

Background: RNA-dependent protein kinase PKR regulates interferon induction.
Results: Knockdown of PKR or mutation of initiation factor eIF-2α results in increased IκB-α protein levels and decreased IFN-β induction in RNA-transfected and virus-infected cells.
Conclusion: PKR amplifies IFN-β induction through an eIF-2α translational control response.
Significance: PKR functions together with additional RNA sensors to modulate signaling leading to interferon gene induction.
Keywords: Interferon, NF-κB (NF-κB), Protein Kinase RNA (PKR), Translation Control, Virus
Abstract
The protein kinase PKR is activated by RNA with double-stranded (ds) structure and subsequently impairs translation through phosphorylation of protein synthesis initiation factor eIF-2α. PKR also mediates activation of signal transduction pathways leading to interferon beta (IFN-β) gene induction following virus-infection or RNA transfection. We previously demonstrated in measles virus-infected cells that PKR is required for the maximal induction of IFN-β gene expression by the interferon promoter stimulator gene 1 (IPS-1) adaptor-dependent cytosolic RNA sensor pathway. While both IPS-1 and PKR are important mediators of IFN-β induction, with PKR contributing to an enhanced NF-κB activation, the mechanism by which PKR enhances NF-κB activity and amplifies IFN-β induction is unresolved. Herein we tested the possibility that PKR could activate signal transduction pathways indirectly through translational control responses. Following transfection with synthetic or natural dsRNAs or infection with measles virus, we observed increased mRNA but decreased protein levels for the inhibitor of NF-κB signaling, IκB-α, that correlated with PKR activation and eIF-2α phosphorylation. Importantly, knockdown of PKR increased IκB-α protein levels and impaired IFN-β induction. Additionally, inhibition of translation by cycloheximide treatment rescued IFN-β induction following PKR knockdown but not IPS-1 knockdown. Mutation of eIF-2α to prevent phosphorylation also impaired IFN-β induction in PKR-sufficient virus-infected cells. These results suggest that an eIF-2α-dependent translation inhibition mechanism is sufficient to explain the PKR-mediated amplification of IPS-1-dependent IFN-β induction by foreign RNA.
Introduction
The innate immune system responds to a diverse array of pathogen-associated molecular patterns (PAMPS),3 one of which is foreign RNA (1–4). Among the cellular sensors of foreign RNAs are the retinoic acid-inducible protein (RIG-I)-like receptor (RLR) and Toll-like receptor (TLR) signaling networks that respond to viral RNA (4–8). RLR- and TLR-mediated responses include the activation of the type I IFN system, a cornerstone of antiviral innate immunity (3, 4, 7, 8). When the RLR and TLR signaling pathways are triggered by viral PAMPS, signaling leads to transcription factor activation and altered gene expression in infected cells. Among the genes up-regulated is interferon β (IFN-β). The IFN-β gene possesses an enhancer element to which factors including IRF3 and NF-κB bind that are necessary for maximal transcriptional activation (9, 10). Following their activation, IRF3/IRF7 and NF-κB enter the nucleus and function together with phosphorylated ATF-2/c-jun to form the IFN-β enhanceosome, thereby recruiting RNA polymerase II to drive IFN-β transcription (10).
IFN induces the expression of several genes (3), and among them is the double-stranded (ds) RNA-dependent protein kinase (PKR) (11). While inducible by type I IFN signaling, PKR also is constitutively expressed and localizes predominantly to the cytoplasm (12). PKR binds dsRNA as well as structured ssRNAs through tandem dsRNA binding domains present in its N-terminal region. RNA binding can lead to PKR dimerization and activation by autophosphorylation of residues including threonine 446 present in the C-terminal kinase catalytic domain (3, 13, 14). While the PKR protein monomer is able to bind a 15-base pair (bp) dsRNA, ∼30-bp are necessary to mediate dimerization of PKR (15), and activation is increased with dsRNA length up to a ∼85-bp that achieves maximal activation (3, 11, 16). The best characterized substrate of activated PKR remains eukaryotic translation initiation factor eIF-2α (17), which when phosphorylated on serine 51 subsequently leads to an inhibition of translation initiation (3, 18, 19).
PKR is reported to modulate the functional activity of signal transduction responses, although it is unclear whether these PKR effects are direct or indirect (19). For example, PKR has been observed to activate NF-κB signaling by activating IκB kinase or NF-κB-inducing kinase (20, 21) and to interact with TRAF family of protein members (22), however conflicting results were found with two different mouse pkr knock-out-derived cell lines regarding NF-κB activation by PKR (23). In addition to a role for PKR in activation of NF-κB signaling, PKR is implicated in the activation of proteins including the phosphatase PP2A, p53, stress-activated JNK and p38 kinases, and IRF-1 (19). The mechanism by which PKR activates these diverse pathways largely remains to be elucidated.
We have shown that PKR is required for maximal IFN-β induction in HeLa cells and that this induction of IFN-β gene expression is dependent upon the RLR interferon promoter signaling 1 (IPS-1) adaptor protein, but not the TLR3 adaptor protein TIR-domain-containing adaptor-inducing IFN-β (TRIF) protein (24). Transcriptional induction of IFN-β is known to require the activation of both IRF and NF-κB transcription factors downstream of the TLRs and RLRs (25, 26). We earlier established that while PKR is not required for IRF3 activation following virus infection or RNA transfection, PKR is required for maximal NF-κB activity (24, 27). The NF-κB family members involved in type I IFN signaling include the p65 (RelA) and p50 subunits. These proteins heterodimerize and are kept in an inactive state in the cytoplasm of cells by a family of inhibitors of NF-κB proteins, among which is IκB-α (26). NF-κB signaling is activated following the phosphorylation, ubiquitination and degradation of IκB proteins by the inhibitor of NF-κB kinase (IκK) complex (28, 29). Following degradation of the inhibitor, NF-κB subunits localize to the nucleus and bind sites present within the promoter region of NF-κB regulated genes. Among these is IκB-α, whose promoter possesses multiple NF-κB binding sites, and following NF-κB activation, IκB-α thereby negatively regulates NF-κB signaling (29, 30).
To probe the mechanism by which PKR amplifies IFN-β induction, we tested the possibility that PKR-mediated inhibition of translation triggered by foreign RNA was sufficient to enhance NF-κB activity and amplify IFN-β transcript induction. We found that the PKR-dependent inhibition of IκB-α protein expression in RNA-transfected and measles virus-infected cells correlated with increased phosphorylation of PKR and eIF-2α. This response was accompanied by enhanced expression of IκB-α and IFN-β RNAs, but reduced levels of IκB-α protein. Knockdown of PKR or expression of a serine-to-alanine mutant initiation factor eIF-2α in PKR-sufficient cells resulted in increased IκB-α protein expression and decreased IFN-β induction following RNA transfection or virus infection. Cycloheximide treatment, which inhibits translation independently of eIF-2α phosphorylation, rescued IFN-β RNA induction in PKR knockdown cells, but importantly did not rescue IFN-β induction following IPS-1 knockdown. These results suggest that PKR amplification of IFN-β RNA induction in response to foreign RNA occurs through a translational control mechanism.
EXPERIMENTAL PROCEDURES
Cells and Virus
PKR-sufficient parental (PKR+) HeLa cells and PKR-deficient HeLa cells stably knocked down for PKR expression (PKRkd), were as previously described (31, 32). Recombinant parental measles virus (MV) MVvac GFP(H) designated wild-type (WT) virus, and isogenic mutant viruses either V-deficient (Vko) or C-deficient (Cko), were generously provided by R. Cattaneo (Mayo Clinic, Rochester, MN) and were as previously described (33). The viruses were constructed based on the Moraten vaccine strain (34, 35), except that a gene encoding green fluorescent protein (GFP) was inserted downstream of the H gene.
For MV infection, PKR+ or PKRkd cells were seeded into 12-well plates 24 h prior to infection. Cell monolayers were washed once with Opti-MEM I and then infected with WT, Vko, or Cko MV at a multiplicity of infection (MOI) of five 50% tissue culture infective doses (TCID50)/cell (35, 36).
Transient Knockdown Experiments
Chemically synthesized siRNAs targeting PKR, IPS-1, and TRIF and a control siRNA targeting firefly luciferase (siLUC) were as previously described (24), and were purchased from Dharmacon. For transient knockdown experiments, a double transfection strategy with siRNA was utilized (27). HeLa cells in 60-mm dishes at ∼60% confluency were transfected with 50 nm of siRNA with Lipofectamine 2000 on day 1 and again on day 3. On day 2, the cells were reseeded in 60-mm dishes for the second siRNA transfection, and on day 4 the cells were seeded in 12-well plates for inducer RNA transfection on day 5.
Double-stranded RNAs and Transfection
Synthetic poly(rI)-poly(rC) was from Sigma. Reovirus genome dsRNA was isolated from purified reovirions as previously described (37). T7 RNA polymerase synthesized 20-bp (Pds20) and 50-bp (Pds50) dsRNAs were made and purified using the Silencer® siRNA construction kit (Ambion) according to the manufacturer's protocol. Further gel purification of the RNAs synthesized in vitro with T7 RNA polymerase was as described by Sambrook et al. (38). The sequence of the Pds20 was the same as the chemically synthesized siLUC; the sense sequence of the T7 transcribed 50-bp Pds50 included this sequence and firefly luciferase sequence 3′ of this region, as previously described (24). For RNA transfections, cells were seeded in 24- or 12-well plates and transfected with the indicated amount of RNA complexed with Lipofectamine 2000 in Opti-MEM I.
DNA Plasmids and Transfection
The expression constructs for human eIF-2α, mutated at serine 51 to either alanine (S51A) or aspartic acid (S51D) in the pMSCV-HA3iresGFP vector, were generously provided by M. Hatzoglou (Case Western University, Cleveland, Ohio). The empty vector (Vec) was generated by deletion of the eIF-2α cDNA insert from the S51D expression plasmid using EcoRI and XhoI. The eIF-2α double mutant construct S48,51A was generated by mutating serine 48 to alanine in the S51A plasmid background using the QuickChange site-directed mutagenesis strategy (Stratagene) and the following primers: forward 5′-GAAGGCATGATTCTTCTTGCTGAATTGGCCAGAAGGCG-3′ and reverse 5′-CGCCTTCTGGCCAATTCAGCAAGAAGAATCATGCCTTC-3′ (mutations are designated by underlined font). Constructs were verified by direct sequencing and restriction enzyme analysis. HeLa cells in 6-well plates were transfected with the indicated eIF-2α expression plasmid or empty vector (1.6 μg/well) using Fugene HD transfection reagent (Roche) according to the manufacturer's protocol. After 20 h the transfected cells were infected with Cko virus at a MOI of 1, maintained in DMEM containing 5% (V/V) fetal bovine serum for 24 h, and then RNA was isolated for qPCR analysis or extracts prepared for Western immunoblot analysis.
Cyclohexamide Treatment
For cycloheximide (CHX) (Sigma) treatment prior to protein or RNA isolation, cells were incubated with 10 μg/ml of CHX as indicated for 4–6 h in Opti-MEM I with or without transfection complexes. In experiments where the time to harvest was greater than 4 h, CHX containing maintenance media was placed on cells following the removal of transfection complexes at 4 h. Cells were then harvested for RNA isolation for qPCR analysis or extract preparation for Western immunoblot analysis.
Real Time PCR Analyses
Total cellular RNA was purified at the indicated time after transfection or infection using an RNeasy mini kit (Qiagen) for 24-well plates or Trizol (Invitrogen) for 12-well plates following the manufacturer's protocol. Random-primed reverse transcription was carried out using 500 ng of RNA and SuperScript II (Invitrogen) according to the manufacturer's protocol. For qPCR reactions, the GAPDH and IFN-β primer pairs were as previously described (27). qPCR reactions were performed in duplicate or triplicate with each RT template, using IQ SYBR Green Supermix (Bio-Rad) and a Bio-Rad MyIQ multicolor real-time qPCR instrument (3-min hot start followed by 30 s at 95 °C, 45 s at 58 °C, 45 s at 72 °C, for 40 to 45 cycles). IFN-β values were normalized to GADPH values.
Western Blot Analyses
Whole cell extracts were prepared with buffer containing protease inhibitors as previously described (39); for measurement of eIF-2α and PKR phosphorylation, 50 nm NaF and 2 nm Na2VO3 (31) or commercial phosphatase inhibitors (Sigma) were also present. Protein fractionation and immunoblot analysis were as described (27, 40) using the following primary antibodies: PKR and IκB-α (Santa Cruz Biotechnology); phosphorylated-PKR threonine 446 (Epitomics); eIF-2α (Cell Signaling Technology) and phosphorylated-eIF-2α serine 51 (Cell Signaling Technology or Epitomics) and α-tubulin (Sigma). Antibody against MV H protein was generously provided by Dr. R. Cattano (Mayo Clinic, Rochester, MN). Blots were visualized and quantified using a Li-Cor Odyssey infrared imager system (Li-Cor Biosciences).
RESULTS
DsRNA Length-dependent Induction of IFN-β and PKR-dependent Reduction of IκB-α
We previously showed that T7 phage RNA polymerase synthesized RNAs (PRNAs) induce IFN-β in a size-dependent manner (24) as also has been shown by others (41, 42). This size-dependent induction is illustrated in Fig. 1A, where the 50-bp dsRNA Pds50 induced IFN-β in HeLa cells as measured by qPCR, while the 20-bp dsRNA Pds20 did not induce IFN-β. The long (>100 bp) synthetic dsRNA pIpC, tested as a positive control, induced IFN-β similar to Pds50.
FIGURE 1.

dsRNA length-dependent induction of IFN-β correlates with PKR-dependent reduction in IκB-α protein and eIF-2α phosphorylation. A, relative IFN-β induction measured by quantitative PCR (qPCR) using RNA extracted 5 h following incubation with lipid alone (NT) or RNA transfection in parental PKR+ HeLa cells. IFN-β mRNA levels were normalized to GAPDH using the ΔΔCT method. Standard deviation determined from three independent experiments. B, Western blot for IκB-α, phosphorylated eIF-2α (P-eIF-2α) and total eIF-2α, and α-tubulin loading control, at 4 h and 8 h following transfection of either parental PKR+ or knockdown PKRkd HeLa cells with the indicated dsRNA. C, quantitation of IκB-α protein level normalized to α-tubulin and compared with cells not transfected (NT). Mean and standard deviation determined from two independent experiments. *, p < 0.05 compared with NT and Pds20 at the corresponding time point in PKR+ cells, **, p < 0.01 compared with NT and Pds20.
Because NF-κB is one of the transcription factors required for IFN-β induction and the activation of NF-κB and induction of IFN-β are enhanced by PKR in measles virus-infected and RNA-transfected cells (24, 27, 43) we hypothesized that differential NF-κB signaling may be responsible in part for the differences in IFN-β expression triggered by the different-sized dsRNAs. We previously observed that the inhibitor of NF-κB signaling, IκB-α, was degraded following Pds50 and pIpC transfection (24). To compare this effect with Pds20, we measured the protein levels of IκB-α following either Pds20 or Pds50 transfection in both parental PKR+ HeLa cells and the PKR stable knockdown cells. We observed that a decrease in IκB-α was only seen following transfection with Pds50 and not Pds20, and that this IκB-α decrease was PKR-dependent (Fig. 1, B and C). Furthermore, the decrease in IκB-α seen in transfected cells correlated with enhanced eIF-2α phosphorylation (Fig. 1B).
Measles Virus Cko Mutant Infection Reduces IκB-α Protein but Increases IκB-α mRNA in a PKR-dependent Manner
Using wild type (WT) measles virus or isogenic mutants lacking either the V (Vko) or C (Cko) viral accessory proteins that impair innate immune system activation (44), we previously established that the Cko virus induces the highest levels of IFN-β (27). We also observed significant PKR activation measured by Thr-446 phosphorylation, but no change in total PKR level following Cko virus infection, and we showed that maximal induction of IFN-β required PKR and IPS-1. Using gel shift and NF-κB luciferase reporter assays, we demonstrated that the measles virus Cko mutant activated NF-κB signaling, and that maximal NF-κB activation was dependent upon both PKR and IPS-1 (27).
To test whether the PKR-dependent decrease in IκB-α protein that we observed following dsRNA transfection (Fig. 1) also occurred in the context of viral infection, we examined the IκB-α level in cells infected with WT, Vko or Cko measles virus at the 24 h time point where we previously observed maximal IFN-β induction, PKR phosphorylation, and NF-κB activation (27, 33). Following infection, Cko virus-infected cells displayed a lower IκB-α protein level than uninfected cells or cells infected with either the WT or Vko virus in a PKR-dependent manner (Fig. 2A).
FIGURE 2.

PKR-dependent reduction in IκB-α protein level inversely correlates with IκB-α mRNA level following MV infection. A, Western blot for IκB-α protein following mock, WT, Vko, and Cko measles virus infection of PKR+ and PKRkd cells. IκB-α protein levels normalized to α-tubulin in each lane are expressed as a percentage of the mock infected lane. B, IκB-α mRNA transcript levels measured by qPCR at 24 h postinfection in PKR+ and PKRkd cells as described in Fig. 1. Mean and standard deviation are shown from two independent experiments analyzed in triplicate. *, p < 0.05 compared with mock treatment in PKR+ cells, **, p < 0.01.
We next examined IκB-α RNA levels by qPCR in order to determine the relationship between the levels of IκB-α protein and IκB-α mRNA, and to assess whether the reduction in IκB-α protein was because of a reduction in IκB-α RNA. While untreated and WT virus-infected cells showed no significant change in IκB-α mRNA transcript levels and the Vko virus only slightly increased IκB-α mRNA in the parental PKR+ cells, the Cko virus significantly increased IκB-α mRNA levels in a PKR-dependent manner (Fig. 2B). IκB-α protein levels were lowest in PKR+ cells following Cko virus infection even though the IκB-α mRNA level was ∼7-fold higher in the PKR+ cells than PKRkd cells. The enhanced induction of IκB-α mRNA in PKR+ cells was not unexpected, because NF-κB is known to induce IκB-α (30) and, as described above, PKR+ cells display enhanced NF-κB signaling. These results suggest that PKR either enhanced IκB-α protein degradation or the inhibition of IκB-α protein synthesis.
PKR-dependent Decrease of IκB-α Protein and Increase of IκB-α mRNA Occurs following Transfection with Naturally Occurring dsRNA
We next examined the PKR dependence of decreased IκB-α protein and the relationship between the level of IκB-α mRNA and IκB-α protein in PKR+ and PKRkd cells transfected with purified reovirus genome RNA. Reovirus genome is composed of ten segments of dsRNA of lengths varying from 1.2 to 3.8 kbp (45), and is not known to include inosine as is present in the synthetic pIpC dsRNA. Reovirus dsRNA activates PKR (37) and both the RIG-I and MDA5 cytosolic RNA signaling pathways which utilize the IPS-1 adaptor protein (42). Following reovirus RNA transfection, we found a significant reduction in IκB-α protein only in the PKR+ cells and not in the PKRkd cells (Fig. 3A). This reduction was observed between 2 and 12 h after transfection and was seen only in PKR+ cells where enhanced eIF-2α phosphorylation was observed (Fig. 3A).
FIGURE 3.
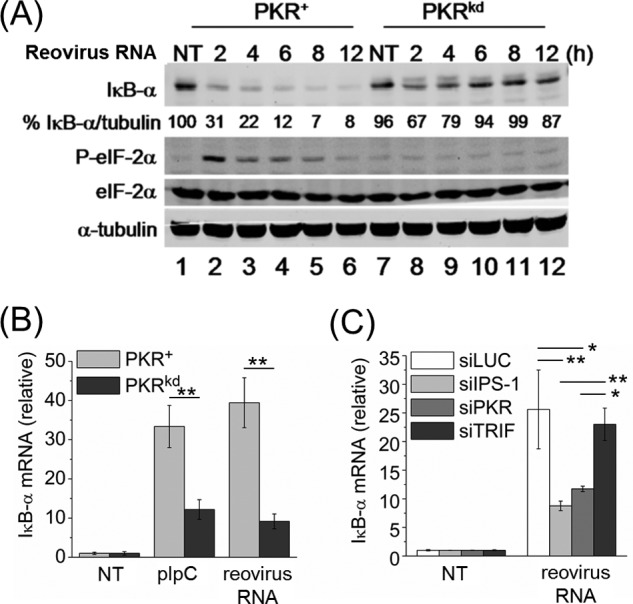
PKR-dependent reduction in IκB-α protein level and increase in IκB-α mRNA level following transfection with reovirus genome dsRNA. A, Western blot for IκB-α, phosphorylated eIF-2α (P-eIF-2α) and total eIF-2α, and α-tubulin loading control, at the indicated times after transfection of PKR+ or PKRkd cells with purified reovirus genome dsRNA. Percentage (%) indicates IκB-α protein level normalized to α-tubulin and compared with cells not transfected. B, IκB-α mRNA transcript level measured by qPCR with RNA isolated from PKR+ and PKRkd cells at 5 h following transfection with reovirus genome dsRNA or pIpC. Error bars represent the standard deviation determined from three independent experiments. C, qPCR measurement of IκB-α mRNA transcript level following transfection with reovirus genome dsRNA of HeLa cells transiently knocked down for PKR, IPS-1 or TRIF or transfected with a control siRNA (siLUC). Standard deviations determined from two independent experiments analyzed in triplicate. *, p < 0.05; **, p < 0.01.
Following transfection with either pIpC or reovirus genome dsRNA, the level of IκB-α mRNA was highest in PKR+ cells and was reduced significantly in PKRkd cells (Fig. 3B), just the opposite of what we observed for IκB-α protein levels (Fig. 3A). We next confirmed this result obtained with PKRkd cells stably deficient in PKR by using cells transiently knocked down for PKR and observed a similar reduction in IκB-α mRNA was seen following reovirus genome RNA transfection (Fig. 3C). We also transiently knocked down IPS-1 to test whether IκB-α mRNA induction was reduced, since IPS-1-dependent signaling has been shown to activate the IκK complex through the NF-κB modulator (NEMO, also known as IκK-γ) protein (46). IPS-1 knockdown but not TRIF knockdown also reduced IκB-α mRNA induction in response to transfected reovirus genome RNA (Fig. 3C), suggesting that NF-κB signaling is activated, or at least enhanced, by PKR and IPS-1-dependent responses.
Inhibition of Translation by Cycloheximide in PKRkd Cells Reduces IκB-α Protein Levels to That Seen in PKR+ Cells following dsRNA Transfection
The mechanism by which PKR enhances NF-κB activation (27) and NF-κB-dependent gene induction (Figs. 2 and 3) could involve the direct activation of NF-κB signaling components. Alternatively, an indirect translational control mechanism also may be operative in which the activity of NF-κB signaling would be affected through a PKR-dependent eIF-2α phosphorylation and subsequent inhibition of synthesis of negative regulatory proteins such as IκB-α. For the latter explanation, IκB-α protein degradation could be initially similar in PKR+ and PKRkd cells (47), but subsequent synthesis of IκB-α protein would be reduced in PKR+ cells due to an inhibition of IκB-α mRNA translation compared with that in PKRkd cells. The observed disparity between IκB-α mRNA and protein levels (Figs. 2 and 3) is consistent with the notion that PKR may enhance activation of NF-κB signaling through a translational control mechanism.
To test the possibility that PKR decreases IκB-α protein through translational control thereby leading to the increase seen in IκB-α mRNA (Fig. 3), we treated PKRkd cells with cycloheximide (CHX), a drug that prevents the elongation step of translation (48), and then examined IκB-α protein levels. In PKRkd cells, CHX treatment significantly decreased IκB-α protein levels (Fig. 4, lanes 11 and 12) to a level comparable to that of PKR+ cells, suggesting that PKR indeed might contribute to NF-κB induction solely by preventing the translation of IκB-α mRNA (47). No changes in eIF-2α phosphorylation were observed in PKRkd cells following CHX treatment.
FIGURE 4.
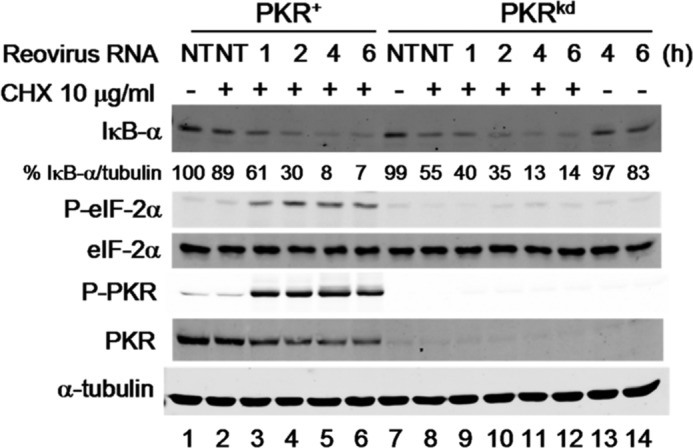
Translation inhibition by cycloheximide prevents IκB-α protein expression in PKRkd cells. PKR+ and PKRkd cells were transfected with reovirus genome dsRNA for the indicated period of time (h) or not transfected (NT), and were treated (+) with cycloheximide (CHX) or left untreated (−) as indicated. Western immunoblot analyses were performed for IκB-α, phosphorylated eIF-2α (P-eIF-2α), total eIF-2α, phosphorylated PKR (P-PKR), and total PKR, and α-tubulin as a loading control. Percentage (%) is calculated for IκB-α protein level normalized to α-tubulin, and compared with PKR+ cells not treated with CHX and not transfected. Extracts from PKRkd cells, transfected with reovirus genome dsRNA but not treated with CHX, are shown in lanes 13 and 14 for comparison.
Cycloheximide Treatment Rescues IFN-β RNA Induction Following PKR-knockdown, but Does Not Rescue Following IPS-1 Knockdown
Most importantly, CHX treatment rescued IFN-β RNA induction in PKRkd cells to a significant extent (p < 0.01) in response to transfected synthetic (pIpC) or natural (reovirus genome) dsRNA (Fig. 5A), indicating that inhibition of synthesis of inhibitors of NF-κB signaling was a likely explanation for the PKR dependence of IFN-β induction. CHX treatment also restored IκB-α RNA induction in PKRkd cells to levels found in PKR+ cells (Fig. 5B).
FIGURE 5.

Translation inhibition by cycloheximide rescues PKR-dependent but not IPS-1-dependent induction of IFN-β transcripts. A, RNA was isolated from PKR+ or PKRkd cells at 5 h after transfection with pIpC or reovirus genome dsRNA in the presence or absence of CHX as indicated and the relative IFN-β mRNA transcript level was determined by qPCR as described in Fig. 1. The standard deviation was determined from two or three independent experiments. B, qPCR measurement of relative IκB-α mRNA transcript levels following treatment conditions described in A. C, relative IFN-β mRNA transcript level in parental PKR+ HeLa cells transiently knocked down for either PKR or IPS-1, or transfected with a control siLUC siRNA prior to transfection of reovirus genome dsRNA in the absence or presence of CHX. Standard deviation is shown for two independent experiments. *, p < 0.05; **, p < 0.01.
While our results are consistent with an amplification of IFN-β RNA induction by PKR-mediated translation inhibition of IκB-α protein synthesis, they do not clearly differentiate between the IPS-1 and PKR contributions to IFN-β signaling. To test whether CHX treatment specifically rescues PKR-dependent but not IPS-1-dependent induction of IFN-β transcripts, we transiently knocked down either PKR or IPS-1 in parental HeLa cells. As we earlier reported (24), transient knockdown of either IPS-1 or PKR significantly reduced IFN-β induction (Fig. 5C). Following CHX treatment, however, IFN-β RNA induction increased to near control levels in the PKR transient knockdown cells, but remained significantly reduced in the IPS-1 knockdown cells (Fig. 5C). These results suggest that the translation inhibition-dependent contribution of PKR to IFN-β induction is independent of the activation of signaling by IPS-1.
Non-phosphorylatable Mutant eIF-2α Reduces IFN-β RNA Induction and Increases IκB-α Protein Levels Following Infection of PKR-sufficient Cells
To further test whether the PKR-dependent enhancement of IFN-β transcript induction was translationally mediated through eIF-2α, the effect of S48,51A, an unphosphorylatable mutant form of eIF-2α in which the serine phosphorylation sites were replaced with alanine, was examined. As shown in Fig. 6, the phosphorylation defective S48,51A mutant resulted in lower levels of both IFN-β transcript (Fig. 6A) and IκB-α transcript (Fig. 6B) following Cko measles virus infection compared with either vector-transfected cells or cells expressing the phospho-mimetic S51D mutant. IFN-β transcript levels in the absence of the Cko infection were comparably low in the vector, S51D and S48,51A-transfected cells (Fig. 6).
FIGURE 6.
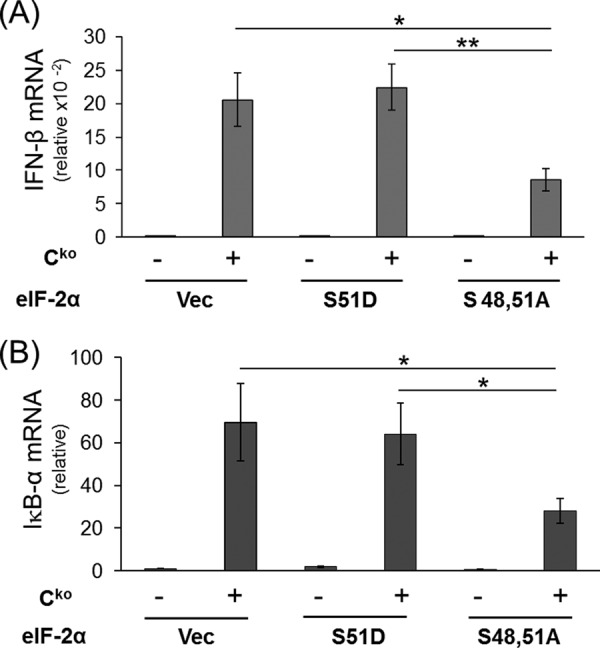
Expression of mutant eIF-2α (S48,51A) in PKR+ cells decreases IFN-β induction by measles virus. Cells (PKR+) were transfected with either an empty vector (Vec) or an expression construct encoding human eIF-2α mutated to prevent phosphorylation (S48,51A) or a phospho-mimetic (S51D) mutant. At 20 h after transfection, cells were either left uninfected (−) or infected (+) with MV Cko virus, and at 24 h after infection total RNA was prepared and transcript levels determined by qPCR. The results shown are normalized to GAPDH transcript level and are relative to uninfected, vector-transfected cells. The mean induction and standard deviation were determined from three independent experiments. *, p < 0.05; **, p < 0.01. A, IFN-β transcript levels; B, IκB-α transcript levels.
Expression of the S48,51A phosphorylation defective mutant form of eIF-2α in PKR+ parental cells also led to enhanced IκB-α protein production in Cko virus-infected cells (Fig. 7A, lane 6), reaching a level of IκB-α comparable to that seen in Cko-infected PKRkd cells (Fig. 7A, lane 8). By contrast, the level of IκB-α in Cko-infected vector- or S51D-transfected cells was 2–3-fold lower (Fig. 7B). Interestingly, PKR+ cells expressing the phosphorylation defective S48,51A mutant also gave rise to increased viral H protein synthesis, as was observed in PKRkd cells, compared with the vector control or S51D-expressing cells (Fig. 7A).
FIGURE 7.
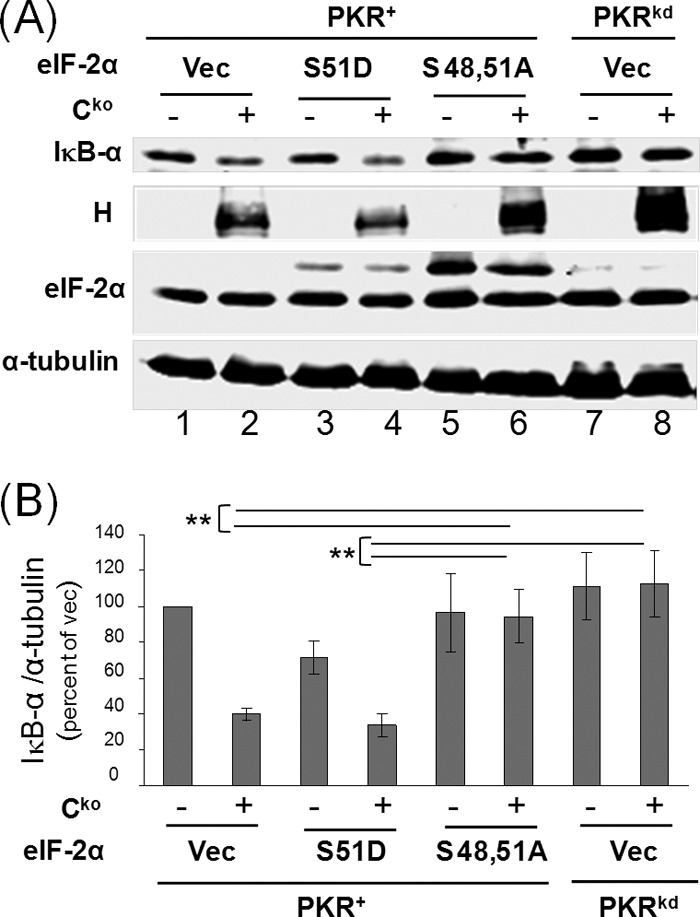
Expression of mutant eIF-2α (S48,51A) increases IκB-α protein levels in Cko measles virus infected PKR+ cells to levels seen in PKRkd-infected cells. A, PKR+ or PKRkd cells were transfected with either an empty vector (Vec) or an expression construct encoding either S48,51A or S51D mutant eIF-2α as indicated. At 20 h after transfection cells were either left uninfected (−) or infected (+) with MV Cko virus, and at 24 h after infection extracts were prepared and western immunoblot analyses performed using antibodies against IκB-α, MV H protein and eIF-2α, and α-tubulin as a loading control. B, quantitation of IκB-α protein levels normalized to α-tubulin; average and standard deviation determined from three independent analyses; **, p < 0.01.
DISCUSSION
PKR has emerged as an important modulator of IFN-β gene induction following infection with RNA viruses, including negative-stranded (27, 43), positive-stranded (49–53) and double-stranded (54) RNA viruses. But, the molecular mechanism by which PKR affects IFN-β induction largely remains unresolved. The objective of our study was to determine the mechanistic basis by which the PKR kinase enhances IFN-β transcript RNA levels following virus infection and RNA transfection. The results reported herein suggest that a translational control mechanism through eIF-2α is likely sufficient to account for the PKR-dependent reduction in IκB-α levels and enhancement of IFN-β induction. In fact, translational control may be broadly important and operative in multiple pathways of IFN signaling as the translational repressors 4E-BP1 and 4E-BP2 also have been shown to negatively regulate the type-I IFN response by inhibiting IRF7 mRNA translation during viral infection (55).
Impaired IFN-β transcript induction in HeLa cells stably knocked down for PKR is known following dsRNA transfection (24) or measles virus infection (27, 43). Using a transient knockdown strategy we earlier had established that, in addition to PKR, the IPS-1 adaptor protein for the RLR cytosolic signaling pathway was required for maximal IFN-β induction by measles virus deficient in C protein, but that the TRIF mediator of TLR3 signaling was not involved. Because both PKR and IPS-1 were necessary for maximal IFN-β transcript induction in response to either dsRNA transfection or MV infection (24, 27), we attempted to further delineate and distinguish the requirements for these proteins. Short structured RNAs without a 5′-triphosphate are known to be weak inducers of IFN-β (42), but they still activate PKR (24, 37), suggesting that PKR activation is not sufficient for IFN-β induction. Although the activation of IRF3 is PKR-independent in both RNA transfected and measles virus infected HeLa cells, the activation of NF-κB is enhanced by PKR (27, 43).
Negative regulators of NF-κB signaling, exemplified by IκB-α, are known to be degraded in response to an NF-κB-activating stimuli, and then resynthesized to subsequently inhibit NF-κB activity (29, 47). Hence, we considered that translation inhibition mediated by PKR might lead to enhanced NF-κB signaling and IFN-β induction. As shown herein, an inverse correlation was found between IκB-α mRNA and protein levels following dsRNA transfection and virus infection. Furthermore, consistent with previous results (24), we observed an inverse correlation between eIF-2α phosphorylation and IκB-α protein levels following these treatments. The steady-state protein expression level of IκB-α was increased and largely rescued when the S49,51A mutant of eIF-2α was expressed, which suggests that the resynthesis rather than the stability of IκB-α is the key parameter affected by PKR activation.
As a further test of the translational basis of the PKR-dependent effect, we used a pharmacologic inhibitor, CHX, to inhibit protein synthesis. Importantly, CHX treatment during RNA transfection rescued PKR-dependent, but not IPS-1-dependent, IFN-β transcriptional induction. This finding, together with our observation that the transient knockdown of either PKR or IPS-1 impaired IFN-β transcript induction, suggests that PKR and IPS-1 differentially function in the signaling response leading to activation of IFN-β transcription.
Our results furthermore demonstrate that the effect of PKR on signaling pathway responses following RNA transfection or virus infection is dependent on PKR enzymatic activity through its eIF-2α kinase activity (40, 56) and hence cannot represent simply a protein adaptor or scaffold role for PKR (22, 57). The observed PKR-dependent decrease in IκB-α following infection or transfection is consistent with results obtained for the endoplasmic reticulum stress-responsive eIF-2α kinase, PERK, where maximal NF-κB signaling following phosphorylation of eIF-2α by PERK likewise correlated with decreased protein levels of IκB-α (56).
Our results taken together support the role of translation control of negative regulators, including IκB-α, in the modulation of NF-κB signaling. Similar to the effects of PKR on IκB-α expression established herein, it is conceivable that other negative regulators of the NF-κB pathway in addition to IκB-α also are modulated in a PKR-dependent manner. One intriguing possibility is the A20 negative regulatory protein, which prevents NF-κB essential modulator (NEMO) activation through its ubiquitin-editing activity (58). Because A20 is induced by NF-κB signaling in a manner similar to IκB-α resulting in inhibition of NF-κB signaling upstream of IκB-α (59), it is reasonable to speculate that A20 would represent an additional potential target for PKR-dependent translational control of NF-κB signaling. Since A20 regulates NEMO activity, and NEMO activity is required for the activation of IRF3 through TBK1 and IκK (60), then PKR-dependent inhibition of A20 translation presumably could also affect IRF activation in some instances, perhaps explaining the PKR-dependent activation of IRF3 observed following mutant vaccinia virus infection (32). In fact, the absence of negative regulators of NF-κB signaling may also explain the PKR-dependence of IκB phosphorylation and activation of IκK shown by others (20, 21). It is also possible that cell type differences in NF-κB signaling, especially in cancer cells (61), may account for the seemingly different effects of PKR on this pathway.
Our results are consistent with a translational inhibition response through eIF-2α phosphorylation for the mechanistic basis of the PKR-dependent amplification of IFN-β expression following transfection or infection (40, 56). First, in PKR-sufficient PKR+ cells as shown herein, a mutant form of eIF-2α that cannot be phosphorylated impaired measles virus-induced activation of IFN-β gene expression and enhanced IκB-α protein production following infection. Secondly, in PKR-deficient cells, catalytically active PKR is necessary in order to complement measles virus-induced PKR-dependent IFN-β transcript induction (40). Our findings, taken together, reveal that PKR functions at least in part through an eIF-2-mediated translational control mechanism to indirectly enhance IFN-β transcript levels, and that IPS-1 functions through direct activation of signaling pathways including initial NF-κB and IRF3 activation as established by others (5).
This work was supported, in whole or in part, by Research Grants AI-12520 and AI-20611 from the NIAID, National Institutes of Health, U.S. Public Health Service.
- PAMPS
- pathogen-associated molecular patterns
- dsRNA
- double-stranded RNA
- Cko
- C knock-out measles virus
- eIF-2α
- eukaryote translation initiation factor-2α
- IFN
- interferon
- MV
- measles virus
- PKR
- RNA-dependent protein kinase
- PKRkd
- PKR knockdown cells
- RLR
- retinoic acid-inducible protein-like receptor
- TLR
- Toll-like receptor.
REFERENCES
- 1. Lampson G. P., Tytell A. A., Field A. K., Nemes M. M., Hilleman M. R. (1967) Inducers of interferon and host resistance. I. Double-stranded RNA from extracts of Penicillium funiculosum. Proc. Natl. Acad. Sci. U.S.A. 58, 782–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Akira S., Takeda K. (2004) Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511 [DOI] [PubMed] [Google Scholar]
- 3. Samuel C. E. (2001) Antiviral actions of interferons. Clin. Microbiol. Rev. 14, 778–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meylan E., Tschopp J. (2006) Toll-like receptors and RNA helicases: Two parallel ways to trigger antiviral responses. Mol. Cell 22, 561–569 [DOI] [PubMed] [Google Scholar]
- 5. Kawai T., Takahashi K., Sato S., Coban C., Kumar H., Kato H., Ishii K. J., Takeuchi O., Akira S. (2005) IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6, 981–988 [DOI] [PubMed] [Google Scholar]
- 6. Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K., Uematsu S., Jung A., Kawai T., Ishii K. J., Yamaguchi O., Otsu K., Tsujimura T., Koh C. S., Reis e Sousa C., Matsuura Y., Fujita T., Akira S. (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105 [DOI] [PubMed] [Google Scholar]
- 7. Uematsu S., Akira S. (2007) Toll-like receptors and type I interferons. J. Biol. Chem. 282, 15319–15323 [DOI] [PubMed] [Google Scholar]
- 8. Yoneyama M., Fujita T. (2010) Recognition of viral nucleic acids in innate immunity. Rev. Med. Virol. 20, 4–22 [DOI] [PubMed] [Google Scholar]
- 9. Wathelet M. G., Lin C. H., Parekh B. S., Ronco L. V., Howley P. M., Maniatis T. (1998) Virus infection induces the assembly of coordinately activated transcription factors on the IFN-β enhancer in vivo. Mol. Cell 1, 507–518 [DOI] [PubMed] [Google Scholar]
- 10. Panne D., Maniatis T., Harrison S. C. (2007) An atomic model of the interferon-β enhanceosome. Cell 129, 1111–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Toth A. M., Zhang P., Das S., George C. X., Samuel C. E. (2006) in Prog. Nucleic Acids Res. Mol. Biol. (Moldave K., ed), pp. 369–434, volume 81 Ed., Academic Press; [DOI] [PubMed] [Google Scholar]
- 12. Thomis D. C., Doohan J. P., Samuel C. E. (1992) Mechanism of interferon action: cDNA structure, expression, and regulation of the interferon-induced, RNA-dependent P1/eIF-2α protein kinase from human cells. Virology 188, 33–46 [DOI] [PubMed] [Google Scholar]
- 13. McCormack S. J., Thomis D. C., Samuel C. E. (1992) Mechanism of interferon action: Identification of a RNA binding domain within the N-terminal region of the human RNA-dependent P1/eIF-2α protein kinase. Virology 188, 47–56 [DOI] [PubMed] [Google Scholar]
- 14. Bevilacqua P. C., George C. X., Samuel C. E., Cech T. R. (1998) Binding of the protein kinase PKR to RNAs with secondary structure defects: role of the tandem A-G mismatch and noncontiguous helixes. Biochemistry 37, 6303–6316 [DOI] [PubMed] [Google Scholar]
- 15. Heinicke L. A., Wong C. J., Lary J., Nallagatla S. R., Diegelman-Parente A., Zheng X., Cole J. L., Bevilacqua P. C. (2009) RNA dimerization promotes PKR dimerization and activation. J. Mol. Biol. 390, 319–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Manche L., Green S. R., Schmedt C., Mathews M. B. (1992) Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol. Cell Biol. 12, 5238–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Samuel C. E. (1979) Mechanism of interferon action: Phosphorylation of protein synthesis initiation factor eIF-2 in interferon-treated human cells by a ribosome-associated kinase processing site specificity similar to hemin-regulated rabbit reticulocyte kinase. Proc. Natl. Acad. Sci. U.S.A. 76, 600–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. García M. A., Gil J., Ventoso I., Guerra S., Domingo E., Rivas C., Esteban M. (2006) Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 70, 1032–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sadler A. J., Williams B. R. (2007) Structure and function of the protein kinase R. Curr. Top. Microbiol. Immunol. 316, 253–292 [DOI] [PubMed] [Google Scholar]
- 20. Zamanian-Daryoush M., Mogensen T. H., DiDonato J. A., Williams B. R. (2000) NF-κB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-κB-inducing kinase and IκB kinase. Mol. Cell Biol. 20, 1278–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kumar A., Haque J., Lacoste J., Hiscott J., Williams B. R. (1994) Double-stranded RNA-dependent protein kinase activates transcription factor NF-κB by phosphorylating I κB. Proc. Natl. Acad. Sci. U.S.A. 91, 6288–6292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gil J., García M. A., Gomez-Puertas P., Guerra S., Rullas J., Nakano H., Alcamí J., Esteban M. (2004) TRAF family proteins link PKR with NF-κB activation. Mol. Cell Biol. 24, 4502–4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baltzis D., Li S., Koromilas A. E. (2002) Functional characterization of PKR gene products expressed in cells from mice with a targeted deletion of the N terminus or C terminus domain of PKR. J. Biol. Chem. 277, 38364–38372 [DOI] [PubMed] [Google Scholar]
- 24. McAllister C. S., Samuel C. E. (2009) The RNA-activated protein kinase enhances the induction of interferon-β and apoptosis mediated by cytoplasmic RNA sensors. J. Biol. Chem. 284, 1644–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thompson A. J., Locarnini S. A. (2007) Toll-like receptors, RIG-I-like RNA helicases and the antiviral innate immune response. Immunol. Cell Biol. 85, 435–445 [DOI] [PubMed] [Google Scholar]
- 26. Vallabhapurapu S., Karin M. (2009) Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 27, 693–733 [DOI] [PubMed] [Google Scholar]
- 27. McAllister C. S., Toth A. M., Zhang P., Devaux P., Cattaneo R., Samuel C. E. (2010) Mechanisms of protein kinase PKR-mediated amplification of beta interferon induction by C protein-deficient measles virus. J. Virol. 84, 380–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alkalay I., Yaron A., Hatzubai A., Orian A., Ciechanover A., Ben-Neriah Y. (1995) Stimulation-dependent IκBα phosphorylation marks the NF-κB inhibitor for degradation via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. U.S.A. 7, 10599–10603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hayden M. S., Ghosh S. (2012) NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 26, 203–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sun S. C., Ganchi P. A., Ballard D. W., Greene W. C. (1993) NF-κB controls expression of inhibitor IκBα: evidence for an inducible autoregulatory pathway. Science 259, 1912–1915 [DOI] [PubMed] [Google Scholar]
- 31. Zhang P., Samuel C. E. (2007) Protein kinase PKR plays a stimulus- and virus-dependent role in apoptotic death and virus multiplication in human cells. J. Virol. 81, 8192–8200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang P., Samuel C. E. (2008) Induction of protein kinase PKR-dependent activation of interferon regulatory factor 3 by vaccinia virus occurs through adapter IPS-1 signaling. J. Biol. Chem. 283, 34580–34587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Toth A. M., Devaux P., Cattaneo R., Samuel C. E. (2009) Protein kinase PKR mediates the apoptosis induction and growth restriction phenotypes of C protein-deficient measles virus. J. Virol. 83, 961–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Devaux P., von Messling V., Songsungthong W., Springfeld C., Cattaneo R. (2007) Tyrosine 110 in the measles virus phosphoprotein is required to block STAT1 phosphorylation. Virology 360, 72–83 [DOI] [PubMed] [Google Scholar]
- 35. Devaux P., Hodge G., McChesney M. B., Cattaneo R. (2008) Attenuation of V- or C-defective measles viruses: Infection control by the inflammatory and interferon responses of rhesus monkeys. J. Virol. 82, 5359–5367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kärber G. (1931) Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Naunyn Schmiedebergs Arch. Pharmacol. 162, 480–483 [Google Scholar]
- 37. Bischoff J. R., Samuel C. E. (1989) Mechanism of interferon action. Activation of the human P1/eIF-2α protein kinase by individual reovirus s-class mRNAs: s1 mRNA is a potent activator relative to s4 mRNA. Virology 172, 106–115 [DOI] [PubMed] [Google Scholar]
- 38. Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, New York [Google Scholar]
- 39. Das S., Ward S. V., Tacke R. S., Suske G., Samuel C. E. (2006) Activation of the RNA-dependent protein kinase PKR promoter in the absence of interferon is dependent upon Sp proteins. J. Biol. Chem. 281, 3244–3253 [DOI] [PubMed] [Google Scholar]
- 40. Taghavi N., Samuel C. E. (2012) Protein kinase PKR catalytic activity is required for the PKR-dependent activation of mitogen-activated protein kinases and amplification of interferon beta induction following virus infection. Virology 427, 208–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Saito T., Gale M., Jr. (2008) Differential recognition of double-stranded RNA by RIG-I-like receptors in antiviral immunity. J. Exp. Med. 205, 1523–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kato H., Takeuchi O., Mikamo-Satoh E., Hirai R., Kawai T., Matsushita K., Hiiragi A., Dermody T. S., Fujita T., Akira S. (2008) Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 205, 1601–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li Z., Okonski K. M., Samuel C. E. (2012) Adenosine deaminase acting on RNA 1 (ADAR1) suppresses the induction of interferon by measles virus. J. Virol. 86, 3787–3794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Randall R. E., Goodbourn S. (2008) Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89, 1–47 [DOI] [PubMed] [Google Scholar]
- 45. Cashdollar L. W., Chmelo R., Esparza J., Hudson G. R., Joklik W. K. (1984) Molecular cloning of the complete genome of reovirus serotype 3. Virology 133, 191–196 [DOI] [PubMed] [Google Scholar]
- 46. Zhao T., Yang L., Sun Q., Arguello M., Ballard D. W., Hiscott J., Lin R. (2007) The NEMO adaptor bridges the nuclear factor-κB and interferon regulatory factor signaling pathways. Nat. Immunol. 8, 592–600 [DOI] [PubMed] [Google Scholar]
- 47. Hoffmann A., Levchenko A., Scott M. L., Baltimore D. (2002) The IκB-NF-κB signaling module: temporal control and selective gene activation. Science 298, 1241–1245 [DOI] [PubMed] [Google Scholar]
- 48. Schneider-Poetsch T., Ju J., Eyler D. E., Dang Y., Bhat S., Merrick W. C., Green R., Shen B., Liu J. O. (2010) Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol. 6, 209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Arnaud N., Dabo S., Maillard P., Budkowska A., Kalliampakou K. I., Mavromara P., Garcin D., Hugon J., Gatignol A., Akazawa D., Wakita T., Meurs E. F. (2010) Hepatitis C virus controls interferon production through PKR activation. PLoS ONE 5, e10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gilfoy F. D., Mason P. W. (2007) West Nile virus-induced interferon production is mediated by the double-stranded RNA-dependent protein kinase PKR. J. Virol. 81, 11148–11158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Garaigorta U., Chisari F. V. (2009) Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe. 6, 513–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Der S. D., Lau A. S. (1995) Involvement of the double-stranded-RNA-dependent kinase PKR in interferon expression and interferon-mediated antiviral activity. Proc. Natl. Acad. Sci. U.S.A. 92, 8841–8845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schulz O., Pichlmair A., Rehwinkel J., Rogers N. C., Scheuner D., Kato H., Takeuchi O., Akira S., Kaufman R. J., Reis e Sousa C. (2010) Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe. 7, 354–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sen A., Pruijssers A. J., Dermody T. S., García-Sastre A., Greenberg H. B. (2011) The early interferon response to rotavirus is regulated by PKR and depends on MAVS/IPS-1, RIG-I, MDA-5, and IRF3. J. Virol. 85, 3717–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Colina R., Costa-Mattioli M., Dowling R. J., Jaramillo M., Tai L. H., Breitbach C. J., Martineau Y., Larsson O., Rong L., Svitkin Y. V., Makrigiannis A. P., Bell J. C., Sonenberg N. (2008) Translational control of the innate immune response through IRF-7. Nature 452, 323–328 [DOI] [PubMed] [Google Scholar]
- 56. Deng J., Lu P. D., Zhang Y., Scheuner D., Kaufman R. J., Sonenberg N., Harding H. P., Ron D. (2004) Translational repression mediates activation of nuclear factor κB by phosphorylated translation initiation factor 2. Mol. Cell Biol. 24, 10161–10168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bonnet M. C., Daurat C., Ottone C., Meurs E. F. (2006) The N-terminus of PKR is responsible for the activation of the NF-κB signaling pathway by interacting with the IKK complex. Cell Signal. 18, 1865–1875 [DOI] [PubMed] [Google Scholar]
- 58. Coornaert B., Carpentier I., Beyaert R. (2009) A20: Central gatekeeper in inflammation and immunity. J. Biol. Chem. 284, 8217–8221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Werner S. L., Kearns J. D., Zadorozhnaya V., Lynch C., O'Dea E., Boldin M. P., Ma A., Baltimore D., Hoffmann A. (2008) Encoding NF-κB temporal control in response to TNF: distinct roles for the negative regulators IκBα and A20. Genes Dev. 22, 2093–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Saitoh T., Yamamoto M., Miyagishi M., Taira K., Nakanishi M., Fujita T., Akira S., Yamamoto N., Yamaoka S. (2005) A20 is a negative regulator of IFN regulatory factor 3 signaling. J. Immunol. 174, 1507–1512 [DOI] [PubMed] [Google Scholar]
- 61. Naugler W. E., Karin M. (2008) NF-κB and cancer-identifying targets and mechanisms. Curr. Opin. Genet. Dev. 18, 19–26 [DOI] [PMC free article] [PubMed] [Google Scholar]