in the affected spinal cord at the symptomatic stage of the disease, a phenomenon that takes place in strict temporal and spatial correlation with degeneration of motor neurons. Furthermore, analyses of isoform-specific target genes and cellular distribution indicate that this peculiar process is driven by motor neuronal PPARγ, possibly through its activation by lipid peroxidation by-products that are generated during the course of the disease.
EXPERIMENTAL PROCEDURES
Transgenic Mice
Transgenic mice expressing the human SOD1G93A (B6SJL-TgN(SOD1-G93A)1Gur) (2) were purchased from The Jackson Laboratories. The colonies were maintained by breeding hemizygote males to wild-type C57Bl6/SJL hybrid females (Charles River Laboratories). Mice genetically engineered to ubiquitously express the luciferase reporter gene under control of the PPAR-responsive elements (hereafter referred to as “PPRE-Luc mice”) were previously described (17). PPRE-Luc+/−; hSOD1G93A+/− double transgenic mice and their PPRE-Luc+/−; hSOD1G93A−/− littermates were obtained by cross-breeding hemizygote hSOD1G93A males with homozygous PPRE-Luc females. The progeny was screened for the presence of the two transgenes using primers 5′-SOD1 (5′-CATCAGCCCTAATCCATCTGA) and 3′-SOD1 (5′-CGCGACTAACAATCAAAGTGA) to detect the hSOD1G93A allele, and 5′-PPRE (5′-GGCAGAAGCTATGAAACGAT) and 3′-PPRE (5′-CGACTGAAATCCCTGGTAAT) to amplify the PPRE-Luc sequence. Genomic DNA extracted from mouse tail lysates was used as DNA template. To reduce the gender-dependent phenotypic variability described in hSOD1G93A ALS mice (18), only animals of a single sex were analyzed in the present study. Females were selected because they show limited variations in PPAR transcriptional activity upon metabolic and hormonal events (17). Fasting experiments were performed by keeping mice for 48 h in the absence of food.
Intracerebroventricular injection of 4-hydroxynonenal (4-HNE) in anesthetized animals was carried out according to specific stereotaxic coordinates (Bregma, −0.25 mm; lateral, 1 mm; depth, 2.25 mm) using a Hamilton syringe rotated on the coronal plate of about 3 degrees from the orthogonal position, as previously described (19).
All experiments described in this study were conducted according to the Guidelines for Care and Use of Experimental Animals. Use of experimental animals was approved by the Italian Ministry of Research and University and controlled by a panel of experts of the Department of Pharmacological Sciences, University of Milan.
Luciferase Enzymatic Assay
Tissue extracts were prepared by homogenization in 200 μl of lysis buffer (100 mm KPO4, 1 mm DTT, 4 mm EGTA, 4 mm EDTA, pH 7.8), one freeze-thaw cycle, and 30 min of centrifugation at 4900 × g (Rotanta 460R, Hettich Zentrifugen). Supernatants containing luciferase were collected and total protein concentration was determined by the Bradford assay (20). Luciferase enzymatic activity in tissue extracts was then assayed with a luciferase assay buffer (470 μm luciferine, 20 mm Tricine, 0.1 mm EDTA, 1.07 mm (MgCO3)4·Mg(OH)2 × 5H2O; 2.67 mm MgSO4 × 7H2O in H2O, pH 7.8, with 33.3 mm DTT and 530 μm ATP). The light intensity was measured with a luminometer (Glomax, Promega) in a 10-s time period and expressed as relative light units/μg of protein.
Histological Analysis
Spines were taken and immersed in 4% buffered paraformaldehyde for 24 h. Spinal cords were extracted, the lumbar tract was removed and paraffin embedded. Serial sections of paraffin-embedded spinal cords were cut at 10 μm and used to perform different immunostainings.
On selected sections, the following immunohistochemical stainings were carried out: PPARγ (rabbit polyclonal antibody, 1:100; Affinity Bioreagents, Pierce), nonphosphorylated neurofilament H (SMI32, mouse monoclonal antibody, 1:500, Sternberger Monoclonals Inc.), and neuronal nuclei (mouse monoclonal antibody, 1:100, Chemicon) as neuronal markers, and glial fibrillary acidic protein (GFAP, mouse Alexa Fluor 488-conjugated monoclonal antibody, 1:1000, Chemicon) as astrocytic marker. Licopersicum esculentum lectin (tomato lectin, 1:200, Sigma) was used to stain microglia. Nuclei were labeled with Hoechst 33342 (Sigma). For quantitative analysis, lumbar spinal cord images (2084 × 1536 pixels) were captured using a ×40 objective on a Zeiss Axioskop 40 microscope equipped with a Nikon Coolpix 9900 digital camera.
The number of nuclei positively stained for PPARγ in the different cell types was determined on images from lumbar spinal cord sections double stained for PPARγ and SMI32, GFAP, or tomato lectin. The results were expressed as percentage of the total number of cells taken into consideration. The nuclear content of PPARγ in motor neurons, other neuronal subtypes, and non-neuronal cells was determined by immunofluorescence quantification on images from lumbar spinal cord and brain sections double stained for PPARγ and SMI32 or neuronal nuclei as follows. Fluorescence intensity of nuclear PPARγ immunostaining was measured in selected areas using ImageJ software (rsb.info.nih.gov/ij). Fluorescence intensity values were then averaged and expressed relative to mean fluorescence intensity obtained from background noise. Because Hoechst 33342 is a fluorescent dye that intercalates into the DNA, we considered its fluorescence intensity as a measure of the DNA content. Thus, the fluorescence intensity of PPARγ immunostaining was normalized to the intensity of Hoechst 33342.
For motor neuron quantification, serial sections were stained with 0.5% cresyl violet (Sigma) to detect motor neuron Nissl substance. Motor neurons were counted for a total of 9 disectors using an unbiased stereologic physical disector technique (21).
For morphometric analysis of skeletal muscles, soleus and tibialis anterior (TA) muscles from 30-day-old and 28-month-old C57Bl/6J mice were fixed in 4% paraformaldehyde for 24 h and subsequently embedded in paraffin. Serial sections of paraffin-embedded muscles were cut at 10 μm and stained for hematoxylin and eosin. The minimal “Feret's diameter” was determined using ImageJ software on a minimum of two randomly chosen fields (corresponding to ∼200 fibers) for each muscle and mouse (n = 3).
Plasmid Constructions
The construction of the PPRE5X-tk-Luc plasmid (hereafter referred to as PPRE-Luc transgene) carrying five tandem repeats of PPRE was previously described (17). The bicistronic pSOD1wt-IRES2-ZsGreen1 expression vector coding for both wild-type human SOD1 and the Zoanthus sp. green fluorescent protein (ZsGreen) under control of the human cytomegalovirus immediate early promoter was generated by inserting the SOD1 open reading frame (ORF) into the multiple cloning site of the pIRES2-ZsGreen1 vector (Clontech). Briefly, a 465-bp PCR product of the wild-type human SOD1 ORF was prepared by using the pcDNA3-Myc-hSOD1wt plasmid (22) as template and primers F1hSOD1ORFEcoR1 (5′-CCGGAATTCACCATGGCGACGAAGGCCGTGTGC) (EcoRI site italicized) and F1hSOD1ORFBamHI (5′-CGCGGATCCTTATTGGGCGATCCCAATTACAC CACAAG) (BamHI site italicized). The amplicon was cleaved with EcoRI and BamHI and cloned into the EcoRI/BamHI-digested pIRES2-ZsGreen1 backbone to yield plasmid pSOD1wt-IRES2-ZsGreen1. The human mutant SOD1G93A ORF was prepared by PCR using the same primers and the pcDNA3-Myc-hSOD1G93A vector (22) as template. The PCR product was then ligated into the pIRES2-ZsGreen1 vector as described above to yield the pSOD1G93A-IRES2-ZsGreen1 construct. The entire ORFs were analyzed by sequencing.
Cell Cultures and Transfections
Primary adult neuronal cultures were prepared as previously described (23). Briefly, pups were sacrificed at 5 days of age. Spinal cords were dissected in HABG (HibernateA, 2% B27 supplement, and 0.5 mm glutamine; Invitrogen). Tissues were dissociated with trypsin (1 mg/ml; Sigma) for 30 min at 37 °C. Neurons were purified on OptiPrep density gradient (Sigma), finally resuspended in Neurobasal®-A, 2% B27 supplement, 0.5 mm glutamine, 50 units/ml of penicillin, 50 μg/ml of streptomycin sulfate, 5 ng/ml of FGF, and plated on 0.1 mg/ml of a poly-d-lysine-coated 6-well plate. To remove waste and debris, the medium was changed on day 4 and then every 7 days. Spinal cord mixed glial cell cultures were prepared as previously described, with modifications (22, 24). Mechanical shaking and treatment with the microglial toxin l-leucine methyl ester were omitted to preserve microglial cells and obtain astrocyte-microglia mixed cultures.
NSC-34 (kindly provided by Prof. Neil R. Cashman, University of British Columbia, Vancouver, Canada) is a mouse-mouse hybrid cell line generated by fusion of neuroblastoma cells with motor neuron-enriched embryonic spinal cord cells. This cell line displays several phenotypic traits of motor neurons (25). P0–17D (kindly provided by Prof. James W. Jacobberger, Case Western Reserve University, Cleveland, OH) is a mouse cell line generated by immortalization of neonatal mouse cortical astrocytes with the SV40 large T antigen. Morphologically, these cells resemble mature primary astrocytes (26). BV-2 (kindly provided by Prof. Rosario Donato, University of Perugia, Perugia, Italy) is a murine cell line generated by immortalization of microglial cells with the J2 virus containing both v-raf and v-myc oncogenes. This cell line exhibits morphological, phenotypical, and functional properties of active microglial cells (27). C2C12 (purchased from Sigma) is a subclone (28) of the mouse myoblast cell line generated by D. Yaffe and O. Saxel (29).
NSC-34, P0–17D, and C2C12 cells were routinely maintained in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, Sigma), 2 mm glutamine, 1 mm sodium pyruvate, and antibiotics (Euroclone). BV-2 cells were cultivated in RPMI 1640 (Invitrogen) supplemented with 10% FBS, 4 mm glutamine, and antibiotics.
Transient transfections of NSC-34, P0–17D, BV-2, and C2C12 cells were performed using FuGENE® HD Transfection Reagent (Promega), according to the manufacturer's instructions. Briefly, cells were plated at a density of 2 × 104 cells/well in 24-well plates. The next day, cells were incubated with a FuGENE HD/DNA mixture containing 300 ng of pSOD1wt-IRES2-ZsGreen1 or pSOD1G93A-IRES2-ZsGreen1 vectors, together with 100 ng of PPRE5X-tk-Luc plasmid, which contains the firefly luciferase gene under control of the PPAR-responsive elements (17), and 2 ng of the normalization plasmid pGL4.74 (Promega) containing the Renilla luciferase gene. To test the responsiveness of endogenous PPARγ to pioglitazone, cells were incubated with 30 μm pioglitazone (Alexis Biochemicals) for 24 h and then lysed. To determine the impact of oxidative agents or lipid peroxidation by-products on PPAR transcriptional activity, cells were transfected with 400 ng of PPRE5X-tk-Luc plasmid and 8 ng of pGL4.74 vector. The next day, cells were exposed to increasing concentrations of hydrogen peroxide (H2O2, Sigma) or 4-HNE (Cayman) for 24 h and then lysed. To perform the luciferase enzymatic assay, cells were lysed using a passive lysis buffer (Promega). Firefly and Renilla luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. Firefly luciferase values were normalized to Renilla luciferase values and data were reported as relative light units.
RT-PCR Gene Expression Analysis
Spinal cords and hindlimb muscles (composed of gastrocnemius and soleus) were snap-frozen in liquid nitrogen immediately after collection. Tissues were homogenized with a Tissue Lyser (Qiagen) and the RNA was purified using RNeasy Mini Kit or RNeasy Fibrous Tissue Mini Kit (Qiagen) accordingly to the manufacturer's instructions. Total RNA from NSC-34 cells, C2C12 myoblasts, primary neurons, and primary mixed glial cultures was prepared using RNeasy Mini Kit following the manufacturer's instructions for the purification of RNA from animal cells. 1 μg of tissue- or cell-extracted total RNA was denatured at 75 °C for 5 min in the presence of 1.5 μg of random primers (Promega) in a 15-μl final volume. Deoxynucleotide triphosphate (GE Healthcare) and Moloney murine leukemia virus reverse transcriptase (RT) (Promega) were added at 0.5 mm and 8 units/μl final concentrations, respectively, in a final volume of 25 μl. The RT reaction was performed at 37 °C for 1 h; the enzyme was inactivated at 75 °C for 5 min. Control reactions without addition of the RT enzyme were performed for each sample. 1 μl of the resulting cDNA was analyzed by semiquantitative PCR. The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (Gapdh) was chosen as a reference. Transcripts were detected using primers 5′-GAAGAGTTGGCGTATGGG and 5′-GCGGAGGGCTCTGTCACACA for Mcad; 5′-GAGGACAGGACAAAGGAGG and 5′-CACGACAATGCCAACCAAAAAG for Acsl6; 5′-CACCGGGAGATGGAGAGCAAA and 5′-CCCAACTCTCATACATTCCC for Lpl; 5′-AAGACTGCCTTGGCAAAAGA and 5′-GCCAGTATCTGTGGCTCCAT for Gsta2; 5′-ATGCTGGCCTCCCTGATGAAT and 5′-CTGGAGCACCTTGGCGAACA for Pparγ; 5′-CACCCAAGGGAACTTGTGCAG and 5′-GGTCGTAGGTGAAGAGAACGG for Adiponectin; and 5′-ACGACCCCTTCATTGACC and 5′-TGCTTCACCACCTTCTTG for Gapdh.
Statistical Analyses
Data are represented as mean ± S.E. and statistical significance was verified using GraphPad Prism® software. Statistical significance was assessed by t test for comparisons between two groups; one-way or two-way ANOVA followed by Bonferroni or Newman-Keuls post hoc tests were used for comparisons of multiple groups.
RESULTS
PPAR Transcriptional Activity Increases in the Spinal Cord of hSOD1G93A ALS Mice at the Symptomatic Stage of the Disease
To analyze the dynamics of PPAR transcriptional activity in hSOD1G93A mice during the progression of the disease, we took advantage of PPRE-Luc mice in which the expression of the luciferase reporter gene is driven by five tandem repeats of PPRE (17). Homozygous PPRE-Luc+/+ females were cross-bred with hemizygote hSOD1G93A+/− males to obtain double transgenic PPRE-Luc+/−; hSOD1G93A+/− or PPRE-Luc+/−; hSOD1G93A−/− mice. Animals of both genotypes were euthanized at the critical stages of ALS disease, i.e. 30 (asymptomatic stage), 100 (onset of motor deficits), and ∼130 days (symptomatic stage). The spinal cord, liver, and kidney were then collected and assayed for luciferase enzymatic activity. Data obtained from these analyses indicate that, in PPRE-Luc+/−; hSOD1G93A+/− mice, PPAR transcriptional activity is constant in the spinal cord until the onset of the clinical symptoms, but significantly increases at the symptomatic stage of disease progression. By contrast, PPRE-Luc+/−; hSOD1G93A−/− animals, which do not express mutant human SOD1, show a fairly constant PPAR transcriptional activity throughout life (Fig. 1). Interestingly, the aberrant effect identified in the hSOD1G93A-expressing animals is specific for the spinal cord, as it was not observed in peripheral organs, such as liver and kidney, which are not affected by the disease (Fig. 1). Considering that PPARs are involved in several metabolic processes, we next investigated whether the increment in PPAR-driven luciferase activity identified in symptomatic PPRE-Luc+/−; hSOD1G93A+/− mice could be ascribed to alterations in the nutritional state of these animals, due to the oral dysphagia mice experience during the active phase of disease progression (30). Thus, we kept PPRE-Luc+/− mice on fasting for 48 h. Animals were then sacrificed and spinal cords were collected and further analyzed for luciferase enzymatic activity. We found that starvation does not influence PPAR transcriptional function in this area of the CNS (Fig. 2), suggesting that the increase in PPAR activity detected at the symptomatic stage of ALS disease strictly depends on the progression of the pathology.
FIGURE 1.

PPAR transcriptional activity increases at the symptomatic stage of ALS disease in the spinal cord of hSOD1G93A mice. The spinal cord (A), liver (B), and kidney (C) of PPRE-Luc+/−; hSOD1G93A−/− (Wild type) and PPRE-Luc+/−; hSOD1G93A+/− (hSOD1G93A) mice were taken at the critical stages of the pathology, i.e. 30 (asymptomatic stage), 100 (onset of motor impairment), and about 130 days of age (symptomatic stage) (n = 5–7 mice per time point per genotype). Luciferase enzymatic activity was determined as surrogate of PPAR transcriptional activity on tissue extracts. Data are expressed as mean ± S.E. **, p < 0.01 versus hSOD1G93A at 30 days of age; ***, p < 0.001 versus hSOD1G93A at 100 days of age, two-way ANOVA followed by Bonferroni post-hoc test. RLU, relative light units.
FIGURE 2.
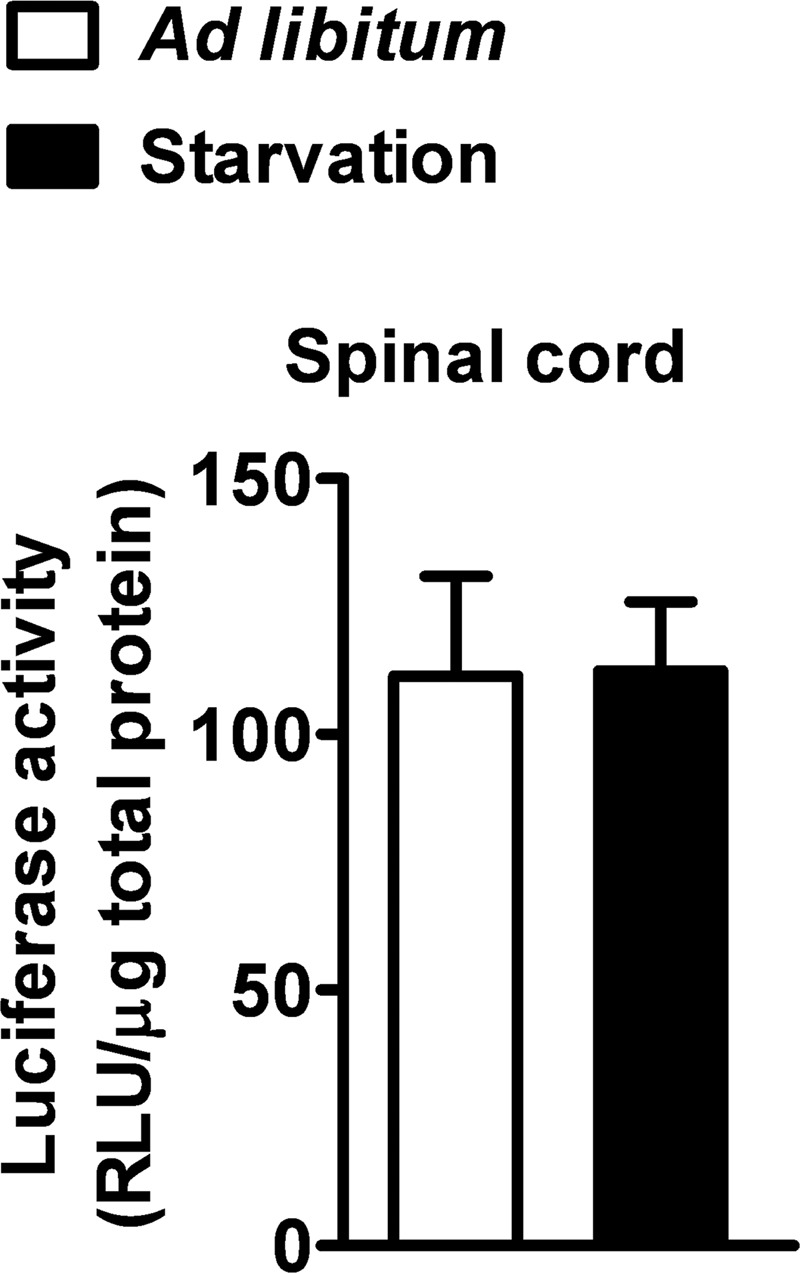
Starvation does not increase PPAR transcriptional activity in the mouse spinal cord. PPRE-Luc+/− mice were fed ad libitum or starved for 48 h. Spinal cords were collected and luciferase enzymatic activity was evaluated (n = 2 mice per each condition). Data are expressed as mean ± S.E. No significant difference was found between the two experimental groups. p = 0.97, unpaired t test. RLU, relative light units.
Enhanced PPARγ-dependent Transcription Correlates with Increased Nuclear Levels of Receptor Selectively in Motor Neurons from Symptomatic ALS Mice
A correlation between the functional data obtained with the reporter luciferase and the specific PPAR isoform implicated in the altered transcriptional process was subsequently addressed by analyzing the expression of isotype-specific endogenous target genes in the spinal cord of hSOD1G93A mice at different ages by semiquantitative RT-PCR. Medium chain acyl-CoA dehydrogenase (Mcad) was selected as a endogenous target gene for PPARα (31), acyl-CoA synthetase 2 (Acsl6) for PPARβ/δ (32), and lipoprotein lipase (Lpl) and glutathione S-transferase α-2 (Gsta2) for PPARγ (13, 33) (Fig. 3). Interestingly, we found that only genes whose expression is driven by PPARγ, such as Lpl and Gsta2, became significantly up-regulated at the symptomatic stage of ALS progression. By contrast, the messenger RNAs (mRNAs) of the target genes for PPARα and PPARβ/δ, Mcad and Acsl6, respectively, did not show significant changes up to 130 days of age, i.e. the time when hSOD1G93A mice manifest full-blown symptoms (Fig. 3). This indicates that PPARγ is the isoform involved in the aberrant transcriptional activity highlighted by the reporter in PPRE-Luc+/−; hSOD1G93A+/− spinal cords.
FIGURE 3.

Enhanced expression of PPARγ and its transcriptional target genes in the spinal cord of hSOD1G93A ALS mice at the symptomatic stage of the disease. Expression of PPAR isoform-specific target genes was determined in the spinal cord of wild-type and hSOD1G93A mice of different ages by semi-quantitative RT-PCR analysis (n = 5–7 mice per time point per genotype). A, Mcad was selected as endogenous target gene for PPARα; B, Acsl6 for PPARβ/δ; C, Lpl; and D, Gsta2 for PPARγ. E, endogenous PPARγ mRNA was quantified in the spinal cord of wild-type and hSOD1G93A mice (n = 5–7 mice per time point per genotype). Results were expressed as fold-change versus 30-day-old wild-type mice. Data are expressed as mean ± S.E. **, p < 0.01 versus hSOD1G93A at 30 days of age; ***, p < 0.001 versus hSOD1G93A at 30 days of age; °, p < 0.05 versus hSOD1G93A at 30 days of age; °°, p < 0.01 versus hSOD1G93A at 100 days of age, two-way ANOVA followed by Bonferroni post-hoc test.
The expression of the receptor itself was subsequently analyzed in the spinal cord of hSOD1G93A mice at different ages during disease progression. Semiquantitative RT-PCR analysis revealed that the PPARγ mRNA is up-regulated at the symptomatic stage of the disease when compared with the earlier time points and with wild-type animals of the same age (Fig. 3).
Thus, we next decided to investigate the cellular population(s) responsible for the PPARγ-dependent anomalous transcriptional response. To this aim, we explored the cellular and subcellular distribution of PPARγ by immunohistochemistry in the spinal cord of hSOD1G93A mice and wild-type littermates at different ages during disease progression. Consistent with previous reports (9), we found that PPARγ is expressed in motor neurons as well as glial cells, particularly astrocytes and microglia (Fig. 4). To determine whether the enhanced PPARγ transcriptional activity detected in the spinal cord of aged hSOD1G93A mice could be ascribed to an increase in the number of cells with nuclear PPARγ, we quantified the proportion of motor neurons, astrocytes, and microglial cells that showed PPARγ-immunopositive nuclei in the relevant time frame, i.e. between 100 and 130 days of age (Fig. 5A). The results obtained from this analysis indicated that 100% of motor neuronal nuclei are immunopositive for PPARγ throughout life. Besides, the number of astrocytes and microglial cells with PPARγ-immunopositive nuclei did not vary significantly at the critical time points (astrocytes, 72 ± 7.9% at 100 days; 58.6 ± 6.2% at 130 days, p = 0.218, unpaired t test; microglia, 64.2 ± 13.8% at 100 days; 71.7 ± 4.4% at 130 days, p = 0.653, unpaired t test). This indicates that the fraction of cells showing PPARγ into the nucleus remains constant throughout disease progression. Because expression of the receptor increases at the advanced stage of the disease (Fig. 3E), we next postulated that PPARγ may accumulate within the nucleus of specific cell populations, and this may affect PPARγ-dependent transcription in such cells. Thus, the nuclear concentration of the receptor was quantitatively investigated on spinal cord sections double labeled for PPARγ and the neuronal nonphosphorylated neurofilament marker SMI32. Immunofluorescence quantification revealed that the amount of nuclear PPARγ is significantly higher in motor neurons from symptomatic hSOD1G93A mice when compared with younger animals or age-matched wild-type mice (Fig. 5B). Such an increment in the nuclear concentration of PPARγ is restricted to motor neurons, because we could not detect a similar increase within spinal cord non-neuronal cells (Fig. 5B) or other neuronal populations present in different areas of the CNS, particularly the cerebral cortex and the hippocampus (Fig. 5C). Thus, we propose that PPARγ becomes activated in the spinal cord of diseased hSOD1G93A mice, mainly within motor neurons.
FIGURE 4.

Persistent nuclear localization of PPARγ in both motor neurons and glial cells in the spinal cord of symptomatic hSOD1G93A ALS mice. Double immunofluorescence staining for PPARγ and SMI32 (A), GFAP (B), or tomato lectin (C) shows intense nuclear staining of PPARγ in motor neurons, astrocytes, and microglial cells, respectively. Scale bars, 50 μm.
FIGURE 5.

The nuclear concentration of PPARγ is quantitatively enhanced in spinal motor neurons from hSOD1G93A mice at the symptomatic stage of the disease. A, the number of motor neurons, astrocytes, or microglial cells showing PPARγ into the nucleus was determined on spinal cord sections from hSOD1G93A mice taken at 100 (100d) or 130 days of age (130d). Data are expressed as percentage of motor neurons, astrocytes, or microglial cells with nuclear PPARγ out of the total number of cells taken into consideration (n = 40 motor neurons per mouse, n = 45 astrocytes per mouse, n = 100 microglial cells per mouse; n = 5–6 mice per time point). B, the intensity of PPARγ immunofluorescence in the nuclei of motor neurons and non-neuronal cells was determined on lumbar spinal cord sections from wild-type and hSOD1G93A mice stained for PPARγ, the neuronal marker SMI32 and the nuclear dye Hoechst 33342 (n = 15 motor neurons per mouse, n = 30 non-neuronal cells per mouse; n = 3–6 mice per time point). C, the intensity of PPARγ immunofluorescence in the nuclei of cortical and hippocampal neurons was determined on brain sections from hSOD1G93A mice stained for PPARγ, the neuronal marker neuronal nuclei and the nuclear dye Hoechst 33342 (n = 30 cortical neurons per mouse, n = 30 hippocampal neurons per mouse; n = 3 mice per time point). Values were normalized relative to fluorescence intensity of Hoechst 33342, as a measure of DNA content. Data are expressed as mean ± S.E. ***, p < 0.001 versus hSOD1G93A motor neurons at 30 and 100 days of age and wild-type at 130 days of age, two-way ANOVA followed by Bonferroni post hoc test.
Skeletal Muscle Atrophy Does Not Affect PPARγ Expression in Spinal Motor Neurons
One crucial and currently debated issue in ALS is whether alterations within motor neurons are induced retrogradely by the atrophic and degenerative processes muscle fibers undergo during disease progression or whether motor neuronal changes occur cell autonomously (and/or with the contribution of glial cells; for review see Ref. 34) (16, 35–37).
Because considerable evidence indicates that normal aging and ALS share similar structural, regional, and molecular alterations at the level of the neuromuscular system (38), we took advantage of a mouse model of age-related sarcopenia to determine whether the increased expression of PPARγ within the nuclei of spinal motor neurons might be influenced by alterations of hindlimb myofibers. Numerous reports in fact indicate that, at the age of 28–29 months, C57Bl/6J wild-type mice exhibit a significant atrophy of skeletal muscle that is associated with increased oxidative stress. Noteworthy, this condition is not accompanied by the loss of motor neuronal cell bodies in the lumbar tract of the spinal cord (39–42). Consistent with these observations, we confirmed that 28-month-old C57Bl/6J mice show a significant reduction in the fiber diameter of hindlimb muscles, particularly the soleus and tibialis anterior muscles, when compared with 30-day-old animals of the same strain (Fig. 6A). This state is not associated with a reduction in the number of motor neuronal somas in the lumbar spinal cord (Fig. 6B). Thus, we next determined the concentration of PPARγ within the nuclei of motor neurons and non-neuronal cells in spinal cord sections from 30-day- and 28-month-old mice double labeled for PPARγ and SMI32. This analysis revealed that the nuclear levels of the receptor do not vary in any cell population (Fig. 6C), suggesting that the deleterious changes of skeletal muscle are unlikely responsible for the increased concentration of PPARγ within the motor neuronal nuclei in the spinal cord of ALS mice.
FIGURE 6.

Muscle atrophy does not increase the nuclear concentration of PPARγ within spinal motor neurons. A, morphometric analysis of soleus and tibialis anterior muscle fibers from C57Bl/6J wild-type mice at 30 days and 28 months of age. Histograms represent the mean ± S.E. of the minimal Feret's diameter of the soleus and tibialis anterior muscle fibers (n = 3 mice for each age). B, quantification of motor neurons in the lumbar tract of the spinal cord from C57Bl/6J mice at 30 days and 28 months of age. Histograms represent the mean ± S.E. of the total number of motor neurons from 9 disectors per mouse (n = 3 mice for each age). C, the intensity of PPARγ immunofluorescence in the nuclei of motor neurons and non-neuronal cells was determined on lumbar spinal cord sections from 30-day-old and 28-month-old mice stained for PPARγ, the neuronal marker SMI32 and the nuclear dye Hoechst 33342 (n = 32 motor neurons per mouse, n = 90 non-neuronal cells per mouse; n = 3 mice per time point). Values were normalized relative to fluorescence intensity of Hoechst 33342, as a measure of DNA content. Data are expressed as mean ± S.E. ***, p < 0.0001 versus 30-day-old C57Bl/6J mice, unpaired t test.
Motor Neuronal PPARγ Displays Enhanced Responsiveness in the Presence of Mutant SOD1 or Lipoperoxidative Products in Vitro
To confirm the cell specificity of PPARγ activation in the different neural cell populations, we switched to the motor neuron-like NSC-34, the astrocytic P0–17D, and the microglial BV-2 cell culture systems (25–27). Cells were transiently co-transfected with the reporter transgene PPRE-Luc together with vectors encoding either the wild-type human SOD1 (hSOD1WT) or the mutant hSOD1G93A proteins. Luciferase activity was then measured 24 h after treating the cells in the absence or presence of the PPARγ agonist pioglitazone (30 μm). We found that both the basal and pioglitazone-induced PPAR transcriptional activities are significantly enhanced in the presence of mutant SOD1 in the NSC-34 cells when compared with the wild-type counterpart (Fig. 7A). Interestingly, no such increases were detected in hSOD1G93A-expressing P0–17D and BV-2 glial cell lines, although they resulted in being susceptible to pioglitazone stimulation and PPARγ activation (Fig. 7A). Consistent with these results, we confirmed an up-regulation of the PPARγ target genes Lpl and Gsta2 in hSOD1G93A-expressing primary neurons, but not in mixed glial cultures, from mouse spinal cords (Fig. 7, B and C).
FIGURE 7.

Mutant SOD1-expressing neuronal cells are highly responsive to PPARγ activation in vitro. A, PPAR transcriptional activity was determined in the motor neuron-like NSC-34, astrocytic P0–17D, and microglial BV-2 cell lines transiently co-transfected with the PPRE5X-tk-Luc plasmid and vectors encoding either hSOD1WT or hSOD1G93A. Renilla luciferase was used as a transfection control. Cells were treated in the absence or presence of 30 μm pioglitazone for 24 h and assayed for luciferase enzymatic activity (n = 3–6 in triplicate). Firefly luciferase levels were normalized to Renilla luciferase values. B and C, expression of the PPARγ target genes Lpl and Gsta2 was determined by semiquantitative RT-PCR analysis in primary neuronal (B) and mixed glial cultures (C) prepared from the spinal cord of wild-type and hSOD1G93A mice. Cells were treated in the absence and presence of 30 μm pioglitazone for 24 h (n = 3 cultures per cell type). Data are expressed as mean ± S.E. *, p < 0.05 versus hSOD1WT; **, p < 0.01 versus hSOD1WT + pioglitazone; §, p < 0.01 versus hSOD1G93A; §§, p < 0.001 versus hSOD1WT; §§§, p < 0.001 versus hSOD1G93A; °, p < 0.05 versus wild type; °°, p < 0.05 versus wild type + pioglitazone; °°°, p < 0.001 versus wild type + pioglitazone, two-way ANOVA followed by Bonferroni post-hoc test.
This raised the question as whether the enhanced activity of PPARγ detected in PPRE-Luc+/−; hSOD1G93A+/− mice at the symptomatic phase of the disease was mainly due to increased expression and sensitivity of the receptor itself, to increased availability of endogenous ligands, or both. To clarify this issue, we reasoned that the symptomatic stage of the disease is characterized by the generation of a plethora of reactive oxygen species in both humans and animal models (43). These agents oxidize various cellular macromolecules, including membrane lipids, and initiate a self-propagating process that results in the production of lipoperoxidation end products, such as 4-HNE and others (44–49). Based on this, we next evaluated the impact of oxidant species and lipid peroxidation by-products, such as H2O2 and 4-HNE, respectively, on PPAR-driven luciferase activity in vitro. NSC-34 cells were transiently transfected with PPRE-Luc and treated with increasing concentrations of H2O2 or 4-HNE for 24 h. Luciferase enzymatic activity of cell lysates was then assayed. As shown in Fig. 8, 4-HNE, but not H2O2, emerged as a powerful inducer of PPAR transcription. Semiquantitative analysis of the mRNAs coding for the isoform-specific target genes Mcad, Acsl6, and Lpl demonstrated that 4-HNE induces the expression of Lpl in a dose-dependent manner (Fig. 9). By contrast, Mcad and Acsl6 expression were not regulated by this agent. These results indicate that PPARγ is the isoform target for lipid peroxidation end products in motor neuron-like cells.
FIGURE 8.

The lipid peroxidation adduct 4-HNE, but not the oxidant hydrogen peroxide, triggers PPAR transcriptional activation in cultured motor neuron-like cells. NSC-34 cells were transiently transfected with the PPRE5X-tk-Luc plasmid and treated with increasing concentrations of H2O2 (A) or 4-HNE (B) for 24 h (n = 3 in triplicate). Pioglitazone (30 μm, Pio) was used as positive control and the vehicle (veh) as negative control. Cell lysates were assayed for luciferase enzymatic activity, and firefly luciferase levels were normalized to the transfection control, Renilla luciferase. Data are expressed as mean ± S.E. °°°, p < 0.001 versus vehicle, H2O2, 10, 50, and 100 μm; **, p < 0.05 versus vehicle; ***, p < 0.01 versus vehicle, 4-HNE, 10 m and 20 μm, one-way ANOVA followed by Bonferroni post hoc test.
FIGURE 9.

4-HNE specifically enhances the expression of the PPARγ target gene Lpl in cultured motor neuron-like cells. Expression of PPAR isoform-specific target genes, i.e. Lpl for PPARγ (A), Mcad for PPARα (B), and Acsl6 for PPARβ/δ (C), was analyzed in NSC-34 cells treated with the vehicle (veh), 4-HNE (40 or 60 μm), or pioglitazone (30 μm) for 24 h (n = 3 in triplicate). Data are expressed as mean ± S.E. *, p < 0.05 versus vehicle; **, p < 0.01 versus vehicle, one-way ANOVA followed by Newman-Keuls post hoc test.
To determine whether the lipid peroxidation end product 4-HNE is effective in activating PPARγ transcriptional function also in vivo, we intracerebroventricularly injected 4-HNE (5 nmol/mouse) in wild-type mice. The expression of the PPARγ target genes Lpl and Gsta2 was then analyzed in the spinal cord 3 h later by semiquantitative RT-PCR analysis. Consistent with the data in vitro, we confirmed that 4-HNE significantly increases the levels of Lpl and Gsta2 mRNAs (Fig. 10).
FIGURE 10.

4-HNE enhances the expression of PPARγ target genes Lpl and Gsta2 in vivo in the spinal cord of wild-type mice. Mice were intracerebroventricularly injected with 4-HNE (5 nmol/mouse) or vehicle and the expression of Lpl and Gsta2 was analyzed in the spinal cord after 3 h by semi-quantitative RT-PCR analysis (n = 4–5 mice per each experimental group). Data are expressed as mean ± S.E. *, p < 0.05 versus vehicle, unpaired t test.
PPARγ Expression and Transcriptional Activity Are Not Increased in Skeletal Muscles from Symptomatic hSOD1G93A ALS Mice
Several lines of evidence indicate that, in both ALS patients and transgenic animal models, skeletal muscles undergo severe atrophy and degeneration, which closely correlate with increased oxidative stress and lipid peroxidation (16, 35–37, 39, 49). To determine whether the occurrence of such events enhanced the expression of PPARγ in these tissues, we next investigated the levels of the receptor mRNA in hindlimb muscles. Semiquantitative RT-PCR analyses revealed that the expression of PPARγ transcripts was not significantly different between symptomatic hSOD1G93A mice and age-matched wild-type animals (supplemental Fig. S1A).
Because PPARγ was consistently reported not to control the expression of scavenger enzymes, such as lipoprotein lipase (LPL), in muscles (50–52), its transcriptional activation was assessed by determining the expression of the tissue-specific PPARγ target gene Adiponectin (52). We found that the levels of Adiponectin mRNA did not change between the two genotypes at the age of 130 days (52), indicating that the nuclear receptor is not activated in muscles of symptomatic mice (supplemental Fig. S1A).
To further confirm the impact of mutant SOD1 on PPARγ transcriptional activity in muscles cells, we transiently co-transfected C2C12 myoblasts with the reporter transgene PPRE-Luc and vectors encoding either hSOD1WT or hSOD1G93A proteins. Consistent with gene expression analyses of skeletal muscles from ALS mice, we did not find significant changes in luciferase activity between the two experimental groups (supplemental Fig. S1B), despite the cells expressed PPARγ (supplemental Fig. S1C).
This suggests that the presence of mutant SOD1 does not impact the expression and activity of the nuclear receptor in muscle cells in vivo and in vitro. Thus, we propose that PPARγ becomes activated in the spinal cord of hSOD1G93A mice to possibly counteract the intense neurodegenerative process that occurs during the symptomatic phase of the pathology.
DISCUSSION
Taking advantage of the PPRE-Luc reporter mouse, which allows to monitor the activity of PPARs during ALS progression, here we show for the first time that PPAR-dependent transcription increases in the spinal cord of hSOD1G93A mice at the symptomatic stage of the disease. This phenomenon clearly depends on the pathology because it is not observed in peripheral organs, such as kidney and liver, which are unaffected by the disorder.
Immunohistochemical and isoform-specific target gene analyses then led to the identification of motor neuronal PPARγ as the subtype involved in the aberrant event. Indeed, we show that Lpl and Gsta2, two genes whose expression is trascriptionally controlled by PPARγ (13, 33), are up-regulated in the spinal cord of symptomatic hSOD1G93A mice. Our findings are consistent with previous gene expression profiling and proteomic analyses that reported increased levels of LPL and Gsta2 in both mutant hSOD1G93A-expressing mouse spinal cords and motor neurons in culture (53, 54).
It is noteworthy that, under conditions of oxidative stress, LPL and Gsta2 were described to limit the propagation of lipid peroxidation events as well as to scavenge cytotoxic lipid hydroperoxides in vitro (55, 56). This suggests that PPARγ has the potential to activate defensive mechanisms against lipoperoxidative reactions and related stress-mediated signaling by controlling the expression of antioxidant enzymes.
Remarkably, in tissues where PPARγ does not control the expression of detoxifying enzymes, such as skeletal muscles (50–52), PPARγ-dependent transcription does not become activated in ALS mice, providing further support to the idea that the protective function of the receptor is mainly within the nervous system.
Although reports from different laboratories indicate that ALS-driven muscle alterations can impact motor neuronal function and survival (35, 36), others demonstrated that reduction in ALS-dependent muscle atrophy does not prevent neurodegeneration (37). Thus, it is possible that motor neurons and muscle fibers can communicate through specific signaling pathways, but not via others. Herein, we found that the expression of PPARγ within motor neurons is not influenced by the occurrence of muscle atrophy in aged mice.
Because of the significant oxygen consumption as well as the high content of easily oxidizable polyunsaturated fatty acids, the CNS results particularly vulnerable to oxidative environments. Thus, in several neurodegenerative disorders, including ALS, the formation of lipid oxidation/peroxidation products is a rather common event in all neural tissues that are exposed to reactive oxidant species (57). In fact, enhanced levels of malondialdehyde and 4-HNE, two reactive aldehydes that are generated endogenously during the process of peroxidation of membrane fatty acids, have been consistently reported in tissues and spinal fluid from both ALS patients and transgenic animal models (45–49). Although the mechanisms of action of such molecules have not been fully clarified, it is clear that these aldehydic compounds exert their biological effects, at least in part, through reaction with cellular proteins. Hence, previous in vitro evidence indicates that 4-HNE impairs glucose and glutamate transport as well as choline acetyltransferase activity in cultured motor neurons, events that precede delayed cell death (58). Furthermore, chronic exposure to high concentrations of 4-HNE was described to trigger motor neuron degeneration in vivo (59). Thus, depending on the concentrations and exposure time, the lipid peroxidation end product 4-HNE appears to be causally involved in multiple pathophysiological events both in vitro and in vivo (60, 61). Because considerable evidence indicates that PPARγ can be activated by a variety of naturally occurring lipids, including oxidized fatty acids (7, 10), here we investigated the impact of 4-HNE toward the transcriptional activity of PPARγ. The fact that 4-HNE specifically activated PPARγ in vitro, in motor neuron-like cultures, and in vivo, in the spinal cord of wild-type mice, suggests that not only the nuclear receptor interferes with lipoperoxidative processes by controlling the expression of scavenger enzymes, but its activation itself is strongly influenced by lipid peroxidation events and metabolites. Thus, we propose that the accumulation of peroxidized lipids in ALS tissues up to a critical threshold is a key determinant for activation of PPARγ-dependent self-defensive mechanisms, which are crucial to remove neurotoxic lipid peroxidation by-products. It is worth mentioning that activation of similar natural protective mechanisms is not unusual for the PPARs. In fact, the PPARβ/δ subtype was also reported to be activated by lipid peroxidation products and, thus, critically control physiopathological events in experimental models of other disorders (62).
Further support to the idea that defective lipid metabolism plays a role in ALS progression was provided by a number of clinical and experimental observations. For instance, abnormalities in sphingolipid and cholesterol metabolism were described in the spinal cords of ALS patients and transgenic mouse models, and these changes were reported to sensitize motor neurons to death (63). In addition, decreased lipid storage and increased peripheral lipid clearance were shown in mutant SOD1-expressing mice at the pre-symptomatic stage of the disease (64–66). Consistently, lower serum lipid levels have been more recently related to respiratory impairment in ALS patients (67), and hyperlipidemia was described to be a predictive factor for a better prognosis (68, 69).
Concerning PPARγ, the implication of this receptor in ALS progression was first suggested by early pharmacological studies on hSOD1G93A mice using the specific agonist pioglitazone (11, 12). The beneficial effect of this agent was initially ascribed to the anti-inflammatory properties of the receptor. However, the results obtained during the course of this study prompt us to postulate that, besides its anti-inflammatory activity, PPARγ can also counteract the process of neurodegeneration directly. This occurs through the positive regulation of gene products that limit the accumulation of neurotoxic lipid peroxidation derivatives. Thus, our study points to PPARγ transcriptional activity as a major player in the defensive response of motor neurons. Furthermore, because treatment with pioglitazone proved effective even when initiated before the onset of the clinical symptoms (11, 12), it is tempting to speculate that anticipating the modulation of PPARγ transcriptional co-activators pharmacologically may boost these natural protective mechanisms, which normally fail because they become activated only when the disease is already in an advanced stage.
On a therapeutic standpoint, no effective pharmacological intervention to halt the progression of ALS is yet available. The only valuable medication to treat this disorder is currently riluzole, which only slightly slows disease progression. Thus, it seems that further improvement of the outcome of ALS treatments may depend on a better understanding of the molecular mechanisms of motor neuron pathology. In this perspective, and considering our and previous results (11, 12), the transcriptional process driven by PPARγ emerges as a very interesting mechanism to target with novel therapeutics.
Acknowledgments
We are grateful to Prof. Neil R. Cashman for providing the NSC-34 motor neuron-like cell line, Prof. James W. Jacobberger for the P0–17D astrocytic cell line, Prof. Rosario Donato for the BV-2 microglial cell line, and Chiara Bergamaschi for experimental support.
This work was supported by Telethon Foundation Grant GGP05244 (to D. R.), the Italian Ministry of Health, European Union's Sixth Framework Programme under grant agreement NoE DIMI LSHBCT-2005-512146 (Diagnostic Molecular Imaging - DiMI) (to A. M.), and Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 278850 (Imaging of Neuroinflammation in Neurodegenerative Diseases - INMiND) (to A. M.).

This article contains supplemental Fig. S1.
- SOD
- superoxide dismutase
- PPARγ
- peroxisome proliferator-activated receptor γ
- PPRE
- PPAR responsive element
- 4-HNE
- 4-hydroxynonenal
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- ANOVA
- analysis of variance
- Mcad
- medium chain acyl-CoA dehydrogenase
- Acs16
- acyl-CoA synthetase 2
- Lpl
- lipoprotein lipase
- Gsta2
- glutathione S-transferase α-2
- Gapdh
- glyceraldehyde-3-phosphate dehydrogenase.
REFERENCES
- 1. Rosen D. R., Siddique T., Patterson D., Figlewicz D. A., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J. P., Deng H. X. (1993) Mutations in Cu/Zn-superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62 [DOI] [PubMed] [Google Scholar]
- 2. Gurney M. E., Pu H., Chiu A. Y., Dal Canto M. C., Polchow C. Y., Alexander D. D., Caliendo J., Hentati A., Kwon Y. W., Deng H. X. (1994) Motor neuron degeneration in mice that express a human Cu,Zn-superoxide dismutase mutation. Science 264, 1772–1775 [DOI] [PubMed] [Google Scholar]
- 3. Bruijn L. I., Becher M. W., Lee M. K., Anderson K. L., Jenkins N. A., Copeland N. G., Sisodia S. S., Rothstein J. D., Borchelt D. R., Price D. L., Cleveland D. W. (1997) ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18, 327–338 [DOI] [PubMed] [Google Scholar]
- 4. Wong P. C., Pardo C. A., Borchelt D. R., Lee M. K., Copeland N. G., Jenkins N. A., Sisodia S. S., Cleveland D. W., Price D. L. (1995) An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14, 1105–1116 [DOI] [PubMed] [Google Scholar]
- 5. Turner B. J., Talbot K. (2008) Transgenics, toxicity, and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 85, 94–134 [DOI] [PubMed] [Google Scholar]
- 6. Bensimon G., Lacomblez L., Meininger V. (1994) A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/riluzole study group. N. Engl. J. Med. 330, 585–591 [DOI] [PubMed] [Google Scholar]
- 7. Ricote M., Glass C. K. (2007) PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta 1771, 926–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heneka M. T., Landreth G. E., Hüll M. (2007) Drug insight. Effects mediated by peroxisome proliferator-activated receptor-γ in CNS disorders. Nat. Clin. Pract. Neurol. 3, 496–504 [DOI] [PubMed] [Google Scholar]
- 9. Heneka M. T., Landreth G. E. (2007) PPARs in the brain. Biochim. Biophys. Acta 1771, 1031–1045 [DOI] [PubMed] [Google Scholar]
- 10. Itoh T., Fairall L., Amin K., Inaba Y., Szanto A., Balint B. L., Nagy L., Yamamoto K., Schwabe J. W. (2008) Structural basis for the activation of PPARγ by oxidized fatty acids. Nat. Struct. Mol. Biol. 15, 924–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kiaei M., Kipiani K., Chen J., Calingasan N. Y., Beal M. F. (2005) Peroxisome proliferator-activated receptor-γ agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 191, 331–336 [DOI] [PubMed] [Google Scholar]
- 12. Schütz B., Reimann J., Dumitrescu-Ozimek L., Kappes-Horn K., Landreth G. E., Schürmann B., Zimmer A., Heneka M. T. (2005) The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J. Neurosci. 25, 7805–7812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao X., Strong R., Zhang J., Sun G., Tsien J. Z., Cui Z., Grotta J. C., Aronowski J. (2009) Neuronal PPARγ deficiency increases susceptibility to brain damage after cerebral ischemia. J. Neurosci. 29, 6186–6195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Song W., Song Y., Kincaid B., Bossy B., Bossy-Wetzel E. (2012) Mutant SOD1(G93A) triggers mitochondrial fragmentation in spinal cord motor neurons. Neuroprotection by SIRT3 and PGC-1α. Neurobiol. Dis., doi: 10.1016/j.nbd.2012.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao W., Varghese M., Yemul S., Pan Y., Cheng A., Marano P., Hassan S., Vempati P., Chen F., Qian X., Pasinetti G. M. (2011) Peroxisome proliferator activator receptor γ coactivator-1α (PGC-1α) improves motor performance and survival in a mouse model of amyotrophic lateral sclerosis. Mol. Neurodegener 6, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liang H., Ward W. F., Jang Y. C., Bhattacharya A., Bokov A. F., Li Y., Jernigan A., Richardson A., Van Remmen H. (2011) PGC-1α protects neurons and alters disease progression in an amyotrophic lateral sclerosis mouse model. Muscle Nerve 44, 947–956 [DOI] [PubMed] [Google Scholar]
- 17. Ciana P., Biserni A., Tatangelo L., Tiveron C., Sciarroni A. F., Ottobrini L., Maggi A. (2007) A novel peroxisome proliferator-activated receptor responsive element-luciferase reporter mouse reveals gender specificity of peroxisome proliferator-activated receptor activity in liver. Mol. Endocrinol. 21, 388–400 [DOI] [PubMed] [Google Scholar]
- 18. Heiman-Patterson T. D., Deitch J. S., Blankenhorn E. P., Erwin K. L., Perreault M. J., Alexander B. K., Byers N., Toman I., Alexander G. M. (2005) Background and gender effects on survival in the TgN(SOD1-G93A)1Gur mouse model of ALS. J. Neurol Sci. 236, 1–7 [DOI] [PubMed] [Google Scholar]
- 19. Benedusi V., Meda C., Della Torre S., Monteleone G., Vegeto E., Maggi A. (2012) A lack of ovarian function increases neuroinflammation in aged mice. Endocrinology 153, 2777–2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 21. Guntinas-Lichius O., Mockenhaupt J., Stennert E., Neiss W. F. (1993) Simplified nerve cell counting in the rat brainstem with the physical disector using a drawing-microscope. J. Microsc. 172, 177–180 [DOI] [PubMed] [Google Scholar]
- 22. Rossi D., Brambilla L., Valori C. F., Roncoroni C., Crugnola A., Yokota T., Bredesen D. E., Volterra A. (2008) Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ. 15, 1691–1700 [DOI] [PubMed] [Google Scholar]
- 23. Brewer G. J., Torricelli J. R. (2007) Isolation and culture of adult neurons and neurospheres. Nat. Protoc. 2, 1490–1498 [DOI] [PubMed] [Google Scholar]
- 24. Rossi D., Brambilla L., Valori C. F., Crugnola A., Giaccone G., Capobianco R., Mangieri M., Kingston A. E., Bloc A., Bezzi P., Volterra A. (2005) Defective tumor necrosis factor-α-dependent control of astrocyte glutamate release in a transgenic mouse model of Alzheimer disease. J. Biol. Chem. 280, 42088–42096 [DOI] [PubMed] [Google Scholar]
- 25. Cashman N. R., Durham H. D., Blusztajn J. K., Oda K., Tabira T., Shaw I. T., Dahrouge S., Antel J. P. (1992) Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 194, 209–221 [DOI] [PubMed] [Google Scholar]
- 26. Frisa P. S., Goodman M. N., Smith G. M., Silver J., Jacobberger J. W. (1994) Immortalization of immature and mature mouse astrocytes with SV40 T antigen. J. Neurosci. Res. 39, 47–56 [DOI] [PubMed] [Google Scholar]
- 27. Blasi E., Barluzzi R., Bocchini V., Mazzolla R., Bistoni F. (1990) Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J. Neuroimmunol. 27, 229–237 [DOI] [PubMed] [Google Scholar]
- 28. Blau H. M., Pavlath G. K., Hardeman E. C., Chiu C. P., Silberstein L., Webster S. G., Miller S. C., Webster C. (1985) Plasticity of the differentiated state. Science 230, 758–766 [DOI] [PubMed] [Google Scholar]
- 29. Yaffe D., Saxel O. (1977) Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 270, 725–727 [DOI] [PubMed] [Google Scholar]
- 30. Lever T. E., Gorsek A., Cox K. T., O'Brien K. F., Capra N. F., Hough M. S., Murashov A. K. (2009) An animal model of oral dysphagia in amyotrophic lateral sclerosis. Dysphagia 24, 180–195 [DOI] [PubMed] [Google Scholar]
- 31. Cullingford T. E., Dolphin C. T., Sato H. (2002) The peroxisome proliferator-activated receptor α-selective activator ciprofibrate up-regulates expression of genes encoding fatty acid oxidation and ketogenesis enzymes in rat brain. Neuropharmacology 42, 724–730 [DOI] [PubMed] [Google Scholar]
- 32. Basu-Modak S., Braissant O., Escher P., Desvergne B., Honegger P., Wahli W. (1999) Peroxisome proliferator-activated receptor β regulates acyl-CoA synthetase 2 in reaggregated rat brain cell cultures. J. Biol. Chem. 274, 35881–35888 [DOI] [PubMed] [Google Scholar]
- 33. Victor N. A., Wanderi E. W., Gamboa J., Zhao X., Aronowski J., Deininger K., Lust W. D., Landreth G. E., Sundararajan S. (2006) Altered PPARγ expression and activation after transient focal ischemia in rats. Eur. J. Neurosci. 24, 1653–1663 [DOI] [PubMed] [Google Scholar]
- 34. Ilieva H., Polymenidou M., Cleveland D. W. (2009) Non-cell autonomous toxicity in neurodegenerative disorders. ALS and beyond. J. Cell Biol. 187, 761–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dobrowolny G., Aucello M., Rizzuto E., Beccafico S., Mammucari C., Boncompagni S., Bonconpagni S., Belia S., Wannenes F., Nicoletti C., Del Prete Z., Rosenthal N., Molinaro M., Protasi F., Fanò G., Sandri M., Musarò A. (2008) Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 8, 425–436 [DOI] [PubMed] [Google Scholar]
- 36. Wong M., Martin L. J. (2010) Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 19, 2284–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Da Cruz S., Parone P. A., Lopes V. S., Lillo C., McAlonis-Downes M., Lee S. K., Vetto A. P., Petrosyan S., Marsala M., Murphy A. N., Williams D. S., Spiegelman B. M., Cleveland D. W. (2012) Elevated PGC-1α activity sustains mitochondrial biogenesis and muscle function without extending survival in a mouse model of inherited ALS. Cell Metab. 15, 778–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Valdez G., Tapia J. C., Lichtman J. W., Fox M. A., Sanes J. R. (2012) Shared resistance to aging and ALS in neuromuscular junctions of specific muscles. PLoS One 7, e34640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Muller F. L., Song W., Jang Y. C., Liu Y., Sabia M., Richardson A., Van Remmen H. (2007) Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R1159–1168 [DOI] [PubMed] [Google Scholar]
- 40. Jang Y. C., Lustgarten M. S., Liu Y., Muller F. L., Bhattacharya A., Liang H., Salmon A. B., Brooks S. V., Larkin L., Hayworth C. R., Richardson A., Van Remmen H. (2010) Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. 24, 1376–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jang Y. C., Liu Y., Hayworth C. R., Bhattacharya A., Lustgarten M. S., Muller F. L., Chaudhuri A., Qi W., Li Y., Huang J. Y., Verdin E., Richardson A., Van Remmen H. (2012) Dietary restriction attenuates age-associated muscle atrophy by lowering oxidative stress in mice even in complete absence of CuZn-SOD. Aging Cell doi: 10.1111/j.1474-9726.2012.00843.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chai R. J., Vukovic J., Dunlop S., Grounds M. D., Shavlakadze T. (2011) Striking denervation of neuromuscular junctions without lumbar motoneuron loss in geriatric mouse muscle. PLoS One 6, e28090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barber S. C., Shaw P. J. (2010) Oxidative stress in ALS. Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 48, 629–641 [DOI] [PubMed] [Google Scholar]
- 44. Simpson E. P., Yen A. A., Appel S. H. (2003) Oxidative stress. A common denominator in the pathogenesis of amyotrophic lateral sclerosis. Curr. Opin. Rheumatol. 15, 730–736 [DOI] [PubMed] [Google Scholar]
- 45. Pedersen W. A., Fu W., Keller J. N., Markesbery W. R., Appel S., Smith R. G., Kasarskis E., Mattson M. P. (1998) Protein modification by the lipid peroxidation product 4-hydroxynonenal in the spinal cords of amyotrophic lateral sclerosis patients. Ann. Neurol. 44, 819–824 [DOI] [PubMed] [Google Scholar]
- 46. Smith R. G., Henry Y. K., Mattson M. P., Appel S. H. (1998) Presence of 4-hydroxynonenal in cerebrospinal fluid of patients with sporadic amyotrophic lateral sclerosis. Ann. Neurol. 44, 696–699 [DOI] [PubMed] [Google Scholar]
- 47. Simpson E. P., Henry Y. K., Henkel J. S., Smith R. G., Appel S. H. (2004) Increased lipid peroxidation in sera of ALS patients. A potential biomarker of disease burden. Neurology 62, 1758–1765 [DOI] [PubMed] [Google Scholar]
- 48. Shibata N., Yamada S., Uchida K., Hirano A., Sakoda S., Fujimura H., Sasaki S., Iwata M., Toi S., Kawaguchi M., Yamamoto T., Kobayashi M. (2004) Accumulation of protein-bound 4-hydroxy-2-hexenal in spinal cords from patients with sporadic amyotrophic lateral sclerosis. Brain Res. 1019, 170–177 [DOI] [PubMed] [Google Scholar]
- 49. Miana-Mena F. J., González-Mingot C., Larrodé P., Muñoz M. J., Oliván S., Fuentes-Broto L., Martínez-Ballarín E., Reiter R. J., Osta R., García J. J. (2011) Monitoring systemic oxidative stress in an animal model of amyotrophic lateral sclerosis. J. Neurol. 258, 762–769 [DOI] [PubMed] [Google Scholar]
- 50. Kageyama H., Hirano T., Okada K., Ebara T., Kageyama A., Murakami T., Shioda S., Adachi M. (2003) Lipoprotein lipase mRNA in white adipose tissue but not in skeletal muscle is increased by pioglitazone through PPAR-γ. Biochem. Biophys. Res. Commun. 305, 22–27 [DOI] [PubMed] [Google Scholar]
- 51. Norris A. W., Chen L., Fisher S. J., Szanto I., Ristow M., Jozsi A. C., Hirshman M. F., Rosen E. D., Goodyear L. J., Gonzalez F. J., Spiegelman B. M., Kahn C. R. (2003) Muscle-specific PPARγ-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. J. Clin. Invest. 112, 608–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Amin R. H., Mathews S. T., Camp H. S., Ding L., Leff T. (2010) Selective activation of PPARγ in skeletal muscle induces endogenous production of adiponectin and protects mice from diet-induced insulin resistance. Am. J. Physiol. Endocrinol. Metab. 298, E28–37 [DOI] [PubMed] [Google Scholar]
- 53. Allen S., Heath P. R., Kirby J., Wharton S. B., Cookson M. R., Menzies F. M., Banks R. E., Shaw P. J. (2003) Analysis of the cytosolic proteome in a cell culture model of familial amyotrophic lateral sclerosis reveals alterations to the proteasome, antioxidant defenses, and nitric oxide synthetic pathways. J. Biol. Chem. 278, 6371–6383 [DOI] [PubMed] [Google Scholar]
- 54. Chen H., Guo Y., Hu M., Duan W., Chang G., Li C. (2010) Differential expression and alternative splicing of genes in lumbar spinal cord of an amyotrophic lateral sclerosis mouse model. Brain Res. 1340, 52–69 [DOI] [PubMed] [Google Scholar]
- 55. Paradis E., Clement S., Julien P., Ven Murthy M. R. (2003) Lipoprotein lipase affects the survival and differentiation of neural cells exposed to very low density lipoprotein. J. Biol. Chem. 278, 9698–9705 [DOI] [PubMed] [Google Scholar]
- 56. Yang Y., Cheng J. Z., Singhal S. S., Saini M., Pandya U., Awasthi S., Awasthi Y. C. (2001) Role of glutathione S-transferases in protection against lipid peroxidation. Overexpression of hGSTA2–2 in K562 cells protects against hydrogen peroxide-induced apoptosis and inhibits JNK and caspase 3 activation. J. Biol. Chem. 276, 19220–19230 [DOI] [PubMed] [Google Scholar]
- 57. Adibhatla R. M., Hatcher J. F. (2010) Lipid oxidation and peroxidation in CNS health and disease. From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 12, 125–169 [DOI] [PubMed] [Google Scholar]
- 58. Pedersen W. A., Cashman N. R., Mattson M. P. (1999) The lipid peroxidation product 4-hydroxynonenal impairs glutamate and glucose transport and choline acetyltransferase activity in NSC-19 motor neuron cells. Exp. Neurol. 155, 1–10 [DOI] [PubMed] [Google Scholar]
- 59. Vigh L., Smith R. G., Soós J., Engelhardt J. I., Appel S. H., Siklós L. (2005) Sublethal dose of 4-hydroxynonenal reduces intracellular calcium in surviving motor neurons in vivo. Acta Neuropathol. 109, 567–575 [DOI] [PubMed] [Google Scholar]
- 60. Esterbauer H., Schaur R. J., Zollner H. (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol. Med. 11, 81–128 [DOI] [PubMed] [Google Scholar]
- 61. Uchida K. (2003) 4-Hydroxy-2-nonenal. A product and mediator of oxidative stress. Prog. Lipid Res. 42, 318–343 [DOI] [PubMed] [Google Scholar]
- 62. Riahi Y., Sin-Malia Y., Cohen G., Alpert E., Gruzman A., Eckel J., Staels B., Guichardant M., Sasson S. (2010) The natural protective mechanism against hyperglycemia in vascular endothelial cells. Roles of the lipid peroxidation product 4-hydroxydodecadienal and peroxisome proliferator-activated receptor δ. Diabetes 59, 808–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cutler R. G., Pedersen W. A., Camandola S., Rothstein J. D., Mattson M. P. (2002) Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Ann. Neurol 52, 448–457 [DOI] [PubMed] [Google Scholar]
- 64. Dupuis L., Oudart H., René F., Gonzalez de Aguilar J. L., Loeffler J. P. (2004) Evidence for defective energy homeostasis in amyotrophic lateral sclerosis. Benefit of a high-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. U.S.A. 101, 11159–11164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fergani A., Oudart H., Gonzalez De Aguilar J. L., Fricker B., René F., Hocquette J. F., Meininger V., Dupuis L., Loeffler J. P. (2007) Increased peripheral lipid clearance in an animal model of amyotrophic lateral sclerosis. J. Lipid Res. 48, 1571–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kim S. M., Kim H., Kim J. E., Park K. S., Sung J. J., Kim S. H., Lee K. W. (2011) Amyotrophic lateral sclerosis is associated with hypolipidemia at the presymptomatic stage in mice. PLoS One 6, e17985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chiò A., Calvo A., Ilardi A., Cavallo E., Moglia C., Mutani R., Palmo A., Galletti R., Marinou K., Papetti L., Mora G. (2009) Lower serum lipid levels are related to respiratory impairment in patients with ALS. Neurology 73, 1681–1685 [DOI] [PubMed] [Google Scholar]
- 68. Dupuis L., Corcia P., Fergani A., Gonzalez De Aguilar J. L., Bonnefont-Rousselot D., Bittar R., Seilhean D., Hauw J. J., Lacomblez L., Loeffler J. P., Meininger V. (2008) Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 70, 1004–1009 [DOI] [PubMed] [Google Scholar]
- 69. Dorst J., Kühnlein P., Hendrich C., Kassubek J., Sperfeld A. D., Ludolph A. C. (2011) Patients with elevated triglyceride and cholesterol serum levels have a prolonged survival in amyotrophic lateral sclerosis. J. Neurol 258, 613–617 [DOI] [PubMed] [Google Scholar]