

Background: Caspase-6 is a critical factor in neurodegeneration, which is regulated by zinc binding.
Results: Caspase-6 is inhibited by zinc and binds one zinc/monomer at an exosite distal from the active site.
Conclusion: Zinc allosterically inhibits caspase-6 by locking it into a naturally occurring, inactive, and extended helical conformation.
Significance: The allosteric inhibition observed in the presence of zinc may aid in the development of allosteric caspase-6 drugs.
Keywords: Alzheimer Disease, Apoptosis, Cysteine Protease, Huntington Disease, Metal Homeostasis, Neurodegeneration, Anomalous Diffraction
Abstract
Zinc and caspase-6 have independently been implicated in several neurodegenerative disorders. Depletion of zinc intracellularly leads to apoptosis by an unknown mechanism. Zinc inhibits cysteine proteases, including the apoptotic caspases, leading to the hypothesis that zinc-mediated inhibition of caspase-6 might contribute to its regulation in a neurodegenerative context. Using inductively coupled plasma optical emission spectroscopy, we observed that caspase-6 binds one zinc per monomer, under the same conditions where the zinc leads to complete loss of enzymatic activity. To understand the molecular details of zinc binding and inhibition, we performed an anomalous diffraction experiment above the zinc edge. The anomalous difference maps showed strong 5σ peaks, indicating the presence of one zinc/monomer bound at an exosite distal from the active site. Zinc was not observed bound to the active site. The zinc in the exosite was liganded by Lys-36, Glu-244, and His-287 with a water molecule serving as the fourth ligand, forming a distorted tetrahedral ligation sphere. This exosite appears to be unique to caspase-6, as the residues involved in zinc binding were not conserved across the caspase family. Our data suggest that binding of zinc at the exosite is the primary route of inhibition, potentially locking caspase-6 into the inactive helical conformation.
Introduction
The human caspases are a family of cysteine-aspartic proteases that post-translationally cleave their substrates with high specificity at exposed aspartate-containing recognition sequences. Caspases were originally identified for inciting the complex process of apoptotic programmed cell death, but they have since been found to play a variety of roles in inflammation, differentiation, and development. Apoptotic caspases are categorized as either initiators (upstream, caspase-2, -8, and -9) or executioners (downstream, caspase-3, -6, and -7). Caspase-6 was originally identified as a mammalian ced-3 homologue, MCH-2, an apoptotic gene from human Jurkat T lymphocytes (1). Caspase-6 differs in several respects from the other executioner caspases. Caspase-6 is much more weakly apoptotic than caspase-3 and -7, although overexpression of caspase-6 does result in apoptosis (2). Caspase-6 also cleaves a different set of cellular substrates (3, 4) and displays different substrate specificity against peptide substrates (5). Because of its lower apoptotic potential, caspase-6 has been the subject of less investigation than other caspases, so new insights into caspase-6 physiological roles continue to be uncovered.
In the recent past, caspase-6 has come into prominence for its role in neurodegeneration. Caspase-6 is expressed in the brain (6–8) and bordering tissue (9) and cleaves several key targets that are critical in neurodegeneration. Caspase-6 cleaves amyloid precursor protein at position Asp-664 leading to production of the toxic APP-C31 fragment that leads to the amyloid-β peptide-independent neuronal death (10), although other caspases may also cleave at this site. Blocking this cleavage is reported to prevent development of the symptoms of Alzheimer disease in a mouse model (11). Caspase-6 has also been implicated in cleavage of the polyglutamine-expanded huntingtin protein (12). Of the caspases, only caspase-6 is activated by the death receptor 6, resulting in neuronal degeneration (13). Perhaps the most compelling evidence for the role of caspase-6 in neurodegeneration is the observation that of the caspases, only caspase-6 activation is observed in neuropil threads, neuritic plaques, and neurofibrillary tangles in both sporadic (6) and familial (14) Alzheimer disease. Cleavage of the Tau protein by active caspase-6 appears to be causal in formation of these disease lesions (7).
Many proteases, including caspase-6, are controlled via zymogen activation, in which proteolytic processing by an upstream protease results in the generation of a cleaved, active protease (for review see Ref. 15). Caspase zymogens (also called procaspases) are maintained in a full-length, unprocessed, and inactive dimeric form prior to induction of apoptosis or other activating stimuli. Processing of dimeric zymogens by an upstream caspase removes the prodomain and cleaves the intersubunit linker to form a heterotetramer, which is then active and competent to cleave cellular targets. Cleavage of the intersubunit linker produces two nascent loops that participate in a four-loop substrate-binding groove bundle. In caspase-6 this region is capable of a strand-to-helix transition that results in a unique helical conformation (16), which cannot be attained by any other caspase (17, 18). Caspase-6 is capable of autoactivation by self-cleavage of the intersubunit linker at position 193 following the sequence TEVD (19). In terms of both zymogen activation and loop control, caspase-6 is distinct from all other caspases.
Because of their cell death-inducing potential, the activities of all apoptotic caspases are tightly regulated. The regulation of each caspase is necessarily unique so that each can perform its independent and nonredundant cellular roles (20, 21). The most prevalent mechanisms of caspase regulation include zymogen activation, binding of inhibitor of apoptosis protein family members, post-translational modification, and metal binding. Among the apoptotic caspases, caspase-6 is uniquely resistant to X-linked inhibitor of apoptosis protein-mediated inhibition at either the active site or the dimer interface (22), underscoring the fact that regulation of caspase-6 is distinctive. To date, only caspase-6 has been reported to be inactivated by phosphorylation at Ser-257 by ARK5 kinase (2), which allosterically prevents the proper conformation of one of the substrate-binding loops (23). The understanding of the unique features of such regulatory pathways improves our ability to utilize caspase-6 as a therapeutic target.
The effect of zinc on caspase-6 has not been explored; however, the relationship of zinc levels to apoptotic potential has been studied. Extremely high zinc concentrations (>500 μm) trigger apoptosis, but at physiologically relevant concentrations, zinc is a suppressor of apoptosis (24, 25). Depletion of zinc also leads to apoptosis. Mouse embryos on a zinc-deficient diet suffer from neural tube defects, retarded development, and apoptotic cell death (26), likely due to loss of zinc-mediated caspase inhibition. The inhibition of caspases by transition metals, including zinc and copper, was reported soon after the discovery of caspases (27–29). Exposure of caspases to metal ions alters their cell death potential. In allergic asthma, the loss of airway epithelial zinc was accompanied by changes in the expression of caspase-3 and apoptosis (30, 31). Treatment of HL-60 cells with zinc chelators increases apoptosis via activation of caspase-3 (32). The small molecule procaspase-3 activator PAC-1 releases procaspase-3 from zinc-mediated inhibition (33–35). The details of caspase-3 zinc-mediated inhibition have not been reported. We have observed that caspase-9 is inhibited by zinc but not by other metals and that caspase-9 binds two zincs per monomer. One zinc binds at the active site, and the second zinc binds at an exosite composed of residues Cys-230, His-224, and Cys-272 (36). Together, these studies suggest that when the zinc concentration in the cell is lowered, zinc-mediated inhibition of caspases is relieved, and an increase in caspase activation leads to apoptotic cell death.
Zinc is an essential element but is not evenly distributed throughout the body. Mounting evidence suggests that zinc is involved in a variety of neurological diseases (37). Zinc is found at extremely high concentrations in brain lesions (plaque rim, 1024 μm; plaque core, 1327 μm; senile plaque, 1055 μm, and Alzheimer neuropil, 786 μm) (38, 39). Zinc accumulation in brain is a prominent feature of advanced Alzheimer disease and is biochemically linked to amyloid-β accumulation and dementia severity. In Alzheimer disease patients, zinc concentrations in post-mortem brain tissue correlated with the severity of the case, whereas the concentrations of other metals did not differ with severity or relative to control tissues (40). Intriguingly, these tissues with zinc accumulation are the same tissues in which caspase-6 activation precedes and leads to the formation of neurofibrillary tangles but does not lead to cell death (7, 42–44).
The spatial overlap between zinc buildup and caspase-6 function in neurodegeneration combined with the new molecular details about caspase-9 inhibition by zinc prompted us to explore the interaction between caspase-6 and zinc as it relates to neurological disorders. In this study, we explore the ability of divalent metals to inactivate caspase-6 and uncover the structural mechanism by which caspase-6 metal binding results in inhibition.
EXPERIMENTAL PROCEDURES
Generation of Caspase-6 Variants
The wild-type caspase-6 constructs consisted of a synthetic gene encoding the Escherichia coli codon-optimized, full-length, C-terminal His6-tagged human CASPASE-6 gene comprising amino acids 1–293 plus His6 ligated into the NdeI/BamHI sites of the pET11a vector (Stratagene) or from a constitutive two-chain (CT)3 version of the synthetic CASPASE-6 gene (Celtek Bioscience) (16). Amino acid substitutions were introduced by the QuikChange® site-directed mutagenesis method (Stratagene) in the full-length construct or the CT construct. All DNA sequences were verified by DNA sequencing.
Caspase-6 Expression and Purification
The caspase-6 expression constructs were transformed into the BL21 (DE3) T7 Express strain of E. coli (New England Biolabs). The cultures were grown in 2× YT media with ampicillin (100 mg/liter, Sigma) at 37 °C until they reached an absorbance at 600 nm of 0.6 (A600 = 0.6). The temperature was reduced to 20 °C, and cells were induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside (Anatrace) to express soluble protein. Cells were harvested after 18 h of expression to ensure complete processing. Cell pellets were stored at −20 °C, freeze-thawed, and lysed in a microfluidizer (Microfluidics, Inc.) in a buffer containing 50 mm Tris, pH 8.5, 300 mm NaCl, 5% glycerol, and 2 mm imidazole. Lysed cells were centrifuged at 18,500 rpm to remove cellular debris. The supernatant was loaded onto a 5-ml HiTrap nickel-affinity column (GE Healthcare). The column was washed with a buffer of 50 mm Tris, pH 8.5, 300 mm NaCl, 5% glycerol, and 50 mm imidazole, and the protein was eluted with the buffer containing 50 mm Tris, pH 8.5, 300 mm NaCl, 5% glycerol, and 250 mm imidazole. The eluted fraction was diluted by 9-fold into a buffer containing 20 mm Tris, pH 8.5, and 2 mm DTT to reduce the salt concentration. This protein sample was loaded onto a 5-ml Macro-Prep High Q column (Bio-Rad). The column was developed with a linear NaCl gradient, and caspase-6 was eluted in a buffer of 20 mm Tris, pH 8.5, 120 mm NaCl, and 2 mm DTT. The eluted protein was stored at −80 °C in the above buffer conditions. The identity of the purified caspase-6 wild type and variants was analyzed by SDS-PAGE to be 98% pure, and ESI-MS was used to confirm mass and purity.
Caspase-3 and -7 Expression and Purification
The CASPASE-3 full-length human gene in the pET23b vector (Addgene) or the caspase-7 C186A variant (zymogen), made by QuikChange® mutagenesis (Stratagene) of the human CASPASE-7 gene in pET23b (gift of Guy Salvesen), was transformed into BL21 (DE3) T7 Express strain of E. coli. Induction of caspase-3 was at 30 °C for 3 h; caspase-7 was at 18 °C for 18 h. These proteins were purified as described previously for caspase-3 (29) and caspase-7 (45). The eluted proteins were stored at −80 °C in the buffer in which they were eluted. The identity of purified caspase-3 and -7 was assessed by SDS-PAGE and ESI-MS to confirm mass and purity.
Activity Assays Using Peptide Substrates
For kinetic measurements of caspase activity, 100 nm freshly purified protein was assayed over the course of 15 min in a caspase-6 activity buffer containing 100 mm HEPES, pH 7.5, 10% sucrose, 0.1% CHAPS, 20 mm TCEP, and 120 mm NaCl. For substrate titrations, a range of 0–500 μm fluorogenic substrates, N-acetyl-Val-Glu-Ile-Asp-7-amino-4-methylcoumarin (VEID-AMC) (Enzo Life Sciences) excitation 365 nm/emission 495 nm was added to initiate the reaction. Assays were performed in triplicate at 25 °C in 100-μl volumes in a 96-well microplate format using a Molecular Devices Spectramax M5 spectrophotometer. Initial velocities versus substrate concentration were fit to a rectangular hyperbola using GraphPad Prism (Graphpad Software) to determine kinetic parameters Km and kcat. Enzyme concentrations were determined by active-site titration with the quantitative inhibitor VEID-CHO (N-acetyl-Val-Glu-Ile-Asp-aldehyde, Enzo Life Sciences). Active-site titrations were incubated over a period of 2 h in 100 mm HEPES, pH 7.5, 10% sucrose, 0.1% CHAPS, 20 mm TCEP, and 120 mm NaCl at nanomolar concentrations of VEID-CHO. Optimal labeling was observed when protein was added to VEID-CHO solvated in DMSO in 96-well V-bottom plates, sealed with tape, and incubated at room temperature in a final volume of 200 μl. 90-μl aliquots were transferred to black-well plates in duplicate and assayed with a 50-fold molar excess of substrate. The protein concentration was determined to be the lowest concentration at which full inhibition was observed. The inhibitory concentrations IC50 for zinc were determined by performing metal titrations from 0.03 to 450 μm ZnCl2 in the presence of 20 nm caspase-6 and 300 μm of the fluorogenic substrate VEID-AMC and fitting the data for initial velocity as a function of zinc using Equation 1.
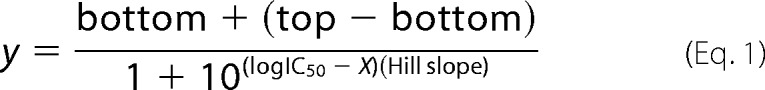 |
The inhibitory constant Ki was fitted from the same titrations using the Origin software with Equation 2 using final enzyme and fluorogenic substrate concentrations of 20 nm and 300 μm, respectively.
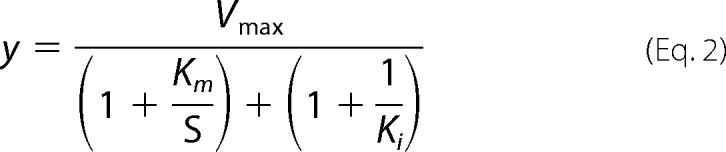 |
Proteolytic Cleavage of Protein Substrates by Active Caspase-6
To test the ability of various metal cations to inhibit caspase-6 activity, we performed proteolytic cleavage assays against full-length procaspases-6 C163S, monitoring cleavage at the intersubunit linker position Asp-193. The full-length procaspase-6 C163S is catalytically inactive and is not capable of self-cleavage at this position, but an active caspase-6 can cleave at the intersubunit linker position Asp-193 and be monitored by assessing migration on a gel. Each metal was tested at a 6-fold molar excess incubated for 30 min at room temperature. For testing cleavage of full-length procaspase-6 C163S, 1 μm full-length procaspase-6 C163S was incubated with 1 μm active caspase-6 in a caspase-6 assay buffer of 100 mm HEPES, pH 7.5, 10% sucrose, 0.1% CHAPS, 20 mm TCEP, and 120 mm NaCl in a 37 °C water bath for 1 h. Samples were analyzed by 16% SDS-PAGE. The inhibitory concentrations (IC50) for zinc against a natural protein substrate were determined by quantification of the cleavage products in the gel. Gel band quantifications were done by measuring band intensities in a SYGENE BIO IMAGING gel doc with the Gene Tools 6.07 software. The full-length procaspase-6 C163S was set at 100%, and quantifications were measured as both the disappearance of the full-length bands and appearance of cleavage bands in the gel.
Zinc Binding Stoichiometry by Inductively Coupled Plasma-Optical Emission Spectroscopy
Purified caspase-6 and variants (10 μm) in 20 mm Tris, pH 8.5, 120 mm NaCl, and 20 mm TCEP were incubated with 6 eq excess of ZnCl2 for 30 min at room temperature. Unbound zinc was removed by incubating samples of 1.5-ml volume in the presence of ∼125 mg of Chelex® 100 for 1 h, mixing every 15 min. A control sample of 1.5 ml of 20 mm Tris, pH 8.5, 120 mm NaCl, and 5 mm TCEP buffer was treated in precisely the same manner to assess the completeness of unbound zinc chelation. Wild-type caspase-6 full-length samples treated with zinc or untreated were also used as controls for metal content of pre- and post-zinc incubation. Zinc content of the sample supernatant was quantified using an Optima 4300 DV (PerkinElmer Life Sciences) inductively coupled plasma-optical emission spectrometer (ICP-OES) equipped with a 40-MHz free-running generator and a segmented-array charge-coupled device detector. The sample introduction system consisted of a concentric nebulizer with a cyclonic spray chamber. The concentration of zinc in each sample was determined at 206.2 nm wavelength and converted from ppm to μm to obtain the zinc to monomer of caspase-6 ratio. Post-ICP-OES analysis, the remaining samples were tested via absorbance at 280 nm to determine the protein concentration.
Crystallization and Data Collection
Purified CT caspase-6 (ΔN D179 CT construct lacking both the prodomain and the intersubunit linker, in which the large and small subunits are independently translated from two translational start sites during overexpression (16)) in 20 mm Tris, pH 8.5, 2 mm DTT, and 120 mm NaCl was concentrated using Amicon Ultrafree 5K NMWL membrane concentrators (Millipore Inc.) to 6 mg/ml. The crystals grew in 0.1 m sodium acetate, pH 4.6, 2 m NaCl, and 3% ethanol (Hampton Research, HR2-112) in 2–3 days at room temperature in a hanging drop setup. The caspase-6 crystals were soaked for 10 min in mother liquor with 10 mm ZnCl2 and extensively washed for 10 min with cryo-protectant 25% ethylene glycol to remove unbound zinc. The crystals were cryo-protected in 25% ethylene glycol and mother liquor and stored in liquid nitrogen. Complete data sets of diffraction images were collected at X6A beamline at Brookhaven National Laboratories National Synchrotron Light Source (Upton, NY).
Structure Determination
An x-ray fluorescence scan was carried out to verify the absorption by zinc in the caspase-6 crystals. A clear absorbance peak indicated the presence of zinc, and an anomalous diffraction data for zinc-soaked caspase-6 was collected above the zinc absorption edge (1.2861 Å). The zinc-bound caspase-6 crystals diffracted to 2.8 Å resolution. The data were processed and scaled in HKL2000 in the primitive monoclinic space group P21 with CCP4i (46). The search model successfully used for molecular replacement using Phaser (47) was a dimer of caspase-6 (Protein Data Bank code 2WDP, 1.6 Å resolution). To avoid phase bias, we omitted the substrate-binding loops comprising residues 53–66 (L1 loop), 164–174 (L2), 184–195 (L2′), 211–254 (L3), and 257–277 (L4). This solution comprising four monomers in the asymmetric unit was built into the electron density by iterative rounds of model building in Coot (48) and NCS restrained refinement using CCP4i (46). The structure was refined using automated refinement in Refmac (49) and TLS to a final R/Rfree of 21.5:23.3% and was validated by Procheck (50) using CCP4i and Molprobity (51). The final model is under Protein Data Bank code 4FXO.
RESULTS
Zinc Is the Only Transition Metal to Inhibit Caspase-6
A number of caspases, including caspase-6, have been reported to be inhibited by zinc (52), but the effects of other transition metals on caspase-6 function have not been reported. To test the ability of caspase-6 to be inhibited by transition metals, we utilized a self-cleavage assay. Whereas a number of caspases autoprocess upon heterologous overexpression in bacteria, only for caspase-6 is a structurally characterized mechanism of autoactivation known (53). This autoactivation occurs by cleavage of the intersubunit linker at position 193 following the sequence TEVD. The uncleaved caspase-6 zymogen is inactive, and cleavage of the intersubunit linker, which generates the two-chain form, is essential for caspase-6 activity (16). The effect of various metal cations on caspase-6 proteolytic activity was assessed by monitoring cleavage of the full-length inactive zymogen (FL C163S, which serves as the substrate), by active, mature caspase-6 (ΔN D179CT, the enzyme) (Fig. 1A). Only zinc and not other metal cations, including iron, nickel, cobalt, copper, manganese, potassium, calcium, and magnesium, was capable of inhibiting caspase-6 (Fig. 1B). This inhibition occurred in a caspase-6 construct that lacks the prodomain and the intersubunit linker, indicating that the inhibition observed occurs in the core domain of caspase-6 and not in the prodomain or intersubunit linker. Caspase-3 shares 33% sequence identity with caspase-6 and has been shown to be sensitive to transition metals like zinc, cupper, cobalt, and iron (33, 52).
FIGURE 1.

Zinc is a potent inhibitor of caspase-6 activity. A, expression constructs showing sites of cleavage (gap) in full-length (FL) wild-type (WT), full-length zymogen (FL C163S), and constitutive two-chain (CT) caspase-6. Protein domains include the prodomain (gray circle), large subunit (gray bar), intersubunit linker (ISL, black line), and small subunit (black bar) with the active site cysteine (black C) mutated to serine (black S) in the large subunit. Residue numbers for each domain are indicated. RBS indicates ribosome-binding sites for initiation of translation in the CT construct. B, caspase-6-mediated cleavage of caspase-6 FL C163S zymogen. Metal inhibition was tested in an electrophoretic mobility gel-based assay as fragments produced from active caspase-6 (ΔND179CT) mediated cleavage of the full-length C163S zymogen that serves as the substrate. Fragments from cleavage include the following: full-length (FL), FL lacking the N-terminal prodomain (ΔN-FL), large (Lg), and small subunits (Sm) with the amino acids present in those bands (subscripts) labeled. Zinc is the only metal cation that inhibits caspase-6 activity. C, Caspase-6-mediated cleavage of caspase-6 FL C163S zymogen was tested at various zinc concentrations in an electrophoretic mobility gel-based assay. The readout for the assay was caspase-6 fragments produced from active caspase-6-mediated cleavage at the intersubunit linker of the zymogen. Proteolytic fragments are labeled as in B. D, in-gel IC50 band quantification measuring band intensities. The full-length procaspase-6 FL C163S was set as 100% (uncleaved). Band quantification was measured as disappearance of these bands and appearance of cleavage bands in the gel. The IC50 value is reported as the zinc concentration at which 50% of FL C163S has disappeared and 50% of the large subunit has appeared. The IC50 value for zinc inhibition is 0.37 μm. E, dose-response of caspase-6 inhibition in the presence of zinc and a fluorogenic substrate (Ac-VEID-AMC). IC50 was fit from independent duplicate metal titration on three separate days.
We were somewhat surprised by the specificity of zinc-mediated inhibition of caspase-6 due to the moderate sequence similarity and reasonably strong structural similarity between these two executioner caspases. To further characterize the inhibitory properties of zinc, we performed kinetic analysis in the presence of increasing concentrations of a real protein substrate (FL C163S) or the fluorogenic tetrapeptide substrate acetyl-valine-glutamate-isoleucine-aspartate-aminomethylcoumarin (Ac-VEID-AMC). Using a protein substrate, caspase-6 is completely inhibited at 1.5 μm zinc (Fig. 1C) with an IC50 of 0.37 μm (Fig. 1D). The result with a fluorogenic tetrapeptide substrate differs, giving an IC50 for zinc of 3.4 ± 0.5 μm and a Ki of 0.15 ± 0.04 μm (Fig. 1E) similar to values reported by Stennicke and Salvesen (29), who also used a benzyloxycarbonyl-Asp-Glu-Val-Asp-AFC fluorogenic tetra-peptide substrate. We note that these differences may be due to the size of the substrates used, as a small tetrapeptide (VEID) binds locally only in the active site, whereas a protein substrate (299 amino acids) may make contact with other regions of the caspase-6 enzyme (54). Although the Ki is in the high nanomolar range, this may in fact be biologically relevant, as the zinc concentration in brain tissue, which is where caspase-6 plays its critical roles in neurodegeneration, can be in the millimolar range (38, 39).
Caspase-6 Binds One Zinc/Monomer
We have recently reported two zinc bindings sites on caspase-9 (36), an initiator caspase that shares 31% amino acid identity in the core domain with caspase-6. The caspase-9 zinc-binding sites were found using a combination of computational prediction of metal-binding sites, site-directed mutagenesis, and zinc quantification by ICP-OES.
One zinc binds in the caspase-9 active site, coordinated by the catalytic dyad residues Cys-287 and His-237 with Cys-239 as a third ligand. The second zinc binds at an exosite composed of His-224, Cys-229, Cys-230, and Cys-272. All caspases contain the same catalytic dyad. In caspase-6 Glu-123 is at an appropriate position to serve a third potential zinc ligand in place of Cys-239. All but one of the amino acids forming the caspase-9 exosite appear in caspase-6, with the exception of the caspase-9 Cys-230 position that is the nonmetal-binding residue Phe-114 in caspase-6. Given that three of the four ligands were present in the exosite and three residues are present in the active site, one might predict that like caspase-9, two binding sites would be observed in caspase-6. We were surprised to find that caspase-6 binds just one zinc/monomer (Table 1), as quantified by ICP-OES.
TABLE 1.
Wild-type caspase-6 binds one zinc/monomer
Zinc binding in caspase-6 was monitored by ICP-OES. S.D. of samples was measured on 4 separate days. Two sites were computationally predicted to bind zinc via HotPatch (41) and ZincPred (55). Zinc binding is at the active site and at the caspase-9 exosite (His-108/Cys-148) in caspase-6 numbering.
| Caspase | Zinc/monomer ratio |
|---|---|
| Caspase-6 | |
| WT | 1.0 ± 0.2 |
| C163S | 0.8 ± 0.1 |
| H108A/C148A | 1.2 ± 0.3 |
| Caspase-9 (36) | 1.8 ± 0.3 |
To interrogate zinc binding at the active site, we quantified zinc bound to the C163S version of caspase-6, which lacks the catalytic cysteine that served as a zinc ligand in caspase-9. Zinc binding was unchanged in C163S, indicating that the sole zinc-binding site does not involve the catalytic dyad at the active site. We designed the double mutant His-108/Cys-148 to knock out the exosite identified in caspase-9. His-108/Cys-148 also retained zinc binding ability (Table 1) suggesting that zinc does not bind at the exosite homologous to that identified in caspase-9. Thus, it appeared that zinc must bind in a site that is unique for caspase-6.
Zinc Binds an Exosite in Caspase-6
Caspase-6 binds just one zinc and that zinc exerts its inhibitory role from a site distinct from the active site or the caspase-9 exosite. To identify the zinc-binding site in caspase-6, we determined the structure of caspase-6 using anomalous diffraction at the zinc edge. To introduce zinc, crystals of wild-type caspase-6 were soaked in a solution of 10 mm ZnCl2 for 10 min and then extensively washed with a zinc-free solution. The x-ray absorbance of these crystals was scanned over the range 9600–9700 eV (Fig. 2). Absorption at the zinc edge (9670 eV) confirmed that zinc had bound to crystallized caspase-6 and was stable to the rigorous washing procedure. X-ray diffraction data were collected above the zinc absorption edge. The structure was solved to 2.83 Å resolution by molecular replacement and refined to Rcryst/Rfree of 21/23% (Table 2). The zinc-bound structure shows four monomers or two identical dimers in the asymmetric unit (Fig. 3A). Each dimer represents one functional biological unit composed of two large and two small subunits. Caspase-6 structures in the unliganded, mature (54, 67), VEID-bound (17) zymogen (17) and zinc-bound form all exhibit the same core fold (Fig. 3B). Caspase-6 is the only caspase known to exist in a latent, highly helical conformation where the 60s and 130s helices are elongated and the 90s helix is rotated out relative to the canonical fold (16, 18). We soaked zinc into crystals of this helical conformation and found that the zinc-bound structure remains in the latent helical form (Fig. 3B).
FIGURE 2.

Caspase-6 crystals bind zinc. An x-ray fluorescence scan measuring intensity (e) as a function of x-ray energy (eV) shows absorption at the zinc energy edge, 9673.92 eV, indicating that zinc is bound in caspase-6 crystals even after 10 min of washing.
TABLE 2.
Crystallographic data collection and statistics
Values in parentheses are for the highest resolution bin. rmsd, root mean square deviation.
| Data collection statistics | |
| Wavelength | 1.28 Å |
| Diffraction resolution | 50.0 -to 2.83 Å (2.92 to 2.83 Å) |
| Measured reflections (n) | 158,964 |
| Unique reflections | 22,033 |
| Completeness | 99.5% (97.4%) |
| Redundancy | 7.2 (5.3) |
| I/σ(I) | 70.7/4.7 (5.2 / 2.6 = 2) |
| Rsym | 0.13 (0.92) |
| Space group | P21 |
| a | 62.7 Å |
| b | 90.9 Å |
| c | 85.8 Å |
| α = γ | 90.0° |
| β | 90.3° |
| Refinement statistics | |
| No. of atoms | 7227 |
| No. waters | 8 |
| Rwork/Rfree | 21.5%/23.3% |
| rmsd bond length | 0.006 Å |
| rmsd bond angle | 0.736° |
| Average B-factor | 36.1 Å2 |
| Ramachandran plot | |
| Core | 94.1% |
| Allowed | 5.5% |
| Disallowed | 0.3% |
FIGURE 3.

Zinc-bound caspase-6 retains fold. A, zinc-bound caspase-6 retains the same fold as zinc-free caspase-6, with one zinc bound per monomer. The zinc (blue sphere), zinc ligands Lys-36, Glu-244, and His-287 (sticks), and a water (red sphere) are shown. B, superposition of available caspase-6 structures underscores that zinc does not change the overall fold in caspase-6. Zinc-bound caspase-6 (4FXO, light blue) is in the helical conformation seen in unliganded, mature caspase-6 (3K7E, orange), which differs from VEID-bound (3OD5, red) caspase-6. The substrate VEID (gray spheres) indicates the substrate-binding region. Only in mature caspase-6 and the zinc-bound form of caspase-6 are the extended helical state of the 60s and 130s helices observed. C, superposition of caspase-6 active site in the apo, mature (3K7E, orange), and zinc-bound (4FXO, light blue) structures. Of the residues in this region, only His-219 undergoes a significant shift in conformation. D, superposition of VEID-bound (3OD5, red) and zinc-bound (4FXO, light blue) caspase-6 highlights the significant conformational change that caspase-6 undergoes during conversion from the helical to canonical structures. The substrate-mimic VEID (gray) is labeled as D1, I2, E3, and V4 to indicate the peptide subsite positions within the peptide.
Structurally, there are several differences near the active site and within the substrate-binding loops that are unique to the zinc-bound structure (Fig. 3, C and D). Specifically, within loop L3 several of the amino acids that form the subsites for substrate binding are present in different rotomeric conformations than in the apo-mature form, which we consider to be a natively populated, inactive structure (Fig. 3C). The cysteine-histidine dyad responsible for peptide bond hydrolysis as well as the L1 and L3 loops is dramatically misaligned in the zinc-bound structure relative to the active VEID-bound conformation (Fig. 3D). Activity of a caspase or any enzyme requires that the enzyme bind substrate in the appropriate orientation to facilitate the chemical reaction and that the side chains involved in catalysis be properly positioned. This misalignment may contribute to the inhibitory effect observed by zinc, but a similar misalignment with different rotomeric conformations has been observed in the apo-mature structure making it difficult to conclude that the zinc binding is entirely responsible for the transition to the inactive conformation.
To unambiguously locate the zinc in the structure, we calculated an anomalous difference electron density map from data collected above the zinc edge. This map showed one strong 5σ peak per monomer, clearly indicating a primary zinc bound at the bottom of the helix that serves as the base of the L4 loop. The amino acids directly involved in zinc binding are Lys-36, Glu-244, His-287, with a water molecule serving as the fourth ligand (Fig. 4A). The ligand composition and distorted tetrahedral coordination geometry suggest that this is a structural zinc. The geometry of the active site, in which the Cys-His dyad are 9 Å apart (Fig. 4B), explains why this conformation of the active site is incapable of binding zinc, whereas a properly ordered active site might be capable of ligating zinc.
FIGURE 4.

Anomalous difference map shows location of zinc binding in caspase-6. A, anomalous difference map calculated from data collected above the zinc absorbance edge is contoured at 5σ (purple), clearly indicating the location of zinc. The side chains and water serving as ligands are drawn as sticks. The 2Fo − Fc electron density map (blue) into which the structure was build is contoured at 1σ. B, caspase-6 dimer. The boxed regions shows active site, which contains residues appropriate for metal binding including His-121, Glu-126, and Cys-163. In this structure, these residues are not properly positioned to coordinate zinc. The caspase-6 zinc-binding exosite is shown within the side chain ligands for zinc in sticks, zinc (blue), and the water molecule (red).
To test the relevance of the zinc-binding site identified by the anomalous diffraction map, we prepared caspase-6 variants in which the zinc-binding residues have been removed. The zinc-binding site variants bound zinc with markedly lower stoichiometry than wild type. In K36A, E244A, H287A, or the double mutant E244A/H287A, zinc binding is ablated (Table 3). The fact that substitution of the zinc ligands by alanine ablates zinc binding confirms that the site occupied with zinc in the crystal structure is also the site occupied in solution and measured by ICP-OES.
TABLE 3.
Catalytic properties and ICP-OES of caspase-6 variants
Substrate titrations to assess Km and kcatvalues were measured from independent duplicate dilutions of substrate on 2 separate days. Zinc binding in caspase-6 was monitored by ICP-OES, and S.D. of samples measured on 4 separate days.
| Zinc/monomer ratio | Km | 10−3 × kcat | 10−3 × kcat/Km | IC50 peptide | |
|---|---|---|---|---|---|
| μm | s−1 | s−1/μm | μm | ||
| WT | 1.0 ± 0.2 | 93 ± 10.1 | 700 ± 1.4 | 7.5 | 3.4 ± 0.5 |
| K36A | 0.4 ± 0.1 | 109 ± 10.5 | 371 ± 25 | 3.4 | 15.8 ± 1.3 |
| E244A | 0.4 ± 0.3 | 82 ± 4.5 | 51 ± 5.8 | 0.6 | 10.7 ± 2.2 |
| H287A | 0.2 ± 0.2 | 140 ± 15.8 | 309 ± 35 | 2.2 | 24.9 ± 2.5 |
| E244A/H287A | 0.3 ± 0.2 | 176 ± 14.2 | 15 ± 15.6 | 0.1 | 25.6 ± 3.5 |
Zinc Allosterically Inhibits Caspase-6
The fact that the active site of caspase-6 does not bind the inhibitory zinc suggests that the exosite for zinc binding should function as an allosteric inhibitory site. To assess this, we tested the caspase-6 variants, K36A, E244A, H287A, and E244A/H287A for inhibition by zinc. Surprisingly, these alanine substitutions were not entirely functionally silent. The function of K36A, E244A, H287A, or the double mutant E244A/H287A is crippled by 2-, 2-, 14-, or 47-fold, respectively, suggesting that this site is important for activity. Nevertheless, the enzymes retained sufficient activity that we expect the overall fold to be unaltered. Titrations of zinc allowed fitting of the inhibitory properties in caspase-6 variants lacking one or more of the zinc ligands. All of the variants in which the zinc-liganding residues are removed have lost a significant amount of sensitivity toward zinc, showing IC50 values about 1 order of magnitude weaker than wild type (Table 3). His-287 appears to be the primary zinc-binding ligand as the alanine mutant H287A shows the worst IC50 at 24.9 ± 2.5 μm. The E244A/H287A double mutant has similar inhibitory properties with an IC50 at 25.6 ± 3.5 μm. K36A IC50 at 15.8 ± 1.3 μm and E244A IC50 at 10.7 ± 2.2 μm have the least effect on inhibition (Table 3), suggesting these ligands are less critical for zinc binding. Overall, these significant decreases in the inhibitory ability suggest that this caspase-6 exosite is the primary zinc-based inhibitory site and that zinc can allosterically inhibit caspase-6 activity.
Caspase-6 Zinc Exosite Is Distinct from the Caspase-9 Exosite
This caspase-6 allosteric site is the second zinc-binding exosite that we have identified in caspases (Fig. 5, A and B). We recently identified another independent exosite for zinc binding in caspase-9 comprising His-224, Cys-229, Cys-230, and Cys-272 (36). The caspase-9 site is nonoverlapping with the caspase-6 zinc exosite. Although overall caspases share a high degree of sequence similarity (caspase-6 shares 37, 33, 29, and 28% identity with caspase-3, -7, -8, and -9, respectively), neither of these sites are strictly conserved.
FIGURE 5.

Caspase-6 exosite is not conserved. A, structure-based sequence alignment of the apoptotic human caspases in the regions of the zinc-binding sites. The active site is conserved but the caspase-6 or -9 exosites are not conserved across the apoptotic caspases. Alignments were performed structurally, and residues are numbered according to the numbering of each individual caspase. The presence of insertions and deletions in the sequences of various caspases explains why the intervals between the sites differ. B, caspase-6 in the canonical conformation shows the active site and zinc-binding exosites circled. Zinc-binding ligands are in sticks and the zincs are shown as gray spheres. The caspase-6 zinc-binding site is shown as in the crystal structure. The zinc-bound conformations of the active site and the caspase-9 exosites were modeled by selecting a new plausible rotomeric conformation for the zinc-liganding side chains and modeling a zinc molecule with proper distance for zinc binding. C, Ki value for wild-type caspase-6 inhibition by zinc is much stronger than caspase-6 variants in which the zinc ligands are deleted, suggesting that the exosite is the primary inhibitory site for zinc. This is in contrast with caspase-9, in which there is no statistical difference in the activity when the caspase-9 exosite is ablated by the C272S substitution.
The caspase-6 exosite is the most poorly conserved of the binding sites, with only one of the ligands (Glu-244) being conserved. This suggests that this site may be unique to caspase-6. The caspase-9 exosite is somewhat more conserved with three of the four ligands present across the apoptotic caspases. Across the caspase family, the most highly conserved zinc-binding site is the active site, which is composed of the conserved catalytic cysteine-histidine dyad (His-121 and Cys-163 in caspase-6) and a third strong metal ligand such as Asp-123 for caspase-3, Glu-146 in caspase-7, or Glu-123 in caspase-6.
In both solution and crystals, the dominant zinc-binding site for caspase-6 is the exosite. This is likely due to the fact that mature (cleaved) caspase-6 rests in the noncanonical helical conformation as we observe in the zinc-bound structure. In this conformation, the active site dyad is not optimally positioned for either catalysis or zinc binding (Fig. 4B). Nevertheless, we wondered whether comparing the zinc inhibitory properties of caspase-6 to those of caspase-9 would provide further insight into the mechanistic details of zinc binding and inhibition. The Ki value for zinc-mediated inhibition of wild-type caspase-6 (0.15 ± 0.04 μm) is substantially stronger than any of the mutants in which zinc-binding residues are removed (Fig. 5C). This loss of inhibitory activity suggests that the unique caspase-6 exosite is the primary inhibitory site. Conversely, wild-type caspase-9 has a Ki for zinc of 1.5 ± 0.3 μm, which does not statistically differ when exosite zinc binding is ablated, as in C272S (Fig. 5C), establishing the active site as the primary site of inhibition in caspase-9.
Given that the constellations of zinc-binding residues in the caspase-6 and caspase-9 active sites are similar, we expect that the Ki value for zinc-mediated inhibition at the active site of caspase-6 could likewise be of similar magnitude. The magnitude of the Ki for zinc in the caspase-6 variants that ablate zinc binding at the exosite is similar to that of caspase-9, which is predominantly inhibited at the active site. This suggests that caspase-6 could potentially be inhibited at the active site with a Ki in the range of 3–7 μm. Active site inhibition of caspase-6 should only be possible after caspase-6 transitions to the canonical conformation, which occurs in the presence of substrate (16, 19). It is possible that zinc could potentially act upon caspase-6 in two ways, through a stronger allosteric mechanism at the exosite and through a weaker competitive mechanism at the active site.
Zinc Binds to Both Initiator and Executioner Caspases
Caspase-6 and caspase-9 are both inhibited by zinc, but at different stoichiometries. This led us to interrogate zinc binding in other caspases. Our findings suggest that each caspase may have its own unique zinc-binding profile. Caspase-3 binds three zincs per monomer and caspase-7 binds just one zinc/monomer (Table 4). A priori, it is tempting to speculate that caspase-7 may bind the single zinc at the active site because it cannot attain the helical conformation that we see in caspase-6 (17). It is difficult to predict the zinc-binding locations in caspase-3. It seems likely that the active site is one of the three caspase-3 zinc-bindings sites. Because neither the caspase-9 nor the caspase-6 exosite is conserved in caspase-3 (Fig. 5A), this analysis would predict two new binding sites should be present in caspase-3. It remains to be seen if these two sites would be of functional significance, similar to the site we have identified in caspase-6.
TABLE 4.
Zinc binds both initiator and executioner caspases at various ratios
Zinc binding in caspases was monitored by ICP-OES. S.D. of samples was measured on 4 separate days.
| Enzyme | Zinc/monomer ratio |
|---|---|
| Caspase-6 | 1.0 ± 0.2 |
| Caspase-3 | 3.2 ± 0.4 |
| Caspase-7 | 0.7 ± 0.1 |
| Caspase-9 (36) | 1.8 ± 0.3 |
DISCUSSION
Because of their critical roles in irreversible processes like apoptosis, inflammation, and neurodegeneration, caspases are highly regulated proteins. It is becoming clear that each caspase is subject to a unique combination of activating and inhibitory processes, including upstream zymogen activation (mediated by an initiator caspase as part of a large multicomponent complex such as the DISC or apoptosome), phosphorylation, and other post-translational modifications. Caspase-6 differs from other family members in that it is not subject to inhibition by inhibitor of apoptosis proteins and can autoactivate under certain conditions. We have shown that caspase-6 can be inhibited by zinc allosterically. The underlying physiological reason as to why caspases evolved to bind zinc is not entirely clear, but we speculate that the native affinity of a cysteine-histidine dyad requires an appropriate response in a zinc-rich environment. Caspase-6, in particular, is known to play a role in cells and tissues with extremely high zinc concentrations (38, 39), so perhaps it was essential that caspase-6 evolve mechanisms to respond to these conditions. Beyond this, why caspase-6 should be subject to allosteric inhibition by zinc remains somewhat unclear. One possible explanation is that zinc can allosterically inhibit caspase-6 regardless of its structural state, zymogen, helical conformation, or mature canonical conformation, and it is thus a global means of caspase-6 inhibition.
Caspase-6 is inhibited by zinc but not by other cationic metals. This is somewhat unusual, given the relative similarity in size and charge of zinc and many other transition metal cations. The coordination sphere in the caspase-6 inhibitory site is composed of Lys-36, Glu-244, His-287, and a water molecule, in a distorted tetrahedral geometry. Glutamate and histidine are common ligands for cations. Coordination of zinc by lysine is not common but is not unheard of, as it has been observed in 1% of structural zinc-binding sites in proteins deposited in the Protein Data Bank (56–61). Although lysine can coordinate zinc, it has not been observed to coordinate other metals. For example, in the type I copper-binding protein amicyanin, the mutation of one of the copper ligands, methionine, to lysine converted the enzyme from copper binding to selectively zinc binding, demonstrating the specificity of lysine as a zinc ligand (62). Thus, the specificity of caspase-6 for inhibition by zinc may be the result of the rare ligand, Lys-36, participating in the coordination sphere.
The caspase-6 zinc-binding exosite is distal from the active site and is capable of leading to enzymatic inhibition, categorizing it as an allosteric site. Allosteric regulation is traditionally viewed as a binding event at one site that indirectly affects the activity at another via inducing a number of linked conformational changes in the protein. Caspases seem to be ripe for allosteric regulation, likely because of the extreme plasticity of the loops that compose the active site groove. We have previously observed how phosphorylation of caspase-6 at Ser-257 results in a steric clash of as few as three atoms in the side chain of Pro-201, which leads to allosteric inhibition by preventing the active conformation of one loop. In caspase-7 (45, 63, 64) or -1 (65, 66), binding small synthetic allosteric inhibitors at the dimer interface leads to a series of side chain conformational changes that ultimately block the substrate-binding groove, preventing proteolytic activity. These examples and a great number of other examples from a large number of proteins are satisfying as it is possible to visualize the series of side chain conformational changes that lead to inactivation. In this structure of zinc-bound caspase-6, we cannot trace a chain of discrete conformational changes in adjacent side chains that lead to inactivation.
A second class of allosteric regulators of enzymes modulate the thermodynamic equilibrium of various folded states that leads to disruption of the enzymatic properties without causing obvious changes along a pathway of side chains linking the allosteric and active sites. Examples of this class include peptide inhibitors of factor VIIa (67, 68) and small molecule inhibitors of β-lactamase (69). Very recently, a novel allosteric peptide, pep419, which binds near the 90s helix in caspase-6 inducing a tetrameric state, has been reported to function via this second class of allosteric mechanism. The authors state the following (70): “The effect of pep419 is not the transmission of a specific conformational change from the peptide-binding site to the active site but rather a more global effect on protein dynamics whereby peptide binding induces tetramerization and, most likely, a structural rigidity that prevents catalytic turnover.” Of course in both classes of allosteric inhibition, the same fundamental thermodynamic properties control the equilibrium, and it is simply the ability to visualize these pathways that differs between the two classes. We posit that zinc falls into this second class of allosteric modulators, locking caspase-6 into the inactive helical conformation by altering the equilibrium ensemble in both the absence and presence of substrate (Fig. 6).
FIGURE 6.

Model for the mechanism of caspase-6 inhibition by zinc. Caspase-6 exists in equilibrium between the helical conformation and the canonical conformation. Prior to substrate binding, the equilibrium strongly favors the helical conformation. To bind substrate, caspase-6 has to reorder the ends of two elongated helices into the strands or loops observed in the canonical form that are compatible with substrate binding. In the presence of zinc, the helical form of caspase-6 binds only one zinc/monomer at the exosite and is allosterically inhibited at this exosite. In the helical conformation, the active site does not bind zinc due to the disorganization of the active site residues, which are also favorable zinc-liganding residues. When caspase-6 is in the canonical conformation, zinc can also bind at the active site so that two zincs per monomer bind and inhibit caspase-6.
We were surprised to observe that substitution of one of the residues in the caspase-6 zinc-binding exosite, Glu-244, had such deleterious effects on enzymatic function. This significant loss in activity may suggest that Glu-244 sits in a sensitive site and leads us to speculate about the role of this site. Only one exosite for substrate binding has been identified in any caspase. In caspase-7 a tetra-lysine sequence at residue 38 (38KKKK) is critical for caspase-7 to bind and distinguish protein substrates (54). Lys-36 is near the location of the caspase-7 exosite, is close to the N terminus of caspase-6, and is one of the first residues to be crystallographically ordered following the disordered prodomain, leading us to ponder whether this zinc-binding site may be involved in regulating substrate recognition. The fact that the Ki value for zinc is stronger for protein substrates than for peptide substrates (Fig. 1, B–E) further supports this idea. Although E244A is an isolated mutation, it may be worthwhile to note that many mysteries about the regulation of caspase-6 remain, including the role of the prodomain and its interaction with other regions of caspase-6. Glu-244 is not involved in any interactions in any caspase-6 structure that would obviously lead to loss of function. Thus, further examination of this mutation may provide new insights into caspase-6 activity.
In the caspase family, only caspase-6 can transition between a canonical and a helical conformation (16). There have been differing opinions about the relevance of this helical conformation (18, 71). We have observed a spectroscopic signature that indicates that caspase-6 is helical in solution at neutral pH in the absence of substrate and is converted to the helical conformation upon binding substrate (16). The active site of caspase-6 presents a constellation of ligands that should be well suited to bind zinc when they are properly positioned for catalysis, as occurs in the canonical conformation. We have shown that deleting the active site zinc-binding ligands has no effect on zinc binding ability, implicating the caspase-6 exosite as the primary zinc-binding and inhibitory site. At the same time, this result provides insight into the solution-based conformation of caspase-6. The fact that caspase-6 does not principally bind zinc in the active site, as does caspase-9, suggests that in solution the active site is not competent for zinc binding. The most likely explanation for this is that caspase-6 exists in the helical conformation in solution so the catalytic cysteine-histidine dyad are far apart from one another and therefore cannot bind zinc at the active site. Thus these data provide a second piece of evidence that caspase-6 exists in the helical conformation in solution in the absence of substrate, which may make the helical conformation an attractive target for inhibitor design.
Acknowledgments
We thank Kristen Huber for many helpful discussions; Michael Maroney, Nitai Giri, and Julius Campecino for assistance with ICP-OES analysis and insights on zinc coordination geometry; and Scott Garman for exceptional help in calculating the anomalous difference map. We thank Vivian Stojanoff for assistance with data collection and acknowledge NSLS X6A, which is funded by the National Institutes of Health Grant GM-0080, and the National Synchrotron Light Source at Brookhaven National Laboratory, which is supported by the United States Department of Energy Grant DE-AC02-98CH10886.
This work was supported, in whole or in part, by National Institutes of Health Grant GM80532.
The atomic coordinates and structure factors (code 4FXO) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- CT
- constitutive two-chain
- VEID
- valine-glutamate-isoleucine-aspartate (a caspase-6 recognition sequence)
- CHO
- aldehyde
- AMC
- aminomethylcoumarin
- ΔN
- N-terminal prodomain deleted
- TCEP
- (tris(2-carboxyethyl)phosphine)
- FL C163S
- full-length inactive zymogen caspase-6 with the catalytic cysteine replaced by serine
- ICP-OES
- inductively coupled plasma-optical emission spectrometer.
REFERENCES
- 1. Fernandes-Alnemri T., Litwack G., Alnemri E. S. (1995) Mch2, a new member of the apoptotic Ced-3/Ice cysteine protease gene family. Cancer Res. 55, 2737–2742 [PubMed] [Google Scholar]
- 2. Suzuki A., Kusakai G., Kishimoto A., Shimojo Y., Miyamoto S., Ogura T., Ochiai A., Esumi H. (2004) Regulation of caspase-6 and FLIP by the AMPK family member ARK5. Oncogene 23, 7067–7075 [DOI] [PubMed] [Google Scholar]
- 3. Ruchaud S., Korfali N., Villa P., Kottke T. J., Dingwall C., Kaufmann S. H., Earnshaw W. C. (2002) Caspase-6 gene disruption reveals a requirement for lamin A cleavage in apoptotic chromatin condensation. EMBO J. 21, 1967–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Takahashi A., Alnemri E. S., Lazebnik Y. A., Fernandes-Alnemri T., Litwack G., Moir R. D., Goldman R. D., Poirier G. G., Kaufmann S. H., Earnshaw W. C. (1996) Cleavage of lamin A by Mch2α but not CPP32. Multiple interleukin 1β-converting enzyme-related proteases with distinct substrate recognition properties are active in apoptosis. Proc. Natl. Acad. Sci. U.S.A. 93, 8395–8400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thornberry N. A., Rano T. A., Peterson E. P., Rasper D. M., Timkey T., Garcia-Calvo M., Houtzager V. M., Nordstrom P. A., Roy S., Vaillancourt J. P., Chapman K. T., Nicholson D. W. (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 272, 17907–17911 [DOI] [PubMed] [Google Scholar]
- 6. Albrecht S., Bourdeau M., Bennett D., Mufson E. J., Bhattacharjee M., LeBlanc A. C. (2007) Activation of caspase-6 in aging and mild cognitive impairment. Am. J. Pathol. 170, 1200–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guo H., Albrecht S., Bourdeau M., Petzke T., Bergeron C., LeBlanc A. (2004) Active caspase-6 and caspase-6-cleaved Tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer disease. Am. J. Pathol. 165, 523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Graham R. K., Deng Y., Carroll J., Vaid K., Cowan C., Pouladi M. A., Metzler M., Bissada N., Wang L., Faull R. L., Gray M., Yang X. W., Raymond L. A., Hayden M. R. (2010) Cleavage at the 586-amino acid caspase-6 site in mutant huntingtin influences caspase-6 activation in vivo. J. Neurosci. 30, 15019–15029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Singh A. B., Kaushal V., Megyesi J. K., Shah S. V., Kaushal G. P. (2002) Cloning and expression of rat caspase-6 and its localization in renal ischemia/reperfusion injury. Kidney Int. 62, 106–115 [DOI] [PubMed] [Google Scholar]
- 10. Sivananthan S. N., Lee A. W., Goodyer C. G., LeBlanc A. C. (2010) Familial amyloid precursor protein mutants cause caspase-6-dependent but amyloid β-peptide-independent neuronal degeneration in primary human neuron cultures. Cell Death Dis. 1, e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Galvan V., Gorostiza O. F., Banwait S., Ataie M., Logvinova A. V., Sitaraman S., Carlson E., Sagi S. A., Chevallier N., Jin K. (2006) Reversal of Alzheimer-like pathology and behavior in human APP transgenic mice by mutation of Asp-664. Proc. Natl. Acad. Sci. U.S.A. 103, 7130–7135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hermel E., Gafni J., Propp S. S., Leavitt B. R., Wellington C. L., Young J. E., Hackam A. S., Logvinova A. V., Peel A. L., Chen S. F., Hook V., Singaraja R., Krajewski S., Goldsmith P. C., Ellerby H. M., Hayden M. R., Bredesen D. E., Ellerby L. M. (2004) Specific caspase interactions and amplification are involved in selective neuronal vulnerability in Huntington disease. Cell Death Differ. 11, 424–438 [DOI] [PubMed] [Google Scholar]
- 13. Nikolaev A., McLaughlin T., O'Leary D. D., Tessier-Lavigne M. (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457, 981–989 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14. Sultana R., Banks W. A., Butterfield D. A. (2010) Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment. Insights into their potential roles for loss of synapses and memory, accumulation of Aβ, and neurodegeneration in a prodromal stage of Alzheimer disease. J. Neurosci. Res. 88, 469–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fuentes-Prior P., Salvesen G. S. (2004) The protein structures that shape caspase activity, specificity, activation, and inhibition. Biochem. J. 384, 201–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vaidya S., Velázquez-Delgado E. M., Abbruzzese G., Hardy J. A. (2011) Substrate-induced conformational changes occur in all cleaved forms of caspase-6. J. Mol. Biol. 406, 75–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vaidya S., Hardy J. A. (2011) Caspase-6 latent state stability relies on helical propensity. Biochemistry 50, 3282–3287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baumgartner R., Meder G., Briand C., Decock A., D'arcy A., Hassiepen U., Morse R., Renatus M. (2009) The crystal structure of caspase-6, a selective effector of axonal degeneration. Biochem. J. 423, 429–439 [DOI] [PubMed] [Google Scholar]
- 19. Wang X. J., Cao Q., Liu X., Wang K. T., Mi W., Zhang Y., Li L. F., LeBlanc A. C., Su X. D. (2010) Crystal structures of human caspase 6 reveal a new mechanism for intramolecular cleavage self-activation. EMBO Rep. 11, 841–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walsh J. G., Cullen S. P., Sheridan C., Lüthi A., Gerner C., Martin S. J. (2008) Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proc. Natl. Acad. Sci. U.S.A. 105, 12815–12819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Slee E. A., Adrain C., Martin S. J. (2001) Executioner caspase-3, -6, and -7 perform distinct, nonredundant roles during the demolition phase of apoptosis. J. Biol. Chem. 276, 7320–7326 [DOI] [PubMed] [Google Scholar]
- 22. Deveraux Q. L., Takahashi R., Salvesen G. S., Reed J. C. (1997) X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388, 300–304 [DOI] [PubMed] [Google Scholar]
- 23. Velázquez-Delgado E. M., Hardy J. A. (2012) Phosphorylation regulates assembly of the caspase-6 substrate-binding groove. Structure 20, 742–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiménez Del Río M., Vélez-Pardo C. (2004) Transition metal-induced apoptosis in lymphocytes via hydroxyl radical generation, mitochondria dysfunction, and caspase-3 activation. An in vitro model for neurodegeneration. Arch. Med. Res. 35, 185–193 [DOI] [PubMed] [Google Scholar]
- 25. Rudolf E., Cervinka M. (2010) Zinc pyrithione induces cellular stress signaling and apoptosis in Hep-2 cervical tumor cells. The role of mitochondria and lysosomes. Biometals 23, 339–354 [DOI] [PubMed] [Google Scholar]
- 26. Rogers J. M., Taubeneck M. W., Daston G. P., Sulik K. K., Zucker R. M., Elstein K. H., Jankowski M. A., Keen C. L. (1995) Zinc deficiency causes apoptosis but not cell cycle alterations in organogenesis-stage rat embryos. Effect of varying duration of deficiency. Teratology 52, 149–159 [DOI] [PubMed] [Google Scholar]
- 27. Chimienti F., Seve M., Richard S., Mathieu J., Favier A. (2001) Role of cellular zinc in programmed cell death. Temporal relationship between zinc depletion, activation of caspases, and cleavage of Sp family transcription factors. Biochem. Pharmacol. 62, 51–62 [DOI] [PubMed] [Google Scholar]
- 28. Schrantz N., Auffredou M. T., Bourgeade M. F., Besnault L., Leca G., Vazquez A. (2001) Zinc-mediated regulation of caspases activity. Dose-dependent inhibition or activation of caspase-3 in the human Burkitt lymphoma B cells (Ramos). Cell Death Differ. 8, 152–161 [DOI] [PubMed] [Google Scholar]
- 29. Stennicke H. R., Salvesen G. S. (1997) Biochemical characteristics of caspases-3, -6, -7, and -8. J. Biol. Chem. 272, 25719–25723 [DOI] [PubMed] [Google Scholar]
- 30. Truong-Tran A. Q., Grosser D., Ruffin R. E., Murgia C., Zalewski P. D. (2003) Apoptosis in the normal and inflamed airway epithelium. Role of zinc in epithelial protection and procaspase-3 regulation. Biochem. Pharmacol. 66, 1459–1468 [DOI] [PubMed] [Google Scholar]
- 31. Truong-Tran A. Q., Ruffin R. E., Foster P. S., Koskinen A. M., Coyle P., Philcox J. C., Rofe A. M., Zalewski P. D. (2002) Altered zinc homeostasis and caspase-3 activity in murine allergic airway inflammation. Am. J. Respir. Cell Mol. Biol. 27, 286–296 [DOI] [PubMed] [Google Scholar]
- 32. Kondoh M., Tasaki E., Takiguchi M., Higashimoto M., Watanabe Y., Sato M. (2005) Activation of caspase-3 in HL-60 cells treated with pyrithione and zinc. Biol. Pharm. Bull. 28, 757–759 [DOI] [PubMed] [Google Scholar]
- 33. Peterson Q. P., Goode D. R., West D. C., Ramsey K. N., Lee J. J., Hergenrother P. J. (2009) PAC-1 activates procaspase-3 in vitro through relief of zinc-mediated inhibition. J. Mol. Biol. 388, 144–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lucas P. W., Schmit J. M., Peterson Q. P., West D. C., Hsu D. C., Novotny C. J., Dirikolu L., Churchwell M. I., Doerge D. R., Garrett L. D., Hergenrother P. J., Fan T. M. (2011) Pharmacokinetics and derivation of an anticancer dosing regimen for PAC-1, a preferential small molecule activator of procaspase-3, in healthy dogs. Invest. New Drugs 29, 901–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peterson Q. P., Hsu D. C., Goode D. R., Novotny C. J., Totten R. K., Hergenrother P. J. (2009) Procaspase-3 activation as an anti-cancer strategy. Structure-activity relationship of procaspase-activating compound 1 (PAC-1) and its cellular co-localization with caspase-3. J. Med. Chem. 52, 5721–5731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huber K. L., Hardy J. A. (2012) Mechanism of zinc-mediated inhibition of caspase-9. Protein Sci. 21, 1056–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li Y., Hawkins B. E., DeWitt D. S., Prough D. S., Maret W. (2010) The relationship between transient zinc ion fluctuations and redox signaling in the pathways of secondary cellular injury. Relevance to traumatic brain injury. Brain Res. 1330, 131–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lovell M. A., Robertson J. D., Teesdale W. J., Campbell J. L., Markesbery W. R. (1998) Copper, iron, and zinc in Alzheimer disease senile plaques. J. Neurol. Sci. 158, 47–52 [DOI] [PubMed] [Google Scholar]
- 39. Popescu B. F., Nichol H. (2011) Mapping brain metals to evaluate therapies for neurodegenerative disease. CNS Neurosci. Ther. 17, 256–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Religa D., Strozyk D., Cherny R. A., Volitakis I., Haroutunian V., Winblad B., Naslund J., Bush A. I. (2006) Elevated cortical zinc in Alzheimer disease. Neurology 67, 69–75 [DOI] [PubMed] [Google Scholar]
- 41. Pettit F. K., Bare E., Tsai A., Bowie J. U. (2007) HotPatch. A statistical approach to finding biologically relevant features on protein surfaces. J. Mol. Biol. 369, 863–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. de Calignon A., Fox L. M., Pitstick R., Carlson G. A., Bacskai B. J., Spires-Jones T. L., Hyman B. T. (2010) Caspase activation precedes and leads to tangles. Nature 464, 1201–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. LeBlanc A., Liu H., Goodyer C., Bergeron C., Hammond J. (1999) Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer disease. J. Biol. Chem. 274, 23426–23436 [DOI] [PubMed] [Google Scholar]
- 44. Rissman R. A., Poon W. W., Blurton-Jones M., Oddo S., Torp R., Vitek M. P., LaFerla F. M., Rohn T. T., Cotman C. W. (2004) Caspase-cleavage of Tau is an early event in Alzheimer disease tangle pathology. J. Clin. Invest. 114, 121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Witkowski W. A., Hardy J. A. (2009) L2′ loop is critical for caspase-7 active site formation. Protein Sci. 18, 1459–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Collaborative Computational Project, Number 4 (1994) The CCP4 suite programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 47. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 49. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 50. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK. A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 51. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity. All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Perry D. K., Smyth M. J., Stennicke H. R., Salvesen G. S., Duriez P., Poirier G. G., Hannun Y. A. (1997) Zinc is a potent inhibitor of the apoptotic protease, caspase-3. A novel target for zinc in the inhibition of apoptosis. J. Biol. Chem. 272, 18530–18533 [DOI] [PubMed] [Google Scholar]
- 53. Klaiman G., Champagne N., LeBlanc A. C. (2009) Self-activation of caspase-6 in vitro and in vivo. Caspase-6 activation does not induce cell death in HEK293T cells. Biochim. Biophys. Acta 1793, 592–601 [DOI] [PubMed] [Google Scholar]
- 54. Boucher D., Blais V., Denault J. B. (2012) Caspase-7 uses an exosite to promote poly(ADP ribose) polymerase 1 proteolysis. Proc. Natl. Acad. Sci. U.S.A. 109, 5669–5674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shu N. (2010) Protein Structure Prediction: Zinc-binding Sites, One-dimensional Structure, and Remote Homology, pp. 47–52, Stockholm University, Elsevier Science Publishers B.V., Amsterdam [Google Scholar]
- 56. Sousa S. F., Lopes A. B., Fernandes P. A., Ramos M. J. (2009) The zinc proteome. A tale of stability and functionality. Dalton Trans., 7946–7956 [DOI] [PubMed] [Google Scholar]
- 57. Vallee B. L., Auld D. S. (1993) Zinc. Biological functions and coordination motifs. Acc. Chem. Res. 26, 543–551 [Google Scholar]
- 58. Vallee B. L., Auld D. S. (1993) New perspective on zinc biochemistry. Cocatalytic sites in multi-zinc enzymes. Biochemistry 32, 6493–6500 [DOI] [PubMed] [Google Scholar]
- 59. Alberts I. L., Nadassy K., Wodak S. J. (1998) Analysis of zinc-binding sites in protein crystal structures. Protein Sci. 7, 1700–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Maret W. (2005) Zinc coordination environments in proteins determine zinc functions. J. Trace Elem. Med. Biol. 19, 7–12 [DOI] [PubMed] [Google Scholar]
- 61. Maret W. (2012) New perspectives of zinc coordination environments in proteins. J. Inorg. Biochem. 111, 110–116 [DOI] [PubMed] [Google Scholar]
- 62. Sukumar N., Choi M., Davidson V. L. (2011) Replacement of the axial copper ligand methionine with lysine in amicyanin converts it to a zinc-binding protein that no longer binds copper. J. Inorg. Biochem. 105, 1638–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hardy J. A., Lam J., Nguyen J. T., O'Brien T., Wells J. A. (2004) Discovery of an allosteric site in the caspases. Proc. Natl. Acad. Sci. U.S.A. 101, 12461–12466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hardy J. A., Wells J. A. (2009) Dissecting an allosteric switch in caspase-7 using chemical and mutational probes. J. Biol. Chem. 284, 26063–26069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Datta D., Scheer J. M., Romanowski M. J., Wells J. A. (2008) An allosteric circuit in caspase-1. J. Mol. Biol. 381, 1157–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Scheer J. M., Romanowski M. J., Wells J. A. (2006) A common allosteric site and mechanism in caspases. Proc. Natl. Acad. Sci. U.S.A. 103, 7595–7600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lee A. W., Champagne N., Wang X., Su X. D., Goodyer C., Leblanc A. C. (2010) Alternatively spliced caspase-6B isoform inhibits the activation of caspase-6A. J. Biol. Chem. 285, 31974–31984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Roberge M., Santell L., Dennis M. S., Eigenbrot C., Dwyer M. A., Lazarus R. A. (2001) A novel exosite on coagulation factor VIIa and its molecular interactions with a new class of peptide inhibitors. Biochemistry 40, 9522–9531 [DOI] [PubMed] [Google Scholar]
- 69. Yoo N. J., Kim M. S., Park S. W., Seo S. I., Song S. Y., Lee J. Y., Lee S. H. (2010) Expression analysis of caspase-6, caspase-9, and BNIP3 in prostate cancer. Tumori 96, 138–142 [DOI] [PubMed] [Google Scholar]
- 70. Stanger K., Steffek M., Zhou L., Pozniak C. D., Quan C., Franke Y., Tom J., Tam C., Elliott J. M., Lewcock J. W., Zhang Y., Murray J., Hannoush R. N. (2012) Allosteric peptides bind a caspase zymogen and mediate caspase tetramerization. Nat. Chem. Biol. 8, 655–660 [DOI] [PubMed] [Google Scholar]
- 71. Müller I., Lamers M. B., Ritchie A. J., Park H., Dominguez C., Munoz-Sanjuan I., Maillard M., Kiselyov A. (2011) A new apo-caspase-6 crystal form reveals the active conformation of the apoenzyme. J. Mol. Biol. 410, 307–315 [DOI] [PubMed] [Google Scholar]