

Background: Cav1.4 provides sustained influx of Ca2+ into photoreceptor terminals, which is required for continuous release of glutamate and normal vision.
Results: We characterize the effects of CaBP4 on Cav1.4.
Conclusion: CaBP4 increases Cav1.4 channel availability. CaBP4 mutations lead to reduced Cav1.4 availability and to retinal disease.
Significance: CaBP4 tailors voltage gating of Cav1.4 for its function in photoreceptor terminals.
Keywords: Calcium-binding Proteins, Calcium Channels, Calmodulin, Patch Clamp Electrophysiology, Retina, CSNB2, CaBP4, Cav1.4
Abstract
Cav1.4 L-type Ca2+ channels are crucial for synaptic transmission in retinal photoreceptors and bipolar neurons. Recent studies suggest that the activity of this channel is regulated by the Ca2+-binding protein 4 (CaBP4). In the present study, we explored this issue by examining functional effects of CaBP4 on heterologously expressed Cav1.4. We show that CaBP4 dramatically increases Cav1.4 channel availability. This effect crucially depends on the presence of the C-terminal ICDI (inhibitor of Ca2+-dependent inactivation) domain of Cav1.4 and is absent in a Cav1.4 mutant lacking the ICDI. Using FRET experiments, we demonstrate that CaBP4 interacts with the IQ motif of Cav1.4 and that it interferes with the binding of the ICDI domain. Based on these findings, we suggest that CaBP4 increases Cav1.4 channel availability by relieving the inhibitory effects of the ICDI domain on voltage-dependent Cav1.4 channel gating. We also functionally characterized two CaBP4 mutants that are associated with a congenital variant of human night blindness and other closely related nonstationary retinal diseases. Although both mutants interact with Cav1.4 channels, the functional effects of CaBP4 mutants are only partially preserved, leading to a reduction of Cav1.4 channel availability and loss of function. In conclusion, our study sheds new light on the functional interaction between CaBP4 and Cav1.4. Moreover, it provides insights into the mechanism by which CaBP4 mutants lead to loss of Cav1.4 function and to retinal disease.
Introduction
Ribbon synapses of retinal photoreceptors and bipolar cells encode graded and sustained changes in membrane potential that are generated during photoreception and maintained over long periods of time throughout the duration of a light stimulus. These graded signals are coupled with high fidelity to tonic glutamate release via permanent Ca2+ influx through presynaptic Cav1.4 L-type Ca2+ channels. Cav1.4 channels are tailored for their function as long-lasting presynaptic Ca2+ entry pathways by a lack of Ca2+-dependent inactivation (CDI)3 and very slow voltage-dependent inactivation (1–6). In Cav1.4 channels, CDI is specifically abolished by a recently discovered inhibitory domain (ICDI: inhibitor of CDI) in the C-terminal tail of the Cav1.4 channel, which binds to upstream C-terminal regulatory motifs (4, 5).
Cav1.4 channels are multiprotein complexes. The core Ca2+ channel complex is composed of the principal pore-forming α1 subunit and the auxiliary β2 and α2δ subunits. Mutations in the gene coding for the α1 subunit of Cav1.4 channels have been identified in patients suffering from congenital stationary night blindness type 2 (CSNB2) (7). This retinal disease is characterized by loss of neurotransmission from rods to second order bipolar cells, which is attributable to a loss of Cav1.4. These findings are corroborated by the analysis of a genetic mouse model deficient for the Cav1.4 α1 subunit (8, 9). In addition, in the mouse, deletion of another component of the Cav1.4 channel complex, the auxiliary β2a subunit, also leads to CSNB2-like phenotype (10).
Recently, it has been shown that mutations in the gene coding for CaBP4 are also associated with CSNB2 and other closely related nonstationary retinal diseases in humans (11–14). In line with these findings, deletion of CaBP4 in the mouse leads to a CSNB2-like phenotype (15). CaBP4 is a neuronal Ca2+ sensor protein with structural homology to calmodulin (CaM). Like CaM, CaBP4 contains two globular domains, the N- and the C-lobe, each containing a pair of EF-hand motifs connected by a central helix (16–18). The second EF-hand in the N-lobe of CaBP4 is nonfunctional. It has been shown that CaBP4 is part of the Cav1.4 channel complex in the retina (15). In addition, there is evidence that CaBP4 interacts with the C terminus of Cav1.4. However, the exact binding motif in Cav1.4 is unknown. CaBP4 has been implicated in the modulation of voltage-dependent Cav1.4 activation (15). Beyond these initial observations, the physiological effects of CaBP4 on Cav1.4 channels remains incompletely understood. The aim of this study was to explore the details of the role of CaBP4 in the regulation of Cav1.4 channel function. We provide evidence that CaBP4 has differential effects on different aspects of Cav1.4 channel function such as voltage-dependent activation and inactivation as well as CDI. We also characterized the functional consequences of two mutations in CaBP4 that are associated with human retinal disease.
EXPERIMENTAL PROCEDURES
Constructs for Electrophysiology
For expression of murine Cav1.4 α subunit wild type channel (1) (accession number AJ579852) and Cav1.4ΔICDI (6), stable HEK293 cell lines were generated using the Flp-InTM system (Invitrogen) according to manufacturer's protocol. Wild type CaBP4 (accession number NM144532, CaBP4 of Mus musculus, corresponds to NM145200, CaBP4 of Homo sapiens) was cloned into pIRES2-EGFP (Clontech) expression vector by using the restriction sites: BglII and SalI. In addition to murine wild type CaBP4, two mutant variants of CaBP4 that are associated with autosomal recessive forms of human congenital retinal disease CSNB2 were cloned (see Fig. 5A). In CaBP4-R216X, a single nucleotide exchange (C646T) replaces an arginine residue at position 216 by a premature stop codon (13). This mutation leads to a truncated CaBP4 protein lacking the C-lobe containing EF-hands 3 and 4. In the second mutation (CaBP4-E267fs), a deletion of two nucleotides causes a frameshift at the last residue of EF-hand 4, which elongates the protein by 91 novel amino acid residues (12). The mutations were introduced into mouse CaBP4 sequence to keep in line with the findings in genetic mouse models. This approach is valid as mouse Cav1.4 and CaBP4 proteins are more than 94 and 83% homologous to their respective human counterparts. Secondly, mutations in the genes coding for Cav1.4 or CaBP4 both lead to a similar phenotype in the mouse and human. Thirdly, functional properties of Cav1.4 as well as regulation by CaM and ICDI domain are also similar in mouse and human. According to the numbering of the mouse Cav1.4 protein sequence, human CaBP4-R216X and human CaBP4-E267fs correspond to murine CaBP4-R212X and CaBP4-E263fs, respectively. Throughout this study, we adhere to human terminology.
FIGURE 5.

FRET between wild type CaBP4 and CaBP4 mutants and C-terminal variants of Cav1.4 channels. A, schematic of the constructs used for FRET experiments. Top, CFP-tagged wild type CaBP4 and CaBP4 mutants. Boxes represent EF-hands 1–4. Gray boxes, functional EF-hands 1, 3, and 4; black box, nonfunctional EF-hand 2. NT, N terminus of CaBP4; CT, C terminus of CaBP4. Middle, C-terminal variants of Cav1.4 tagged by YFP or double-labeled by YFP and CFP. Boxes represent EF-hand, IQ motif, and ICDI domain; 5A represents the mutated IQ motif. 1.4DL, C terminus of Cav1.4 double-labeled by YFP and CFP. Bottom, N terminus of Cav1.4 (1.4 NT). B, interaction of CFP-CaBP4 with YFP-tagged fragments of the C terminus or with the N terminus of Cav1.4 as indicated. C, interaction of the ICDI domain with the C terminus of Cav1.4 lacking the ICDI domain (ΔICDI) in the absence and the presence of CaBP4 variants (group of four bars from the left). Intramolecular FRET for the C terminus of Cav1.4 double-labeled by YFP and CFP (1.4 DL) in the absence and presence of CaBP4 (group of 4 bars from the right). D, Western blot demonstrating the expression of mutant and wild type CaBP4 coupled to CFP. control: nontransfected HEK cells. E, interaction of CFP-tagged wild type and mutant variants of CaBP4 with the C terminus of Cav1.4. FR: FRET ratio; the number of cells is given in parentheses. Error bars indicate S.E. *, p ≤ 0.05, ***, p ≤ 0.001. Statistical significance is given in comparison with 1.4/5A and 1.4 NT (B), -WT (C), and NT (D).
Electrophysiology
HEK293 cells stably expressing Cav1.4 α subunits (wild type and Cav1.4ΔICDI) were transiently transfected with expression vectors encoding the calcium channel β2a (accession number: X64297) and α2δ1 (accession number: M21948) as described in Ref. 1. In some cases, wild type CaBP4 or CaBP4 variants were also transfected (1.5 μg from each construct). ICa and IBa were measured by using the following solutions: pipette solution, 112 mm CsCl, 3 mm MgCl2, 3 mm Mg-ATP, 10 mm EGTA, 5 mm HEPES, adjusted to pH 7.4 with CsOH; standard bath solution, 102 mm NaCl, 10 mm BaCl2 or 10 mm CaCl2, 5.4 mm CsCl2, 1 mm MgCl2, 20 mm tetraethylammonium chloride, 5 mm HEPES, 10 mm glucose, adjusted to pH 7.4 with NaOH. For experiments carried out in 2 mm Ca2+ in the extracellular solution, Ca2+ was equimolarily replaced by Na+. ICa and IBa were measured from the same cell. Bath solution was changed by a local solution exchanger. Currents were recorded at room temperature 1–3 days after transfection by using whole-cell patch clamp technique. Data were analyzed by using Origin 7.5 software (MicroCal).
The peak I/V relationship was measured by applying 350-ms voltage pulses to potentials between −80 and +70 mV in 10-mV increments from a holding potential of −80 mV (see Fig. 2A). For determination of half-maximum activation voltage (V0.5), the chord conductance (G) was calculated from the current voltage curves by dividing the peak current amplitude by its driving force at that respective potential G = I/(Vm − Vrev), where Vrev is the interpolated reversal potential, Vm is the membrane potential, and I is the peak current. The chord conductance was then fitted with a Boltzmann equation G = Gmax/(1 + e(V0.5 − Vm)/kact), where Gmax is the maximum conductance, V0.5 is the half-maximum activation voltage, Vm is the membrane potential, kact is the slope factor of the activation curve and e is the Euler's number.
FIGURE 2.

Voltage-dependent activation and inactivation of Cav1.4 in the absence and presence of CaBP4. A, voltage protocol used for the determination of activation properties (top left) and representative family of Ca2+ current traces for Cav1.4 in the absence (middle left) and presence of CaBP4 (bottom left). Currents were recorded in bath solution containing 10 mm Ca2+ as charge carrier. Similar experiments were used for the determination of I/V relationships (panels C and D) and activation curves (Figs. 3, 4, and 6). B, voltage protocol used for the determination of pseudo-steady-state inactivation properties (top right) and representative family of Ca2+ current traces for Cav1.4 in the absence (middle right) and presence of CaBP4 (bottom right). Similar experiments were used for the determination of pseudo-steady-state inactivation curves (Figs. 3, 4, and 6). C, I/V relationship for Cav1.4. D, I/V relationship for Cav1.4 in the presence of CaBP4. In C and D, currents were recorded in bath solution containing 10 mm Ca2+ (circles) or 10 mm Ba2+ (squares) as charge carrier. Currents were normalized to peak Ba2+ current.
Cav1.4 channel inactivation was quantified by calculating the fraction of peak Ba2+ and Ca2+ currents remaining after 350 ms of depolarization (R350) as described in Ref. 4. R350 is used to quantify CDI.
Half-maximum inactivation voltage (V0.5,inact) was determined using a pseudo-steady-state inactivation protocol (19–21) rather than true steady-state inactivation for the following reasons. For the induction of complete inactivation, a prepulse length of more than 30 s and a recovery interval between individual trials of more than 10–20 s are required. For the recovery from ultraslow inactivation, even more than 10 min have been reported (22). To efficiently record inactivation, we adjusted the protocol and decreased prepulse lengths and intertrial interval as follows. From a holding potential of −80 mV, a series of 2.5-s conditioning prepulses to various voltages between −100 mV and +50 mV was used. The conditioning pulse was followed by a 10-ms return to the holding potential and a 150-ms test pulse to +10 mV. The individual trials of the protocol were applied at a time interval of 10 s (see Fig. 2B).
The conditioning prepulse duration of 2.5 s was sufficient to induce considerable open state inactivation, which was comparable with the 5-s prepulse used in Ref. 1. The time interval between trials (10 s) was sufficient to recover the majority of inactivated channels. Ultraslow inactivation of Cav1.4, which needs more than 10 min to recover (22), was not addressed in the present study. Therefore, we cannot fully exclude the possibility that a small fraction of channels did not completely recover from ultraslow inactivation, leading to some cumulative inactivation that may sequentially add up in subsequent trials of the inactivation protocol.
The family of current traces obtained by applying the pseudo-steady-state inactivation protocol was analyzed by normalizing tail currents immediately after the test pulse to +10 mV to maximum current amplitude and plotted as a function of the preceding membrane potential. The data points were fitted with the Boltzmann function: I = 1/(1 + e(Vm − V0.5,inact)/kinact) where Vm is the test potential, V0.5,inact is the half-maximum voltage for pseudo-steady-state inactivation, kinact is the slope factor of the curve and e is the Euler's number. The liquid junction potential calculated according to Ref. 23 was 5.4 mV for extracellular solution containing 10 mm Ca2+ and 5.0 mV for extracellular solutions containing 2 mm Ca2+. Unless stated otherwise, the liquid junction potential was not corrected.
Constructs for Photometric FRET Experiments
For all FRET constructs, we used monomeric enhanced YFP (A206K) or enhanced CFP (A206K) (24, 25) to avoid homo- and heterodimerization. For simplicity, we refer to the different fluorescent proteins as “YFP” and “CFP” throughout the text. All FRET constructs were cloned into the pcDNA3 expression vector (Invitrogen).
For CaBP4 interaction assays, the following YFP-tagged C-terminal fragments of Cav1.4 were used: the complete C terminus of Cav1.4, 1.4 (Asp-1445–Leu-1984); the C terminus of Cav1.4 lacking the ICDI domain, ΔICDI (Asp-1445–Thr-1883); the EF-hand motif, EF (Asp-1445–Ile-1492); a fragment extending from the beginning of the C terminus to the end of the IQ motif, EF-IQ (Asp-1445–Gly-1609); and a variant of the complete C terminus in which the CaM preassociation site is disrupted by mutating Ile-1592–Phe-1596 of the IQ motif to alanines, respectively, 1.4/5A (Asp-1445–Leu-1984). In all CaBP4 fusion constructs, CFP is fused N-terminally to CaBP4 by an AAA linker. In addition, the YFP-tagged N terminus of Cav1.4 was used as control (1.4 NT, Met-1-Lys-92).
For intramolecular FRET experiments, the complete C terminus of Cav1.4 was N-terminally fused to YFP and C-terminally fused to CFP separated by a triple alanine linker, 1.4 DL (Asp-1445–Leu-1984). For ICDI interaction assays, YFP-tagged 1.4 or YFP-tagged ΔICDI was used. The ICDI domain (Leu-1885–Leu-1984) was C-terminally fused to CFP.
Photometric FRET Measurements
HEK293 cells were grown in cell culture dishes (ibiTreat, ibidi, Martinsried, Germany) and transiently transfected using GeneExpresso Max (Excellgen, Rockville, MD). 1–2 days later, the cells were washed and maintained in buffer solution composed of 140 mm NaCl, 5 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 10 mm glucose, 10 mm Na-HEPES, pH 7.4, at room temperature. Cells were placed on an Axiovert200 inverted epifluorescence microscope equipped with an oil immersion 40× objective (Fluar 40× OIL/1.3, Carl Zeiss, Inc.) and a dual emission photometric system (Till Photonics, Gräfelfing, Germany). Excitation was done at 440 ± 7.5 nm or 505 ± 7.5 nm, applied with a frequency of 2 Hz with light from a Polychrome V (Till Photonics). Fluorescence was recorded from entire single cells. For recording of CFP and YFP emission, a photometric system (Till Photonics) was used consisting of the following filter cubes (in parentheses, excitation band of the excitation filter; long pass beam splitter; emission band of the emission filter; company): CFP (438 ± 12 nm; 458 nm; 483 ± 16 nm; AHF Analysentechnik, Tübingen, Germany); YFP (500 ± 12 nm; 520 nm; 542 ± 16.5 nm; AHF Analysentechnik) and FRET (438 ± 12 nm; 458 nm; 542 ± 16.5 nm, AHF Analysentechnik). Signals detected by amplified photodiodes (Till Photonics) were digitized using an analog digitizer (MINI DIGI1B; Molecular Devices) and stored on a personal computer using Clampex 10 software (Molecular Devices).
Measurements of single-cell FRET based on aggregate (nonspatial) fluorescence recordings were performed using three-cube FRET as described previously (26, 27). The notation and abbreviations used follow the definitions of Refs. 26 and 27. The degree of FRET in an individual cell was quantified using the FRET ratio (FR), which is defined as the fractional increase in YFP emission caused by FRET. The FR was calculated using
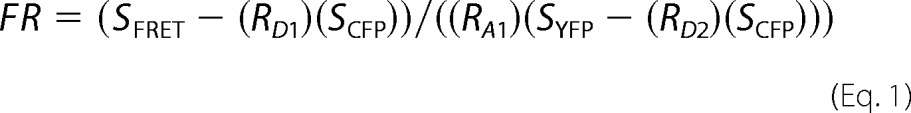 |
Fluorescence measurements for the determination of SFRET, SCFP, and SYFP were performed in cells coexpressing CFP-tagged and YFP-tagged peptides or intramolecular FRET constructs dually labeled by CFP and YFP using the following parameters: SCFP, excitation at 440 ± 7.5 nm and emission at 483 ± 16 nm (donor excitation; donor emission); SFRET, excitation at 440 ± 7.5 nm and emission at 542 ± 13.5 nm (donor excitation; acceptor emission); and SYFP, excitation at 505 ± 7.5 nm and emission at 542 ± 13.5 nm (acceptor excitation; acceptor emission). RD1, RA1, and RD2 are experimentally predetermined constants from measurements applied to single cells expressing only CFP- or YFP-tagged molecules. These constants are used to correct for bleed-through of CFP into the YFP channel (RD1), direct excitation of YFP by CFP excitation (RA1), and the small amount of CFP excitation at the YFP excitation wave-length (RD2) as described by Refs. 26 and 27. Throughout all FRET experiments, the CFP:YFP ratio was between 1:10 and 20:1, which can be expected to yield reliable results (supplemental Fig. S1) (5, 28).
Western Blots of HEK293 Cells
Recombinant CFP-tagged human CaBP4 mutations (CaBP4 R216X and CaBP4 E267fs) and wild type CaBP4 were expressed in HEK293 cells using the calcium phosphate transfection method. Western blot protocol was performed as published previously (29) using an antibody against GFP (Clontech).
Data Analysis and Statistics
Electrophysiology data were analyzed using Clampfit 10.0 (Molecular Devices) and Origin 7.5 (MicroCal). FRET data were analyzed using Microsoft® Office Excel® software (Microsoft®). Data plotting, curve fitting, and statistical analysis were performed using Origin 7.5 (MicroCal). All values are presented as mean ± S.E. for the indicated number n of experiments. An unpaired Student's t test was performed for the comparison between two groups. Significance was tested by analysis of variance followed by Dunnett's test if multiple comparisons were made. Values of p < 0.05 were considered significant. The determination of molar CFP/YFP ratios and the determination of the window conductances are described in supplemental methods.
RESULTS
To characterize the functional effect of CaBP4 on Cav1.4 channels, we first tested the effect on CDI. We compared Ca2+ and Ba2+ currents through Cav1.4 in transfected HEK293 cells in the absence of CaBP4 (Fig. 1A), which is not endogenously expressed (supplemental Fig. S2), and in the presence of overexpressed CaBP4 (Fig. 1B). Wild type channels did not display any CDI under both conditions (Fig. 1, A and B). Truncation of the ICDI domain in the distal C terminus of Cav1.4 unmasked CDI, which is caused by endogenous CaM (4) (Fig. 1C). In contrast, CaBP4 completely abolished CDI in Cav1.4ΔICDI channels (Fig. 1D). This finding indicated that both CaBP4 and the ICDI domain have equivalent functions with regard to their effect on CDI. Furthermore, these observations suggested that CaM and CaBP4 stabilize the channel in a different conformational state that gives rise to the absence of CDI in the case of CaBP4 and to the presence of CDI in the case of CaM. To study the effect of CaBP4 on voltage-dependent gating of Cav1.4, we determined activation and inactivation curves for Ca2+ and Ba2+ currents in the absence and presence of CaBP4 (Figs. 2 and 3). From these experiments, it was evident that CaBP4 shifted the activation curve of Cav1.4 to more hyperpolarized potentials (Fig. 3A, Table 1) and increased the steepness of the activation curve (lower kact; Table 1). Voltage-dependent inactivation gating was characterized using a pseudo-steady-state inactivation protocol (Fig. 2B) rather than true steady-state inactivation for reasons outlined in detail under “Experimental Procedures”. The protocol is well suited to characterize overall Cav1.4 channel inactivation consisting of open state inactivation and a small amount of closed state inactivation. These two components were not further dissected. Using this protocol, CaBP4 dramatically reduced pseudo-steady-state inactivation (Fig. 3, B and C). Together the effects of CaBP4 on activation and inactivation lead to a pronounced increase of the window conductance caused by the overlap of the activation and the inactivation curve (Fig. 3C, Tables 1 and 2). We determined the window conductance for Cav1.4 channels in extracellular recording solution containing 2 mm Ca2+, which is close to the physiological extracellular Ca2+ concentrations (supplemental Fig. S3). As compared with standard extracellular recording solution containing 10 mm Ca2+, the window conductance was shifted to more hyperpolarized potentials. We used the experimentally determined voltage shift to correct the window conductances obtained in experiments using standard recording solutions (10 mm extracellular Ca2+; supplemental Fig. S4). A comparison revealed that in the presence of CaBP4, the predicted window conductance for the physiological conditions of 2 mm Ca2+ was more than 2-fold larger than in the absence of CaBP4 (supplemental Figs. S3–S5). To account for the physiological voltage range observed in photoreceptors (less than −30 mV (30–34)), we also calculated window conductances at −30 and −40 mV (supplemental Fig. S5). We found that the window conductance was 2.2- or 1.7-fold higher in the presence of CaBP4 at −30 or −40 mV, respectively. Together our findings indicated that CaBP4 markedly increased overall Cav1.4 channel availability. This effect was equally pronounced for Ca2+ and Ba2+ currents, indicating that this effect is not Ca2+ dependent (Fig. 3C).
FIGURE 1.

CaBP4 abolishes CDI in Cav1.4 channels lacking the ICDI domain. A–D, representative traces of ICa (red traces) and IBa (black traces) through Cav1.4 (A), Cav1.4 + CaBP4 (B), Cav1.4ΔICDI, (C), and Cav1.4ΔICDI + CaBP4 (D) recorded in bath solution containing 10 mm Ca2+ (red trace) or 10 mm Ba2+ (black trace) as charge carrier. Currents were evoked by stepping from a holding potential of −80 mV to +10 mV (pulse duration: 350 ms). Current traces were normalized to peak current. Throughout, Ba2+ traces for CDI are scaled to match Ca2+ traces (scale bar). E and F, quantification of CDI. Fractional inactivation of ICa (E) or IBa (F) during a 350-ms test pulse to Vmax was detected. R350 corresponds to the fraction of ICa or IBa remaining after 350 ms. The number of experiments is given in parentheses. ***, p < 0.001. Statistical significance is given in comparison with the other constructs of this panel.
FIGURE 3.

CaBP4 affects voltage-dependent gating of Cav1.4 and dramatically increases channel availability. A, activation curves for Cav1.4 in the absence (red) and in the presence of CaBP4 (black). Throughout the figure, in the left panel, Ba2+ (10 mm) was the charge carrier, and in the right panel, Ca2+ (10 mm) was the charge carrier. B, pseudo-steady-state inactivation curve for Cav1.4 in the absence (red) and in the presence of CaBP4 (black). C, the overlay of activation curves presented in A and pseudo-steady-state inactivation curves presented in B demonstrates an increase in availability of Cav1.4 in the presence of CaBP4.
TABLE 1.
Voltages for half-maximum activation (V0.5,act) and slope values (kact) from the measurements shown in Figs. 2–4 and 6 and supplemental Fig. S3, respectively
* = significantly different compared to measurements of Cav1.4 (with 10 mm Ca2+ or 10 mm Ba2+ in the extracellular solution). # = significantly different compared to measurements of Cav1.4 in presence of CaBP4 (with 10 mm Ca2+ or 10 mm Ba2+ in the extracellular solution). p < 0.05 for one symbol (* or #), p < 0.01 for two symbols, p < 0.001 for three symbols. n = number of cells.
| Concentration of charge carrier [mm] | V0.5,act Ca2+ |
V0.5,act Ba2+ |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| V0.5 | S.E. | Slope (kact) | S.E. | n | V0.5 | S.E. | Slope (kact) | S.E. | n | ||
| Cav1.4 | 10 mm | −9.41## | ± 1.88 | 7.88### | ± 0.23 | 7 | −13.39### | ± 0.60 | 7.00### | ± 0.17 | 18 |
| Cav1.4 | 2 mm | −15.09* | ± 1.36 | 6.88** | ± 0.14 | 9 | |||||
| Cav1.4 | 10 mm | −18.38** | ± 2.15 | 5.26*** | ± 0.50 | 4 | −18.89*** | ± 1.53 | 5.39*** | ± 0.36 | 6 |
| + CaBP4 | |||||||||||
| Cav1.4 | 10 mm | −9.47## | ± 0.84 | 7.77## | ± 0.21 | 5 | −15.16 | ± 1.36 | 6.18 | ± 0.47 | 9 |
| + R216X | |||||||||||
| Cav1.4 | 10 mm | −3.62*,### | ± 1.07 | 7.88## | ± 0.43 | 5 | −10.45**,### | ± 0.51 | 6.51*,### | ± 0.10 | 14 |
| + E267fs | |||||||||||
| Cav1.4ΔICDI | 10 mm | −16.75** | ± 0.95 | 6.83** | ± 0.29 | 10 | −21.30*** | ± 0.64 | 5.42*** | ± 0.14 | 23 |
| Cav1.4ΔICDI | 10 mm | −17.09** | ± 1.47 | 5.51*** | ± 0.22 | 13 | −21.75*** | ± 0.92 | 4.75*** | ± 0.23 | 17 |
| + CaBP4 | |||||||||||
TABLE 2.
Voltages for half-maximum inactivation (V0,5,inact) and slope values (kinact) from the measurements shown in Figs. 2–4 and 6 and supplemental Fig. S3, respectively
* = significantly different compared to measurements of Cav1.4 (with 10 mm Ca2+ or 10 mm Ba2+ in the extracellular solution). # = significantly different compared to measurements of Cav1.4 in presence of CaBP4 (with 10 mm Ca2+ or 10 mm Ba2+ in the extracellular solution). p < 0.05 for one symbol (* or #), p < 0.01 for two symbols, p < 0.001 for three symbols. n = number of cells.
| Concentration of charge carrier [mm] |
V0.5,act Ca2+ |
V0.5,act Ba2+ |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| V0.5 | S.E. | Slope (kinact) | S.E. | n | V0.5 | S.E. | Slope (kinact) | S.E. | n | ||
| Cav1.4 | 10 mm | −2.77 | ± 5.00 | 17.41 | ± 1.62 | 4 | −23.63## | ± 2.00 | 15.16 | ± 0.83 | 5 |
| Cav1.4 | 2 mm | −5.12 | ± 2.23 | 16.93 | ± 1.22 | 12 | |||||
| Cav1.4 | 10 mm | −13.27 | ± 4.67 | 20.24 | ± 3.37 | 3 | −13.55** | ± 2.72 | 17.42 | ± 2.26 | 8 |
| + CaBP4 | |||||||||||
| Cav1.4 | 10 mm | −13.47 | ± 3.29 | 15.45 | ± 1.42 | 4 | 9.48***,### | ± 2.71 | 19.57* | ± 1.23 | 6 |
| + R216X | |||||||||||
| Cav1.4 | 10 mm | 13.55# | ± 6.35 | 19.37 | ± 2.66 | 6 | 14.25***,### | ± 2.26 | 20.21* | ± 1.07 | 12 |
| + E267fs | |||||||||||
| Cav1.4ΔICDI | 10 mm | −20.50** | ± 2.12 | 14.97 | ± 2.48 | 4 | −11.55** | ± 2.75 | 21.72* | ± 1.77 | 12 |
| Cav1.4ΔICDI | 10 mm | −22.95** | ± 2.07 | 11.64 | ± 2.34 | 5 | −16.45 | ± 3.02 | 10.54*,# | ± 0.85 | 11 |
| + CaBP4 | |||||||||||
Similar experiments were carried out for Cav1.4ΔICDI channels. Surprisingly, in the absence of the ICDI domain, CaBP4 had no effect on voltage-dependent activation and inactivation and window currents (Fig. 4, A and B, supplemental Fig. S4, C and D, Tables 1 and 2). Moreover, voltage-dependent activation and inactivation of Cav1.4ΔICDI channels were very similar to that of wild type Cav1.4 channels in the presence of CaBP4. In contrast, in the absence of CaBP4, Cav1.4ΔICDI channels displayed CDI (Fig. 1C). In summary, in the absence of the ICDI domain, CaBP4 has no effect on voltage-dependent gating (Fig. 4) but blocks CDI (Fig. 1D). Together these observations indicate that CaBP4 modulates the functional impact of the ICDI domain in a complex fashion. With respect to voltage-dependent gating of Cav1.4 channels, the ICDI domain and CaBP4 have opposing functional effects. The ICDI domain shifts the activation curve of Cav1.4 to more depolarized potentials and increases inactivation, whereas CaBP4 shifts the activation curve of Cav1.4 to more hyperpolarized potentials and decreases inactivation. With respect to the inhibition of the CDI, the ICDI domain and CaBP4 have equivalent functional effects.
FIGURE 4.

In the absence of the ICDI domain, the effect of CaBP4 on voltage-dependent gating is abolished. A and B, overlay of activation curves and pseudo-steady-state inactivation curves for Ba2+ (10 mm; A) or Ca2+ (10 mm; B) currents through Cav1.4ΔICDI channels in the absence (red) and the presence (black) of CaBP4.
To further examine the interaction between CaBP4 and Cav1.4, FRET experiments were carried out using YFP-tagged variants of Cav1.4 C terminus and CFP-CaBP4 (Fig. 5). We found that CaBP4 interacts with the C terminus of both Cav1.4 wild type and Cav1.4ΔICDI (Fig. 5, A and B). Truncation of the C terminus downstream of the IQ motif (EF-IQ) did not significantly change FRET when coexpressed with CaBP4 as compared with the complete C terminus of Cav1.4. In contrast, there was no FRET between the EF-hand motif and CaBP4. One important possibility would be that CaBP4 binds to the IQ motif in the C-terminal tail of Cav1.4. Binding of other CaBPs and of CaM to the IQ motif of high voltage-activated and low voltage-activated Ca2+ channels including Cav1.4 has been reported (6, 27, 35–37). To test this possibility in our experimental setting, we performed FRET experiments using a mutant of the C terminus of Cav1.4, in which five amino acid residues within the IQ motif are replaced by alanines (1.4/5A). The FRET between CaBP4 and 1.4/5A was very low. The most likely interpretation of this finding is that CaBP4 does not interact with 1.4/5A, indicating that CaBP4 and CaM share the same binding domain on the C terminus of Cav1.4 (Fig. 5B).
We next tested the possibility whether CaBP4 also associates with the N terminus of Cav1.4 channels. For Cav1.2 and Cav1.4 channels, CaM or CaBP1 binding to the N terminus has been reported (14, 39, 40). However, we found that the FRET signal between CaBP4 and the N terminus of Cav1.4 was very low (Fig. 5B), indicating weak or no interaction. Alternatively, the positioning or the angle between the fluorescent tags may be unfavorable for FRET.
To assess whether CaBP4 interferes with binding of the ICDI domain, we compared the FRET signal between the ICDI domain and the C terminus of Cav1.4 lacking the ICDI domain in the absence and in the presence of CaBP4 (Fig. 5C). These experiments revealed that binding of the ICDI domain was impaired in the presence of CaBP4. In agreement with these coexpression experiments, CaBP4 significantly decreased intramolecular FRET for the C terminus of Cav1.4 that was N- and C-terminally tagged with YFP and CFP, respectively (Fig. 5C). A direct interaction between CaBP4 and the ICDI domain is not likely because the FRET signal between CaBP4 and 1.4/5A was very low (Fig. 5B). This finding indicates that besides the IQ motif, other CaBP4 binding regions within the C terminus including the ICDI domain are unlikely.
We next set out to analyze the functional consequences of two mutations in CaBP4, which are associated with autosomal recessive forms of human congenital retinal disease CSNB2 (Fig. 5A). The first mutation (CaBP4-R216X) results in a truncated CaBP4 protein lacking the C-lobe containing EF-hands 3 and 4 (13). In the second mutation (CaBP4-E267fs), the last residue of EF-hand 4 (Glu-267) is exchanged to valine followed by a frameshift elongating the protein (12). Western blot experiments confirmed the expression of mutant CaBP4 variants in HEK cells (Fig. 5D). In FRET experiments, we found that CaBP4-E267fs and CaBP4-R216X interact with the C terminus of Cav1.4 (Fig. 5E). The FRET signal of both mutant CaBP4s was even higher than that of wild type CaBP4. The finding that CaBP4-R216X interacts with the C terminus of Cav1.4 is very interesting because it shows that the N-lobe of CaBP4 is sufficient to interact with Cav1.4 with a FRET ratio (FR) even higher than wild type CaBP4. Furthermore, the presence of CaBP4-E267fs and CaBP4-R216X partially impairs binding of the ICDI domain (Fig. 5C).
The functional impact of the CaBP4 mutations was analyzed in electrophysiological experiments (Fig. 6). After coexpression of CaBP4-R216X or CaBP4-E267fs together with wild type Cav1.4 channels, Ca2+ currents did not display CDI (not shown). In contrast, in coexpression experiments of mutated CaBP4s together with Cav1.4ΔICDI, slowly developing CDI could be observed, which was pronounced for CaBP4-R216X and subtle but detectable for CaBP4-E267fs (Fig. 6, A–C). The appearance of slow CDI in coexpression experiments of Cav1.4ΔICDI channels and mutant CaBP4 suggests that CaM can access the effector site for CDI at least in some of the channels. We next analyzed the effect of CaBP4 mutations on voltage-dependent gating of Cav1.4 (Fig. 6, D and E). In contrast to wild type CaBP4, both CaBP4 mutations did not shift voltage-dependent activation to more hyperpolarized potentials. For CaBP4-R267fs, the activation curve was even shifted by about 6 mV to more depolarized potentials in experiments using Ca2+ as charge carrier (Fig. 6E, Tables 1 and 2). Slope factors of both CaBP4 mutations were significantly higher than those observed in the presence of CaBP4 and very similar to that observed in the absence of CaBP4 (Table 1). CaBP4 mutants, like wild type CaBP4, functionally antagonize the inactivation process (Fig. 6, D and E). However, the antagonizing effect of mutant CaBP4s was significantly less pronounced as compared with wild type CaBP4. For CaBP4-R216X, this effect was only observed using Ba2+ as charge carrier (Fig. 6D). Together the observed effects in CaBP4 mutants significantly decreased the predicted window conductance as compared with wild type CaBP4 (2- or 3-fold for CaBP4-R216X at −40 or −30 mV, respectively and 3- or 4-fold for CaBP4-E267fs at −40 or −30 mV, respectively; supplemental Figs. S4 and S5). These observations indicate that changes in Cav1.4 channel availability are significantly less pronounced in the presence of mutant CaBP4 proteins than in the presence of wild type CaBP4.
FIGURE 6.

Mutant CaBP4 variants reduce Cav1.4 channel availability as compared with WT CaBP4. A and B, representative traces of ICa (10 mm; red traces) and IBa (10 mm; black traces) through Cav1.4ΔICDI coexpressed with CaBP4-R216X (A) or CaBP4-E267fs (B). Currents were evoked by stepping from a holding potential of −80 mV to +10 mV (pulse duration: 350 ms). Current traces were normalized to peak current C, quantification of CDI. Fractional inactivation of ICa is given as R350 as outlined in Fig. 1. The number of experiments is given in parentheses. ***, p < 0.001. Statistical significance is given in comparison with WT. D and E, overlay of activation and pseudo-steady-state inactivation curves for Cav1.4 coexpressed with CaBP4-R216X (D) and CaBP4-E267fs (E). For comparison, activation and pseudo-steady-state inactivation curves for Cav1.4 in the absence (red) and presence of CaBP4 (gray) are indicated. Graphs for Ba2+ (10 mm) are shown on the left, and those for Ca2+ (10 mm) are shown on the right.
DISCUSSION
Here, we describe the functional effects of CaBP4 on wild type Cav1.4 channels. The main effect of CaBP4 is a profound increase in Cav1.4 channel availability. We propose the following mechanism to explain this finding (Fig. 7). In wild type Cav1.4 channels, the ICDI domain shifts the activation curve to the right and induces voltage-dependent inactivation. CaBP4 antagonizes these effects. In line with this hypothesis, in the presence of CaBP4, the ICDI domain is functionally silent. Furthermore, in Cav1.4 channels lacking the ICDI domain, CaBP4 has no effects on Cav1.4 voltage dependence. These observations suggest that Cav1.4 channels lacking the ICDI domain are locked in a functional state resembling wild type Cav1.4 channels in the presence of CaBP4. Our FRET experiments provide evidence that the effect of CaBP4 is brought about by structural interference with the binding of the ICDI domain to the C terminus of Cav1.4. On the functional level, CaBP4 selectively abolishes the effects of the ICDI domain on Cav1.4 channel availability. As a consequence, the voltage dependence of channel availability is pushed toward physiological operating voltage range in photoreceptors (30–34). Besides the effects on voltage-dependent channel availability, CaBP4 also blocks CDI. This effect is only evident in Cav1.4 channels lacking the ICDI domain. With respect to its inhibitory effect on CDI, CaBP4 and the ICDI domain are functionally equivalent.
FIGURE 7.

Schematic of CaBP4 and ICDI effects on Cav1.4 channel activation and inactivation. A, in the absence of CaBP4, the activation curve of Cav1.4 channels (black curve: −CaBP4 +CaM) is at depolarized potentials. CaBP4 shifts the activation curve of wild type Cav1.4 channels to the left (green curve: +CaBP4, −CaM). The same left shift is observed in Cav1.4ΔICDI channels (green curve: −ICDI). B, in the absence of CaBP4, there is pronounced inactivation of Cav1.4 channels (black curve: −CaBP4). CaBP4 decreases Cav1.4 channel inactivation (green curve: +CaBP4). The same decrease in Cav1.4 inactivation is observed in Cav1.4ΔICDI channels (green curve: −ICDI). The ICDI domain and CaBP4 have opposing functional effects on voltage-dependent gating of Cav1.4 channels. The ICDI domain shifts the activation curve of Cav1.4 to more depolarized potentials and increases inactivation (black curve: +ICDI), whereas CaBP4 shifts the activation curve of Cav1.4 to more hyperpolarized potentials and decreases inactivation (green curve: +CaBP4). In addition, with respect to the inhibition of the CDI, ICDI and CaBP4 are functionally equivalent. The voltage axis is in arbitrary units; the physiological voltage range of operation in photoreceptor cells is shown in green. The intensity of interaction between the ICDI domain and Cav1.4 is represented by the gray triangle. The black activation curve corresponds to intermediate interaction strength of the ICDI in the presence of endogenous CaM. The green activation curve indicates weak interaction between the ICDI and Cav1.4. The purple activation curve represents tight interaction between the ICDI and the channel, which may be observed in the absence of CaBP4 and endogenous CaM. Boxes summarize the conditions under which the respective activation curve is observed. +ICDI refers to the wild type Cav1.4 channel; −ICDI refers to truncated channels lacking the ICDI domain. Furthermore, in the boxes, the presence or absence of endogenous CaM or CaBP4 is given.
In FRET experiments, we find that CaBP4 tightly associates with the IQ motif of Cav1.4 channels and that CaBP4 is able to displace CaM from binding to the IQ motif at physiological conditions. In line with this interpretation, we find that CaBP4 can very efficiently regulate the functional properties of Cav1.4 channels in HEK293 cells in which CaM is endogenously expressed at high levels. It is very likely that binding of CaBP4 induces a conformation different from the conformation in the presence of CaM. This difference could be the reason for differential effects of CaBP4 and CaM on CDI in Cav1.4ΔICDI channels.
How can the functional effects of CaBP4 on Cav1.4 voltage gating mechanistically and structurally be explained? Our FRET experiments demonstrate that CaBP4 decreases binding of the ICDI domain to the proximal C terminus of Cav1.4. In structural terms, one possible explanation could be that CaBP4 partially displaces the ICDI domain. In line with this idea is the observation that the FRET signal between the ICDI domain and C terminus of Cav1.4 is only reduced but does not equal zero (FR = 1). Partial departure of the ICDI domain could selectively abolish the effects of the ICDI domain on voltage-dependent Cav1.4 channel gating. Our results that CaBP4 mutants both interact with the C terminus of Cav1.4 but lack most of the effects of wild type CaBP4 suggest that both lobes of CaBP4 need to be present and act in concert to produce a conformational change that regulates voltage-dependent activation and inactivation.
Our model also gives insight into the pathomechanism of CaBP4 mutants identified in CSNB2 patients. A common feature of CaBP4 mutants is a significant reduction of overall channel availability as compared with wild type CaBP4. We find that in CaBP4 mutants, there was no negative shift of the voltage-dependent activation curve and no change in the slope of the activation curve. This suggests that CaBP4 mutants, although bound to the channel, cannot antagonize the effect of the ICDI domain on voltage-dependent activation. In CaBP4-R216X, this could be because the functional C-lobe is missing. In the absence of the C-lobe, CaBP4-R216X could indeed interact with the channel, but would not induce the conformational change of Cav1.4-CaBP4 complex, which is required for the interference with the ICDI domain. In CaBP4-E267fs, both lobes are present, but the function of the C-lobe could be affected by the exchange of the last amino acid residue in the fourth EF-hand and the additional nonsense sequence. Interestingly, for CaBP4-E267fs, even a significant shift of the activation curve toward more positive potentials was observed. A possible explanation is that the CaBP4 mutant stabilizes the channel in a conformational state that intensifies the interaction with the ICDI domain, shifting the activation curve even toward more positive potentials. Alternatively, the interaction of the ICDI domain and the channel could be intensified by completely displacing endogenous CaM (Fig. 7). In both CaBP4 mutants, voltage-dependent inactivation was more pronounced as compared with wild type CaBP4. This indicates that mutant CaBPs can only partially induce the conformational change required for the full wild type CaBP4 effect. In total, the overall window currents and the physiological relevant window currents are both significantly reduced. These effects are more pronounced in CaBP4-R216X. The appearance of slow CDI in Cav1.4ΔICDI channels coexpressed with mutant CaBP4 suggests that CaM can access the effector site for CDI at least in some of the channels.
Finally, the effect of CaBP4 on CDI may be relevant in a Cav1.4 variant lacking the ICDI domain. It has been shown that Cav1.4 splice variants lacking the ICDI domain exist under physiological conditions (38). In these channels, CDI is predicted to be switched off by the action of CaBP4. Similar regulation has been observed for Cav1.3 channels in inner hair cells of the auditory system (35).
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) and by the Bayerische Forschungsstiftung.

This article contains supplemental Methods and Figs. S1–S5.
- CDI
- Ca2+-dependent inactivation
- ICDI
- inhibitor of CDI
- CSNB2
- congenital stationary night blindness type 2
- CaM
- calmodulin
- CaBP4
- Ca2+-binding protein 4.
REFERENCES
- 1. Baumann L., Gerstner A., Zong X., Biel M., Wahl-Schott C. (2004) Functional characterization of the L-type Ca2+ channel Cav1.4α1 from mouse retina. Invest. Ophthalmol. Vis. Sci. 45, 708–713 [DOI] [PubMed] [Google Scholar]
- 2. Koschak A., Reimer D., Walter D., Hoda J. C., Heinzle T., Grabner M., Striessnig J. (2003) Cav1.4α1 subunits can form slowly inactivating dihydropyridine-sensitive L-type Ca2+ channels lacking Ca2+-dependent inactivation. J. Neurosci. 23, 6041–6049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McRory J. E., Hamid J., Doering C. J., Garcia E., Parker R., Hamming K., Chen L., Hildebrand M., Beedle A. M., Feldcamp L., Zamponi G. W., Snutch T. P. (2004) The CACNA1F gene encodes an L-type calcium channel with unique biophysical properties and tissue distribution. J. Neurosci. 24, 1707–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wahl-Schott C., Baumann L., Cuny H., Eckert C., Griessmeier K., Biel M. (2006) Switching off calcium-dependent inactivation in L-type calcium channels by an autoinhibitory domain. Proc. Natl. Acad. Sci. U.S.A. 103, 15657–15662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singh A., Hamedinger D., Hoda J. C., Gebhart M., Koschak A., Romanin C., Striessnig J. (2006) C-terminal modulator controls Ca2+-dependent gating of Cav1.4 L-type Ca2+ channels. Nat. Neurosci. 9, 1108–1116 [DOI] [PubMed] [Google Scholar]
- 6. Griessmeier K., Cuny H., Rotzer K., Griesbeck O., Harz H., Biel M., Wahl-Schott C. (2009) Calmodulin is a functional regulator of Cav1.4 L-type Ca2+ channels. J. Biol. Chem. 284, 29809–29816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Striessnig J., Bolz H. J., Koschak A. (2010) Channelopathies in Cav1.1, Cav1.3, and Cav1.4 voltage-gated L-type Ca2+ channels Pflugers Arch. 460, 361–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Specht D., Wu S. B., Turner P., Dearden P., Koentgen F., Wolfrum U., Maw M., Brandstatter J. H., tom Dieck S. (2009) Effects of presynaptic mutations on a postsynaptic Cacna1s calcium channel colocalized with mGluR6 at mouse photoreceptor ribbon synapses. Invest. Ophthalmol Vis. Sci. 50, 505–515 [DOI] [PubMed] [Google Scholar]
- 9. Mansergh F., Orton N. C., Vessey J. P., Lalonde M. R., Stell W. K., Tremblay F., Barnes S., Rancourt D. E., Bech-Hansen N. T. (2005) Mutation of the calcium channel gene Cacna1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retina. Hum. Mol. Genet. 14, 3035–3046 [DOI] [PubMed] [Google Scholar]
- 10. Ball S. L., Powers P. A., Shin H. S., Morgans C. W., Peachey N. S., Gregg R. G. (2002) Role of the β2 subunit of voltage-dependent calcium channels in the retinal outer plexiform layer. Invest Ophthalmol Vis. Sci. 43, 1595–1603 [PubMed] [Google Scholar]
- 11. Aldahmesh M. A., Al-Owain M., Alqahtani F., Hazzaa S., Alkuraya F. S. (2010) A null mutation in CABP4 causes Leber's congenital amaurosis-like phenotype. Mol. Vis. 16, 207–212 [PMC free article] [PubMed] [Google Scholar]
- 12. Zeitz C., Kloeckener-Gruissem B., Forster U., Kohl S., Magyar I., Wissinger B., Matyas G., Borruat F. X., Schorderet D. F., Zrenner E., Munier F. L., Berger W. (2006) Mutations in CABP4, the gene encoding the Ca2+-binding protein 4, cause autosomal recessive night blindness. Am. J. Hum. Genet. 79, 657–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Littink K. W., van Genderen M. M., Collin R. W., Roosing S., de Brouwer A. P., Riemslag F. C., Venselaar H., Thiadens A. A., Hoyng C. B., Rohrschneider K., den Hollander A. I., Cremers F. P., van den Born L. I. (2009) A novel homozygous nonsense mutation in CABP4 causes congenital cone-rod synaptic disorder. Invest Ophthalmol Vis. Sci. 50, 2344–2350 [DOI] [PubMed] [Google Scholar]
- 14. Zhou H., Yu K., McCoy K. L., Lee A. (2005) Molecular mechanism for divergent regulation of Cav1.2 Ca2+ channels by calmodulin and Ca2+-binding protein-1. J. Biol. Chem. 280, 29612–29619 [DOI] [PubMed] [Google Scholar]
- 15. Haeseleer F., Imanishi Y., Maeda T., Possin D. E., Maeda A., Lee A., Rieke F., Palczewski K. (2004) Essential role of Ca2+-binding protein 4, a Cav1.4 channel regulator, in photoreceptor synaptic function. Nat. Neurosci. 7, 1079–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haeseleer F., Imanishi Y., Sokal I., Filipek S., Palczewski K. (2002) Calcium-binding proteins: intracellular sensors from the calmodulin superfamily. Biochem. Biophys. Res. Commun. 290, 615–623 [DOI] [PubMed] [Google Scholar]
- 17. Nejatbakhsh N., Feng Z. P. (2011) Calcium binding protein-mediated regulation of voltage-gated calcium channels linked to human diseases. Acta Pharmacol. Sin. 32, 741–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Haeseleer F., Sokal I., Verlinde C. L., Erdjument-Bromage H., Tempst P., Pronin A. N., Benovic J. L., Fariss R. N., Palczewski K. (2000) Five members of a novel Ca2+-binding protein (CABP) subfamily with similarity to calmodulin. J. Biol. Chem. 275, 1247–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dong Y., White F. J. (2003) Dopamine D1-class receptors selectively modulate a slowly inactivating potassium current in rat medial prefrontal cortex pyramidal neurons. J. Neurosci. 23, 2686–2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Takahashi S. X., Mittman S., Colecraft H. M. (2003) Distinctive modulatory effects of five human auxiliary β2 subunit splice variants on L-type calcium channel gating. Biophys. J. 84, 3007–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yasuda T., Lewis R. J., Adams D. J. (2004) Overexpressed Cavβ3 inhibits N-type (Cav2.2) calcium channel currents through a hyperpolarizing shift of ultra-slow and closed-state inactivation. J. Gen. Physiol. 123, 401–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peloquin J. B., Doering C. J., Rehak R., McRory J. E. (2008) Temperature dependence of Cav1.4 calcium channel gating. Neuroscience 151, 1066–1083 [DOI] [PubMed] [Google Scholar]
- 23. Barry P. H. (1994) JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial, and bilayer measurements and for correcting junction potential measurements. J. Neurosci. Methods 51, 107–116 [DOI] [PubMed] [Google Scholar]
- 24. Zacharias D. A., Violin J. D., Newton A. C., Tsien R. Y. (2002) Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296, 913–916 [DOI] [PubMed] [Google Scholar]
- 25. Tsien R. Y. (1998) The green fluorescent protein. Annu. Rev. Biochem. 67, 509–544 [DOI] [PubMed] [Google Scholar]
- 26. Erickson M. G., Alseikhan B. A., Peterson B. Z., Yue D. T. (2001) Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron 31, 973–985 [DOI] [PubMed] [Google Scholar]
- 27. Erickson M. G., Liang H., Mori M. X., Yue D. T. (2003) FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron 39, 97–107 [DOI] [PubMed] [Google Scholar]
- 28. Berney C., Danuser G. (2003) FRET or no FRET: a quantitative comparison. Biophys. J. 84, 3992–4010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schieder M., Rötzer K., Brüggemann A., Biel M., Wahl-Schott C. A. (2010) Characterization of two pore channel 2 (TPCN2)-mediated Ca2+ currents in isolated lysosomes. J. Biol. Chem. 285, 21219–21222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Corey D. P., Dubinsky J. M., Schwartz E. A. (1984) The calcium current in inner segments of rods from the salamander (Ambystoma tigrinum) retina. J. Physiol. 354, 557–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Witkovsky P., Schmitz Y., Akopian A., Krizaj D., Tranchina D. (1997) Gain of rod to horizontal cell synaptic transfer: relation to glutamate release and a dihydropyridine-sensitive calcium current. J. Neurosci. 17, 7297–7306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moriondo A., Pelucchi B., Rispoli G. (2001) Calcium-activated potassium current clamps the dark potential of vertebrate rods. Eur. J. Neurosci. 14, 19–26 [DOI] [PubMed] [Google Scholar]
- 33. Schneeweis D. M., Schnapf J. L. (2000) Noise and light adaptation in rods of the macaque monkey. Vis. Neurosci. 17, 659–666 [DOI] [PubMed] [Google Scholar]
- 34. Thoreson W. B., Rabl K., Townes-Anderson E., Heidelberger R. (2004) A highly Ca2+-sensitive pool of vesicles contributes to linearity at the rod photoreceptor ribbon synapse. Neuron 42, 595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang P. S., Alseikhan B. A., Hiel H., Grant L., Mori M. X., Yang W., Fuchs P. A., Yue D. T. (2006) Switching of Ca2+-dependent inactivation of Cav1.3 channels by calcium binding proteins of auditory hair cells J. Neurosci. 26, 10677–10689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liang H., DeMaria C. D., Erickson M. G., Mori M. X., Alseikhan B. A., Yue D. T. (2003) Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron 39, 951–960 [DOI] [PubMed] [Google Scholar]
- 37. Zhou H., Kim S. A., Kirk E. A., Tippens A. L., Sun H., Haeseleer F., Lee A. (2004) Ca2+-binding protein-1 facilitates and forms a postsynaptic complex with Cav1.2 (L-type) Ca2+ channels. J. Neurosci. 24, 4698–4708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tan G. M., Yu D., Wang J., Soong T. W. (2012) Alternative splicing at C terminus of CaV1.4 calcium channel modulates calcium-dependent inactivation, activation potential, and current density. J. Biol. Chem. 287, 832–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oz S., Tsemakhovich V., Christel C. J., Lee A., Dascal N. (2011) CaBP1 regulates voltage-dependent inactivation and activation of CaV1.2 (L-type) calcium channels. J. Biol. Chem. 286, 13945–13953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dick I. E., Tadross M. R., Liang H., Tay L. H., Yang W., Yue D. T. (2008) A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature 451, 830–834 [DOI] [PMC free article] [PubMed] [Google Scholar]