

Background: The function of RPA and RPA phosphorylation in the activation of ATR is unknown.
Results: RPA with phosphomimetic mutations cannot support ATR kinase function yet maintains functions in nucleotide excision repair.
Conclusion: These results reveal a RPA separation of function for checkpoint activation and excision repair.
Significance: RPA phosphorylation may modulate ATR checkpoint signaling while maintaining other cellular functions of RPA.
Keywords: Checkpoint Control, DNA Binding Protein, DNA Nucleotide Excision Repair, DNA-Protein Interaction, p53, Phosphorylation, Protein Kinases, Protein phosphorylation, Rad17-RFC, TopBP1
Abstract
Replication protein A (RPA) plays essential roles in DNA metabolism, including replication, checkpoint, and repair. Recently, we described an in vitro system in which the phosphorylation of human Chk1 kinase by ATR (ataxia telangiectasia mutated and Rad3-related) is dependent on RPA bound to single-stranded DNA. Here, we report that phosphorylation of other ATR targets, p53 and Rad17, has the same requirements and that RPA is also phosphorylated in this system. At high p53 or Rad17 concentrations, RPA phosphorylation is inhibited and, in this system, RPA with phosphomimetic mutations cannot support ATR kinase function, whereas a non-phosphorylatable RPA mutant exhibits full activity. Phosphorylation of these ATR substrates depends on the recruitment of ATR and the substrates by RPA to the RPA-ssDNA complex. Finally, mutant RPAs lacking checkpoint function exhibit essentially normal activity in nucleotide excision repair, revealing RPA separation of function for checkpoint and excision repair.
Introduction
Replication protein A (RPA)2 is a heterotrimeric protein made up of RPA1 (p70), RPA2 (p34), and RPA3 (p14) that has high affinity for single-stranded DNA (ssDNA) and performs essential functions in replication, repair, recombination, and DNA damage checkpoints in human cells (1, 2). These roles for RPA in DNA metabolism are essential for the maintenance of chromosomal stability and tumor suppression (3, 4). In the cell, RPA maintains ssDNA in extended structures and interacts with proteins involved in replication, repair, and checkpoint activation (5). However, the relative contributions of these interactions to chromosome maintenance have not been clearly delineated.
The N-terminal domain of RPA1 directly interacts with several checkpoint proteins, including ATR (through ATRIP), Rad17-Replication Factor C (RFC), p53, and Rad9 (6–19). In addition, eight serine and threonine residues in the N-terminal domain of RPA2 have been shown to be phosphorylated in a cell cycle- and DNA damage-dependent manner (20–25). However, it is unclear whether the phosphorylation plays a role in all RPA functions and in particular its checkpoint functions (5).
Recently, we developed an in vitro system with defined protein/DNA constituents for studying the human ATR-mediated DNA damage checkpoint response (26–29). With this system, we established that the phosphorylation of Chk1 signal transducing kinase by the damage sensor kinase ATR, under appropriate reaction conditions, was dependent on ssDNA, RPA, and TopBP1 (30). Here, we have investigated the requirement for phosphorylation of two other ATR substrates, p53 and Rad17, in this system. We find that these two substrates exhibit the same requirements. We also found that RPA is phosphorylated during the reaction and that, unexpectedly, the p53 and Rad17 substrates, whose phosphorylation depended on RPA, strongly inhibited phosphorylation of RPA2 by ATR. Furthermore, phosphomimetic RPA2 mutations abolished ATR kinase activity in this system, whereas the non-phosphorylatable RPA2 mutant functioned similar to wild-type, and the effects of various RPA mutations on ATR kinase paralleled their abilities to recruit substrates to the RPA-ssDNA complex. Finally, we demonstrate that RPA mutants with no detectable checkpoint activity have essentially normal excision repair activity, indicating that RPA plays different roles in the assembly of checkpoint and excision repair proteins on DNA and in executing its functions in these reactions.
EXPERIMENTAL PROCEDURES
Antibodies and DNA
p53 phospho-Ser15 antibodies (9284) and Rad17 phospho-Ser645 antibodies (3421) were purchased from Cell Signaling Technology, RPA2 phospho-Ser33 (A300–246A) and RPA1 (A300–241A) antibodies were from Bethyl Laboratories, GST (sc-138) antibodies were purchased from Santa Cruz Biotechnology, anti-FLAG M2 antibodies (F3165) were from Sigma, anti-RPA2 (NA-18) was purchased from Calbiochem, and mouse monoclonal antibodies against Rad17 were a kind gift from Lan Bo Chen (31). The ϕX174 ssDNA was purchased from New England Biolabs (N3023).
Purification of Checkpoint Proteins
Native ATR-ATRIP (30), GST-TopBP1-His (29), full-length GST-p53 (28), GST-p531–102 (32), HisFLAG-ATRIP (33), and RPA wild-type (34) and mutants (35, 36) were purified as described previously. FLAG-Rad17 was expressed in 293 FlpIn T-REX cells as described in the manufacturer's instructions (Invitrogen) and purified with anti-FLAG agarose (Sigma) as described previously (37, 38).
Kinase Assays
The procedure was essentially as described previously (30). Briefly, kinase assay reactions contained 14 mm Hepes, pH 7.9, 30 mm KCl, 1 mm MgCl2, 0.1 mm ATP, 0.5 mm of DTT, 2% glycerol, and 1 μm microcystin in a 12 μl final volume. Purified ATR-ATRIP (0.2 nm) and TopBP1 (2.5 nm) were incubated in reaction buffer for 10 min at 30 °C with the indicated amounts of DNA and recombinant RPA, GST-p53, GST-p531–102, or FLAG-Rad17-RFC. The experiments in Fig. 2C contained 20 μCi of radiolabeled [γ-32P]ATP and analyzed as described previously (26). The reactions were terminated by the addition of 3 μl of 5× SDS-PAGE loading buffer and separated by 4–15% TGX-PAGE (Bio-Rad). Rad17, p53, and RPA2 phosphorylation was detected by immunoblotting using the indicated phospho-specific antibodies, and the level of total protein was subsequently detected by immunoblotting the same membrane with the indicated antibodies. Levels of phosphorylation were quantified using ImageQuant software (version 5.2, GE Healthcare) after scanning the immunoblots. The highest level of phosphorylation in each experiment was set equal to 100, and the levels of phosphorylation in the other lanes were determined relative to this value. The averages from at least three independent experiments were graphed and presented as means ± S.E.
FIGURE 2.

Competition between ATR substrates. A. Quantitative analysis of p53 inhibition of RPA2 phosphorylation. Titration of p53 (0, 6.25, 12.5, 25, 50, or 100 nm) in kinase reactions containing ATR-ATRIP, TopBP1, RPA, and single-stranded ϕX174 DNA as in Fig. 1A. The graph on the right shows the mean inhibition of RPA2 phosphorylation ± S.E. (n ≥ 3). B, quantitative analysis of Rad17-RFC inhibition of RPA2 phosphorylation. Titration of Rad17-RFC (0, 1.56, 3.125, 6.25, 12.5, or 25 nm) in kinase reactions as described in A. The graph on the right shows the mean inhibition of RPA2 phosphorylation ± S.E. (n ≥ 3). C, p53 and Rad17-RFC are noncompetitive inhibitors of RPA2 phosphorylation by ATR. Rates of phosphorylation of RPA2 (10–75 nm) by ATR in the absence (triangles) and the presence of 12.5 nm p53 (circles) or Rad17-RFC (squares) are plotted. Under these conditions, the maximum RPA2 phosphorylation obtained was 2 fmol Pi/fmol of RPA. Apparent Km and Vmax values for ATR phosphorylation of RPA2 (with or without p53 and Rad17-RFC) were determined by nonlinear regression using GraphPad Prism software (version 5).
DNA Binding Assay
A biotinylated 30-mer oligonucleotide annealed to ϕX174 ssDNA (29) was bound to streptavidin-beads according to the manufacturer's instructions (Dynal). Beads containing 50 ng of ϕX174 ssDNA (30 fmol) were incubated with 1 pmol of RPA or RPA mutants in 50 μl of binding buffer (10 mm Tris-Cl, pH 7.5, 100 mm NaCl, 0.01% Nonidet P-40, 10% glycerol, 10 μg/ml BSA) at room temperature for 15 min. The beads were retrieved, washed three times with binding buffer, and then incubated with ∼0.5 pmol HisFLAG-ATRIP, GST-p53, GST-p531–102, or FLAG-Rad17-RFC in 50 μl of binding buffer at room temperature for 30 min. The beads were retrieved and washed three times with binding buffer, and the bound proteins were eluted by boiling in SDS-loading buffer. The eluted proteins were separated on 4–15% TGX-PAGE and analyzed by immunoblotting as described above.
Excision Repair Assay
The repair assay was performed as described previously (39). Internally 32P-labeled DNA substrate (140 bp) containing a single (6–4) UV photoproduct (5–7 fmol) was incubated in a 12.5-μl reaction containing the core excision repair factors (MBP-XPA (72.5 ng), XPC-hR23b (17.5 ng), XPF-ERCC1 (7.5 ng), XPG (4 ng), TFIIH (300 ng), and 150 ng of RPA or RPA mutants) as indicated. The final reactions contained 23 mm Hepes-KOH (pH 7.9), 44 mm KCl, 2.5 mm MgCl2, 2 mm ATP, 2.5% glycerol, 0.04 mm EDTA, and 0.2 mm DTT. After 90 min at 30 °C, the excision reactions were stopped by addition of SDS (0.34%) and proteinase K (20 μg). After incubation at 50–60 °C for 20 min, excision products were purified by phenol-chloroform extraction and then precipitated in ethanol. Excision products were separated on urea-containing DNA sequencing gels and then detected with a PhosphorImager. Excision repair activity was quantified using Image Quant software (version 5.2) by dividing the signal intensity from the small ∼30-mer products by the total signal from both the full-length substrate and ∼30-mer products.
RESULTS
Phosphorylation of RPA, p53, and Rad17 by ATR in a Defined Minimal Checkpoint System
RPA-covered ssDNA is commonly accepted to be the major signal for activating the ATR-mediated DNA damage checkpoint (40). We have previously shown that RPA-ssDNA+TopBP1 are necessary and sufficient to activate the ATR kinase to phosphorylate Chk1 (30). We wished to find out whether other proteins known to be phosphorylated by ATR in vivo during the checkpoint response would be phosphorylated in a similar manner in our in vitro system. We chose to test p53 and Rad17 because these are well characterized substrates (41). In addition, we monitored phosphorylation of Ser33 of RPA2. Eight Ser/Thr residues in the N terminus of RPA2 are phosphorylated in S-phase and during the checkpoint response (20–25). Of these eight residues, Ser33 is specifically phosphorylated by ATR during the DNA damage response (42–44); however, it was not known whether phosphorylation of RPA2 affects the ATR kinase activity or plays a role in the checkpoint response in vivo. Therefore, we tested the phosphorylation of RPA2 at Ser33 in these reactions to gain some insight into the potential significance of this phosphorylation. The results are shown in Fig. 1. As is evident from Fig. 1, all three proteins, p53, Rad17 (included in the reaction is the physiologically relevant Rad17-RFC complex), and the RPA2 subunit of RPA were phosphorylated in a manner dependent on ssDNA, TopBP1, RPA, and ATR (Fig. 1, A and B) under our reaction conditions. Interestingly, in reactions containing p53 or Rad17-RFC, it seemed that phosphorylation of RPA2 was inhibited compared with the reaction with RPA alone (Fig. 1A, compare lane 3 with lanes 8 and 13). Quantitative analysis of the data revealed that under these reaction conditions, the phosphorylation of RPA2 was reduced by 20 to 40% (Fig. 1C).
FIGURE 1.

RPA-ssDNA-dependent phosphorylation of p53 and Rad17 by ATR. A, TopBP1-dependent stimulation of ATR kinase activity by RPA-ssDNA. ATR kinase reactions were carried out with ATR-ATRIP, TopBP1, RPA, single-stranded DNA, p53, and Rad17-RFC, as indicated. 32 nm RPA was pre-incubated with 0.6 ng ϕX174 ssDNA (145 nm of nucleotides), and 0.2 nm ATR-ATRIP, 2.5 nm TopBP1, and 12 nm p53 or Rad17-RFC were then added to the reaction and incubated. Reactions were analyzed by immunoblotting for phospho-p53 (Ser15), phospho-Rad17 (Ser645), and phospho-RPA2 (Ser33). The blots were also analyzed for GST (p53), Rad17, and RPA2 to control for loading. B, the relative levels of phosphorylated p53 and Rad17 from identical repeats of the experiment shown in A were quantified and presented as mean ± S.E. (n ≥ 3). C, the relative levels of RPA2 phosphorylation in reactions with or without p53 or Rad17-RFC from A were quantified and presented as mean ± S.E. (n ≥ 3).
To investigate the effects of p53 and Rad17-RFC on RPA phosphorylation in more detail, we carried out titration reactions. Under conditions of constant RPA-ssDNA, increasing concentrations of either p53 (Fig. 2A) or Rad17-RFC (Fig. 2B) RPA2 phosphorylation was correspondingly inhibited, and at saturating concentrations of these proteins, RPA2 phosphorylation was drastically reduced. To determine the inhibition mechanism, we conducted the kinase assay under constant concentration of p53 or Rad17-RFC and increasing concentrations of RPA-ssDNA. For quantitative analysis, the reaction was performed with [γ-32P]ATP, and the rates of 32P incorporation into RPA2 were measured. The results are shown in Fig. 2C. As evident, p53 and Rad17-RFC inhibited RPA2 phosphorylation similarly. Analysis of the data by Michaelis-Menten formalism revealed that, somewhat surprisingly, inhibition was essentially non-competitive with no (Rad17-RFC) or only minor (p53) effect on Km and 5–7-fold reduction in Vmax (Fig. 2C, bottom). In fact, RPA recruits both ATR-ATRIP and these two substrates to RPA-ssDNA and in a sense reduced the Km values for ATR actions on these substrates while presenting a new substrate (RPA2) for ATR. Clearly, the reaction is more complex than a simple competition reaction and may encompass substrate recruitment by RPA, conformational change of the substrates (RPA versus p53 and RPA versus Rad17-RFC) imposed on one another when in a complex as well as competition for substrate binding sites on ATR-ATRIP. This point, to some extent, is discussed below.
Recruitment of p53 to RPA-ssDNA Is Necessary for Its Phosphorylation by ATR
Many of the ATR substrates bound directly to RPA. To determine whether RPA binding was necessary for the substrate to be phosphorylated by ATR in a manner dependent on RPA-ssDNA, we used a p53 fragment, p531–102, which carries the ATR phosphorylation site (Ser15) but lacks strong RPA binding activity (12), in our kinase assay. As seen in Fig. 3A, the full-length p53 bound weakly to ssDNA as expected because p53 itself had intrinsic DNA binding activity (Fig. 3A, lane 3). This binding was strongly stimulated when the ssDNA was coated with RPA (Fig. 3A, lane 4). In contrast, p531–102 did not bind to either ssDNA (Fig. 3A, lane 7) or to the RPA-ssDNA complex (Fig. 3A, lane 8). Having established these properties of p53 and its N-terminal fragment, we proceeded to carry out kinase reactions with the two forms of p53. The results are shown in Fig. 3B: p53 was weakly phosphorylated by ATR in the presence of RPA alone (Fig. 3B, lane 7), and this phosphorylation was strongly stimulated by ssDNA, presumably in the form of RPA-ssDNA complex (Fig. 3B, lanes 4, 6, and 8). In contrast, the p531–102 fragment, which carries the ATR phosphorylation site but does not bind RPA-ssDNA, was phosphorylated only when present at high concentrations and even then only weakly (Fig. 3B, lanes 11 and 12). Importantly, this phosphorylation was not affected by the presence or absence of ssDNA. These results support the idea that an efficient substrate for ATR must be recruited to ATR on RPA-ssDNA by binding itself to RPA-ssDNA. This conclusion was further studied by the next series of experiments.
FIGURE 3.
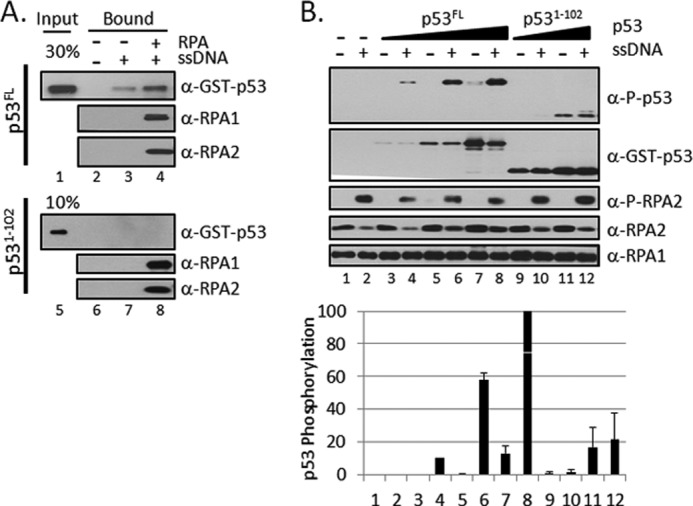
An N-terminal fragment of p53 (p531–102) does not bind to RPA-ssDNA and is not phosphorylated by ATR in a RPA-ssDNA-dependent manner. A, full-length p53FL recruitment to ssDNA by RPA but not the N-terminal fragment of p531–102. Streptavidin-beads with or without single-stranded ϕX174 DNA annealed to a biotinylated 30-mer oligonucleotide (1 pmol) were incubated with or without 3 pmol of RPA. The beads were retrieved, washed, and incubated with 5 pmol of GST- p53FL or fragment p531–102. The beads were then isolated and washed, and bound proteins were separated on SDS-PAGE and analyzed by immunoblotting with anti-GST, anti-RPA1, or anti-RPA2 antibodies. B, the N-terminal fragment of p53 was phosphorylated by ATR, but the phosphorylation was not stimulated by RPA-ssDNA. Full-length p53FL (0, 6.25, 12.5, 25 nm) or the N-terminal fragment, p531–102 (25 or 50 nm), were included in kinase reactions containing ATR-ATRIP, TopBP1, RPA, and with or without single-stranded ϕX174 DNA as in Fig. 1A. The graph at the bottom shows the mean p53 phosphorylation ± S.E. (n ≥ 3).
Effects of RPA1 and RPA2 Mutations on in Vitro Checkpoint Activity of RPA
Genetic screens in yeast and structure-based site-directed mutagenesis identified two RPA1 mutants with normal replication function but defective checkpoint activity (36, 45). In addition, because eight Ser/Thr residues in the RPA2 subunit are phosphorylated in S-phase, and in response to DNA damage (20–25), mutants of this subunit were generated in which the target Ser/Thr residues were replaced by either the phosphomimetic Asp or the non-phosphorylatable Ala residues (11) to understand the physiological significance of this phosphorylation. We purified all of these mutant forms of RPA (Fig. 4A) and tested them in our in vitro kinase assay. The t11 mutant in RPA1 (R41E,T42F) is the human counterpart of the t11 mutant in yeast, which is known to cause checkpoint defects (46). The ΔF mutant in RPA1 had a deletion of the N terminus of RPA1 (ΔN(1–168)), which removes the “F” DNA binding OB-fold (oligonucleotide/oligosaccharide binding fold) domain. With respect to RPA2, it has been shown that RPA2D and RPA2A mutants support mismatch repair (47) and simian virus 40 DNA replication in vitro (48), but it is not known whether they affect ATR activation. With this background as a reference point, then, we tested all four mutants in our in vitro checkpoint assay (Fig. 4B). In agreement with the in vivo data, t11 and ΔF mutants failed to promote the phosphorylation of p53 and Rad17 by ATR. Significantly, in these RPA1 mutants, even RPA2 was not phosphorylated by ATR, even though both mutants have normal DNA binding activity. Interestingly, the RPA2 phosphorylation site mutants (RPA2D and RPA2A), both of which have normal mismatch repair and in vitro replication activities, exhibited opposite phenotypes in our assay: RPA2D mutant lost its ability to promote ATR kinase activity entirely, whereas RPA2A had essentially normal checkpoint activity. As expected, because the ATR kinase target residue Ser33 in these mutants was replaced by non-phosphorylatable Ala or Asp residues, RPA2 was not phosphorylated by ATR.
FIGURE 4.

Mutant RPAs are defective for in vitro checkpoint activation. A, schematic of RPA indicating OB-fold domains A–C and F in RPA1, D in RPA2, and E in RPA3. The N terminus of RPA2, which is heavily phosphorylated, is indicated by P. The locations of point and deletion mutations in RPA1 (RPA1t11(R41E,Y42F) and RPA1ΔF (deletion of amino acids 1–168)) and RPA2 (RPA2D (Ser-8/11/12/13/23/29/33 and Thr21 are replaced by aspartate) and RPA2A (the same mutated amino acids plus Ser2 and Ser4 are replaced with alanine)) are indicated. Purified RPA proteins were analyzed by 4–15% TGX-PAGE and visualized by silver staining. Shown are WT, RPA2D (D); RPA2A (A); RPA1t11 (t11); RPA1ΔF (ΔF). B, ATR kinase reactions were performed as in Fig. 1A, except with the indicated RPA proteins. The kinase reactions in lanes 1–6 contain p53 and those in lanes 7–12 contain Rad17-RFC. The relative levels of phosphorylated p53 and Rad17 from identical repeats were quantified and presented as mean ± S.E. (n ≥ 3). C, ATR kinase reactions were performed as in Fig. 4B, except with various amounts of purified RPAWT, RPA2D, RPA2A (18, 36, or 72 nm). Lane 11 contains 36 nm of both WTWT and RPA2D. The relative levels of phosphorylated p53 from identical repeats of the experiment were quantified and presented as mean ± S.E. (n ≥ 3).
Because the lack of ATR kinase activity in the presence of RPA2D mutation was rather unexpected, we titrated this mutant in the kinase assay. As seen in Fig. 4C, at up to 100 nm concentration, the RPA2D showed no activity, whereas under identical reaction conditions, wild-type RPA and the RPA2A mutant reached saturation at ∼50 nm. Furthermore, the lack of activity in RPA2D was not due to the presence of an inhibitor in the protein preparation because when RPA2D was mixed with wild-type RPA, it did not inhibit the phosphorylation of either p53 or the RPA2 subunit of the wild-type RPA (Fig. 4C, lane 11).
Recruitment of ATR Substrates by RPA
It has been shown that the N-terminal domain of RPA1 (F domain, see Fig. 4A) interacts with many checkpoint proteins. Therefore, we wished to ascertain whether the RPA mutants that were found to be defective in the in vitro checkpoint assay failed to activate ATR kinase because of failure to recruit the ATR substrates to the RPA-ssDNA complex. The binding of the small subunit of ATR, ATRIP, of p53, and of Rad17-RFC to RPA-ssDNA have been well characterized (6–19, 30, 40). Hence, we tested the binding of these proteins to RPA-ssDNA complexes made with wild-type and mutant RPAs to find out whether recruitment of the substrates to ssDNA is necessary and sufficient to target them for phosphorylation by ATR. Immobilized ssDNA was incubated with RPA, the beads were washed to remove free RPA, the immobilized RPA-ssDNA was incubated with the target protein and the protein-protein, protein-DNA interactions were probed by conventional pulldown assays (Fig. 5). Several conclusions can be drawn from this experiment. First, as expected, all forms of RPA (as measured by the amounts of RPA1 and RPA2 retained on ssDNA beads) bind equally well to ssDNA. Second, the binding of ATRIP, p53, and Rad17-RFC to RPA-ssDNAs with RPA1t11 and RPA1ΔF either was reduced drastically or non-detectable, consistent with the reports that the N-terminal domain of RPA1 is involved in protein-protein interactions (8, 35). This would explain the inability of these mutants to activate ATR kinase on these substrates. Third, the RPA2A mutant bound all three substrates as well as the wild-type RPA, whereas the binding of RPA2D was 2–3-fold reduced relative to the wild-type. However, a comparison of the reduced binding of the mutants with substrates observed in Fig. 5 with a total lack of ATR-stimulating activity seen in Fig. 4C strongly suggests that the lack of substrate recruitment cannot entirely explain the inability of RPA2D to activate ATR. It has been proposed that phosphorylation of the target S/T residues in RPA2 may change its interaction with the N terminus of RPA1 (11, 25, 49–51), and as a consequence, even if the substrates are recruited to RPA-ssDNA, this form of RPA cannot activate the ATR kinase. Alternatively, even though the recruitment of substrates by RPA-ssDNA was only partially reduced with mutant RPAs, the recruitment of ATRIP and thus of ATR was also reduced, and thus, the combination of reduced substrate and reduced enzyme on RPA-ssDNA filament may be enough to reduce the ATR-mediated phosphorylation to an undetectable level.
FIGURE 5.

ATRIP, p53, and Rad17-RFC recruitment to ssDNA by RPA and RPA mutants. The binding reactions were performed as in Fig. 3A, and bound proteins were separated by SDS-PAGE and analyzed by immunoblotting with anti-FLAG, anti-GST, anti-Rad17, anti-RPA1, or anti-RPA2 antibodies as indicated. The percent protein bound in identical repeats of the experiment were quantified and presented as mean ± S.E. (n ≥ 3).
Effect of RPA Mutations on Nucleotide Excision Repair
To ascertain that the observed effects of RPA mutations was due to the unique role RPA plays in checkpoint and not to overall decrease in RPA activity because of improper folding, we tested the mutant RPAs for nucleotide excision repair activity. RPA is one of the six core factors of nucleotide excision repair and, without it, the dual incisions characteristic of this repair system do not take place. When the human excision nuclease was reconstituted with the five core repair factors plus wild-type or mutant RPAs, the results shown in Fig. 6 were obtained. Although the checkpoint defective RPA1t11 and RPA1ΔF mutants have somewhat reduced activities, the most striking finding in this figure was that the RPA2A mutant with normal checkpoint activity and the RPA2D mutant with no detectable checkpoint activity exhibited excision repair functions indistinguishable from wild-type RPA. Thus, the RPA2D mutant represents a clear separation of function of RPA with respect to checkpoint and nucleotide excision repair.
FIGURE 6.
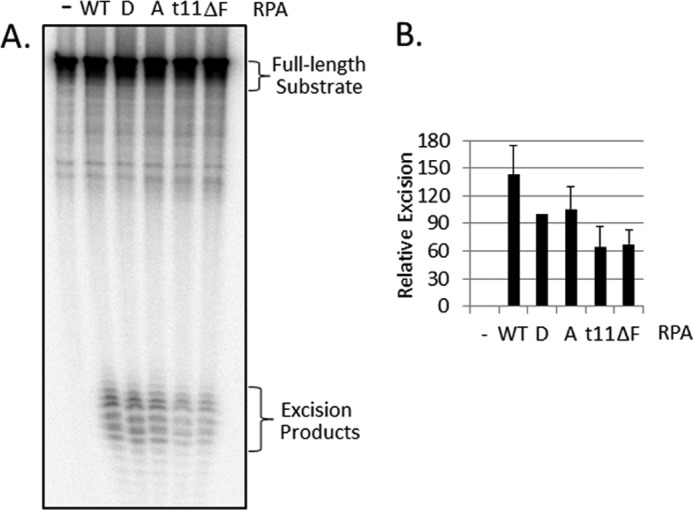
Effect of RPA mutations on nucleotide excision repair. A, image of a representative gel for excision reconstituted with 5–7 fmol of (6–4) UV photoproduct-containing substrate, MBP-XPA, XPC-hR23b, TFIIH, XPF-ERCC1, XPG, and the indicated RPA. For this experiment, percentage excision values were 17.0 (WT), 11.8 (8D), 14.3 (10A), 7.1 (t11), and 7.0 (ΔF). B, graphic presentation of excision values expressed relative to the values for the RPA2D mutant in the same experiment; percentage excision for RPA2D was 13.0 ± 6.8 (n = 6).
DISCUSSION
RPA is the major ssDNA-binding protein in eukaryotic cells (1). It participates in all DNA transactions, including replication, recombination, nucleotide excision repair, mismatch repair, and transcription. In addition, extensive data indicate that it also plays a crucial role in DNA damage checkpoint response. However, attempts to define the role of RPA in checkpoint responses in vitro have had limited success (6, 7, 16, 30). In particular, the function of RPA in the ATR-mediated checkpoint response remains to be delineated by in vitro experiments. In addition, during the checkpoint response in vivo, RPA is phosphorylated (23, 25, 42–44), and currently, it is unclear whether this phosphorylation is part of the checkpoint response. Here, we have attempted to address some of these questions.
An in Vitro ATR Checkpoint System
There are a number of studies using Xenopus or mammalian cell-free extracts or purified mammalian and yeast proteins that have reconstituted certain aspects of the ATR (Mec1)-mediated DNA damage checkpoint. In virtually all of these studies, liberal reaction conditions, such as Mn2+ as the bivalent cation, macromolecular crowding agents in the reactions, and non-physiological substrates were employed (33, 52, 53). Although these studies provided valuable insight into the ATR-mediated checkpoint, they fell short of reconstituting a checkpoint system that incorporates all of the genetically identified constituents of the ATR-mediated DNA damage checkpoint. Here, we have attempted to develop a defined system with purified proteins that more closely approximates the in vivo reaction. In particular, by systematic adjustment of the reaction conditions and enzyme and substrate concentrations, we have been able to establish a defined system consisting of ATR-ATRIP+TopBP1+RPA+ssDNA (30). Using this system, we were able to demonstrate the phosphorylation of p53 and Rad17, two well known substrates of ATR whose phosphorylation is required for ATR-mediated checkpoint response in vivo. We note, however, that our reconstituted system falls short of being a complete representation of the in vivo reaction: The in vivo ATR-mediated checkpoint response is dependent on the Rad17-RFC/9-1-1 checkpoint clamp loader/checkpoint clamp, but in our in vitro system, these are not required even though the Rad17 subunit of Rad17-RFC is phosphorylated by ATR. Clearly, further work is needed to better define the requirement for the 9-1-1 clamp and how this requirement is circumvented under special reaction conditions.
Phosphorylation of RPA and Its Role in the Checkpoint
RPA2 is multiply phosphorylated during S-phase and upon activation of the DNA damage response (20–25). Studies suggest that phosphorylated RPA2 may compete with binding of RPA-interacting proteins to the N terminus of RPA1 (11, 25, 49–51). However, neither the significance of RPA2 phosphorylation for RPA activity during ATR activation nor the effect of other checkpoint proteins on RPA phosphorylation was known. Previous studies with immunoaffinity purified ATR concluded that, provided Mn2+ was used as the bivalent cation, ATR was capable of phosphorylating RPA2 in the RPA-ssDNA complex in the absence of any other factors (33, 52, 53). Here, we show that under the physiologically relevant condition of Mg2+ as the bivalent cation, RPA2 phosphorylation by ATR is dependent on ssDNA and TopBP1, as is the case for other ATR substrates.
The role of RPA phosphorylation in the checkpoint response remains unclear. Multiple studies have implicated phosphorylated forms of RPA in coordinating DNA replication with the cell cycle after DNA damage (5, 44, 54), but the targets and mechanism of regulation remain unknown. Here, we demonstrate that the non-phosphorylatable RPA2A is as efficient as wild-type in promoting phosphorylation of p53 and Rad17 by ATR (Fig. 7). Conversely, RPA2D with phosphomimetic amino acid substitutions fails to activate ATR. This finding would suggest that RPA phosphorylation may play a role in modulating the checkpoint response. RPA recruits both ATR (through its interactions with ATRIP) and target proteins to RPA-ssDNA promoting efficient ATR phosphorylation. Phosphorylation of RPA modulates RPA interactions with checkpoint and other proteins (8, 10, 55). Our results indicate that phosphorylated RPA, by its interaction with substrates and/or ATR may cause conformational changes in both the enzyme and the substrate that modulate activity. This suggests that RPA is playing an active role, rather than just serving as a scaffold, in checkpoint activation.
FIGURE 7.

Model of wild-type and mutant RPA interactions with DNA and checkpoint and repair proteins. RPA is represented in brown, and the N-terminal domain of RPA1 is indicated (F). The N terminus of RPA2, which contains the ATR phosphorylation site at Ser33, is indicated with a brown line, and in the RPA2D mutant, where this domain is heavily charged with aspartic acid residues, the domain is represented in red (bottom panels). The checkpoint proteins ATR-ATRIP, Rad17-RFC, and p53 interact with the N-terminal F domain of RPA1 in wild-type and RPA2A. Mutations in or deletion of the F domain (left middle panel), or phosphomimetic mutations of RPA2, which stabilize the interactions of RPA2 with RPA1 (left bottom panel) abolish checkpoint activation. In contrast, the nucleotide excision repair factors XPA, XPF-ERCC1, and XPG interact with RPA independent of the F domain, and as a consequence, mutations in or deletion of the F domain of RPA1 (right middle panel) or phosphomimetic mutations in RPA2 that stabilize an RPA1-RPA2 interaction (right bottom panel), do not affect nucleotide excision repair.
RPA is an essential factor for nucleotide excision repair (56) and interacts directly with three other essential repair factors, XPA, XPF-ERCC1, and XPG (57–61). Here, we show that mutant RPAs lacking ATR activation exhibit essentially normal activity in nucleotide excision repair. In fact, previous studies have shown that RPA2 phosphorylation does not greatly affect excision repair (55, 62). Together, these results reveal a RPA separation of function for checkpoint and excision repair and suggest that controlling access to the N terminus of RPA1 by phosphorylation of RPA2 after DNA damage may be a mechanism for regulating cell cycle progression without impairing DNA repair.
This work was supported by National Institutes of Health Grants GM32833 (to A. S.) and GM44721 (to M. S. W.).
- RPA
- replication protein A
- RFC
- Replication Factor C
- OB-fold
- oligonucleotide/oligosaccharide binding fold.
REFERENCES
- 1. Wold M. S. (1997) Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 66, 61–92 [DOI] [PubMed] [Google Scholar]
- 2. Fanning E., Klimovich V., Nager A. R. (2006) A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 34, 4126–4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hass C. S., Gakhar L., Wold M. S. (2010) Functional characterization of a cancer causing mutation in human replication protein A. Mol. Cancer Res. 8, 1017–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang Y., Putnam C. D., Kane M. F., Zhang W., Edelmann L., Russell R., Carrión D. V., Chin L., Kucherlapati R., Kolodner R. D., Edelmann W. (2005) Mutation in Rpa1 results in defective DNA double-strand break repair, chromosomal instability, and cancer in mice. Nat. Genet. 37, 750–755 [DOI] [PubMed] [Google Scholar]
- 5. Oakley G. G., Patrick S. M. (2010) Replication protein A: directing traffic at the intersection of replication and repair. Front Biosci. 15, 883–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ball H. L., Myers J. S., Cortez D. (2005) ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol. Biol. Cell 16, 2372–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ball H. L., Ehrhardt M. R., Mordes D. A., Glick G. G., Chazin W. J., Cortez D. (2007) Function of a Conserved Checkpoint Recruitment Domain in ATRIP Proteins. Mol. Cell. Biol. 27, 3367–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu X., Vaithiyalingam S., Glick G. G., Mordes D. A., Chazin W. J., Cortez D. (2008) The basic cleft of RPA70N binds multiple checkpoint proteins, including RAD9, to regulate ATR signaling. Mol. Cell Biol. 28, 7345–7353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bochkareva E., Kaustov L., Ayed A., Yi G. S., Lu Y., Pineda-Lucena A., Liao J. C., Okorokov A. L., Milner J., Arrowsmith C. H., Bochkarev A. (2005) Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc. Natl. Acad. Sci. U.S.A. 102, 15412–15417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Majka J., Binz S. K., Wold M. S., Burgers P. M. (2006) Replication protein A directs loading of the DNA damage checkpoint clamp to 5′-DNA junctions. J. Biol. Chem. 281, 27855–27861 [DOI] [PubMed] [Google Scholar]
- 11. Binz S. K., Lao Y., Lowry D. F., Wold M. S. (2003) The phosphorylation domain of the 32-kDa subunit of replication protein A (RPA) modulates RPA-DNA interactions. Evidence for an intersubunit interaction. J. Biol. Chem. 278, 35584–35591 [DOI] [PubMed] [Google Scholar]
- 12. Dutta A., Ruppert J. M., Aster J. C., Winchester E. (1993) Inhibition of DNA replication factor RPA by p53. Nature 365, 79–82 [DOI] [PubMed] [Google Scholar]
- 13. He Z., Brinton B. T., Greenblatt J., Hassell J. A., Ingles C. J. (1993) The transactivator proteins VP16 and GAL4 bind replication factor A. Cell 73, 1223–1232 [DOI] [PubMed] [Google Scholar]
- 14. Wu X., Shell S. M., Zou Y. (2005) Interaction and colocalization of Rad9/Rad1/Hus1 checkpoint complex with replication protein A in human cells. Oncogene 24, 4728–4735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zou L., Elledge S. J. (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548 [DOI] [PubMed] [Google Scholar]
- 16. Zou L., Liu D., Elledge S. J. (2003) Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc. Natl. Acad. Sci. U.S.A. 100, 13827–13832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Namiki Y., Zou L. (2006) ATRIP associates with replication protein A-coated ssDNA through multiple interactions. Proc. Natl. Acad. Sci. U.S.A. 103, 580–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Furuya K., Miyabe I., Tsutsui Y., Paderi F., Kakusho N., Masai H., Niki H., Carr A. M. (2010) DDK phosphorylates checkpoint clamp component Rad9 and promotes its release from damaged chromatin. Mol. Cell 40, 606–618 [DOI] [PubMed] [Google Scholar]
- 19. Jacobs D. M., Lipton A. S., Isern N. G., Daughdrill G. W., Lowry D. F., Gomes X., Wold M. S. (1999) Human replication protein A: global fold of the N-terminal RPA-70 domain reveals a basic cleft and flexible C-terminal linker. J. Biomol. NMR 14, 321–331 [DOI] [PubMed] [Google Scholar]
- 20. Din S., Brill S. J., Fairman M. P., Stillman B. (1990) Cell cycle-regulated phosphorylation of DNA replication factor A from human and yeast cells. Genes Dev. 4, 968–977 [DOI] [PubMed] [Google Scholar]
- 21. Dutta A., Stillman B. (1992) cdc2 family kinases phosphorylate a human cell DNA replication factor, RPA, and activate DNA replication. EMBO J. 11, 2189–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fotedar R., Roberts J. M. (1992) Cell cycle-regulated phosphorylation of RPA-32 occurs within the replication initiation complex. EMBO J. 11, 2177–2187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carty M. P., Zernik-Kobak M., McGrath S., Dixon K. (1994) UV light-induced DNA synthesis arrest in HeLa cells is associated with changes in phosphorylation of human single-stranded DNA-binding protein. EMBO J. 13, 2114–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu V. F., Weaver D. T. (1993) The ionizing radiation-induced replication protein A phosphorylation response differs between ataxia telangiectasia and normal human cells. Mol. Cell Biol. 13, 7222–7231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shao R. G., Cao C. X., Zhang H., Kohn K. W., Wold M. S., Pommier Y. (1999) Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J. 18, 1397–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lindsey-Boltz L. A., Serçin O., Choi J. H., Sancar A. (2009) Reconstitution of human claspin-mediated phosphorylation of Chk1 by the ATR (ataxia telangiectasia-mutated and rad3-related) checkpoint kinase. J. Biol. Chem. 284, 33107–33114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Choi J. H., Sancar A., Lindsey-Boltz L. A. (2009) The human ATR-mediated DNA damage checkpoint in a reconstituted system. Methods 48, 3–7 [DOI] [PubMed] [Google Scholar]
- 28. Choi J. H., Lindsey-Boltz L. A., Sancar A. (2009) Cooperative activation of the ATR checkpoint kinase by TopBP1 and damaged DNA. Nucleic Acids Res. 37, 1501–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Choi J. H., Lindsey-Boltz L. A., Sancar A. (2007) Reconstitution of a human ATR-mediated checkpoint response to damaged DNA. Proc. Natl. Acad. Sci. U.S.A. 104, 13301–13306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Choi J. H., Lindsey-Boltz L. A., Kemp M., Mason A. C., Wold M. S., Sancar A. (2010) Reconstitution of RPA-covered single-stranded DNA-activated ATR-Chk1 signaling. Proc. Natl. Acad. Sci. U.S.A. 107, 13660–13665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chang M. S., Sasaki H., Campbell M. S., Kraeft S. K., Sutherland R., Yang C. Y., Liu Y., Auclair D., Hao L., Sonoda H., Ferland L. H., Chen L. B. (1999) HRad17 colocalizes with NHP2L1 in the nucleolus and redistributes after UV irradiation. J. Biol. Chem. 274, 36544–36549 [DOI] [PubMed] [Google Scholar]
- 32. Lee J. H., Paull T. T. (2004) Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304, 93–96 [DOI] [PubMed] [Google Scholar]
- 33. Unsal-Kaçmaz K., Sancar A. (2004) Quaternary structure of ATR and effects of ATRIP and replication protein A on its DNA binding and kinase activities. Mol. Cell Biol. 24, 1292–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Henricksen L. A., Umbricht C. B., Wold M. S. (1994) Recombinant replication protein A: expression, complex formation, and functional characterization. J. Biol. Chem. 269, 11121–11132 [PubMed] [Google Scholar]
- 35. Binz S. K., Wold M. S. (2008) Regulatory functions of the N-terminal domain of the 70-kDa subunit of replication protein A (RPA). J. Biol. Chem. 283, 21559–21570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haring S. J., Mason A. C., Binz S. K., Wold M. S. (2008) Cellular functions of human RPA1. Multiple roles of domains in replication, repair, and checkpoints. J. Biol. Chem. 283, 19095–19111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kang T. H., Lindsey-Boltz L. A., Reardon J. T., Sancar A. (2010) Circadian control of XPA and excision repair of cisplatin-DNA damage by cryptochrome and HERC2 ubiquitin ligase. Proc. Natl. Acad. Sci. U.S.A. 107, 4890–4895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lindsey-Boltz L. A., Sancar A. (2011) Tethering DNA damage checkpoint mediator proteins topoisomerase IIbeta-binding protein 1 (TopBP1) and Claspin to DNA activates ataxia-telangiectasia mutated and RAD3-related (ATR) phosphorylation of checkpoint kinase 1 (Chk1). J. Biol. Chem. 286, 19229–19236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reardon J. T., Sancar A. (2006) Purification and characterization of Escherichia coli and human nucleotide excision repair enzyme systems. Methods Enzymol. 408, 189–213 [DOI] [PubMed] [Google Scholar]
- 40. Cimprich K. A., Cortez D. (2008) ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim S. T., Lim D. S., Canman C. E., Kastan M. B. (1999) Substrate specificities and identification of putative substrates of ATM kinase family members. J. Biol. Chem. 274, 37538–37543 [DOI] [PubMed] [Google Scholar]
- 42. Vassin V. M., Anantha R. W., Sokolova E., Kanner S., Borowiec J. A. (2009) Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA replication stress. J. Cell Sci. 122, 4070–4080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Anantha R. W., Vassin V. M., Borowiec J. A. (2007) Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. J. Biol. Chem. 282, 35910–35923 [DOI] [PubMed] [Google Scholar]
- 44. Olson E., Nievera C. J., Klimovich V., Fanning E., Wu X. (2006) RPA2 is a direct downstream target for ATR to regulate the S-phase checkpoint. J. Biol. Chem. 281, 39517–39533 [DOI] [PubMed] [Google Scholar]
- 45. Umezu K., Sugawara N., Chen C., Haber J. E., Kolodner R. D. (1998) Genetic analysis of yeast RPA1 reveals its multiple functions in DNA metabolism. Genetics 148, 989–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim H. S., Brill S. J. (2001) Rfc4 interacts with Rpa1 and is required for both DNA replication and DNA damage checkpoints in Saccharomyces cerevisiae. Mol. Cell Biol. 21, 3725–3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guo S., Zhang Y., Yuan F., Gao Y., Gu L., Wong I., Li G. M. (2006) Regulation of replication protein A functions in DNA mismatch repair by phosphorylation. J. Biol. Chem. 281, 21607–21616 [DOI] [PubMed] [Google Scholar]
- 48. Vassin V. M., Wold M. S., Borowiec J. A. (2004) Replication protein A (RPA) phosphorylation prevents RPA association with replication centers. Mol. Cell Biol. 24, 1930–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Abramova N. A., Russell J., Botchan M., Li R. (1997) Interaction between replication protein A and p53 is disrupted after UV damage in a DNA repair-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 94, 7186–7191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Oakley G. G., Patrick S. M., Yao J., Carty M. P., Turchi J. J., Dixon K. (2003) RPA phosphorylation in mitosis alters DNA binding and protein-protein interactions. Biochemistry 42, 3255–3264 [DOI] [PubMed] [Google Scholar]
- 51. Oakley G. G., Tillison K., Opiyo S. A., Glanzer J. G., Horn J. M., Patrick S. M. (2009) Physical interaction between replication protein A (RPA) and MRN: involvement of RPA2 phosphorylation and the N terminus of RPA1. Biochemistry 48, 7473–7481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Barr S. M., Leung C. G., Chang E. E., Cimprich K. A. (2003) ATR kinase activity regulates the intranuclear translocation of ATR and RPA following ionizing radiation. Curr. Biol. 13, 1047–1051 [DOI] [PubMed] [Google Scholar]
- 53. Liu J. S., Kuo S. R., Melendy T. (2006) Phosphorylation of replication protein A by S-phase checkpoint kinases. DNA Repair 5, 369–380 [DOI] [PubMed] [Google Scholar]
- 54. Anantha R. W., Sokolova E., Borowiec J. A. (2008) RPA phosphorylation facilitates mitotic exit in response to mitotic DNA damage. Proc. Natl. Acad. Sci. U.S.A. 105, 12903–12908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Patrick S. M., Oakley G. G., Dixon K., Turchi J. J. (2005) DNA damage induced hyperphosphorylation of replication protein A. 2. Characterization of DNA binding activity, protein interactions, and activity in DNA replication and repair. Biochemistry 44, 8438–8448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mu D., Park C. H., Matsunaga T., Hsu D. S., Reardon J. T., Sancar A. (1995) Reconstitution of human DNA repair excision nuclease in a highly defined system. J. Biol. Chem. 270, 2415–2418 [DOI] [PubMed] [Google Scholar]
- 57. Matsunaga T., Park C. H., Bessho T., Mu D., Sancar A. (1996) Replication protein A confers structure-specific endonuclease activities to the XPF-ERCC1 and XPG subunits of human DNA repair excision nuclease. J. Biol. Chem. 271, 11047–11050 [DOI] [PubMed] [Google Scholar]
- 58. Bessho T., Sancar A., Thompson L. H., Thelen M. P. (1997) Reconstitution of human excision nuclease with recombinant XPF-ERCC1 complex. J. Biol. Chem. 272, 3833–3837 [DOI] [PubMed] [Google Scholar]
- 59. He Z., Henricksen L. A., Wold M. S., Ingles C. J. (1995) RPA involvement in the damage recognition and incision steps of nucleotide excision repair. Nature 374, 566–569 [DOI] [PubMed] [Google Scholar]
- 60. de Laat W. L., Appeldoorn E., Sugasawa K., Weterings E., Jaspers N. G., Hoeijmakers J. H. (1998) DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev. 12, 2598–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fisher L. A., Bessho M., Wakasugi M., Matsunaga T., Bessho T. (2011) Role of interaction of XPF with RPA in nucleotide excision repair. J. Mol. Biol. 413, 337–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pan Z. Q., Park C. H., Amin A. A., Hurwitz J., Sancar A. (1995) Phosphorylated and unphosphorylated forms of human single-stranded DNA-binding protein are equally active in simian virus 40 DNA replication and in nucleotide excision repair. Proc. Natl. Acad. Sci. U.S.A. 92, 4636–4640 [DOI] [PMC free article] [PubMed] [Google Scholar]