

Background: HpaI is a metal-dependent pyruvate aldolase bridging aromatic hydrocarbon degradation and central metabolism.
Results: Crystal structures of enzyme·substrate complexes reveal the catalytic mechanism.
Conclusion: Two water molecules, one of them Co2+-bound, are acid-base catalysts, whereas an arginine residue stabilizes transient negative charges.
Significance: Results provide a framework for engineering pyruvate aldolases with improved biocatalysis of C–C bond formation.
Keywords: Crystal Structure, Enzyme Kinetics, Enzyme Mechanisms, Enzyme Structure, X-ray Crystallography, Charge Stabilization, Cryotrapping, Metal Ion Catalysis, Pyruvate Class II Aldolase, Substrate Trapping
Abstract
Crystal structures of divalent metal-dependent pyruvate aldolase, HpaI, in complex with substrate and cleavage products were determined to 1.8–2.0 Å resolution. The enzyme·substrate complex with 4-hydroxy-2-ketoheptane-1,7-dioate indicates that water molecule W2 bound to the divalent metal ion initiates C3–C4 bond cleavage. The binding mode of the aldehyde donor delineated a solvent-filled capacious binding locus lined with predominantly hydrophobic residues. The absence of direct interactions with the aldehyde aliphatic carbons accounts for the broad specificity and lack of stereospecific control by the enzyme. Enzymatic complex structures formed with keto acceptors, pyruvate, and 2-ketobutyrate revealed bidentate interaction with the divalent metal ion by C1-carboxyl and C2-carbonyl oxygens and water molecule W4 that is within close contact of the C3 carbon. Arg70 assumes a multivalent role through its guanidinium moiety interacting with all active site enzymatic species: C2 oxygen in substrate, pyruvate, and ketobutyrate; substrate C4 hydroxyl; aldehyde C1 oxygen; and W4. The multiple interactions made by Arg70 stabilize the negatively charged C4 oxygen following proton abstraction, the aldehyde alignment in aldol condensation, and the pyruvate enolate upon aldol cleavage as well as support proton exchange at C3. This role is corroborated by loss of aldol cleavage ability and pyruvate C3 proton exchange activity and by a 730-fold increase in the dissociation constant toward the pyruvate enolate analog oxalate in the R70A mutant. Based on the crystal structures, a mechanism is proposed involving the two enzyme-bound water molecules, W2 and W4, in acid/base catalysis that facilitates reversible aldol cleavage. The same reaction mechanism promotes decarboxylation of oxaloacetate.
Introduction
Aldolases catalyze reversible stereospecific carbon-carbon bond formation and play critical roles in carbon cycling in nature. They are also useful biocatalysts for synthesis of organic molecules for use as pharmaceuticals due to their carbon-carbon joining activity (1–3). They exhibit diversity in terms of their sequences, structures, substrate specificities, and nature of catalytic residues. For example, class I aldolases utilize a lysine residue that forms a Schiff base with the keto function of the substrate, whereas class II aldolases contain divalent metal ions that stabilize the carbanion intermediate. Within class II aldolases, the dihydroxyacetone phosphate-specific enzymes generally contain a tetrahedral coordinated Zn2+ ion as cofactor (4–7), whereas the pyruvate-specific enzymes contain an octahedral coordinated divalent metal cofactor, which is usually Mg2+, Mn2+, or Co2+ (8–12).
HpaI (also known as HpcH) (4-hydroxy-2-ketoheptane-1,7-dioate; EC 4.1.3) is a class II (metal-dependent) pyruvate aldolase involved in the last step of 3- and 4-hydroxyphenylacetate catabolism in Escherichia coli strains W and C (13). The physiological substrate for HpaI is 4-hydroxy-2-ketoheptane-1,7-dioate (HKHD),3 which it transforms through a retro-aldol reaction to pyruvate and succinic semialdehyde. The principal steps in the chemical mechanism comprise abstraction of a proton from C4-OH of the substrate (1) catalyzed by a base, aldol cleavage resulting in the formation of a pyruvate enolate intermediate (2) that is stabilized by the divalent metal ion, and proton donation at the C3 carbon of pyruvate enolate to form pyruvate (3) completing the reaction (Scheme 1).
SCHEME 1.
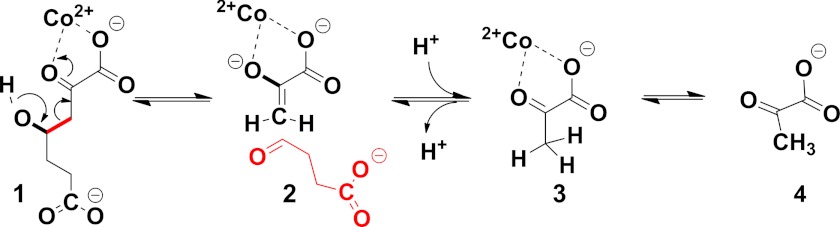
Intermediates (1–4) of the catalytic mechanism in pyruvate class II aldolases.
HpaI displays broad substrate specificity as it is able to utilize other 4-hydroxy-2-oxoacid derivatives as substrates (11). For example, it can convert 4-hydroxy-2-oxopentanoate (HOPA) to pyruvate and acetaldehyde (14). It is also able to utilize both pyruvate and 2-ketobutyrate as carbonyl donors in the aldol addition reaction, although the enzyme lacks stereospecific control, producing equal mixtures of 4R and 4S enantiomers (15).
The three-dimensional structures of HpaI in its apo form and containing the pyruvate structural analog oxamate have been determined (9). The enzyme adopts a triose-phosphate isomerase barrel fold with helix 8 of the (αβ)8 barrel in each subunit, forming a domain-swapped dimer with a neighboring subunit in an overall hexameric structure. This structure resembles that of its homologs, 2-dehydro-3-deoxygalactarate aldolase (8) and 2-dehydro-3-deoxy-l-rhamnonate aldolase (YfaU) (16), which are involved in sugar metabolism in microorganisms. An enzyme-bound phosphate molecule was found in the structure of apo-2-dehydro-3-deoxygalactarate aldolase and has been proposed to be the acid that donates a proton to the pyruvate enolate (8). However, in phosphate-free buffer, HpaI is able to catalyze the half-reaction of pyruvate C3 proton exchange (11), thus discounting the role of phosphate in the catalytic mechanism of the enzyme. Instead, an arginine residue (Arg70) in the active site appears to be important for catalysis because substitution of this residue in HpaI with alanine led to inactive enzyme that was unable to catalyze pyruvate C3 proton exchange (11). Replacements of histidine 45 in HpaI to alanine and glutamine on the other hand led to a decrease in kcat of the enzyme by 78- and 2059-fold, respectively, and the dependence of the enzyme activity on the concentration of hydroxide ions (17). Lacking among currently available structures of HpaI and its homologs are structures showing substrate binding as well as that of the aldehyde product. How the enzyme recognizes its substrates and products provides the requisite structural information that delineates the roles of pertinent active site residues and the reaction mechanism by which the enzyme promotes catalysis.
A crystallographic study of HpaI in complex with reaction products pyruvate, 2-ketobutyrate, and succinic semialdehyde was undertaken. HpaI crystals cocrystallized with pyruvate and then soaked with excess succinic semialdehyde trapped the physiological substrate 4-hydroxy-2-ketoheptane-1,7-dioate. We also present crystal structures of R70A and D42A variants of HpaI aldolase. The availability of the first structures of a class II pyruvate aldolase containing the full substrate and the cleavage products significantly advances our understanding of how substrate binding and catalysis occur in this important group of enzymes.
MATERIALS AND METHODS
Chemicals
l-Lactate dehydrogenase (rabbit muscle) and Chelex 100 were from Sigma-Aldrich. Restriction enzymes and Pfu Ultra DNA polymerase were from Invitrogen or New England Biolabs (Pickering, Ontario, Canada). (S)-4-Hydroxy-2-ketopentanoate used in kinetic analysis was generated enzymatically from catechol using catechol dioxygenase, 2-hydroxy-6-oxohepta-2,4-dienoate hydrolase, and 2-hydroxypent-2,4-dienoate hydratase (11). 4-Hydroxy-3-methyl-2-ketopentanoate on the other hand was synthesized using Mn2+-HpaI according to a previous method (15) that generated a racemic mixture. All other chemicals were of analytical grade and were obtained from Sigma-Aldrich and Fisher Scientific.
DNA Manipulation
The R70A variant of HpaI has been constructed previously (11). The QuikChange method (Stratagene) for site-specific mutagenesis was used to create the R70K and D42A variants of HpaI. The sequences of the sense oligonucleotides used in the mutagenesis procedure are CCGGTGGTAGCTCCGTCGTGGAACG and GACTGGTTATTGATCGCCGGTGAGCAC, respectively (the altered codons are underlined). The reactions contained 30 ng of wild-type hpaI in the vector pT7-7, 115 ng of each mutagenic oligonucleotide, 0.2 mm dNTPs, and 2.5 units of Pfu Ultra DNA polymerase (Stratagene). Mutants were screened by direct DNA sequencing.
Expression and Purification of Recombinant Wild-type and Mutant Enzymes
The overexpression and purification of wild-type and mutant HpaI were performed according to previous methods (11).
Enzymatic Assay
Kinetics assays were performed in 100 mm sodium HEPES, pH 8.0 with 0.5 mm CoCl2 as described previously (11). Solvent isotope effects were determined using the normal enzyme activity assays except that assay solutions and substrate were dissolved in D2O, evaporated to dryness two times, and then redissolved in the appropriate amount of D2O. The assay buffer was adjusted to pD 8.0 using NaOD according to the equation pD = pHmeter reading + 0.4 (18).
Protein Fluorescence Measurements
Protein fluorescence titration was used to determine the equilibrium dissociation constant of substrate analogs in wild-type and mutant enzymes as described previously (11). Tryptophan quenching of about 50% was observed when the enzymes were saturated with ligands. The concentration of enzymes in all measurements was fixed at 90 μg/ml. Metal-free apoenzyme was prepared using Chelex 100 (Sigma) as described previously (11).
α-Proton Exchange of 2-Keto Acids Catalyzed by Wild-type and Mutant HpaI
The α-proton exchange rates of pyruvate and 2-ketobutyrate catalyzed by the enzymes were determined in deuterated buffer by 1H NMR (15). The effects of pD on the proton exchange rate of pyruvate catalyzed by enzyme were studied in deuterated constant ionic strength buffer containing 100 mm Tris, 50 mm acetic acid, 50 mm MES, and 1.0 mm CoCl2 with pD ranging from 5.4 to 9.4. The signal of acetate methyl protons (1.80 ppm) was chosen as an internal reference to assess the pyruvate α-proton exchange. The pD data were fitted to the following equation by non-linear regression in the program Leonora (19),
where C is the pD-independent plateau value of the α-proton exchange rate of pyruvate, D is the deuterium concentration, and Ka1 is the ionization constant of the deprotonation group involved in the reaction.
Crystallization and Data Collection
The wild-type enzyme and D42A and R70A mutants were crystallized by the hanging drop vapor diffusion technique. Each drop consisted of 2 μl of protein solution (15 and 10 mg/ml, respectively) and 2 μl of reservoir solution (0.5 m ammonium dihydrogen phosphate, 30% glycerol, 2 mm CoCl2, pH 5 for the native enzyme and D42A mutant and 22% PEG 2000 monomethyl ether, 10 mm NiCl2, 25 mm HEPES, pH 8 for the R70A mutant). Ligand concentrations were set to 10 mm in the crystallization solution. Harvested crystals were soaked in mother liquor solutions containing fresh ligand at the same concentration as that in the crystallization solution prior to their harvesting. Crystals were cryoprotected in mother liquor made up with 15% (v/v) glycerol and where appropriate 10 mm ligand and then flash cooled in liquid nitrogen. The data were collected at beamlines X25 and X29 of the National Synchrotron Light Source at Brookhaven National Laboratories.
A fluorescence energy scan performed at beamline X29 by energy scanning about the CoKα edge (1.6083 Å) demonstrated that crystals of R70A variants contained cobalt, although no exogenous metal had been added during purification or recrystallization of the protein. Different from the R70A mutant, cobalt was a constituent of the crystallization buffer used for the native enzyme. To corroborate the presence of cobalt ion in the active site in native enzyme cocrystallized with ligand, full anomalous data sets were collected for native enzyme cocrystallized with pyruvate and 2-ketobutyrate with the wavelength set to 1.6 Å near the CoKα edge. Data reduction was performed using HKL2000 (20) or XDS (21), and final statistics are shown in Table 1.
TABLE 1.
Wild-type and mutant HpaI aldolase data collection statistics
| Data collection | Crystal structure |
|||||
|---|---|---|---|---|---|---|
| WT·pyruvate complex | WT·ketobutyrate complex | WT·pyruvate + succinic semialdehyde complex | WT·4-hydroxyl-2-ketoheptane-1,7-dioate complex | R70A·pyruvate complex | D42A | |
| Resolution (Å) | 45.7–1.68 | 47.8–1.92 | 41.9–1.91 | 37.9–2.04 | 38.5–1.79 | 30.86–1.80 |
| (1.72–1.68)a | (2.03–1.92) | (1.96–1.91) | (2.09–2.04) | (1.85–1.79) | (1.90–1.80) | |
| Wavelength (Å) | 1.075 | 0.9795 | 0.9795 | 0.9795 | 1.1 | 1.0809 |
| Unique reflections/multiplicity | 131,767/21.4 | 78,453/23.6 | 87,289/21.8 | 72,826/19.4 | 127,694/6.2 | 103,465/17.8 |
| (9,675/19.15) | (5,707/17.5) | (5,314/4.9) | (4,391/4.2) | (11,986/5.4) | (13,728/7.8) | |
| Completeness (%) | 99.9(100) | 99.7 (98.1) | 98.6 (81.7) | 98.5 (80.9) | 98.9 (93.8) | 97.5 (87.0) |
| Average I/σ(I) | 19.2(2.0) | 12.9 (2.8) | 21.1 (2.9) | 18.5 (2.0) | 25.5 (4.8) | 16.8(1.8) |
| Rpimb | 0.02 (0.41) | 0.03 (0.24) | 0.03 (0.24) | 0.03 (0.29) | 0.05 (0.23) | 0.02(0.38) |
| Space group | P213 | P213 | P213 | P213 | P212121 | P213 |
| Unit cell parameters a, b, c (Å) | 151.74 | 151.74 | 151.13 | 151.74 | 81.35, 119.14, 140.56 | 151.17 |
a All values in parentheses are given for the highest resolution shell.
b Rpim = Σhkl√(1/N − 1)Σi|Ii(hkl) − ¯Ii(hkl)|/Σhkl ΣiIi(hkl) with i running over the number of independent observations of reflection hkl and N representing the number of replicate observations.
Structure Determination
Structures were solved by molecular replacement with the PHENIX (22) automr strategy using a homology model generated by MODELLER (23) based on the structure of E. coli 2-dehydro-3-deoxygalactarate aldolase in complex with pyruvate (Protein Data Bank code 1DXF). Refinement was performed using PHENIX software suites and model building with Coot (24). Ligand modeling was based on interpretation of the electron density shapes of 2Fo − Fc and Fo − Fc annealed omit maps; the phenix.elbow command was used for generation of topology and parameters.
Final model statistics calculated with PHENIX, Molprobity (25), and SFCHECK (26) are shown in Table 2. The atomic coordinates and structure factors for the native enzyme in complex with pyruvate, ketobutyrate, pyruvate and succinate semialdehyde, and HKHD as well as the R70A mutant in complex with pyruvate and D42A mutant have been deposited with the Protein Data Bank (see Table 2 for Protein Data Bank accession codes). The final structure models have Rcryst (Rfree) values of 15.2% (17.9%), 15.5% (18.5%), 13.0% (15.4%), 15.3% (17.9%), 14.4% (18.4%), and 16.5% (19.3%), respectively. All figures were prepared using the program PyMOL (27).
TABLE 2.
Refinement statistics
| Crystal structure |
||||||
|---|---|---|---|---|---|---|
| WT·pyruvate complex | WT·ketobutyrate complex | WT·pyruvate + succinic semialdehyde complex | WT·4-hydroxyl-2-ketoheptane-1,7-dioate complex | R70A·pyruvate complex | D42A | |
| Number of atoms | ||||||
| Protein | 3,828 | 3,800 | 3,806 | 3,828 | 11,473 | 3,808 |
| Water | 1,091 | 815 | 808 | 803 | 1,858 | 896 |
| Hetero | 62 | 60 | 100 | 132 | 65 | 68 |
| Rcryst (%)a | 15.2 | 15.5 | 13.0 | 15.3 | 14.4 | 16.5 |
| Rfree (%)b | 17.9 | 18.5 | 15.4 | 17.9 | 18.4 | 19.3 |
| Root mean square deviation | ||||||
| Bond length (Å) | 0.018 | 0.011 | 0.014 | 0.004 | 0.010 | 0.017 |
| Bond angle (°) | 1.65 | 1.32 | 1.39 | 0.868 | 1.25 | 1.46 |
| Average B-factor (Å2) | 24.2 | 32.4 | 23.9 | 35.9 | 21.2 | 24.6 |
| Ramachandran analysisc (%) | ||||||
| Favored regions | 99.6 | 99.5 | 99 | 99 | 98.8 | 98.6 |
| Allowed regions | 0.4 | 0.5 | 1 | 1 | 1.2 | 1.4 |
| Atomic coordinate error (Å) | 0.16 | 0.17 | 0.13 | 0.24 | 0.16 | 0.19 |
| Protein Data Bank accession codes | 4B5S | 4B5T | 4B5U | 4B5V | 4B5W | 4B5X |
a Rcryst = Σhkl‖Fo(hkl)| − |Fc(hkl)‖/Σhkl|Fo(hkl)|.
b Rfree = ΣhklÎT‖Fo(hkl) − |Fc(hkl)|/ΣhklÎT|Fo(hkl)| where T is a test data set randomly selected from the observed reflections prior to refinement. The test data set was not used throughout refinement and contained 7.5–10% of the total unique reflections.
c Analyzed by Molprobity.
For all structures, electron density was observed that was consistent with cobalt metal binding. However, the use of a fully occupied cobalt binding site during structure refinement resulted in a negative peak in the Fo − Fc electron density difference map for the cobalt atom. Analysis of the anomalous scattering data sets collected near the cobalt Kα absorption edge (1.6 Å) revealed an electron density peak in the anomalous difference map that was coincident with the metal binding site and confirmed the presence of bound cobalt (supplemental Fig. S1). Refinement of the anomalous data sets indicated partial occupancy by the cobalt cofactor; however; full occupancy was indicated for the ligands. Refinement was continued with a mixture of cobalt and magnesium ions whose combined populations were consistent with full occupancy of the metal ion binding site.
Structure alignment among selected pyruvate aldolases was generated with the PyMOL “super” command using the Cα atomic positions in each subunit of the asymmetric unit cell. After formatting of the aligned Cα coordinates, r.m.s.d. was computed using the PyMOL “rms_cur” command. Interatomic distances calculated in non-crystallographically related subunits differed less than the atomic coordinate error shown in Table 2.
Superimposition of evolutionary related structures P-enolpyruvate binding domain of enzyme I for bacterial P-enolpyruvate-dependent carbohydrate:phosphotransferase systems (Protein Data Bank code 2BG5, residues 261–573) and pyruvate, phosphate dikinase (Protein Data Bank code 1KC7 residues 534–874) was conducted using DaliLite (28). Superimposition of non-homolog class II pyruvate aldolases with functional commonality consisted of matching the atomic positions of pyruvate atoms and the divalent metal ion in the pyruvate-bound HpaI aldolase structure against equivalent atoms in the target structures. In the bacterial 4-hydroxy-2-oxopentanoate (DmpG) aldolase structure (Protein Data Bank code 1NVM) (10), the target atoms were pyruvate atoms and the divalent manganese atom, respectively. For the bacterial oxaloacetate decarboxylase (Protein Data Bank code 3B8I) (30), target atoms included oxalate atoms and the divalent magnesium atom, respectively. In bacterial HMG/CHA aldolase (Protein Data Bank code 3NOJ) (12), target atoms were pyruvate atoms and the divalent magnesium atom, respectively. In the absence of a bound active site ligand in human 3-hydroxy-3-methylglutaryl-CoA lyase (Protein Data Bank code 2CW6) (31), a structural alignment was performed using HpaI aldolase as the target structure.
RESULTS
Intermediate Trapping and General Structure
Initial electron density maps of HpaI aldolase crystal soaked with ligands, especially aldehydes, did not always show full occupancies in the active site. To ensure highest occupancy of intermediates, the cryoprotection buffer consisting of mother liquor (pH 5.0), 15% (v/v) glycerol, and 1.0 mm CoCl2 was adjusted to include a 10 mm concentration of the appropriate ligands wherever feasible.
A modified trapping strategy used for active site binding of HKHD as HpaI crystals soaked with an excess of pyruvate and succinic semialdehyde did not reveal electron density consistent with aldol condensation. Although loss of catalytic activity under crystallization conditions would explain the absence of trapped substrate in the electron density map, the kinetic parameters toward HOPA, a surrogate HpaI aldolase substrate, indicated turnover under crystallization conditions even though kcat was reduced 500-fold to 0.69 ± 0.04 s−1 compared with HpaI activity in 100 mm sodium HEPES buffer, pH 8.0. For HpaI crystals soaked in the presence of 10 mm substrate prior to cryotrapping, this would result in a number of turnovers on a minute time scale. Although substrate binding occurs at 10 mm concentrations under crystallization conditions as Km for HOPA increased only 10-fold to 3.4 ± 0.6 mm from the reference condition, turnover would result in not only HOPA but also pyruvate and acetaldehyde trapped in the active site and make interpretation of the composite electron density map challenging. To minimize the number of potential intermediates trapped in the active site, crystals of HpaI aldolase grown in the presence of pyruvate were soaked with excess succinic semialdehyde (10 mm) to chase all 2-keto acid into substrate. Under these soaking conditions, electron density was observed in the active site of HpaI crystals whose shape was consistent with HKHD binding.
Ligands and metal were modeled into the electron density map in the last stages of refinement, and the quality of the fit to the electron density was validated by inspection of simulated annealing and kicked omit maps (32). The high quality of the electron density map enabled interpretation of all bound ligands and metal ions in the active site.
Each HpaI aldolase subunit exhibits a triose-phosphate isomerase barrel fold containing a repeated 8-fold (β/α) motif with the active site located on the C-terminal side of the β barrel, which is consistent with the apo- and oxamate-bound HpaI structures determined previously (9). The asymmetric unit contains one HpaI aldolase dimer, and the 3-fold crystallographic symmetry generates a hexameric assembly with 32 symmetry, which is the physiologically relevant quaternary structure, as corroborated by size exclusion chromatography (9). The crystal structure of the R70A variant possessed a hexamer exhibiting 32 non-crystallographic symmetry in the asymmetric unit cell, confirming that the HpaI quaternary structure is indeed a trimer of dimers (supplemental Fig. S2). The asymmetric unit of the D42A contains a dimer and together with the 3-fold crystallographic symmetry also generates a hexameric assembly with 32 symmetry.
Metal and 2-Keto Acid Binding
The active site of HpaI aldolase recognizes pyruvate and 2-ketobutyrate identically (Fig. 1, A and B), which is consistent with the similar dissociation constants (Kd) observed for the two compounds (0.32 ± 0.02 and 0.47 ± 0.02 mm, respectively). Backbone amides of conserved residues Ala174 (3.00 Å) and Asp175 (3.04 Å) form hydrogen bonds and direct the carboxylate oxygens of the 2-keto acid ligand, whereas conserved residues Gln147 (3.11 Å) and Arg70 (2.90 Å) interact with the keto oxygen. The 2-keto and carboxylate oxygens of the two ligands also coordinate with the divalent metal ion. This coordination plays an important role for 2-keto acid binding because the Kd for pyruvate binding decreased by 60–70-fold in comparison with apoenzyme (Table 3). The carboxylate side chains of two conserved residues, Glu149 (2.27 Å) and Asp175 (2.22 Å), and two water molecules, W1 (2.30 Å) and W2 (2.20 Å), complete the octahedral shell of the bound metal ion. W1 and W2 positions are in turn stabilized through hydrogen bonds with Glu44 and the Val118′ backbone carbonyl of a symmetry-related subunit, respectively (Fig. 1C).
FIGURE 1.

Trapped 2-keto acids in active site of HpaI pyruvate aldolase. A, electron density of pyruvate bound in the active site of the native enzyme. B, electron density of ketobutyrate trapped in the active site of the native enzyme. C, stereoview showing active site interactions when pyruvate binds to HpaI aldolase. In A and B, water molecules occupying the aldehyde binding site (presented in Fig. 3) are shown in green. Magenta dashes depict coordination of Co2+ made by the two conserved water molecules (W1 and W2), pyruvate (blue), and residues Glu149 and Asp175. The backbone nitrogens of Ala174 and Glu175 direct the carboxylate oxygens of the pyruvate molecule, and Gln147 and Arg70 interact with the keto oxygen; these interactions are shown by black dashes. These interactions are used to specifically orient pyruvate in the active site. Hydrogen bonding interactions not directly contacting the metal ion or pyruvate (gray dots) organize remaining active site residues and water molecules. Interactions from Val118′ and Asp84′ of an adjacent contacting subunit (blue) complete the active site binding locus. The consistency in hydrogen bonding interactions made with W3 implies that Glu44 and Glu172 are hydrogen bond acceptors, whereas His45 is a hydrogen bond donor. Electron density encompassing the ligands was calculated from a simulated annealing Fo − Fc omit map and contoured at 3.5 σ.
TABLE 3.
Dissociation constants of pyruvate and oxalate in HpaI aldolase determined by protein fluorescence quenching titration
All measurements were performed in 100 mm sodium HEPES buffer, pH 8.0.
| Enzymes |
Kd |
||
|---|---|---|---|
| Pyruvate | Pyruvate and 0.5 mm Co2+ | Oxalate and 0.5 mm Co2+ | |
| mm | mm | μm | |
| ApoHpaI | 21.2 ± 2.4 | 0.32 ± 0.022 | 10.4 ± 0.75 |
| ApoR70K | 18.8 ± 1.3 | 0.32 ± 0.004 | 8.2 ± 0.3 |
| ApoR70A | 26.9 ± 3.1 | 0.47 ± 0.001 | 7601 ± 552 |
| ApoD42A | 32.5 ± 1.7 | 31.2 ± 1.6 | a |
a No fluorescence quenching was observed for sodium oxalate at concentrations up to 40 mm.
W2 is positioned 4.5 Å from C3 of pyruvate, and the position would not be favorable to act as a base/acid for pyruvate α-proton exchange as proposed previously for YfaU aldolase (16). However, another well ordered water molecule, W4, which is positioned 3.00 Å from the C3 carbon (Fig. 1C) and hydrogen bonds with the η2 nitrogen of Arg70 (3.09 Å) and W2 (2.85 Å), would be well positioned to catalyze proton exchange at the pyruvate C3 carbon. This water molecule, W4, would be displaced by aldehyde binding in HpaI structures in complex with pyruvate and aldehydes as shown below. Superimposition of HpaI aldolase onto 2-dehydro-3-deoxygalactarate aldolase and YfaU aldolases (8, 16) corroborates a similar spatial disposition of active site residues that interact with their 2-keto acid moieties.
In the 2-ketobutyrate-liganded structure, although exhibiting binding identical to pyruvate, the position of the C4 methyl carbon is spatially disordered as indicated by weak electron density for this moiety compared with the rest of its structure, and its position was arbitrarily modeled to avoid potential steric clashes, including those with well defined water molecules (Fig. 1B). The absence of active site interactions with the C4 methyl group would allow configurational mobility about the 2-ketobutyrate C2–C3 bond, thereby smearing out the electron density for the C4 methyl group. The bulkier shape of the electron density of 2-ketobutyrate, consistent with an overall B-factor increased by 25% compared with pyruvate, indicates greater residual configurational disorder upon binding presumably due to the mobility of the C4 methyl group.
Electrostatic surfaces of the HpaI enzyme calculated according to the Poisson-Boltzmann equation (33) reveal an active site with a significant negative charged electrostatic field situated at the bottom of a deep cavity in the apoenzyme (Fig. 2A). In the holoenzyme, cobalt ion binding screens the negative charges, resulting in the cavity bottom of the pyruvate binding locus acquiring a slight positive charge (Fig. 2B).
FIGURE 2.

The electrostatic surface of an HpaI pyruvate aldolase evaluated by adaptive Poisson-Boltzmann solver. Views of the pyruvate aldolase active site (shown by yellow arrow) and of pyruvate aldolase in complex with substrates, pyruvate (cyan) and succinic semialdehyde (yellow) are shown. The color code has blue as positive charged and red as negative charged. A, electrostatic surface of apoenzyme where divalent cobalt ion and ligands were not included in electrostatic surface calculations. B, the positive charge in the active site in native holoenzyme with cobalt facilitates the binding of pyruvate and the aldehyde moiety of succinic semialdehyde, shown as stick models. Crystallographic symmetry operations were used to generate a complete active site. C, close-up of the active site cavity depicting bound pyruvate and succinic semialdehyde as stick models and showing the large neutral cavity (white) adjacent to the aldehyde ligand. W2, which hydrogen bonds the aldehyde carbonyl oxygen, and the divalent cobalt are also shown as semitransparent spheres. The electrostatic surface at W2 has a slight negative charge.
The Structure of HpaI Aldolase in Complex with HKHD
The structure resulting from HpaI aldolase crystals in complex with pyruvate and soaked in 10 mm succinic semialdehyde exhibited continuous electron density that was interpreted as HKHD substrate (Fig. 3A and inset). The electron density for the HKHD substrate could be fitted equally with either R or S conformation at its C4 hydroxyl. The racemic binding modes support the discovery of lack of stereospecificity in this enzyme (15). Substrate binding in the dimeric subunits in the asymmetric unit cell was consistent with HKHD bound with partial occupancies of 0.66 in both subunits. The residual electron density was modeled and refined as HKHD cleavage product, pyruvate, and water molecules as shown in Fig. 1A. Occupancies were determined such that HKHD B-factors did not differ significantly with respect to B-factors of interacting active site residues. The occupancy determined for HKHD is reasonable as occupancy for the surrogate substrate HOPA at 10 mm is 0.75, which would be expected under the crystallization conditions given a Michaelis constant of 3.4 ± 0.6 mm for HOPA.
FIGURE 3.

Substrate and cleavage products trapped in HpaI pyruvate aldolase active site. A, electron density of substrate HKHD trapped in the native enzyme. HKHD orientation corresponding to R chirality at C4 is shown in yellow, and that of S chirality is shown in red. The inset shows the same electron density looking down and corresponds to a counterclockwise rotation in the vertical plane of ∼90°. B, active site interactions implicating the metal cofactor, HKHD, Arg70, and W2. C, electron density of cleavage products pyruvate and succinic semialdehyde (re face orientation in yellow; si face in pink). D, interactions highlighting succinic semialdehyde binding and recognition. Hydrogen bonds are shown as short dashes, whereas long dashes connect atoms that are chemically bound upon aldol condensation. Water molecules in alternate conformation with the aldehyde binding site are shown in green. Upon pyruvate binding, Arg70 and the conserved water W2 (red) orient the carbonyl moiety of the aldehyde substrate. In B and D, the angle subtended by either C4 hydroxyl or aldehyde oxygen, W2, and Val118′ corresponds to tetrahedral hybridization geometry. Electron density encompassing the ligands was calculated from a simulated annealing Fo − Fc omit map and contoured at 3.5 σ. The divalent cobalt ion is depicted by a salmon sphere.
Active site interactions made by HKHD are shown in Fig. 3B. The substrate coordinates the cobalt ion through its keto (2.69 Å) and carboxylate oxygen atoms (2.76 Å). The 2-carbonyl oxygen forms a weak hydrogen bond with the η1 guanidinium nitrogen of Arg70 (3.13 Å), which in turn makes a salt bridge with Asp42. This electrostatic interaction will raise the pKa of Arg70 to ∼17 and maintain its positive charge at physiological pH based on computational pKa calculations (34). The η2 guanidinium nitrogen of Arg70 contacts the C4-OH of HKHD (3.28 Å), and its positive charge would facilitate C4-OH deprotonation by stabilizing the resultant negative charge. Moreover, water molecule W2, coordinated to the Co2+ metal ion, is also proximal to the substrate C4-OH (2.12 Å) and interacts with the backbone oxygen of Val118′ (2.13 Å) and His45 (3.07 Å). His45 further forms an ion pair interaction with Asp84′ of the symmetry-related neighboring subunit. Small torsional rotations of the His45 imidazole side chain could readily position it within hydrogen bonding distance of W2 and without breaking its interaction with Asp84′ would make W2 the likely candidate mediating proton abstraction at the C4 hydroxyl. The W2-cobalt ion distance of 2.12 Å in this structure and 2.22 Å in the structure bound by the cleavage products described below is consistent with W2 coordination by Co(II) metal ion (35).
Aldehyde Binding and Recognition
The structure derived from the HpaI crystals soaked with 10 mm pyruvate and 10 mm succinic semialdehyde showed electron density that was modeled based on the structures of the two cleavage products and is shown in Fig. 3C. Pyruvate was refined as full occupancy and yielded B-factors similar to those refined for the 2-keto acid in the pyruvate-bound HpaI structure, whereas occupancy of the aldehyde binding site was set to 0.66 with water molecules (shown in Fig. 1A) used to fill the residual electron density at an occupancy of 0.33. Refinement of succinic semialdehyde yielded B-factors similar to those for the nascent aldehyde moiety of HKHD.
The interpretation of the electron density map of HpaI in complex with both pyruvate and succinic semialdehyde resulted in two distinct aldehyde configurations as shown in Fig. 3C. These two prochiral binding modes of each aldehyde (shown in Fig. 3D), positioned si or re face to the attacking pyruvate enolate, are consistent with the absence of stereospecificity in this enzyme (15). In each binding mode, the plane of the aldehyde moiety was oriented vicinal and roughly perpendicular to the putative trajectory of the attacking pyruvate enolate.
Anchoring of the aldehyde donor molecule in the active site occurs through hydrogen bonding of its aldehyde oxygen with the η2 guanidinium nitrogen of Arg70 (2.66 Å) and water molecule W2 (2.86 Å) for both prochiral orientations of succinic semialdehyde (Fig. 3D). Additional interactions with the semialdehyde molecule occur between its carboxylate moiety and backbone amide dipoles of the enzyme (not shown) that form a collar of positive charge at the active site entrance (Fig. 2, B and C).
HpaI exhibits broad specificity toward a variety of aldehyde donors with diverse lengths and substituents (15). In the presence of the divalent metal ion and pyruvate, the aldehyde binding site is effectively neutral except for the collar of positive charge at the active site cavity entrance (Fig. 2B). Hydrophobic residues line this binding locus: Trp19, Ala174, Leu212, and Val234 as well as Val118′, Ala121′, and Leu122′ from the neighboring subunit at 5–6-Å distance from the aliphatic atoms of the aldehyde molecule, forming a large cavity. As none of these residues makes close contact with the ligand, water molecules would solvate the aldehyde substrates. Succinic semialdehyde binding entails displacement of at least six well ordered water molecules with respect to the pyruvate-liganded structure, whereas acetaldehyde would displace fewer such water molecules. The facile displacement of water molecules in the structures indicates an aldehyde donor binding locus that readily accommodates aldehyde molecules of different lengths.
To further investigate the potential substrate diversity of HpaI aldolase, compounds cleaved by YfaU, such as 2-keto-3-deoxy-l-lyxonate, 2-keto-3-deoxy-l-mannonate, 2-keto-3-deoxy-l-rhamnonate (16), and HOPA, were modeled into the active site using the HKHD binding locus as a template and then further refined on the basis of their fit to the electron density omit map shown in Fig. 3A. These substrates were readily threaded onto the HKHD template (supplemental Fig. S3) and resulted in no close contacts or steric clashes in the active site, indicating that they are potential substrates for HpaI aldolase.
Active Site Binding Geometries of Reaction Intermediates
Superposition of the HKHD structure onto the cleavage product structure yielded an overall r.m.s.d. value of 0.12 Å with no significant conformational differences between the two structures. The resultant superposition of the active site geometries of the reaction intermediates is shown in Fig. 4. Comparison between the two structures on the basis of interatomic distances reveals the 2-keto moiety of the HKHD substrate not as strongly bound in the active site as is the respective cleavage product, pyruvate. Although Arg70 makes potentially charged interactions in both structures, its interactions with the respective nascent pyruvate and aldehyde atoms in the HKHD structure are slightly longer and are no longer considered hydrogen bonds. The weaker active site affinity for HKHD explains the inability to trap significant concentrations of substrate in the presence of 10 mm pyruvate and 10 mm succinic semialdehyde in HpaI crystals. Only when pyruvate concentrations were low (stoichiometric with respect to enzyme concentration in HpaI crystals such that most pyruvate could be chased into HKHD with excess aldehyde) was it possible to trap HKHD in the active site.
FIGURE 4.

Superposition of the active site binding geometries of HKHD and resultant cleavage products trapped in the active site of HpaI pyruvate aldolase. Substrate HKHD (orange) is depicted for both R and S conformations at its C4 hydroxyl, whereas the aldehyde (dark gray) is shown in re and si face orientations. Hydrogen bonds are shown as short dashes. Close van der Waals contacts made by Arg70 with HKHD O3 and O4 atoms are shown by long dashes (yellow). The divalent cobalt ion (Co) is depicted by a salmon sphere. The composite image corresponds to superposition of one HpaI subunit bound with HKHD and the equivalent subunit in complex with its cleavage products. The interactions by HKHD with Co(II) and Arg70 are weaker, indicated by longer interatomic distances, compared with the equivalent interactions made with Co(II) and Arg70 by the oxygen atoms of the 2-keto moiety, O2 and O3, as well as aldehyde C1 oxygen in the structure binding the cleavage products. Ald, aldehyde; Pyr, pyruvate.
Conformation Changes following Ligand Binding
Similar conformational responses were observed in all structures containing 2-keto acid moieties. Superimpositions between the structure of the apoenzyme (8) and each of the liganded complexes yielded an overall r.m.s.d. value of 0.73 ± 0.03 Å, which is slightly greater than the calculated r.m.s.d. value based on superimposition of only ligand complexes (0.20 ± 0.05 Å). The difference can be attributed largely to conformational changes from the loop region defined by residues 116–124. Indeed, the overall r.m.s.d. value computed using only these residues increased to 2.94 ± 0.10 Å between apoenzyme and holoenzyme complexes and was reduced to 0.47 ± 0.05 Å for all structures if residues 116–124 were excluded from the calculation. Interestingly, the movement of this polypeptide region (116–124) permits the interaction of the backbone oxygen of Val118′ with water molecule W2, which is coordinated to the active site metal ion (Figs. 1C, 3B and 3D). Binding by the aldehyde acceptor molecule on the other hand does not require a conformational change in the presence of the bound 2-keto acid moiety.
Arg70 and Asp42 Mutant Structures
We successfully trapped pyruvate in the crystal structure of the R70A variant of HpaI (supplemental Fig. S4A); however, trapping of the aldehyde or the condensation products was unsuccessful under the experimental conditions. The loss of the charged interaction between the pyruvate C2 carbonyl and the guanidinium group of Arg70 only slightly perturbed the 2-keto acid binding locus, resulting in a slight displacement of pyruvate in its plane with respect to its position in the native structures (0.79 ± 0.23 Å). However, the C2 carbonyl retains its hydrogen bonding interaction with Gln147 (supplemental Fig. S4B). This residue also interacted with Arg70 in the wild-type enzyme, indicating that Arg70 is not mandatory for ensuring alignment of the 2-keto acid moiety in the active site. Further support comes from similar Kd values for pyruvate binding in the wild-type and mutant proteins (R70K and R70A) in the presence or absence of Co2+ (Table 3). However, the loss of the charged interaction destabilized binding with water molecule W4, consistent with its absence in the mutant structure.
Superimposition of the D42A mutant structure onto the pyruvate-bound native structure (r.m.s.d., 1.20 ± 0.02 Å) shows two loop regions, residues 42–56 and 116–124, that are displaced by 3.09 ± 0.04 and 3.85 ± 0.02 Å, respectively, and whose movement disrupts the structural integrity of the active site (supplemental Fig. S5). The active site disruption results in a concomitant loss of the metal ion in the D42A mutant structure. In the liganded native enzyme, residues Glu44 and His45 in one loop interact with water molecules W1 and W3, whereas Val118′ in the second loop of the symmetry-related subunit interacts through its backbone carbonyl with water molecule W2. The repositioning of these active site residues in the D42A mutant induces loss of the three active site water molecules, two of which, W1 and W2, directly coordinated the metal ion cofactor, and underscores the essential structural role of Asp42 in maintaining active site integrity in the native enzyme.
Kinetic Analysis
The kcat value for HOPA cleavage in deuterated buffer was reduced by 30% with a solvent isotope effect of kcatD and (kcat/Km)D of ∼1.5, implying an involvement of water in catalysis (Table 4). A similar solvent isotope effect was shown for another divalent metal ion-dependent C–C bond lyase, α-isopropylmalate synthase (1.8–2.2), which also implicated a catalytic water molecule (36). The pD profile for the pyruvate C3 proton exchange reaction in the native enzyme showed deprotonation with a pK1 of 6.5 ± 0.1 (Fig. 5), which could correspond to the base that abstracts a proton from the C3 methyl of pyruvate. Although a slight shift in pKa value can occur in deuterated buffer (∼0.4 pH units) (37), this pK1 value is not significantly different from the pKa of 6.5 associated with a catalytic base that abstracts the proton from C4 hydroxyl in the HOPA cleavage reaction (17), suggesting two chemical steps involving the same base/acid. HpaI aldolase is also able to efficiently catalyze the aldol cleavage of 2-keto-3-methyl-4-hydroxypentanoate to form 2-ketobutyrate and acetaldehyde with a Km value of 0.05 ± 0.01 mm and kcat of 14.0 ± 1.1 s−1. Thus, it is not surprising that the aldolase can catalyze C3 proton exchange of 2-ketobutyrate (15). A single exponential decay is observed for 1H NMR signal loss at C3 of 2-ketobutyrate (Fig. 6). The absence of a biphasic α-proton exchange profile implied no spatial discrimination for the two C3 hydrogens, which is not inconsistent with rotational disorder about the C2–C3 bond in the 2-ketobutyrate-bound crystal structure.
TABLE 4.
Solvent isotope effect of the HOPA cleavage reaction catalyzed by HpaI aldolase in presence of 0.5 mm Co2+
| Solvent | kcat | Km | kcat/Km |
|---|---|---|---|
| s−1 | mm | mol−1·s−1 | |
| H2O | 361.5 ± 24.7 | 0.35 ± 0.06 | (1.0 ± 0.1) × 106 |
| D2O | 259.0 ± 19.1 | 0.39 ± 0.04 | (6.6 ± 0.8) × 105 |
| Ratio (H2O/D2O) | 1.40 | 0.90 | 1.52 |
FIGURE 5.

pD profile of pyruvate C3 proton exchange reaction in HpaI aldolase. Assays were performed in deuterated constant ionic strength buffer containing 100 mm Tris, 50 mm acetic acid, 50 mm MES, and 1.0 mm CoCl2 with pD ranging from 5.4 to 9.4. The data were fitted to Equation 1 by non-linear regression using Leonora and showed a pK1 of 6.5 ± 0.1.
FIGURE 6.

α-Proton exchange of 2-ketobutyrate catalyzed by Co2+-HpaI aldolase. The proton exchange was monitored in 20 mm deuterated MOPS buffer, pD 8.0 by 1H NMR. The loss of proton signal at the C3 carbon of 2-ketobutyrate followed a single exponential decay model.
Arg70 and Asp42 form an ion pair in the native HpaI structure. The replacement of Asp42 with alanine resulted in a complete loss of aldolase activity and pyruvate α-proton exchange activity (Table 5), consistent with the active site disruption observed in this HpaI mutant structure. Replacement of Arg70 with Lys reduced catalytic efficiency (kcat/Km) by 270-fold but resulted in only a 2-fold reduction in the pyruvate α-proton exchange rate (Table 5), indicating that pyruvate α-proton exchange is not rate-limiting.
TABLE 5.
Steady-state kinetic parameters for HOPA cleavage and pyruvate α-proton exchange by wild-type and mutant HpaI enzymes
Aldolase activity assays were performed as described under “Material and Methods” with (S)-4-hydroxy-2-ketopentanoate as substrate. The pyruvate α-proton exchange assay was carried out using 600-μl assay solutions containing 30 mm pyruvate, 0.5 mm CoCl2, and 20 mm deuterated MOPS buffer, pD 8.0 in the presence of 5.25 μg of wild-type and R70K enzyme and 200 μg of R70A and D42A enzyme, respectively, at 25 °C using a 5.0-mm NMR tube.
| Enzymes | Km | Ksia | kcat | kcat/Km | Vexchange |
|---|---|---|---|---|---|
| mm | mm | s−1 | mol−1·s−1 | s−1 | |
| HpaI | 0.38 ± 0.01 | 4.56 ± 0.14 | 353.5 ± 5.7 | (9.41 ± 0.28) × 105 | 359.8 ± 10.2 |
| R70K | 3.32 ± 0.04 | b | 11.8 ± 0.08 | (3.55 ± 0.05) × 103 | 191.2 ± 7.2 |
| R70A | NDc | ND | |||
| D42A | ND | ND | |||
a The substrate inhibition constant derived from analysis of the kinetic data at high substrate concentrations.
b No substrate inhibition was observed.
c ND, no activity was detectable.
Pyruvate binding was similar for mutants and native enzyme in the metal-free apoenzymes (Table 3); however, in the presence of 0.5 mm Co2+, the Kd of pyruvate binding in D42A remains unchanged, being 100-fold weaker compared with wild-type enzyme and Arg70 mutants. In the presence of oxalate, a pyruvate enolate analog, the measured Kd is significantly higher for the R70A mutant as compared with the R70K mutant and native enzyme (Table 3). The 750-fold weaker Kd for the R70A mutant supports an important role for the positive charged Arg70 in pyruvate enolate intermediate binding and stabilization. For D42A, no oxalate binding was detectable up to a concentration of 40 mm.
DISCUSSION
The ability to cryotrap enzymatic intermediates in the crystalline state provided a unique opportunity to delineate the essential mechanistic features associated with substrate specificity and proton transfers during substrate turnover in class II pyruvate aldolases. A key facet was the successful trapping of the aldol condensation product HKHD, the natural substrate of the enzyme, in the active site under crystallization conditions. The conversion of the enzyme-bound pyruvate reactant complex into substrate in the presence of excess aldehyde reactant demonstrates crystallization of a catalytically competent conformer and validates that the enzyme complexes cryotrapped in the active site are genuine reaction intermediates.
The virtual positional invariance of active site residues in HpaI upon ligand binding alludes to a highly preorganized active site, which does not implicate significant conformational changes during the catalytic cycle. The absence of subunit cooperativity in the enzyme kinetics, even though each active site implicates residues from an adjacent subunit of the HpaI hexamer, supports the structural interpretation that ligand binding in the active site does not entail conformational remodeling of subunit interactions.
Structural Basis for Substrate and Stereospecificity
Pyruvate, 2-ketobutyrate, and the 2-keto acid moiety of substrate are extensively stabilized by multiple electrostatic interactions and hydrogen bonds with active site residues and metal ion cofactor (Figs. 1C, 3B and 3D). The 2-keto acid binding locus is located at the bottom of the active site whose shape is narrower and is lined with more conserved residues compared with the aldehyde binding site (Fig. 2C). This feature underpins the strict substrate specificity for the carbonyl donor by the enzyme, which is catalytically competent only with pyruvate and 2-ketobutyrate (15).
The diversity of catalytically competent aldehyde donors arises from the nature of its large binding pocket that is lined by non-directional hydrophobic residues and is filled with more than 15 water molecules that can be readily displaced by an incoming aldehyde. Aldol condensation of diverse electrophiles with pyruvate is supported by the different cleavable substrates that can be modeled into the HpaI aldolase active site without steric clash (supplemental Fig. S3). From the same modeling, small chemical substituents on the nascent aldehyde moiety would also be tolerated in the active site cavity.
The C1 carbon of succinic semialdehyde is positioned within close contact of the pyruvate enolate and with the plane containing the C1 carbon of each aldehyde oriented nearly co-planar with respect to the pyruvate molecule (Fig. 3D). Anchoring of the aldehyde within the active site is minimal, entailing two interactions by its C1 oxygen with the guanidinium nitrogen of Arg70 and W2. Additional weak interactions can occur, depending on the chemical structure of the aldehyde, with primarily backbone amide dipoles as was observed with the carboxylate tail of succinic semialdehyde. The orientation of the aldehyde moiety perpendicular to the plane containing the nucleophile is consistent with an attack trajectory for incipient C–C bond formation with the enolate (shown as long dashed lines in Fig. 3D). The capacious aldehyde binding locus further allows the plane containing the electrophile to rotate without steric hindrance, enabling the pyruvate enolate to attack si or re face to yield racemic products with R and S configurations at C4 (15).
Catalytic Mechanism
A catalytic mechanism to promote cleavage of the C–C bond in pyruvate aldolases integrating enzymology and structure data is outlined in Scheme 2. The reaction chemistry catalyzed by HpaI aldolase entails a water molecule that is bound to the divalent metal ion, which ionizes the water molecule, thereby enabling the metal-bound hydroxide to act as a base catalyst in mediating proton transfers. A key feature is an arginine residue that directs and stabilizes formation of reaction intermediates to facilitate proton transfers.
SCHEME 2.

Proposed catalytic mechanism (reactions a–d and intermediates 1–4) of C–C bond cleavage in pyruvate aldolase. R = CH2CH2COOH.
In the C3–C4 bond cleavage of HKHD, a catalytic base is required to deprotonate C4-OH, and a catalytic acid is required to donate a proton to pyruvate enolate. His45 was proposed previously to be the catalytic base (9); however, this is unlikely due to the following considerations. The identical hydrogen bonding pattern by the His45 Nϵ nitrogen in pyruvate aldolase structures crystallized over the pH range 5–8 indicates a basic pKa for His45 Nϵ nitrogen that places it outside the pH-dependent range attributed to C3 proton exchange and HKHD substrate cleavage. Basic pKa values have been observed from NMR studies in proteins where raised pKa values of histidines (7.6–7.9) have been attributed to their interaction with an aspartate or glutamate residue (38–40) as compared with lower pKa for histidines in comparable environments and that are not engaged in charged interactions (40). Indeed, in HpaI aldolase, Asp84′ of the adjacent subunit forms a hydrogen bond with His45 Nδ nitrogen (2.74 Å) that would argue in favor of a basic pKa shift by the His45 Nϵ nitrogen. Furthermore, slight repositioning of the His45 imidazole side chain without breaking its hydrogen bond to Asp84′ would allow it to donate a hydrogen bond to W2 and promote geometry where W2 donates a hydrogen bond to the backbone carbonyl of the neighboring Val118′, generating the geometry conducive for in-line proton transfer between W2 and W4 or C4 hydroxyl of the substrate. Although the η2 guanidinium nitrogen of Arg70 interacts with C4-OH of HKHD (3.28 Å), the possibility of Arg70 acting as general base/acid in catalysis is also unlikely due to its high pKa by virtue of its salt bridge with Asp42 that ensures structural integrity of the active site. Here, we propose a well ordered and conserved water, W2, as the catalytic base. The pKa of a water molecule coordinated by a divalent cobalt metal ion ranges from 6.2 to 9.8 for metal complexes in an aqueous medium (41) and decreases to ∼5.8 in cobalt-substituted carboxypeptidase A (42). In HpaI aldolase, W2 is bound to the cobalt metal ion and donates a hydrogen bond to the Val118′ backbone carbonyl. It can also be further stabilized by accepting a hydrogen bond from His45, resulting in an orientation that would allow W2 as a metal-bound hydroxide ion to catalyze in-line proton transfer with W4, which hydrogen bonds with W2. The analogous water in the structure of YfaU, a homolog of HpaI, has also been proposed to be a catalytic base (16). Base catalysis by a metal-bound hydroxide ion represents an efficient mechanism to generate a transient nucleophile in a solvated environment. The proposed mechanism of rate-limiting base catalysis mediated by the W2 hydroxide molecule would explain the pH profile observed for proton exchange (Fig. 5). The involvement of water molecules W2 and W4 in the proposed mechanism is further supported by the solvent kinetic isotope effect observed.
As for the catalytic acid, W2 was suggested previously to serve as the proton donor to protonate pyruvate enolate (11, 16). But a direct role of W2 mediating pyruvate α-proton exchange is unlikely as W2 is unfavorably positioned to promote direct proton transfer to the pyruvate C3 carbon. Rather, W4, positioned by Arg70 perpendicular to the plane of the pyruvate molecule, is within close contact of the C3 carbon of pyruvate, allowing catalysis of proton exchange with the pyruvate enolate. In the 2-ketobutyrate-liganded structure, the W4 binding site is also present in a position similar to that in the pyruvate-bound structure, and Arg70 interacts with W4 (seen in Fig. 1C), consistent with their roles in promoting C3 proton exchange in 2-ketobutyrate. In the R70K mutant, only a slight shift by W4 (∼1 Å) would be required for the Lys70 side chain to orient W4 for nucleophilic attack by the enolate.
Through its guanidinium moiety interactions with substrate C2 and C4 oxygens, pyruvate and ketobutyrate C2 oxygens, aldehyde C1 oxygen, and W4, Arg70 plays multiple roles in HpaI catalysis by facilitating deprotonation of substrate C4 hydroxyl, stabilizing the enolate, and mediating W4 protonation of the enolate. This is supported by kinetic evidence showing the loss of pyruvate α-proton exchange and aldol cleavage activities in the R70A mutant (11) and the significant decrease in catalytic efficiency for the R70K mutant (Table 5) that would result from reduced stabilization of reaction intermediates by the shorter lysine side chain. A significant Kd increase for binding by the pyruvate enolate analog oxalate in the R70A mutant compared with the wild type reinforces its critical role in pyruvate enolate stabilization in HpaI (Table 5). The primary role of Arg70 is thus to stabilize in the transition state the resultant negative charge created at the C4 hydroxyl and indirectly at the enolate by virtue of its electrostatic interaction.
Therefore, the catalytic cycle (Scheme 2) begins by HKHD binding in the active site cavity whereby its coordination with the metal ion creates an electron sink at the C2 keto carbonyl in intermediate 1 and simultaneously induces a small conformational adjustment in loop region (116–124) to position W2, enabling it to hydrogen bond with the HKHD C4 hydroxyl. The positive charged side chain of Arg70 interacts with both the 2-keto oxygen and the C4 hydroxyl of HKHD. Proton abstraction from the C4 hydroxyl by the W2 hydroxide ion induces C3–C4 bond cleavage to yield intermediate 2 (reaction a). Reaction a is reversible as the pyruvate enolate and succinic semialdehyde are bound proximally to Arg70 and can recombine by aldol addition. Exchange with W4 releases the aldehyde in intermediate 2 and initiates a proton relay from W2 to W4 to the C3 methyl of the pyruvate enolate intermediate 3 (reaction b). Protonation of pyruvate enolate by W4 yields the keto product 4 and regenerates the hydroxide ion at W2 (reaction c). Finally, exchange in intermediate 4 with HKHD releases the keto product, pyruvate, and recommences the catalytic cycle (reaction d). At low reactant concentrations, the enzyme would function efficiently in the direction of aldol addition with the tighter binding pyruvate displacing HKHD upon synthesis.
The catalytic mechanism is consistent with a trajectory of least atomic motion requiring minimal atomic librations and imposes no stereochemical constraint at C4 to select either the R or S isomer of HKHD. In the condensation direction, the catalytic mechanism allows for a random binding order with respect to pyruvate and aldehyde donor.
The decarboxylation of oxaloacetate observed with HpaI aldolase (11) would use a mechanism identical to the proposed cleavage mechanism. As oxaloacetate and HKHD are isostructural, differing at the C5 position of HKHD, which in oxaloacetate becomes the carboxylic oxygen atom, superimposition of oxaloacetate in acid form onto the HKHD binding site in HpaI aldolase results in an intermediate isostructural with 1. As the pKa of the 4-carboxylate group of oxaloacetate is 4–5, the conjugate base would be the dominant species at physiological pH. In the decarboxylation of β-keto acids, the bond undergoing cleavage by Schiff base catalysis must be out of the plane of the imine π bond (43). Similarly, in HpaI aldolase, the close contact between the W2 hydroxyl and C4 carboxyl oxygen would force the C4 carboxyl out of the O2-C2-C3-C4-O4 plane and favor the expected geometry for decarboxylation.
Evolutionary Relationships with Other Enzymes
Using the Dali server, the HpaI structure was shown to be closely related to that of 2-dehydro-3-deoxygalactarate aldolase, YfaU, macrophomate synthase, citrate lyase, and pyruvate kinase as reported previously (9). Intriguingly, the enzyme was also structurally related to the P-enolpyruvate binding domain of enzyme I (residues 261–573) in bacterial P-enolpyruvate-dependent carbohydrate:phosphotransferase systems (Protein Data Bank code 2HWG) (44) and of pyruvate, phosphate dikinase (residues 534–874) (Protein Data Bank code 1DIK) (45) with both structures yielding identical r.m.s.d. values of 2.7 Å (Fig. 7A). The residues used in metal binding (Glu149 and Asp175 for HpaI, Glu431 and Asp455 for enzyme I, and Glu745 and Asp769 for PPDK) and the residue equivalent to Arg70 of HpaI (Arg332 in enzyme I and Arg617 in PPDK) were spatially superimposable in the enzyme structures (Fig. 7A). Although these enzymes catalyze different overall reactions, they all share a common partial chemical reaction step of pyruvate enolate formation and protonation of the enolate to form pyruvate. In enzyme I and PPDK, a cysteine was proposed as the residue that donates a proton to pyruvate enolate from its re face (Cys831 for PPDK and Cys502 for enzyme I). In HpaI, this residue would correspond to Leu212, which cannot function as a proton donor, implying the use of a different functional entity in HpaI aldolase. A binding role was proposed for Arg617 in terms of charge interactions with the phosphoryl group in PPDK (46). The structural analogy with Arg70 in HpaI suggests an additional role of Arg617 in PPDK and Arg332 in enzyme I in pyruvate enolate stabilization.
FIGURE 7.

Active site architecture comparison of evolutionary related enzymes and non-homologous pyruvate lyases. A shows superposition of the HpaI·pyruvate complex structure with the P-enolpyruvate binding domain of enzyme I for bacterial P-enolpyruvate-dependent carbohydrate:phosphotransferase systems (Protein Data Bank code 2BG5, residues 261–573) and pyruvate, phosphate dikinase (Protein Data Bank code 1KC7, residues 534–874). B shows superposition of HpaI pyruvate aldolase (white), human HMG-CoA lyase (Protein Data Bank code 2CW6) (yellow), DmpG aldolase (Protein Data Bank code 1NVM) (cyan), bacterial oxaloacetate decarboxylase (Protein Data Bank code 3B8I) (magenta), and HMG/CHA aldolase (Protein Data Bank code 3NOJ) (green). Structures are presented from the same perspective, taking the pyruvate analog and the metal ion (large sphere) as reference except for human HMG-CoA lyase as noted under “Material and Methods.” Succinic semialdehyde in HpaI aldolase is shown in white, and 3-hydroxyglutarate, a cleavage product of HMG-CoA lyase, is shown in yellow. For HpaI, the condensing aldehyde will approach from the distal side of the pyruvate, whereas for DmpG aldolase, HMG-CoA lyase, HMG/CHA aldolase, and oxaloacetate decarboxylase it will approach from the proximal side. Water molecules identified as W2′ and W4′, equivalent to W2 and W4, which are described as part of the catalytic machinery in HpaI aldolase, also show the same spatial disposition as would their respective condensing aldehydes. The guanidinium moieties of adjacent arginine residues cluster tightly (identified as Arg) and interact with their respective metal-binding ligand and water molecule identified as W4/W4′ where present. Water molecules within hydrogen bonding distance of W4/W4′ and bound to the metal ion are identified as W2/W2′. Note that the Arg70-Asp42 pair has a direct analog in HMG-CoA lyase (Arg41-Glu72), HMG/CHA aldolase (Arg123-Glu36), and DmpG aldolase (Arg17-Glu48), whereas only in oxaloacetate decarboxylase is the equivalent arginine residue, Arg159, not ion-paired with a carboxylate group of an amino acid. The carboxylate groups of the interacting aspartate or glutamate are identified (Asp or Glu).
Arginine residues are also found in spatially similar positions in the active sites of other evolutionarily unrelated yet analogous pyruvate lyases, including DmpG (10), HMG/CHA aldolase (12), and oxaloacetate decarboxylase (30) (Fig. 7B). The arginine residue in DmpG was directed through its electrostatic interaction with a glutamate residue (Glu48) whose carboxylate group is positioned isostructurally with Asp42 in HpaI aldolase. In HMG/CHA aldolase, a similar glutamate was located distally to the metal-binding ligand. Furthermore, Arg159 from oxaloacetate decarboxylase superposes with Arg70 of HpaI aldolase such that its guanidinium group also points toward the pyruvate; however, Arg159 orientation is not directed by an interaction with a proximal carboxylate group. Replacement of the respective arginine residues by a neutral residue in HMG/CHA aldolase, BphI aldolase, and 3-hydroxy-3-methylglutaryl-CoA lyase also severely compromised catalytic activities (12, 29, 31). The analysis of the non-homologous pyruvate lyases further identified water molecules equivalent to catalytic water molecules W2 and W4; however, these water molecules were located on the distal side of the pyruvate substrate in DmpG and HMG/CHA aldolases as seen in Fig. 7B.
The structural comparison of the active sites of evolutionarily distant and non-homologous enzymes (shown in Fig. 7, A and B) has highlighted a highly conserved constituent in their active sites, an arginine residue, which in HpaI aldolase is central to proton exchange with the pyruvate enolate and to cleavage of the C–C bond. The conservation of this single catalytic entity indicates significant commonality in the reaction chemistry catalyzed by these enzymes and is reinforced by similar spatial orientation of putative catalytic water molecules in the pyruvate aldolases. However, as the aldehyde or keto acid acceptor specificities of these pyruvate aldolases are different, extensive sequence divergence appears to have occurred to accommodate recognition of diverse substrates.
Thus, HpaI pyruvate aldolase epitomizes an exceedingly simple yet highly conserved catalytic mechanism to promote C–C bond cleavage of non-phosphorylated compounds. Our results provide an important framework for future utilization and engineering of class II pyruvate aldolases as improved biocatalysts. Moreover, the substrate/product trapping methods described here applied to other aldolases should shed further insight into the catalytic mechanism of these enzymes.
Acknowledgment
Work was carried out in part at the National Synchrotron Light Source, Brookhaven National Laboratory, which is supported by the United States Department of Energy, Division of Materials Sciences, and Division of Chemical Sciences under Contract DE-AC02-98CH10886.
This work was supported by discovery grants from the National Science and Engineering Research Council of Canada (to S. S. and J. S.) and by a grant from the Canadian Institutes for Health Research (to J. S.).

This article contains supplemental Figs. S1–S5.
- HKHD
- 4-hydroxy-2-ketoheptane-1,7-dioate
- CHA
- 4-carboxy-4-hydroxy-2-oxoadipate
- HMG
- 4-hydroxy-4-methyl-2-oxoglutarate
- HOPA
- 4-hydroxy-2-oxopentanoate
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1. Takayama S., McGarvey G. J., Wong C. H. (1997) Microbial aldolases and transketolases: new biocatalytic approaches to simple and complex sugars. Annu. Rev. Microbiol. 51, 285–310 [DOI] [PubMed] [Google Scholar]
- 2. Samland A. K., Sprenger G. A. (2006) Microbial aldolases as C–C bonding enzymes—unknown treasures and new developments. Appl. Microbiol. Biotechnol. 71, 253–264 [DOI] [PubMed] [Google Scholar]
- 3. M. J., Floriano B., J. J., Santero E. (2002) Identification of a hydratase and a class II aldolase involved in biodegradation of the organic solvent tetralin. Appl. Environ. Microbiol. 68, 4841–4846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Joerger A. C., Gosse C., Fessner W. D., Schulz G. E. (2000) Catalytic action of fuculose 1-phosphate aldolase (class II) as, derived from structure-directed mutagenesis. Biochemistry 39, 6033–6041 [DOI] [PubMed] [Google Scholar]
- 5. Kroemer M., Merkel I., Schulz G. E. (2003) Structure and catalytic mechanism of l-rhamnulose-1-phosphate aldolase. Biochemistry 42, 10560–10568 [DOI] [PubMed] [Google Scholar]
- 6. Blom N. S., S., Coulombe R., Sygusch J. (1996) Novel active site in Escherichia coli fructose 1,6-bisphosphate aldolase. Nat. Struct. Biol. 3, 856–862 [DOI] [PubMed] [Google Scholar]
- 7. Cooper S. J., Leonard G. A., McSweeney S. M., Thompson A. W., Naismith J. H., Qamar S., Plater A., Berry A., Hunter W. N. (1996) The crystal structure of a class II fructose-1,6-bisphosphate aldolase shows a novel binuclear metal-binding active site embedded in a familiar fold. Structure 4, 1303–1315 [DOI] [PubMed] [Google Scholar]
- 8. Izard T., Blackwell N. C. (2000) Crystal structures of the metal-dependent 2-dehydro-3-deoxy-galactarate aldolase suggest a novel reaction mechanism. EMBO J. 19, 3849–3856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rea D., Fülöp V., Bugg T. D., Roper D. I. (2007) Structure and mechanism of HpcH: a metal ion dependent class II aldolase from the homoprotocatechuate degradation pathway of Escherichia coli. J. Mol. Biol. 373, 866–876 [DOI] [PubMed] [Google Scholar]
- 10. Manjasetty B. A., Powlowski J., Vrielink A. (2003) Crystal structure of a bifunctional aldolase-dehydrogenase: sequestering a reactive and volatile intermediate. Proc. Natl. Acad. Sci. U.S.A. 100, 6992–6997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang W., Seah S. Y. (2005) Purification and biochemical characterization of a pyruvate-specific class II aldolase, HpaI. Biochemistry 44, 9447–9455 [DOI] [PubMed] [Google Scholar]
- 12. Wang W., Mazurkewich S., Kimber M. S., Seah S. (2010) Structural and kinetic characterization of 4-hydroxy-4-methyl-2-oxoglutarate (HMG)/4-carboxy-4-hydroxy-2-oxoadipate (CHA) aldolase: a protocatechuate degradation enzyme evolutionarily convergent with the HpaI and DmpG pyruvate aldolases. J. Biol. Chem. 285, 36608–36615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Díaz E., Ferrández A., Prieto M. A., García J. L. (2001) Biodegradation of aromatic compounds by Escherichia coli. Microbiol. Mol. Biol. Rev. 65, 523–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fish D. C., Blumenthal H. J. (1966) 2-Keto-3-deoxy-d-glucarate aldolase. Methods Enzymol. 9, 529–535 [Google Scholar]
- 15. Wang W., Baker P., Seah S. Y. (2010) Comparison of two metal-dependent pyruvate aldolases related by convergent evolution: substrate specificity, kinetic mechanism and substrate channelling. Biochemistry 49, 3774–3782 [DOI] [PubMed] [Google Scholar]
- 16. Rea D., Hovington R., Rakus J. F., Gerlt J. A., Fülöp V., Bugg T. D., Roper D. I. (2008) Crystal structure and functional assignment of YfaU, a metal ion dependent class II aldolase from Escherichia coli K12. Biochemistry 47, 9955–9965 [DOI] [PubMed] [Google Scholar]
- 17. Wang W., Seah S. Y. (2008) The role of a conserved histidine residue in a pyruvate-specific Class II aldolase. FEBS Lett. 582, 3385–3388 [DOI] [PubMed] [Google Scholar]
- 18. Lumry R., Smith E. L., Glantz R. R. (1951) Kinetics of carboxypeptidase action. Effect of various extrinsic factors on kinetic parameters. J. Am. Chem. Soc. 73, 4330–4340 [Google Scholar]
- 19. Cornish-Bowden A. (1995) Analysis of Enzyme Kinetic Data, Oxford University Press, New York [Google Scholar]
- 20. Otwinowski Z., Minor W. (1999) in Methods in Enzymology (Carter C. W., Jr., Sweet R. M., eds) Vol. 276 pp. 307–326, Academic Press, New York: [DOI] [PubMed] [Google Scholar]
- 21. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 23. Eswar N., Eramian D., Webb B., Shen M. Y., Sali A. (2008) Protein structure modeling with MODELLER. Methods Mol. Biol. 426, 145–159 [DOI] [PubMed] [Google Scholar]
- 24. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 25. Lovell S. C., Davis I. W., Arendall W. B., 3rd, de Bakker P. I., Word J. M., Prisant M. G., Richardson J. S., Richardson D. C. (2003) Structure validation by C-α geometry: φ, ψ and C-β deviation. Proteins 50, 437–450 [DOI] [PubMed] [Google Scholar]
- 26. Vaguine A. A., Richelle J., Wodak S. J. (1999) SFCHECK: a unified set of procedure for evaluating the quality of macromolecular structure-factor data and their agreement with atomic model. Acta Crystallogr. D Biol. Crystallogr. 55, 191–205 [DOI] [PubMed] [Google Scholar]
- 27. DeLano W. L. (2004) The PyMOL Molecular Graphics System, Schrödinger, LLC, New York [Google Scholar]
- 28. Holm L., Park J. (2000) DaliLite workbench for protein structure comparison. Bioinformatics 16, 566–567 [DOI] [PubMed] [Google Scholar]
- 29. Baker P., Carere J., Seah S. Y. (2011) Probing the molecular basis of substrate specificity, stereospecificity, and catalysis in the class II pyruvate aldolase, BphI. Biochemistry 50, 3559–3569 [DOI] [PubMed] [Google Scholar]
- 30. Narayanan B. C., Niu W., Han Y., Zou J., Mariano P. S., Dunaway-Mariano D., Herzberg O. (2008) Structure and function of PA4872 from Pseudomonas aeruginosa, a novel class of oxaloacetate decarboxylase from the PEP mutase/isocitrate lyase superfamily. Biochemistry 47, 167–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fu Z., Runquist J. A., Forouhar F., Hussain M., Hunt J. F., Miziorko H. M., Kim J. J. (2006) Crystal structure of human 3-hydroxy-3-methylglutaryl-CoA lyase: insights into catalysis and the molecular basis for hydroxymethyl-glutaric aciduria. J. Biol. Chem. 281, 7526–7532 [DOI] [PubMed] [Google Scholar]
- 32. Pražnikar J., Afonine P. V., Guncar G., Adams P. D., Turk D. (2009) Averaged kick maps: less noise, more signal and probably less bias. Acta Crystallogr. D. Biol. Crystallogr. 65, 921–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bashford D., Karplus M. (1990) pKa of ionizable groups in proteins: atomic detail from a continuum electrostatic model. Biochemistry 29, 10219–10225 [DOI] [PubMed] [Google Scholar]
- 35. Zheng Y. O., Kong Z. P., Lin J. L. (2002) Supramolecular assemblies via hydrogen bonding and π-π stacking interactions: crystal structures of [Co(phen)(H2O)4]L·0.5H2O and [CO2(phen)2(H2O)2L2] with H2L = adipic acid. J. Coord. Chem. 55, 1249–1257 [Google Scholar]
- 36. de Carvalho L. P., Blanchard J. S. (2006) Kinetic and chemical mechanism of α-isopropylmalate synthase from Mycobacterium tuberculosis. Biochemistry 45, 8988–8999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Quinn D. M., Sutton L. D. (1991) in Enzyme Mechanism from Isotope Effects, (Cook P. F., ed) pp. 73–126, CRC Press, Boca Raton, FL [Google Scholar]
- 38. McNutt M., Mullins L. S., Raushel F. M., Pace C. N. (1990) Contribution of histidine residues to the conformational stability of ribonuclease T1 and mutant Glu58 → Ala. Biochemistry 29, 7572–7576 [DOI] [PubMed] [Google Scholar]
- 39. Steyaert J., Hallenga K., Wyns L., Stanssens P. (1990) Histidine-40 of ribonuclease T1 acts as base catalyst when the true catalytic base, glutamic acid-58, is replaced by alanine. Biochemistry 29, 9064–9072 [DOI] [PubMed] [Google Scholar]
- 40. Liu T., Ryan M., Dahlquist F. W., Griffith O. H. (1997) Determination of pKa values of the histidine side chains of phosphatidylinositol-specific phospholipase C from Bacillus cereus by NMR spectroscopy and site-directed mutagenesis. Protein Sci. 6, 1937–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bertini I., Luchinat C. (1994) in Bioinorganic Biochemistry (Bertini I., Gray H. B., Lippard S. J., Selverstone Valentine J., eds) p. 41, University Science Books, Mill Valley, CA [Google Scholar]
- 42. Mock W. L., Tsay J. T. (1988) pK values for active site residues of carboxypeptidase A. J. Biol. Chem. 263, 8635–8641 [PubMed] [Google Scholar]
- 43. O'Leary M. H. (1992) in Mechanisms of Catalysis (Sigman D. S., ed) p. 239, Academic Press, New York [Google Scholar]
- 44. Teplyakov A., Lim K., Zhu P. P., Kapadia G., Chen C. C., Schwartz J., Howard A., Reddy P. T., Peterkofsky A., Herzberg O. (2006) Structure of phosphorylated enzyme I, the phosphoenolpyruvate:sugar phosphotransferase system sugar translocation signal protein. Proc. Natl. Acad. Sci. U.S.A. 103, 16218–16223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Herzberg O., Chen C. C., Kapadia G., McGuire M., Carroll L. J., Noh S. J., Dunaway-Mariano D. (1996) Swiveling-domain mechanism for enzymatic phosphotransfer between remote reaction sites. Proc. Natl. Acad. Sci. U.S.A. 93, 2652–2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Herzberg O., Chen C. C., Liu S., Tempczyk A., Howard A., Wei M., Ye D., Dunaway-Mariano D. (2002) Pyruvate site of pyruvate phosphate dikinase: crystal structure of the enzyme-phosphonopyruvate complex, and mutant analysis. Biochemistry 41, 780–787 [DOI] [PubMed] [Google Scholar]