

Background: ATP7B trafficking from Golgi to vesicles is essential for copper homeostasis.
Results: Mutating Ser-340/341 alters the inter-domain contacts, diminishes protein phosphorylation, and shifts ATP7B localization to vesicles.
Conclusion: Spatial arrangement of functional domains determines ATP7B readiness to traffic, whereas phosphorylation may maintain the trafficking-compatible state.
Significance: The proposed mechanism explains how unrelated signals trigger similar trafficking responses from ATP7B.
Keywords: ATPases, Copper Transport, Golgi, Phosphorylation, Trafficking, ATP7A, ATP7B, P-type ATPase, TGN, Copper
Abstract
The copper-transporting ATPase ATP7B has a dual intracellular localization: the trans-Golgi network (TGN) and cytosolic vesicles. Changes in copper levels, kinase-mediated phosphorylation, and mutations associated with Wilson disease alter the steady-state distribution of ATP7B between these compartments. To identify a primary molecular event that triggers ATP7B exit from the TGN, we characterized the folding, activity, and trafficking of the ATP7B variants with mutations within the regulatory N-terminal domain (N-ATP7B). We found that structural changes disrupting the inter-domain contacts facilitate ATP7B exit from the TGN. Mutating Ser-340/341 in the N-ATP7B individually or together to Ala, Gly, Thr, or Asp produced active protein and shifted the steady-state localization of ATP7B to vesicles, independently of copper levels. The Ser340/341G mutant had a lower kinase-mediated phosphorylation under basal conditions and no copper-dependent phosphorylation. Thus, negative charges introduced by copper-dependent phosphorylation are not obligatory for ATP7B trafficking from the TGN. The Ser340/341A mutation did not alter the overall fold of N-ATP7B, but significantly decreased interactions with the nucleotide-binding domain, mimicking consequences of copper binding to N-ATP7B. We propose that structural changes that specifically alter the inter-domain contacts initiate exit of ATP7B from the TGN, whereas increased phosphorylation may be needed to maintain an open interface between the domains.
Introduction
Copper homeostasis plays an important role in human physiology. Copper is both a catalytic and a structural cofactor for a variety of enzymes essential for human cellular metabolism (cytochrome c oxidase, superoxide dismutase, dopamine-β-hydroxylase, etc). Under physiologic conditions, copper cycles between two oxidation states, Cu(I) and Cu(II); and copper levels must be tightly regulated to avoid participation in unwanted reactions affecting an intracellular redox status (1). Copper homeostasis is primarily maintained by the copper-transporting ATPases ATP7A and ATP7B. Both ATP7A and ATP7B are P-type ATPases that use the energy of ATP hydrolysis to transport copper across membranes; mutations in these proteins result either in copper accumulation in the body (Wilson's disease) or copper deficiency (Menkes disease) (2–7).
ATP7B is highly expressed in the liver and has a dual function in hepatocytes. Under basal conditions, it transports copper into the TGN, where copper is incorporated into cuproenzymes. Elevation of intracellular copper triggers ATP7B trafficking from the TGN to vesicles, which accumulate copper and then fuse with the apical plasma membrane to excrete copper out of the cell. Thus, ATP7B trafficking is essential for regulation of cellular copper levels. Mechanistic link between copper sensing by ATP7B and recognition of ATP7B by trafficking machinery is not yet understood, although involvement of such proteins as COMMD1, dynactin, clusterin, and glutaredoxin have been proposed (8–15).
In addition to regulating ATP7B localization, copper modulates ATP7B phosphorylation by kinases. In low copper, ATP7B has a basal level of phosphorylation. Copper elevation results in a hyperphosphorylation of ATP7B, and this event is coupled to trafficking of ATP7B from the TGN to vesicles, whereas dephosphorylation is associated with the return of ATP7B from vesicles to the TGN (16). The basal and copper-induced phosphorylation sites are different and involve different kinases (16). Mass spectrometry studies revealed a large number of potential phosphorylation sites (17, 18); simultaneous mutation of four such sites disrupts ATP7B trafficking (19). However, the cumulative effects of this multi-site mutation on either ATP7B structure, activity, or phosphorylation level have not been explored, thus specific reason for the disrupted trafficking remains uncertain.
Biochemical and cellular studies also revealed the important role of the cytosolic N-terminal domain of ATP7B (N-ATP7B)3 in both copper sensing and kinase-mediated phosphorylation. N-ATP7B is composed of six metal binding domains (MBDs) that are connected by flexible loops. Each MBD binds one copper (Fig. 1A) (20, 21). Copper binding to all 6 sites induces significant conformational changes in N-ATP7B (22) that are accompanied by the rearrangements of the loops connecting MBD's (17) and decreased interactions with the nucleotide-binding domain, NBD (23). The recombinant N-ATP7B is phosphorylated in vitro and in cells (17), and in the full-length ATP7B, deletion of the N-terminal region results in the loss of copper-dependent phosphorylation (16). Together, these observations suggested that copper binding to N-ATP7B induces structural changes favorable for kinase-mediated phosphorylation and led to the hypothesis that phosphorylation may act as a signal for ATP7B exit from the TGN.
FIGURE 1.
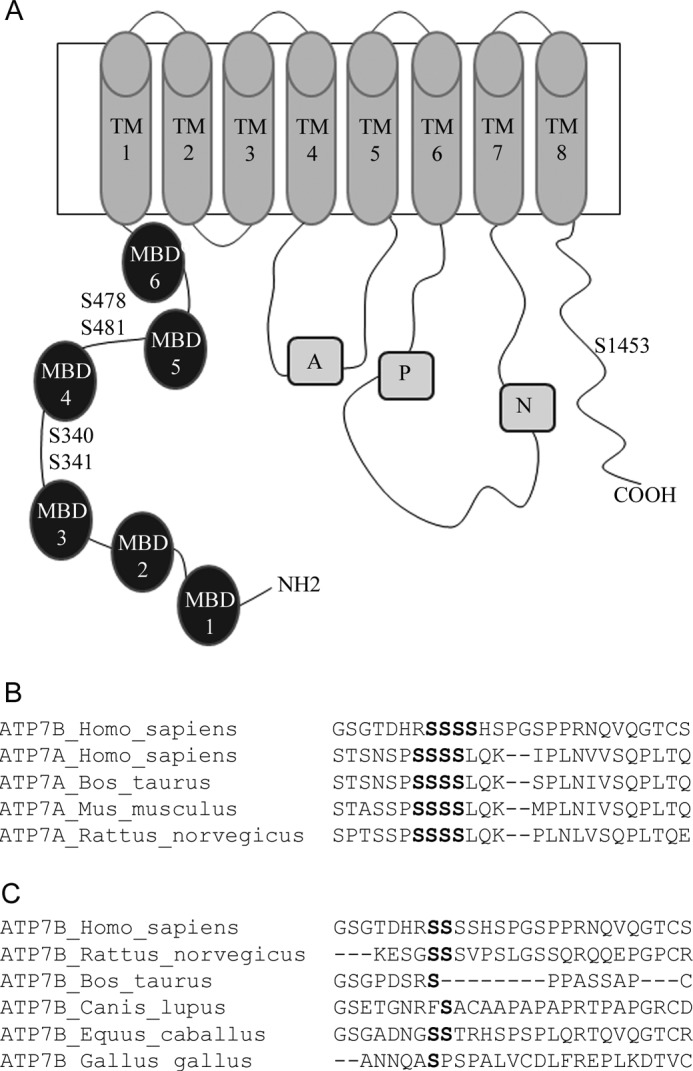
Schematic representation of ATP7B showing main functional domains and sites of kinase-mediated phosphorylation. A, filled circles represent the N-terminal MBDs and are numbered 1 to 6. Transmembrane segments are numbered TM1 to TM8. The other domains are as follows: A: actuator domain; P: phosphorylation domain; N: nucleotide-binding domain. The position of serine residues mutated in this study is indicated by S and the residue number. B, sequence similarity of the Ser-rich cluster (shown in bold) in human ATP7B (Ser-340–Ser-343) and mammalian ATP7A orthologs. C, sequence alignment in the region containing Ser-340/341 in ATP7B orthologs; residues that align with human ATP7B Ser-340 and Ser-341 are shown in bold.
However, other reports indicate that ATP7B exit from the TGN can be induced by mutation of residues that are not directly involved in either copper binding or phosphorylation. The N41S mutation results in a poor TGN retention and mis-targeting to the basolateral membrane even when copper is low (24). A triple mutant 858TGE860→AAA is found in vesicles in either basal or high copper (25). This latter mutant is catalytically inactive and trapped in a hyperphosphorylated state (although in this case the increased phosphorylation could be due to increased stability of a catalytic acyl-phosphate intermediate (25). These observations suggest an alternative model according to which stabilization of ATP7B in a distinct conformational state is sufficient to cause its traffic from the TGN (25).
To gain insight into the mechanism of ATP7B exit from the TGN, we have focused on the Ser-rich region in N-ATP7B, which we previously identified as a region of a kinase-mediated phosphorylation (17). We demonstrate that mutating Ser-340/341 in this region results in ATP7B acquiring conformation that resembles its copper saturated state. Although Ser-340/341 mutant has reduced levels of kinase-mediated phosphorylation, it shows poor retention in the TGN and preferential localization to vesicles. Our data suggest that exit of ATP7B from the TGN is predominantly governed by a conformational state of the protein and requires copper transfer from the cytosol.
EXPERIMENTAL PROCEDURES
Cell Lines
Hek293TRex cells were maintained in MEM supplemented with 10% FBS, 1% pen/strep, 1% non-essential amino acids, 12.5 μg/ml blasticidin, and 100 μg/ml zeocin. During experiments, media without blasticidin and zeocin was used. YSTT cells were maintained in DMEM supplemented with 10% FBS, 1% pen/strep, 1% non essential amino acids, 200 μg/ml G418, and 0.5 μg/ml puromycin. Hek293 cells were cultured in DMEM containing 10% FBS, 1% pen/strep, and 1% non-essential amino acids.
Site-directed Mutagenesis in ATP7B
The previously generated construct of N-terminally Flag-tagged ATP7B in pcDNA 5.1 FRT/TO vector was used as a template (26). The Ser→Ala, Ser→Gly, Ser→Thr, and Ser→Asp mutations were introduced by PCR using the Quickchange XL mutagenesis kit (Stratagene). All primers were obtained from Invitrogen, and the primer sequences are available upon request. The presence of the mutation and the fidelity of the cDNA sequence were verified by DNA sequencing of the coding region.
Tyrosinase Activation Assay
YSTT cells grown on glass coverslips were transfected with either 2 μg of pcTYR (a plasmid encoding apo-tyrosinase) or with 2 μg of pcTYR and 2 μg of Flag-ATP7B plasmids using Turbofect (Fermentas). Positive control (wt GFP-ATP7B, known as pYG7 (9)) and negative control (only pcTYR) were used to ensure the specificity of the assay. 24 h post-transfection, cells were washed with PBS and fixed in cold acetone-methanol for 30 s. The cells were then incubated for 4 h at 37 °C in 0.1 m Na-phosphate buffer (pH 6.8) containing 0.15% (w/v) levo-3,4-dihydroxy-l-phenylalanine (l-DOPA). Coverslips were mounted, and the formation of the black DOPA-chrome pigment was detected by phase microscopy. Pigment color intensity was quantified using Image J.
Analysis of Kinase-mediated Phosphorylation
Hek293TRex cells were grown in 6 cm dishes until confluent. The cells were transiently transfected with either wt Flag-ATP7BR875 or Flag-ATP7BR875,S340/341G plasmids using Turbofect (Fermentas). Protein expression was induced with 40 ng/ml DOX (Sigma) for 16 h in basal conditions. Cells were rinsed with PBS and kept in the phosphate-free DMEM for 3 h. Then, media was changed to fresh phosphate-free DMEM (Invitrogen) containing 0.3 mCi of 32P (Perkin Elmer). After 3 h, media was removed, and cells were treated with 100 μm BCS or 100 μm CuCl2 (both pre-dissolved in FBS) added to a phosphate-free DMEM for 2 h. Cells were harvested, homogenized in a homogenizing buffer (25 mm imidazole, 0.25 m sucrose, 1 mm AEBSF, and protease inhibitor tablets (Roche)) using a Dounce homogenizer with 40 strokes of a tight-fitting pestle. The lysate was centrifuged at 700 × g for 10 min at 4 °C to pellet cell debri and nuclei. The postnuclear supernatant was centrifuged at 3,000 × g for 10 min at 4 °C to pellet mitochondria. The resulting supernatant was then centrifuged at 20,000 × g for 30 min at 4 °C to pellet down the microsomal membranes. The pellet was resuspended in 100 μl of IP buffer (150 mm NaCl, 10 mm Tris-HCl, 2 mm EDTA (pH 7.5), 1% n-dodecyl β-d-maltoside (DDM) (Sigma), protease inhibitor tablets (Roche)). 200 μl of blocking buffer (3% BSA in PBST) was added. After 5 min, 2.5 μl of anti-Flag antibody (Sigma) was added and rotated for 1 h at room temperature. 55 μl of pre-equilibrated protein G resin (Thermo Scientific) was added and rotated for 1 h at room temperature. The resin was washed with 20 column volumes of PBS and the bound protein was eluted using 60 μl of elution buffer (0.17 m Tris, pH 6.8, 6.8% SDS, 2.7 m urea, 1:100 β-mercaptoethanol). 24 μl of the eluted protein was loaded to a 4–20% TGX gel (Bio-Rad) (2 gels were used). One of the gels was dried for determining the phosphorylation level. The proteins in the second gel were transferred to PVDF membrane using a 10 mm CAPS, pH 11 transfer buffer. Immunoblotting was done using rat polyclonal anti-N-ATP7B antibody (1:5000) as primary antibody, and HRP-conjugated goat anti-rat IgG (1:10,000) (Santa Cruz Biotechnology) The average intensity of protein bands was determined using densitometry (Alpha Imager 2200). The level of phosphorylation was normalized to protein levels detected in the immunoblot.
Cloning, Expression, and Purification of N-ATP7B Variants
The cDNA fragment encoding the N-ATP7B in a pTYB12 vector was used for expression of N-ATP7B-intein fusion protein. The Ser>Ala replacement was introduced by PCR using the Quickchange XL mutagenesis kit. 1 liter of Escherichia coli BL21 (DE3) cells transformed with the appropriate plasmids were grown at 37 °C to an A600 of 0.7. Protein expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside (IPTG). Protein was expressed at 25 °C for 4 h. Cells were harvested by centrifugation, disrupted using French press, recombinant proteins were purified on chitin beads according to manufacturer's protocol (NEB) and eluted with 50 mm DTT for 36 h at room temperature. The proteins were concentrated using ultracel-30K (Millipore) and the purity of recombinant N-ATP7B was verified by SDS-PAGE.
Limited Proteolysis with Trypsin
5 μg of purified N-ATP7B was diluted to 1 μg/μl in 40 mm Na-phosphate, pH 7.5, containing 400 mm NaCl, 1 mm BCS and mixed with 0.05 μg TPCK-treated bovine pancreatic trypsin (Sigma) at room temperature in a total volume of 12 μl. The reaction was quenched for 10 min by adding 1.2 μl of 50 mm AEBSF. Proteins were precipitated using 1.2 μl of 20% TCA, and mixed with a loading dye and 1 μl of 1 m NaOH. Digestion patterns were determined using 12% Tricine gels and staining of protein bands with Coomassie Blue.
For limited proteolysis of Flag-ATP7B, confluent Hek293TRex cells in 10 cm dishes were transiently transfected with either wt Flag-ATP7BR875 or Flag-ATP7BR875,S340/341G plasmids using Turbofect. Protein expression was induced with 10 ng/ml DOX for 4 h in basal conditions and cells were kept overnight in media containing 50 μm BCS. Cells were harvested, homogenized in a homogenizing buffer (25 mm imidazole, 0.25 m sucrose), and the microsomal fractions were isolated as described above. The pellet containing the microsomal membranes was resuspended in 100 μl of trypsin buffer containing 40 mm Na-phosphate, pH 7.5, 400 mm NaCl, and 0.25 m sucrose. Equal amounts of total protein were loaded (15 μg); when necessary levels of ATP7B were also equalized by using membranes from non-transfected cells. The microsomal fractions were treated with 0.15 μg of TPCK-treated trypsin at 37 °C in a total volume of 20 μl. The reaction was quenched by adding 2 μl of 100 mm AEBSF. Proteins were analyzed by 8% SDS gel, transferred to PVDF membrane using a 10 mm CAPS transfer buffer, pH 11 and immunostained using rabbit polyclonal anti-NBD-ATP7B antibody (1:2500) as primary antibody, and HRP-conjugated goat anti-rabbit IgG (1:10,000) (Santa Cruz Biotechnology). The average intensity of protein bands was determined by densitometry (Alpha Imager 2200).
Domain-Domain Interactions
Nucleotide-binding domain (NBD) cloned in a pTYB12 vector (27) was expressed in E. coli BL21 strain in 500 ml of culture medium using 1 mm IPTG. N-ATP7B (wt and S340/341A) fused with maltose-binding protein (MBP) (∼105 kD) was expressed as previously described (20). Bacterial cells were harvested by centrifugation at 5000 rpm at 4 °C, lysed by French press (3 passes) followed by centrifugation at 17,000 rpm at 4 °C to remove cell debris and collect supernatants. 100 μl of supernatant were analyzed by 10% SDS-PAGE to quantitate amount of expressed recombinant proteins by densitometry. Supernatants were then mixed to reach an equimolecular ratio of NBD and N-ATP7B and incubated for 1 h in a rotator at 4 °C. The mixture was applied to amylose beads and incubated for 1 h in the rotator at room temperature. The beads were then washed with 20 column volumes of 50 mm Na-phosphate buffer containing 500 mm NaCl (pH 7.0) and complete EDTA-free protease inhibitor mixture (Roche). Interacting proteins were eluted with the same buffer containing 10 mm maltose. 10 μl of the eluate was ran on a 15% SDS-PAGE, transferred to PVDF membrane and the presence of NBD was detected with rabbit polyclonal anti-NBD antibody. To quantify N-ATP7B, the membrane was stripped with 10× PBS for 1 h and re-probed with rat polyclonal anti-N-ATP7B Ab. The bands intensity was quantified by densitometry using Alpha Imager 2200.
NMR Analysis of N-ATP7B
The 15N-isotopic labeling of N-ATP7B for NMR was performed using published protocol (27). Following purification by chitin affinity chromatography, N-ATP7B was additionally purified by ion-exchange chromatography on a Hi Trap Q column (GE Healthcare) using linear concentration gradient of NaCl (0–0.5 m) for elution, dialyzed to remove salt, and concentrated by membrane filtration. Samples for NMR contained 20–90 μm protein, 50 mm HEPES-Na, pH 7.4, 50 mm NaCl, 5 mm Tris(2-carboxyethyl)phosphine, 5% (v/v) D2O, and 0.5 mm 2,2-dimethyl-2-sila-pentane-5-sulfonate (DSS) for chemical shift referencing. 1N,15N-TROSY (28) experiments were recorded on a 900 MHz Varian NMR spectrometer at 298 K.
Immunofluorescence Microscopy
Hek293TRex cells on coverslips were transiently transfected with the wt or mutant Flag-ATP7B using Turbofect. Protein expression was induced with 40 ng/ml DOX for 16 h in basal conditions. Cells were treated with 100 μm BCS or 100 μm CuCl2 for 2 h, rinsed with PBS, and fixed with in cold acetone-methanol mixture (1:1) for 30 s. The cells were blocked in blocking buffer (1% gelatin, 1% BSA in PBS) and then incubated with appropriate primary and secondary antibodies diluted in blocking buffer. (Primary antibody: mouse anti-Flag antibody 1:250 (Sigma) and rabbit anti-TGN38 1:100 (Santa Cruz Biotechnology). Secondary antibody: Alexa Fluor 488 Goat anti mouse antibody 1:500 (Invitrogen) and Alexa Fluor 555 goat anti rabbit 1:500 (Invitrogen)). Coverslips were mounted onto glass slides using Vectashield w/DAPI (Vector Laboratories). Images were taken using a Zeiss PALM confocal microscope and a 100× oil immersion objective.
Immuno-electron Microscopy
HEK293 cells transfected with either wt or mutant Flag-ATP7B were fixed with a mixture of 4% paraformaldehyde and 0.05% glutaraldehyde, labeled with rabbit polyclonal anti-Flag antibody (Sigma) using the gold-enhance protocol as described previously (29). Then, cells were fixed in uranyl acetate and in OsO4, dehydrated through a graded series of ethanol and embedded in the epoxy resin (Epon 812). Thin sections were cut with Leica EM UC6 ultramicrotome (Leica Microsystems, Wetzlar, Germany). Images from sections were acquired using a JEM-1011 electron microscope (JEOL, Tokyo, Japan) equipped with an MORADA CCD digital camera (Soft Imaging Systems GmbH, Munster, Germany).
RESULTS
The Loop Connecting MBD3 and MBD4 of ATP7B Has Several Putative Sites for Kinase-mediated Phosphorylation
To test whether inhibiting phosphorylation would affect the ATP7B exit from the TGN we examined the Ser-rich cluster in the loop between MBD3 and MBD4, which we previously identified as a region of a kinase-mediated phosphorylation (17). This cluster is conserved between human ATP7A and ATP7B (Fig. 1B), and in ATP7A, several Ser residues in this region are phosphorylated (30). Bioinformatic analysis of ATP7B using NetPhos or GPS 2.1 program predicts 4–5 potential sites within that loop. Consistent with these predictions, mutating any single serine within the Ser340-Ser348 region to alanine decreases the in vitro phosphorylation of a recombinant N-ATP7B by 25–40%, however none of these mutations completely abolishes phosphorylation (our data, not shown). To determine if any of the serines was more important than the others we compared the ATP7B orthologs (Fig. 1C). None of the residues was invariant; however Ser-340 and Ser-341 showed highest conservation; consequently, we focused our studies on these residues.
Mutating Ser-340 and/or Ser-341 Shifts the Steady-state Localization of ATP7B toward Vesicles
The Ser340/341>A mutation was introduced into Flag-ATP7B cloned into the pcDNA5/FRT-TO plasmid to allow for a tet-regulated expression (We have previously observed that high expression often leads to ATP7B mis-localization. Consequently, expression of the Flag-ATP7B or the S340/341A mutant was controlled by doxycycline, and only cells with a low/medium Flag signal were imaged). As expected, in basal conditions the wild-type Flag-ATP7B was localized to TGN (Fig. 2, A and B, upper panels). We expected that Flag-ATP7BS340/341A would also be targeted to the TGN but might be unable to traffic. Instead, the mutant showed predominantly vesicular localization (Fig. 2, A and B, lower panels). In cell with high expression, the TGN localization was partially retained, resulting in both vesicular and TGN staining of Flag-ATP7BS340/341A (Fig. 2C).
FIGURE 2.

Mutating Ser-340 and/or Ser-341 to Ala shifts the steady-state localization of ATP7B in Hek293TRex cells from TGN to vesicles. A, Hek293TRex cells transiently transfected with Flag-ATP7B or Flag-ATP7BS340/341A were immunostained with anti-Flag antibody (left, green) and anti-TGN38 (middle, red) and the patterns were compared (merge, right). Scale bar: 5 μm. B, magnified images demonstrating good colocalization between the wt Flag-ATP7B and TGN 38 (upper panel), and the vesicular localization of Flag-ATP7BS340/341A and the loss of colocalization with TGN38 (lower panel). C, number of cells with the following predominant pattern of Flag-ATP7B or Flag-ATP7BS340/341A localization: TGN, TGN+vesicles, or vesicles; data for the representative experiment are shown.
In hepatocytes, ATP7B traffics from TGN to vesicles in response to 10 μm copper, and copper chelation with BCS causes ATP7B to return to TGN. In renal cells and in HEK 293 cells, ATP7B does not traffic in response to 10 μm copper (31). Trafficking can only be seen upon treatment with very high copper (100 μm) and only small fraction of protein leaves the TGN under these conditions (data not shown). Therefore, finding the S340/341A mutant in vesicles in Hek293TRex cells was particularly striking and suggested that this mutation facilitated conversion of ATP7B from a “TGN retained” form to a “trafficking-compatible” state. This phenotype also argued against the essential role of Ser-340/341 phosphorylation in ATP7B exit from the TGN.
ATP7BS340/341A Does Not Return to the TGN in Response to Copper Depletion
One reason for vesicular localization of Flag-ATP7BS340/341A could be an increased sensitivity to basal copper levels induced by the mutation. If this were the case, then decreasing intracellular copper using the chelator BCS would induce trafficking of the Flag-ATP7BS340/341A back to the TGN. However, after prolonged treatment with 100 μm of BCS, the localization of mutant was unchanged, i.e. Flag-ATP7BS340/341A remained in vesicles (low expression) (Fig. 3A) or in vesicles and TGN (higher expression). We generated and observed similar results for single mutants S340A, S340T, and S341A (Fig. 3C illustrates the results for ATP7BS340A. Other single mutants had similar vesicular localization).
FIGURE 3.

ATP7BS340/341A and ATP7BS340A do not return to TGN with copper depletion. A, Hek293TRex cells transiently transfected with wt Flag-ATP7B or Flag-ATP7BS340/341A were treated with 100 μm BCS for 2 h. Cells were immunostained using anti-Flag (green) and anti-TGN 38 antibodies (red). Merged image is shown in the right panel. Scale bar: 5 μm. B, copper depletion was done using 200 μm BCS for 3 h, and cells were processed for immuno-gold EM. The wt Flag-ATP7B (left panel) was detected over the tubular-vesicular profiles (arrows) at the trans-side of the Golgi stacks (asterisks). Arrowheads indicate large vesicles which were devoid of ATP7B signal. Flag-ATP7BS340/341A (right panel) decorated membrane profiles (arrows) at the trans-side of the Golgi stacks (asterisk) as well as large vesicles (arrowheads). C, left: representative confocal image of Flag-ATP7BS340A (green) with a TGN marker (red) in copper-depleted conditions (100 μm BCS for 2 h). Right: electron microscopy of GFP-ATP7BS340A under copper-depleted conditions (200 μm BCS for 3 h). GFP-ATP7BS340A is localized at the trans-side (arrows) of the Golgi complex (asterisk) and in large “vesicles” (arrowheads).
The differences in the compartmentalization of wt ATP7B, the S340/341A mutant, and the S340A mutant were verified by immuno-EM. Gold labeling showed the wt ATP7B to be distributed along cisterns and tubular profiles at the trans face of the Golgi, whereas little of S340/341A mutant was detected in similar structures (Fig. 3B; arrows). Instead, the mutant was primarily localized in large vesicles (300–1000 nm in size). Reciprocally, wt ATP7B was almost completely excluded from such vesicular profiles (Fig. 3B, arrowheads). Similar to ATP7BS340/341A, the single mutant GFP-ATP7BS340A appeared in large “vesicles” (arrowheads) (Fig. 3C, right panel). Thus, the presence of serine residues in positions 340/341 is important for ATP7B retention in the TGN, whereas modification of these residues facilitates exit.
Mutation of Ser-340/341 Mimics Consequences of Copper Binding
The shift in the steady-state distribution of the S340/341A mutant from TGN to vesicles in low copper suggested that the mutant acquired a conformation resembling that of ATP7B under high copper conditions. To test this hypothesis, we took advantage of a naturally occurring ATP7B variant ATP7BR875, which has a low apparent affinity to copper compared with a common ATP7BG875 variant and a very distinct cellular behavior. In basal or low copper, ATP7BR875 localizes in the endoplasmic reticulum (ER), but it traffics to the TGN and then to vesicles when copper is increased in a stepwise fashion (26). Stabilizing the “copper-bound” state for the ATP7BR875 variant is expected to change its localization from ER to TGN and/or vesicles under basal conditions. To test this prediction, we mutated Ser-340/341 to Ala-340/341 in ATP7BR875 and examined the localization of this mutant in Hek293TRex cells. In basal medium, the control ATP7BR875 showed expected ER localization, and it trafficked to vesicles upon copper treatment (Fig. 4A). In contrast, the mutant was localized exclusively to vesicles mimicking the behavior of ATP7BR875 in high copper (Fig. 4B). Copper chelation treatment did not alter the localization of ATP7BR875,S340/341A (Fig. 4B). To further minimize structural perturbations (side chains of glycine are smaller compared with alanine), we mutated S340/341 to G340/341 in ATP7BR875. The ATP7BR875,S340/341G variant was also localized exclusively to vesicles in both basal and copper depleted conditions (Fig. 4C).
FIGURE 4.

ATP7BR875,S340/341A and ATP7BR875,S340/341G behave as ATP7BR875 in a copper-saturated state. Hek293TRex cells transiently transfected with Flag-ATP7BR875, Flag-ATP7BR875,S340/341A, or Flag-ATP7BR875,S340/341G were kept in basal media, copper depleted using 100 μm BCS for 2 h or treated with 100 μm CuCl2 for 2 h. Cells were immunostained using Flag antibody (green) and TGN 38 (red); merged images are shown (Scale bar: 5 μm). A, Flag-ATP7BR875 is localized in ER in basal (left panel) and copper-depleted conditions (middle panel), and moves to vesicles upon copper treatment (right panel). B and C, both ATP7BR875,S340/341A and ATP7BR875,S340/341G are localized in vesicles in basal (left panel) and copper-depleted (right panel) conditions.
To determine whether on its way to vesicles the Ser-340/341 mutant follows a normal intracellular route of ATP7B trafficking via the TGN, we utilized the tyrosinase activation assay. The assay uses Menkes fibroblasts expressing the copper-dependent enzyme tyrosinase (YSTT cells). The YSTT cells lack active copper ATPases and thus produce inactive tyrosinase. Transfection of YSTT cells with a wt ATP7B, which is targeted to the TGN, restores copper transport to the TGN lumen and activates tyrosinase resulting in a black chromogenic precipitate (Fig. 5, left panel) (26). The negative control, Flag-ATP7BR875 localized in the ER, does not activate tyrosinase (Fig. 5, middle panel) (26). Expression of Flag-ATP7BR875,S340/341G induces pigment formation, indicating that the mutant followed the normal trafficking itinerary and delivered copper to tyrosinase, presumably when passing through the TGN (Fig. 5, right panel).
FIGURE 5.
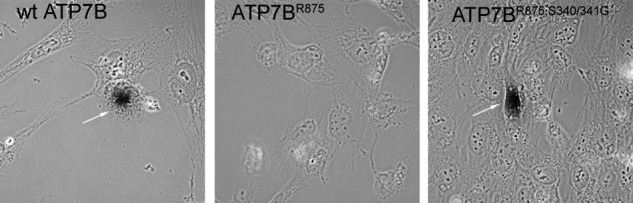
ATP7BR875,S340/341G has copper transport activity and activates tyrosinase in the YSTT cells. Tyrosinase assay shows pigment formation for wt GFP-ATP7B (arrow, left panel) and Flag-ATP7BR875,S340/341G (arrow, right panel) but no pigment formation for Flag-ATP7BR875 (middle panel) in YSTT cells in basal growth conditions.
ATP7BR875,S340/341G Has a Reduced Kinase-mediated Phosphorylation
Normally, ATP7B exit from the TGN is stimulated by high copper, which also stimulates ATP7B phosphorylation by a kinase (16). Given our observations that the S340/340A mutation mimics behavior of ATP7B in elevated copper, we were wondering whether this mutation instead of eliminating phosphorylation induced structural changes that exposed sites critical for kinase-mediated phosphorylation in high copper.
To clarify consequences of Ser-340/341 mutation we compared kinase-mediated phosphorylation of ATP7BR875 and ATP7BR875,S340/341G using in vivo metabolic labeling with radioactive orthophosphate. (The ATP7BR875 variant was particularly useful for these studies because both ATP7BR875 and ATP7BR875,S340/341G have uniform intracellular localization, ER, and vesicles, respectively). In low copper, the mutant had ∼30% lower phosphorylation compared with ATP7BR875 (Fig. 6). Copper treatment did not affect the phosphorylation level of the mutant and difference between ATP7BR875 and ATP7BR875,S340/341G became larger when metabolic labeling was carried out in the presence of copper (Fig. 6). Thus, Ser-340/341 are important for normal phosphorylation of ATP7B, particularly in high copper, however the loss of phosphorylation caused by mutations is not detrimental for the ability of ATP7B to exit the TGN.
FIGURE 6.
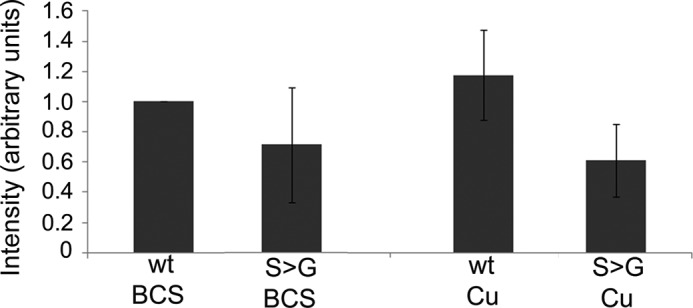
ATP7BR875,S340/341G has a decreased level of kinase mediated phosphorylation. Hek293TRex cells were transiently transfected with wt Flag-ATP7BR875 or Flag-ATP7BR875,S340/341G, treated with 100 μm BCS or 100 μm CuCl2 for 2 h and in vivo metabolically labeled with radioactive orthophosphate. The kinase-mediated phosphorylation of Flag-ATP7BR875 (wt) and Flag-ATP7BR875,S340/341G (S→G) was compared following immunoprecipitation, gel-separation and densitometry. Error bars indicate S.D. across four independent experiments.
To reconcile these observations, we hypothesized that in a wild-type ATP7B phosphorylation of Ser-340 and/or Ser-341 induces structural changes similar to those induced by Ser mutations. To test this prediction we generated and characterized a phospho-mimetic mutation S340/341D. This variant behaved indistinguishably from the S340/341A i.e. it was targeted to vesicles (Fig. 7A).
FIGURE 7.

The shift to vesicles for the Ser-340/341 mutant is also induced by negative charge and is selective. A, phospho-mimetic mutant Flag-ATP7BR875,S340/341D is localized to vesicles in basal (upper panel) and copper-depleted (100 μm BCS for 2 h) conditions (lower panel). B, Flag-ATP7BR875,S478A, Flag-ATP7BR875,S481A, and Flag-ATP7BR875,S1453A behave as wt Flag-ATP7BR875. They are located in the ER in basal conditions, and traffic to vesicles upon treatment with 100 μm CuCl2 for 2 h (Scale bar: 5 μm).
The Effect of Ser340/341 Mutations on Trafficking Is Selective
We were also curious whether vesicular targeting observed by mutating Ser-340 and/or Ser-341 was an effect specific for these sites or manipulating any phosphorylation site(s) in ATP7B would induce changes in localization. Consequently, using ATP7BR875 as a template we mutated to alanine Ser-478, Ser-481, and Ser-1435, which were previously suggested as additional sites of phosphorylation in ATP7B (18). No effect on localization was observed for all three mutants. Similarly to ATP7BR875, these mutants were found in the ER in basal condition, were targeted to the TGN in low copper, and trafficked to vesicles in response to higher copper (Fig. 7B). We concluded that Ser-340/341 play a specific role in the TGN retention/exit of ATP7B.
The S340/341A Mutation Does Not Markedly Alter Folding of N-ATP7B
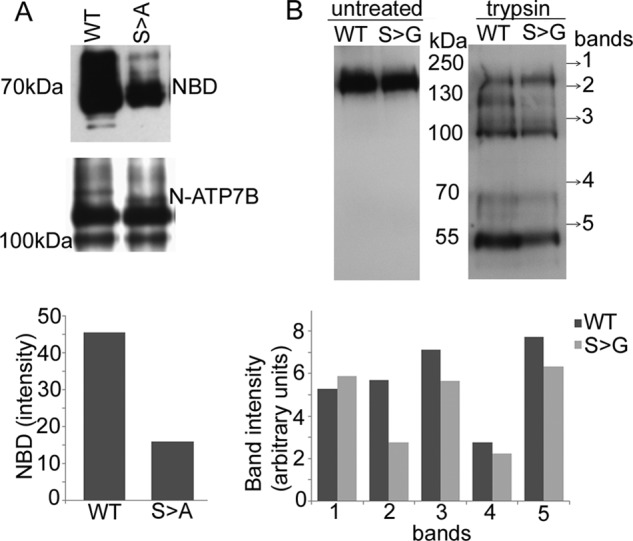
Our data suggested that the S340/341 substitution induced structural changes in ATP7B, which could either be local or transmitted via inter-domain interactions to affect entire ATP7B. Ser-340/341 residues are located in the N-terminal domain of ATP7B (N-ATP7B). We first investigated the effect of Ser-340/341 mutation on a recombinant N-ATP7B. The folding of N-ATP7B and N-ATP7BS340/341A was compared using limited proteolysis with varying concentrations of trypsin at different time intervals. No difference in proteolytic patterns was detected under all tested conditions (Fig. 8A). Treatment with different protease, papain, yielded similar results (our data, not shown). We concluded that the S340/341A substitution did not markedly alter the N-ATP7B fold. To verify this conclusion, we examined the effect of Ser mutations on N-ATP7B structure in more detail. The recombinant N-ATP7B and N-ATP7BS340/341A were metabolically labeled with N15, purified in a non-tagged form, and their folding was compared by NMR (Fig. 8B). The spectra of wt N-ATP7B and N-ATP7BS340/341A showed excellent chemical shift dispersion, characteristic of the well-folded proteins. The differences in spectra were very minor, demonstrating that the S340/341A mutation does not significantly change the fold of N-ATP7B.
FIGURE 8.

The Ser340/341A mutation does not disrupt protein folding of N-ATP7B. A, proteolytic patterns obtained following treatment of 5 μg of purified recombinant N-ATP7B (WT) and N-ATP7BS340/341A (S→A) with 0.05 μg of trypsin. Peptides were resolved using 12% Tricine gel and stained with Coomassie Blue. B, 900 MHz 1H,15N-TROSY spectrum of the S340/341A mutant MBD1–6 (left panel) and an overlay (right panel) of the wild type (black) and the S340/341A mutant (red).
Mutation of Ser-340/341 Induces Conformational Changes within ATP7B by Reducing Interactions between N-ATP7B and Nucleotide-binding Domain
The lack of significant effects on N-ATP7B structure suggested that the Ser mutations may alter the inter-domain interactions in ATP7B. It was previously shown that N-ATP7B interacted with the nucleotide-binding domain (NBD) and that copper binding to N-ATP7B decreased this interaction (23). We hypothesized that the S340/341A mutation might mimic this effect and decrease interactions between N-ATP7B and NBD. To test this hypothesis, we performed a co-purification experiment, using cell lysates containing the recombinant maltose fusion N-ATP7B (WT and S340/341A) and the intein fusion of NBD. N-ATP7BS340/341A showed a ∼50% reduction of inter-domain interaction compared with N-ATP7B (Fig. 9A). Thus, similarly to copper binding, S340/341 mutation decreases interactions between the N-terminal domain and NBD.
FIGURE 9.
The Ser340/341A mutation reduces interactions between N-ATP7B and nucleotide-binding domain (NBD) and alters conformation of ATP7B. A, cell lysates containing maltose-binding fusions of N-ATP7B (WT) or N-ATP7BS340/341A (S→A) were mixed with lysates containing recombinant NBD and passed over amylose resin. Purified NBD (upper panel) and N-ATP7B (middle panel) were detected using corresponding antibodies and proteins were quantified by densitometry (lower panel). D, top: proteolytic pattern of Flag-ATP7BR875 (WT) or Flag-ATP7BR875,S340/341G (S→G) following 15 min treatment with trypsin. Fragments were resolved using 8% SDS gel and immunoblotted with anti-NBD-ATP7B antibody. Bottom: bands intensity was compared by densitometry.
In the full-length ATP7B, the change of inter-domain interactions is expected to influence the kinetics and/or pattern of limited proteolysis. Consistent with this prediction, treatment with trypsin reproducibly yielded different rates of digestion for the membrane-bound Flag-ATP7BR875 and Flag-ATP7BR875,S340/341G. This was evident from differences in the relative intensities of proteolytic bands (Fig. 9B). We concluded that Ser-340/341 influences the interdomain interface and overall conformation of ATP7B.
Copper Transport Activity Is Necessary for the Localization of ATP7BR875,S340/341G to Vesicles
Copper binds to ATP7B in at least 8 sites (6 in the cytosol and likely 2 sites in the trans-membrane portion). Occupation of the cytosolic MBDs with copper and changes in interdomain contacts may be sufficient to trigger trafficking of ATP7B to vesicles. Alternatively, binding of copper at both the cytosolic and membrane/lumenal sites of the transporter might be necessary to initiate ATP7B exit from the TGN. To discriminate between these two possibilities, we generated a catalytically inactive mutant D1027A and a triple mutant D1027A,S340/341G (all on the R875 background). The D1027 mutation is expected to have no effect on the ability of ATP7B to bind copper at the cytosolic MBDs, but it would prevent transfer of copper to the lumenal side due to inability to hydrolyze ATP. Analysis of intracellular targeting showed that the D1027A mutant had a mixed ER/TGN targeting; a higher proportion of TGN targeting compared with the ER-localized parent ATP7BR875 (Fig. 10) implied that the mutant might have increased protein stability. Significantly, the D1027A mutation completely blocked trafficking of the Ser340/341G variant, i.e. the localization of ATP7BS340/341G,D1027A mutant was no longer shifted toward vesicles (Fig. 10). We concluded that both domain/domain dissociation and copper transfer into the lumen are necessary for exiting the TGN and trafficking to vesicles.
FIGURE 10.
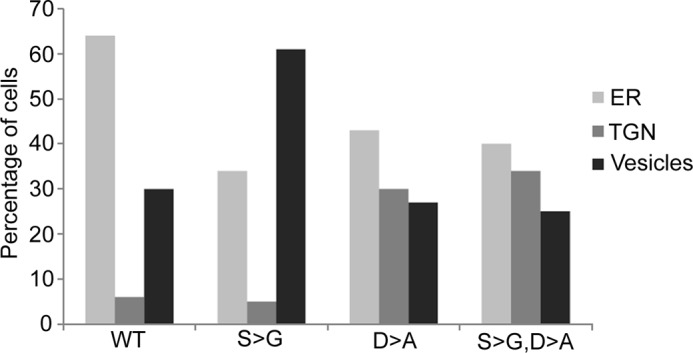
Copper transport activity is necessary to shift the intracellular localization of ATP7BR875,S340/341G to vesicles. Hek293TRex cells were transiently transfected with one of the Flag-tagged ATP7B variants: ATP7BR875 (WT), ATP7BR875,D1027A (D→A), ATP7BR875,S340/341G (S→G), or ATP7BR875,S340/341G,D1027A (S→G, D→A) and immunostained under basal conditions using the anti-Flag antibody and TGN 38. Random fields of view were chosen and the total number of cells and the fraction of cells having ER, TGN, or vesicles localization was calculated for each of the variants. Whereas the S→G mutation causes an expected shift to vesicles when compared with WT; the D→A mutation increases a fraction of the TGN-localized ATP7B compared with WT, but prevents the S→G-induced shift to vesicles.
DISCUSSION
In this study, we demonstrate that the N-terminal Ser-rich region of ATP7B is an important regulator of protein exit from the TGN. The residues Ser-340/341 contribute to a regulatory phosphorylation of ATP7B, particularly in high copper, and modulate the inter-domain interactions (Figs. 6 and 9A). Significantly, mutations inhibiting phosphorylation (S→A, S→G) and mutation mimicking the phosphorylated state (S→D) all facilitate ATP7B trafficking from the TGN. Thus, the structural change that alters inter-domain contacts in ATP7B is a key trigger of ATP7B trafficking to vesicles. The role of copper-dependent phosphorylation is likely to maintain this “trafficking-compatible” state.
N-ATP7B acts as a copper sensor; and copper binding to MBDs alters the conformation of the entire N-ATP7B (17, 22). ATP7B with non-functional MBDs (which cannot bind copper) does not traffic in response to copper elevation (32). Residues S340/341 are located within the long loop connecting MBD3 and MBD4 and are not expected to have any effect on copper binding capacity of N-ATP7B, although this remains to be experimentally verified. It is also unlikely that S340/341 interact directly with the cellular trafficking machinery; because a deletion mutant of ATP7B that lacks the N-terminal residues 63–443 exhibits normal TGN localization and copper-dependent trafficking (9). Instead, the Ser-340/341 region appears to play an important role in interactions between N-ATP7B and the nucleotide-binding domain. This role could be direct because the effect of S340/341A mutation on overall structure of N-ATP7B is minimal (Fig. 8, A and B), whereas the effect on inter-domain interaction is significant (Fig. 9A).
Phosphorylation of other membrane proteins was shown to expose sorting signals (33) or itself serve as a signal for trafficking by facilitating interaction with the components of cellular trafficking machinery (34). Our data are more consistent with a scenario, where phosphorylation alone is not a required factor in the exit of ATP7B from the TGN; rather it stabilizes the trafficking ready copper bound state of ATP7B. We speculate that in low copper Ser-340/341 are protected from a kinase by inter-domain interactions. Domain dissociation in response to copper binding to N-ATP7B exposes Ser-340/341 to phosphorylation by a kinase. The phosphorylation would then keep domains apart, thus stabilizing ATP7B is a state that has a poor TGN retention (Fig. 11).
FIGURE 11.

The proposed sequence of events that triggers ATP7B trafficking. In basal copper, N-ATP7B has a partial copper occupancy and ATP7B pumps copper into the lumen of TGN, where released copper is incorporated into available apo-proteins (green beads and brown half-spheres). In high copper, MBDs are saturated with copper and N-ATP7B dissociates from the N-domain resulting in increased pump activity. The Ser-340/341 sites situated in the MBD3-MBD4 loop are phosphorylated and that keeps the domains apart (dotted red arrow). Because of higher pump activity, the acceptor proteins become saturated, copper release is diminished, and ATP7B is stabilized is a conformation favorable for TGN exit. In this conformation, ATP7B interacts with the trafficking machinery (green rhomboid). Similar effect on ATP7B conformation is achieved by mutating Ser-340/341. The domains are denoted as, A: actuator; N: nucleotide-binding domain; P: phosphorylation domain. The six metal-binding sites on N-ATP7B are shown in purple beads.
The fact that even a mild mutation such as S340T produces the loss of TGN retention indicates that this region of ATP7B is not merely an exposed loop, but a site critical for the conformational state of entire ATP7B. From this perspective, it is interesting to consider that mutations in another N-terminal region 37FAFDNVGYE45 also result in the loss of TGN retention (24). It is tempting to speculate that in a three-dimensional structure of ATP7B, the two N-terminal regions can be in vicinity to each other. Copper binding and subsequent structural rearrangements that expose Ser-340/341 for phosphorylation may simultaneously expose the Phe-37–Glu-45 region for interaction with the apical trafficking machinery (24).
Mass spectrometry analysis of the recombinant ATP7B demonstrated that Ser-478, Ser-481, and Ser-1453 can be phosphorylated and that quadruple mutant (Ser-478, 481, 1121, 1453A) fails to traffic in response to copper (18, 19). In our studies, individual mutations of these residues do not alter trafficking pattern (Fig. 7B). Difference in the phenotypes could be due to global conformational changes induced by a quadruple mutation versus a single mutation. These observations emphasize the role of ATP7B structure in its localization.
Finally, the phenotype of the Ser-340/341 mutant resembles a phosphatase-deficient ATP7B mutant, 858TGE860→AAA (25). This mutant is trapped in a so called E2P state (25). Similar E2P state would be stabilized under conditions of copper saturation, when copper release into the TGN lumen is diminished. Thus, although 858TGE860 and Ser-340/341 residues are located in different domains of ATP7B, the mutation of these sites are likely to have similar effects on the overall conformation of ATP7B.
The following model (Fig. 11) is consistent with our results as well as many experimental observations reported in the literature. Under basal conditions, N-ATP7B has a partial copper occupancy and ATP7B pumps copper into the lumen of TGN, where the released copper is incorporated into available apo-proteins. In high copper, more copper binds to N-ATP7B; the binding relieves the autoinhibitory interaction between N-ATP7B and the nucleotide-binding domain and increases the pump activity. Higher transport activity would saturate the copper binding capacity of the acceptor proteins and eventually inhibit copper release from ATP7B into the lumen. Altogether, these events would lead to ATP7B acquiring a “copper-saturated” conformation, which is a target of a kinase-mediated phosphorylation. Phosphorylation of serine residues may stabilize the “TGN-exit ready” conformation, which is posed to interact with the cellular machinery responsible for an anterograde trafficking of ATP7B. The mutations that stabilize this specific form would facilitate the TGN exit and result in vesicular localization of ATP7B.
Acknowledgments
We thank Dr. Mee Bartee for contribution to the initial phase of these studies. We would also like to acknowledge Telethon Electron Microscopy Core Facility (IBP, CNR) and Integrated Microscopy Facility (IGB, CNR) for the assistance with the EM analysis and Giancarlo Chesi for the help with EM specimen preparation. We are grateful to Dr. Marco Tonelli of the National Magnetic Resonance Facility at Madison (NMRFAM) for recording the NMR spectra.
This work was supported, in whole or in part, by National Institutes of Health Grants DK071865 and P01GM067166 (to S. L.) and by Telethon Grants GTF08001 and TRPCB4TELD (to R. P.).
- N-ATP7B
- N-terminal domain of ATP7B
- NBD
- nucleotide binding domain
- MBD
- metal-binding domain
- TGN
- trans-Golgi network
- CAPS
- 3-(cyclohexylamino) propane-sulfonic acid
- BCS
- bathocuproine disulfonate
- AEBSF
- 4- (2-aminoethyl) benzenesulfonyl fluoride hydrochloride
- TCA
- trichloroacetic acid
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- MOPS
- 3-(N-morpholino) propanesulfonic acid
- TPCK
- tosylphenylalanyl chloromethyl ketone
- DOX
- doxycycline hyclate.
REFERENCES
- 1. Gupta A., Lutsenko S. (2009) Human copper transporters: mechanism, role in human diseases and therapeutic potential. Future Med. Chem. 1, 1125–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vulpe C., Levinson B., Whitney S., Packman S., Gitschier J. (1993) Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat. Genet. 3, 7–13 [DOI] [PubMed] [Google Scholar]
- 3. Tanzi R. E., Petrukhin K., Chernov I., Pellequer J. L., Wasco W., Ross B., Romano D. M., Parano E., Pavone L., Brzustowicz L. M. (1993) The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat. Genet. 5, 344–350 [DOI] [PubMed] [Google Scholar]
- 4. Voskoboinik I., Brooks H., Smith S., Shen P., Camakaris J. (1998) ATP-dependent copper transport by the Menkes protein in membrane vesicles isolated from cultured Chinese hamster ovary cells. FEBS Lett. 435, 178–182 [DOI] [PubMed] [Google Scholar]
- 5. Payne A. S., Kelly E. J., Gitlin J. D. (1998) Functional expression of the Wilson disease protein reveals mislocalization and impaired copper-dependent trafficking of the common H1069Q mutation. Proc. Natl. Acad. Sci. U.S.A. 95, 10854–10859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. La Fontaine S. L., Firth S. D., Camakaris J., Englezou A., Theophilos M. B., Petris M. J., Howie M., Lockhart P. J., Greenough M., Brooks H., Reddel R. R., Mercer J. F. (1998) Correction of the copper transport defect of Menkes patient fibroblasts by expression of the Menkes and Wilson ATPases. J. Biol. Chem. 273, 31375–31380 [DOI] [PubMed] [Google Scholar]
- 7. Bull P. C., Thomas G. R., Rommens J. M., Forbes J. R., Cox D. W. (1993) The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat. Genet. 5, 327–337 [DOI] [PubMed] [Google Scholar]
- 8. Bartee M. Y., Lutsenko S. (2007) Hepatic copper-transporting ATPase ATP7B: function and inactivation at the molecular and cellular level. Biometals 20, 627–637 [DOI] [PubMed] [Google Scholar]
- 9. Guo Y., Nyasae L., Braiterman L. T., Hubbard A. L. (2005) NH2-terminal signals in ATP7B Cu-ATPase mediate its Cu-dependent anterograde traffic in polarized hepatic cells. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G904–G916 [DOI] [PubMed] [Google Scholar]
- 10. Hung I. H., Suzuki M., Yamaguchi Y., Yuan D. S., Klausner R. D., Gitlin J. D. (1997) Biochemical characterization of the Wilson disease protein and functional expression in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 272, 21461–21466 [DOI] [PubMed] [Google Scholar]
- 11. Lim C. M., Cater M. A., Mercer J. F., La Fontaine S. (2006) Copper-dependent interaction of dynactin subunit p62 with the N terminus of ATP7B but not ATP7A. J. Biol. Chem. 281, 14006–14014 [DOI] [PubMed] [Google Scholar]
- 12. Lim C. M., Cater M. A., Mercer J. F., La Fontaine S. (2006) Copper-dependent interaction of glutaredoxin with the N termini of the copper-ATPases (ATP7A and ATP7B) defective in Menkes and Wilson diseases. Biochem. Biophys. Res. Commun. 348, 428–436 [DOI] [PubMed] [Google Scholar]
- 13. Tao T. Y., Liu F., Klomp L., Wijmenga C., Gitlin J. D. (2003) The copper toxicosis gene product Murr1 directly interacts with the Wilson disease protein. J. Biol. Chem. 278, 41593–41596 [DOI] [PubMed] [Google Scholar]
- 14. Roelofsen H., Wolters H., Van Luyn M. J., Miura N., Kuipers F., Vonk R. J. (2000) Copper-induced apical trafficking of ATP7B in polarized hepatoma cells provides a mechanism for biliary copper excretion. Gastroenterology 119, 782–793 [DOI] [PubMed] [Google Scholar]
- 15. Materia S., Cater M. A., Klomp L. W., Mercer J. F., La Fontaine S. (2012) Clusterin and COMMD1 independently regulate degradation of the mammalian copper ATPases ATP7A and ATP7B. J. Biol. Chem. 287, 2485–2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vanderwerf S. M., Cooper M. J., Stetsenko I. V., Lutsenko S. (2001) Copper specifically regulates intracellular phosphorylation of the Wilson's disease protein, a human copper-transporting ATPase. J. Biol. Chem. 276, 36289–36294 [DOI] [PubMed] [Google Scholar]
- 17. Bartee M. Y., Ralle M., Lutsenko S. (2009) The loop connecting metal-binding domains 3 and 4 of ATP7B is a target of a kinase-mediated phosphorylation. Biochemistry 48, 5573–5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pilankatta R., Lewis D., Adams C. M., Inesi G. (2009) High yield heterologous expression of wild-type and mutant Cu+-ATPase (ATP7B, Wilson disease protein) for functional characterization of catalytic activity and serine residues undergoing copper-dependent phosphorylation. J. Biol. Chem. 284, 21307–21316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pilankatta R., Lewis D., Inesi G. (2011) Involvement of protein kinase D in expression and trafficking of ATP7B (copper ATPase). J. Biol. Chem. 286, 7389–7396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lutsenko S., Petrukhin K., Cooper M. J., Gilliam C. T., Kaplan J. H. (1997) N-terminal domains of human copper-transporting adenosine triphosphatases (the Wilson's and Menkes disease proteins) bind copper, selectively in vivo and in vitro with stoichiometry of one copper per metal-binding repeat. J. Biol. Chem. 272, 18939–18944 [DOI] [PubMed] [Google Scholar]
- 21. Lutsenko S., LeShane E. S., Shinde U. (2007) Biochemical basis of regulation of human copper-transporting ATPases. Arch. Biochem. Biophys. 463, 134–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. DiDonato M., Hsu H. F., Narindrasorasak S., Que L., Jr, Sarkar B. (2000) Copper-induced conformational changes in the N-terminal domain of the Wilson disease copper-transporting ATPase. Biochemistry 39, 1890–1896 [DOI] [PubMed] [Google Scholar]
- 23. Tsivkovskii R., MacArthur B. C., Lutsenko S. (2001) The Lys-1010–Lys-1325 fragment of the Wilson's disease protein binds nucleotides and interacts with the N-terminal domain of this protein in a copper-dependent manner. J. Biol. Chem. 276, 2234–2242 [DOI] [PubMed] [Google Scholar]
- 24. Braiterman L., Nyasae L., Guo Y., Bustos R., Lutsenko S., Hubbard A. (2009) Apical targeting and Golgi retention signals reside within a 9-amino acid sequence in the copper-ATPase, ATP7B. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G433–G444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cater M. A., La Fontaine S., Mercer J. F. (2007) Copper binding to the N-terminal metal-binding sites or the CPC motif is not essential for copper-induced trafficking of the human Wilson protein (ATP7B). Biochem. J. 401, 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gupta A., Bhattacharjee A., Dmitriev O. Y., Nokhrin S., Braiterman L., Hubbard A. L., Lutsenko S. (2011) Cellular copper levels determine the phenotype of the Arg-875 variant of ATP7B/Wilson disease protein. Proc. Natl. Acad. Sci. U.S.A. 108, 5390–5395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dmitriev O., Tsivkovskii R., Abildgaard F., Morgan C. T., Markley J. L., Lutsenko S. (2006) Solution structure of the N-domain of Wilson disease protein: distinct nucleotide-binding environment and effects of disease mutations. Proc. Natl. Acad. Sci. U.S.A. 103, 5302–5307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pervushin K., Riek R., Wider G., Wüthrich K. (1997) Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. U.S.A. 94, 12366–12371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Polishchuk E. V., Di Pentima A., Luini A., Polishchuk R. S. (2003) Mechanism of constitutive export from the Golgi: bulk flow via the formation, protrusion, and en bloc cleavage of large trans-Golgi network tubular domains. Mol. Biol. Cell 14, 4470–4485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villén J., Haas W., Sowa M. E., Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barnes N., Bartee M. Y., Braiterman L., Gupta A., Ustiyan V., Zuzel V., Kaplan J. H., Hubbard A. L., Lutsenko S. (2009) Cell-specific trafficking suggests a new role for renal ATP7B in the intracellular copper storage. Traffic 10, 767–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cater M. A., Forbes J., La Fontaine S., Cox D., Mercer J. F. (2004) Intracellular trafficking of the human Wilson protein: the role of the six N-terminal metal-binding sites. Biochem. J. 380, 805–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bonifacino J. S., Traub L. M. (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395–447 [DOI] [PubMed] [Google Scholar]
- 34. Hinners I., Tooze S. A. (2003) Changing directions: clathrin-mediated transport between the Golgi and endosomes. J. Cell Sci. 116, 763–771 [DOI] [PubMed] [Google Scholar]