

Background: The role of TRIM56 in innate antiviral immunity is unclear.
Results: Knockdown of TRIM56 impairs TLR3-dependent IRF3 activation, interferon-stimulated gene expression and induction of antiviral responses, while overexpression of TRIM56 enhances TLR3-mediated innate immune responses.
Conclusion: TRIM56 interacts with TRIF and promotes TLR3 signaling.
Significance: This study identifies TRIM56 as an essential component of the TLR3 antiviral signaling pathway.
Keywords: Double-stranded RNA, Innate Immunity, Interferon, Toll-like Receptors (TLR), TRIF, IRF3, TLR3, TRIM56, Interferon-stimulated Gene, Virus
Abstract
Members of the tripartite motif (TRIM) proteins are being recognized as important regulators of host innate immunity. However, specific TRIMs that contribute to TLR3-mediated antiviral defense have not been identified. We show here that TRIM56 is a positive regulator of TLR3 signaling. Overexpression of TRIM56 substantially potentiated extracellular dsRNA-induced expression of interferon (IFN)-β and interferon-stimulated genes (ISGs), while knockdown of TRIM56 greatly impaired activation of IRF3, induction of IFN-β and ISGs, and establishment of an antiviral state by TLR3 ligand and severely compromised TLR3-mediated chemokine induction following infection by hepatitis C virus. The ability to promote TLR3 signaling was independent of the E3 ubiquitin ligase activity of TRIM56. Rather, it correlated with a physical interaction between TRIM56 and TRIF. Deletion of the C-terminal portion of TRIM56 abrogated the TRIM56-TRIF interaction as well as the augmentation of TLR3-mediated IFN response. Together, our data demonstrate TRIM56 is an essential component of the TLR3 antiviral signaling pathway and reveal a novel role for TRIM56 in innate antiviral immunity.
Introduction
Host cells are equipped with elaborate intrinsic mechanisms, known as the innate immune response, to combat invading microbial pathogens. Upon infection by viruses, the innate immune system launches an immediate defense program that is characterized by the rapid induction of antiviral cytokines/chemokines including type I interferons (IFN-β and IFN-α).4 These soluble factors act in an autocrine/paracrine fashion to induce hundreds of IFN-stimulated genes (ISGs), thereby establishing an antiviral state that curbs viral replication and dissemination. IFNs and cytokines/chemokines also regulate the activation and/or trafficking of immune cells to the infection sites, shaping the development of antigen-specific adaptive immunity (1, 2).
Induction of the IFN response is initiated upon cellular sensing of pathogen-associated molecular patterns (PAMPs) from intruding viruses by the so-called pattern recognition receptors (PRRs). Viral nucleic acids, most notably dsRNAs, are sensed by and engaged with at least two classes of PRRs, i.e. Toll-like receptor-3 (TLR3) and retinoic-inducible gene-I (RIG-I)-like receptors (RLRs, RIG-I, and MDA5), in endolysosomes and cytoplasm, respectively (3, 4). These PRRs recruit their cognate adaptor proteins (TRIF for TLR3, while MAVS for the RLRs) (5,6), triggering signaling pathways converging on a common set of IKK-related kinases (TBK1 and IKKϵ)(7,8) with the assistance of several shared scaffold proteins such as tumor necrosis factor receptor-associated factor 3 (TRAF3)(9,10) and NF-κB essential modulator (NEMO) (11). Activated TBK1 or IKKϵ directly phosphorylate serine residues in the C-terminal region of a latent transcription factor, IFN regulatory factor-3 (IRF3), resulting in IRF3 nuclear translocation and transcriptional activation of promoters for IFN-β and a subset of ISGs (12). Because the early virus-host interplay may be decisive in determining the outcome of viral infections, defining the mechanisms of innate immune signaling will provide useful clues that inform novel approaches to antiviral therapy.
The tripartite motif (TRIM) protein family comprises over 70 members that are implicated in a wide array of cellular processes, including cell growth, differentiation, development, apoptosis, and immunity, etc. (13, 14). Characterized by their N-terminal signature tripartite motif, i.e. RING, B-Box, and coiled-coil domains (14, 15), the TRIMs constitute a new class of single RING-finger E3 ubiquitin ligases that promote post-translational modifications of various substrates, including themselves (16). The TRIM proteins also multimerize by means of self-association via the coiled-coil domains, acting as scaffolds for assembly of multi-protein complexes that identify subcellular compartments (17). Over the past decade, much attention has been garnered in exploring the role of TRIM proteins in innate immunity to viral infections, in particular, against retroviruses (18). More recently, a subset of TRIMs were shown to be involved in orchestrating broader immune responses by regulating innate signaling pathways downstream of the PRRs (19). However, among this group of TRIMs most have been found to behave as negative regulators (such as TRIM21 (20), TRIM30α (21), TRIM27 (22), TRIM28 (23), and TRIM38 (24) etc), and only a handful of TRIMs have been reported to promote PRR signaling. As a prime example for the positively regulatory TRIMs, TRIM25 catalyzes ubiquitination of the caspase recruitment domain of RIG-I, thereby promoting the interaction of RIG-I with its adaptor MAVS, culminating in IRF3 activation and subsequent IFN induction (25).
We recently demonstrated that TRIM56 is an IFN- and virus-inducible E3 ubiquitin ligase which acts to restrict replication of bovine viral diarrhea virus (BVDV), an economically important positive-strand RNA virus and that both the E3 ubiquitin ligase activity and the C-terminal structural integrity is crucial for TRIM56's antiviral activity (26). Others found that TRIM56 associates with and promotes the ubiquitination of stimulator of IFN genes (STING), an innate immune signaling adaptor residing in the endoplasmic reticulum (ER), thereby enhancing cytosolic dsDNA-induced IFN production (27). However, whether TRIM56 participates in innate immune signaling pathways that recognize RNA viruses remains unclear. In this study, we provide molecular and virological evidence that TRIM56 is an essential component of the TLR3 signaling pathway, revealing a novel role of TRIM56 in antiviral innate immunity.
EXPERIMENTAL PROCEDURES
Plasmids
Conventional PCR and mutagenesis techniques were used to construct N-terminal 2×HA-tagged, wild-type (WT) and various mutant forms of TRIM56-encoding plasmids in the pcDNA5/FRT/TO backbone (Invitrogen). pFlag-TRIF-mRHIM contained a cDNA insert encoding a Flag-tagged, constitutively active form of human TRIF (here referred to as Flag-TRIFSA), in which the C-terminal 4-amino acid RIP homotypic interaction motif (RHIM) has been mutated to four alanines (28). When overexpressed, Flag-TRIFSA strongly activates IRF3, but it no longer induces apoptosis as does WT TRIF (28). pCX4bsr-Flag-TLR3 has been described (29). pCX4pur-FlagACE2 were constructed by inserting a cDNA fragment encoding Flag-tagged human angiotensin-converting enzyme-2 (ACE2), the receptor for SARS coronavirus, into the pCX4pur vector.
Cells
HeLa, Human embryo kidney (HEK) 293, 293-TLR3 (HEK293 cells stably expressing Flag-tagged human TLR3) were cultured using conventional techniques. Human hepatoma Huh7.5 cells stably reconstituted for TLR3 expression (7.5-TLR3 cells) were cultured as described (29).
We adopted the Flp-IN T-Rex expression system (Invitrogen) to create isogenic, stable HeLa cell lines exhibiting tet-inducible expression of WT or mutant HA-TRIM56 by a Saccharomyces cerevisiae-derived Flp recombinase-dependent DNA recombination mechanism. HeLa Flp-IN T-Rex cells (30) were cultured in DMEM supplemented with 4 μg/ml of blasicidin and 50 μg/ml of Zeocin. Initially aimed at creating a cell system to study host cell-SARS coronavirus interactions, HeLa Flp-IN T-Rex cells were stably transduced with a replication incompetent retrovirus encoding human ACE2 (packaged from pCX4pur-FlagACE2), followed by selection with puromycin. The resultant cell line was designated as HeLa-FitA2, which retained all features of HeLa Flp-IN T-Rex cells (including the intact innate immune signaling pathways) and additionally became permissive for SARS coronavirus infection (data not shown). We then co-transfected HeLa-FitA2 cells with pOG44 (encoding the Flp recombinase) and pcDNA5/FRT/TO-HA-TRIM56 (or HA-mutant TRIM56) at a ratio of 9:1 and selected stable cell pools for double resistance to Blasticin (4 μg/ml) and Hygromycin (200 μg/ml).
Viruses and Miscellaneous Reagents
Sendai virus (SeV) (strain Cantell) was purchased from Charles River Laboratories and used to infect cells at 100 HAU/ml for the indicated time periods. VSV-Luc is a recombinant firefly luciferase-encoding vesicular stomatitis virus, the replication of which in infected cells can be conveniently monitored by luciferase assay (31). A cell culture-derived hepatitis C virus (HCV, strain JFH-1) was propagated in Huh7.5 cells as described (29, 32). For infection of 7.5-TLR3 cells with HCV, an MOI of 0.5 was used. Poly-I:C (a dsRNA surrogate) and poly-dA:dT (a dsDNA surrogate) were obtained from Sigma. For stimulation of cells, poly-I:C was added directly into culture medium (pIC) at a final concentration of 50 μg/ml, or transfected into cells (T-pIC, 4 μg/35-mm dish) after being complexed with Lipofectamine at 1:1 (μg:μl) ratio. Poly-dA:dT was administered to cells by transfection at 4 μg/35-mm dish after being complexed with Lipofectamine at 1:1 (μg:μl) ratio. IFN-α(con) was used at a final concentration of 100 units/ml for cell stimulation.
Reporter Gene Assay
IFN-β promoter activities in transfected cells following various treatments were determined using luciferase reporter assay as described (33–35).
RNA Interference
For stable knockdown of TRIM56, HeLa cells were transfected with linearized pRevTRE-T56–097shRNA plasmid (26), followed by selection in the presence of 200 μg/ml of Hygromycin. After ∼3 weeks, individual cell colonies were picked, expanded, and screened for reduced TRIM56 expression (in comparison with control HeLa cells) by Western blot. For transient knockdown of TRIM56 in HeLa, 293-TLR3, and Huh7.5-TLR3 cells, we used two independent synthetic siRNAs (Invitrogen) targeting distinct regions of human TRIM56 (target sequences described in ref (26).) and a scrambled negative control (Ambion). The siRNAs were transfected into cells at a final concentration of 100 nm using Lipofectamine 2000 (Invitrogen).
RNA Analyses
Total RNA was isolated from cells following the indicated treatments using Trizol reagent (Invitrogen) and quantified by spectrometry at 260 nm. Northern blotting was performed using traditional procedures. Briefly, equal amount of RNA samples (15 μg) were fractionated on 1% denaturing agarose gel, transferred to Hybond-N+ Nylon membranes (GE Healthcare) and hybridized with digoxigenin-labeled DNA probes specific for human ISG56. After extensive washing, bound DNA probes were detected using anti-digoxigenin-alkaline phosphatase-conjugate (Roche), followed by reaction with CSPD and exposure to x-ray films. As loading controls, ethidium bromide-stained 18S and 28S rRNAs were used.
For quantitative realtime PCR (qPCR), 1 μg of total RNA was programmed for synthesis of cDNA by MMLV reverse transcriptase in a 20-μl reaction. The abundance of ISG56, RANTES, TLR3, OASL, IFN-β, TRIM56, IP-10, and 28S (internal control) transcripts and HCV RNA were then analyzed using gene-specific primers (see below) and GoTaq qPCR Master Mix (Promega) on an iCycler IQ5 real-time PCR system (Bio-Rad). The primer sequences for detection of TRIM56 (26), 28S, RANTES, and HCV RNA (36) have been described. Other primers used were: IFN-β, 5′-gtctcctccaaattgctctc-3′ (forward) and 5′-acaggagcttctgacactga-3′ (reverse); ISG56, 5′-ccacaaaaaatcacaagccattt-3′ (forward) and 5′-cagggcaaggagaaccttaatatatc-3′(reverse); TLR3, 5′-gccctttgggatgctgtgt-3′ (forward) and 5′-acttcaggtggctgcagtca-3′ (reverse); OASL, 5′-tcttctcccacactcacatctatctg-3′ (forward) and 5′-caccatcaggattcttcacgaa-3′ (reverse); and IP-10, 5′-tgaaaaagaagggtgagaagagatg-3′ (forward) and 5′-cctttccttgctaactgctttcag-3′ (reverse).
Protein Analyses
Whole cell lysates were prepared by lysing cells in a buffer containing 50 mm HEPES (pH 7.4), 1.5 mm EDTA, 150 mm NaCl, 10% glycerol, 10 mm NaF, 1 mm Na3VO4, 0.5 mm DTT, 1% Triton X-100, and protease inhibitor mixture (Sigma). After clarification by centrifugation, cell lysates were quantified for protein concentration and subjected to immunoprecipitation (IP) and immunoblot analyses as previously described (26, 33, 34). In some experiments, cytoplasmic and nuclear extracts were isolated using a Nuclear Extract kit from Active Motif. The following polyclonal (pAb) and monoclonal (mAb) antibodies were used: anti-TRIM56 pAb (26), anti-ISG56 pAb (29); anti-ISG15 pAb (a gift from Arthur Haas), anti-IRF3, anti-IRF1 and anti-tubulin pAbs (Santa Cruz Biotechnology), anti-IRF3-p396 rabbit mAb (Cell Signal), anti-SeV pAb (a gift from Ilkka Julkunen), anti-Flag M2 mAb (Sigma), anti-HA mAb (Invivogen), anti-Lamin A/C (636) mAb (Santa Cruz Biotechnology); anti-actin mAb (Sigma); and peroxidase-conjugated secondary anti-rabbit and anti-mouse pAbs (Southern Biotech). Protein bands were visualized by enhanced chemiluminescence (Millipore), followed by exposure to x-ray films.
RESULTS
Conditional Expression of TRIM56 Enhances Induction of Type I Interferon Response through the TLR3 Pathway
To explore the role of TRIM56 in antiviral innate immune signaling, we created HeLa cells with tet-regulated conditional expression of HA-tagged TRIM56 (designated as “HeLa-FitA2-T56”). HeLa cells were selected for this purpose because they possess intact TLR3 and RLR signaling pathways as well as that recognizes dsDNA (35, 37, 38). When cultured in the absence of tetracycline, HeLa-FitA2-T56 cells expressed very low levels of endogenous TRIM56 protein, and no detectable exogenous HA-TRIM56 (Fig. 1A, lane 2). However, upon addition of tetracycline into culture medium, these cells exhibited robust expression of HA-TRIM56 (lane 1), which migrated slightly more slowly than endogenous TRIM56 (lane 2). Turning on HA-TRIM56 expression in the absence of other stimuli did not induce a detectable IFN response, as evidence by a lack of induction of ISG56 and ISG15 proteins (Fig. 1, A–D, compare lanes 1 versus 2), ISG56 and RANTES mRNAs (Fig. 1E, compare mock-treated, +tet versus -tet), and IFN-β promoter (Fig. 1G, compare mock-treated, +tet versus -tet). This is consistent with our previous observation (26), and suggests that TRIM56 expression per se does not activate IFN signaling.
FIGURE 1.
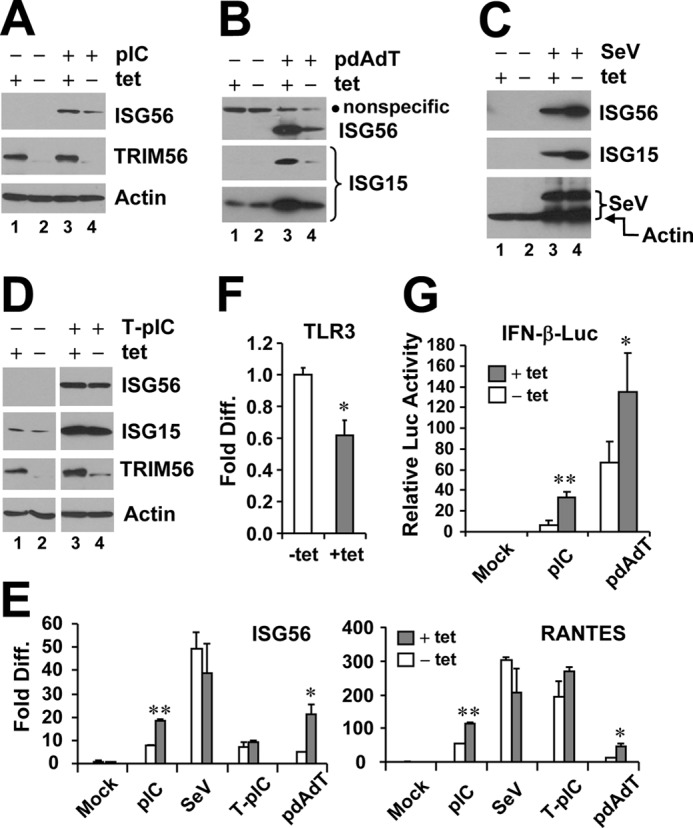
Effects of tet-inducible expression of TRIM56 on activation of the IFN response via different signaling pathways. HeLa-FitA2-T56 cells with tet-regulated conditional expression of WT HA-TRIM56 were manipulated to repress (−tet) or induce (+tet) HA-TRIM56 expression, followed by stimulation by extracellular poly-I:C (pIC, panel A), transfected poly-dA:dT (pdAdT, panel B), SeV infection (panel C), and transfected poly-I:C (T-pIC, panel D) for 12 h. Cell lysates were harvested for immunoblot analysis of ISG56, ISG15, TRIM56, and actin expression. In panel B, a nonspecific band (marked by filled circle) detected by the anti-ISG56 antibody served as a loading control. Two different exposures (short- and long-exposure) of the ISG15 blot are shown. In panel C, the blot at the bottom was detected by a mixture of anti-SeV and anti-actin antibodies. The anti-SeV antibody (Keskinen et al., Ref. 43) mainly reacted with two viral proteins, i.e. hemagglutinin-neuraminidase and nucleocapsid, with the latter having a molecular mass similar to actin. Note that in panels A and D, the anti-TRIM56 antibody detected only the endogenous TRIM56 protein in cells cultured in the absence of tet (lanes 2 and 4), which migrated slightly faster than the HA-TRIM56 detected in induced cells (+tet, lanes 1 and 3). E, HeLa-FitA2-T56 cells were manipulated and treated similarly to those in panels A–D, but cells were harvested for RNA extraction and qPCR analysis of ISG56 and RANTES mRNA expression. * and ** indicate statistical differences exist between −tet and +tet cells with a p value of <0.05 and <0.01, respectively. F, qPCR analysis of TLR3 mRNA levels in HeLa-FitA2-T56 cells repressed and induced for HA-TRIM56 expression. G, reporter gene assay of pIC- or pdAdT-induced IFN-β promoter activities in HeLa-FitA2-T56 cells without (−tet) or with (+tet) HA-TRIM56 expression.
Notably, expression of HA-TRIM56 substantially augmented the induction of ISG56 by extracellular administered poly-I:C, which activates the TLR3 signaling pathway (Fig. 1A, compare lanes 3 versus 4). HA-TRIM56 also boosted the ISG induction in response to transfected dsDNA (poly-dA:dT) (Fig. 1B, compare lanes 3 versus 4). However, HA-TRIM56 slightly decreased SeV-induced ISG protein expression (which is mediated by the RIG-I pathway), without affecting SeV replication (Fig. 1C, compare lanes 3 versus 4). Furthermore, TRIM56 expression had no appreciable impact on ISG protein induction by intracellular dsRNA (Fig. 1D, transfected poly-I:C, which activates RIG-I and MDA5 signaling). qPCR analysis showed that induction of ISG56 and RANTES mRNAs by extracellular dsRNA or intracellular dsDNA was significantly enhanced in the presence of HA-TRIM56 (Fig. 1E). The potentiation of TLR3-mediated responses did not result from increased TLR3 expression upon HA-TRIM56 induction (Fig. 1F). Reporter gene assay of the IFN-β promoter indicated that the enhancing effects of TRIM56 on TLR3- and dsDNA-signaling occurred at level of gene transcription (Fig. 1G). Taken collectively, these data demonstrate that in addition to promoting dsDNA-mediated innate immune response (27), TRIM56 also positively regulates the TLR3 signaling pathway.
The C-terminal Portion of TRIM56 Governs the Ability to Promote TLR3 Signaling
TRIM56 has intrinsic E3 ubiquitin ligase activity that lies in the N-terminal RING domain (26). In addition, the C-terminal structural integrity of TRIM56 is critical for mediating protein-protein interactions, as exemplified by that between TRIM56 and the N-terminal protease of BVDV (26). To delineate the molecular determinants dictating TRIM56 capacity to facilitate TLR3 signaling, we compared several TRIM56 deletion mutants (Fig. 2A) with WT TRIM56 for the capability of regulating extracellular dsRNA-induced IFN response. In 293-TLR3 cells, overexpression of WT TRIM56 significantly augmented poly-I:C induced activation of the IFN-β promoter. A TRIM deletion mutant lacking the entire RING domain (mutant A) was as effective as the WT protein (Fig. 2B), suggesting that the E3 ubiquitin ligase activity is dispensable for the enhancement of TLR3 signaling by TRIM56. In contrast, such capacity was completely lost when the C-terminal portion of TRIM56 was deleted. Because deletion of aa 621–695 (M mutant) and aa 693–750 (N mutant) were equally effective in nullifying the ability of TRIM56 to potentiate TLR3-mediated IFN induction (Fig. 2B), we conclude that the structural integrity rather than the primary amino acid sequence of the C-terminal region of TRIM56 is important for the positive regulation of TLR3 signaling.
FIGURE 2.
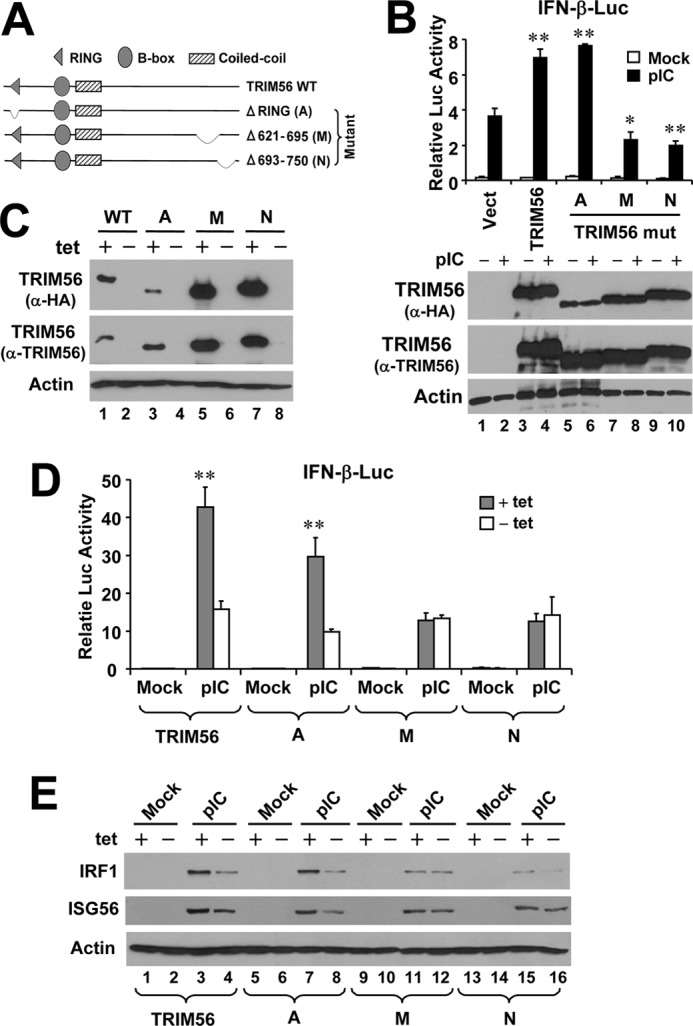
The C-terminal portion, but not the RING domain, controls the ability of TRIM56 to promote TLR3 signaling. A, schematic representation of the domain structures of WT TRIM56 and the individual TRIM56 deletion mutants. B, upper panel: activation of the IFN-β promoter by extracellular poly-I:C in HEK293-TLR3 cells transiently transfected with vectors encoding WT or the indicated mutant TRIM56. Vect: empty vector. * and ** indicate statistical differences exist with a p value of <0.05 and <0.01, respectively, when compared with cells transfected with empty vector and stimulated by poly-I:C. Expression of WT/mutant TRIM56 proteins in cell lysates was analyzed by immunoblotting using either anti-HA or anti-TRIM56 antibody and shown in lower panel. Note that the A mutant was expressed at a level similar to that of WT or the M or N mutant TRIM56 when detected by anti-TRIM56 antibody. The lower expression level of this mutant revealed by immunoblotting with anti-HA antibody likely reflects the different folding of this mutant from WT and other mutant forms of TRIM56 that partially masks the HA epitope introduced to the N terminus of the protein. C, immunoblot analysis of TRIM56 and actin expression in HeLa-FitA2-derived cell lines conditionally expressing WT or the indicated TRIM56 mutant. Note that the exogenous introduced HA-TRIM56 (WT or mutants) could be detected in +tet cells by either anti-TRIM56 or anti-HA tag antibodies. D, activation of the IFN-β promoter by extracellular poly-I:C in HeLa-FitA2-derived cell lines conditionally expressing WT or the indicated TRIM56 mutant. ** indicates statistical difference exists between −tet and +tet cells with a p value of <0.01. E, immunoblotting of IRF1, ISG56 and actin expression in HeLa-FitA2-derived cell lines conditionally expressing WT or the indicated TRIM56 mutant following mock-stimulation or stimulation by extracellular poly-I:C for 12 h.
To corroborate this finding in a more physiologic setting, we developed HeLa cell lines with tet-inducible expression of HA-tagged individual TRIM56 mutants (A, M, and N, respectively). As shown in Fig. 2C, in these cell lines expression of the different TRIM56 mutants was tightly regulated by tetracycline and could be detected in de-repressed (+tet) cells by either anti-TRIM56 antibody or an antibody against the HA tag introduced to the N terminus. The individual mutants also had their expected molecular masses. Having characterized these TRIM56 mutant cell lines, we manipulated them, along with cells conditional expressing WT TRIM56, to repress (−tet) or induce (+tet) TRIM56 (or mutant TRIM56) expression, then compared their responses to poly-I:C stimulation by determining activation of the IFN-β promoter using reporter gene assay (Fig. 2D) and ISG induction by immunoblotting of IRF1 and ISG56 (Fig. 2E and supplemental Fig. S1). The results were in agreement with those obtained from transiently transfected 293-TLR3 cells (Fig. 2B) and demonstrate that WT and RING-deleted (mutant A) TRIM56, but not the C-terminal deletion mutants (M and N), were capable of promoting dsRNA-induced IFN response via the TLR3 pathway.
Knockdown of TRIM56 Impairs TLR3-mediated Induction of ISGs by dsRNA
We next investigated whether the endogenous TRIM56 protein contributes to TLR3 signaling. HeLa cells were stably transfected with a shRNA construct specifically targeting human TRIM56. After hygromycin selection, two independent cell clones, i.e. T56i-7 and T56i-12, were found to express TRIM56 protein at ∼20% of that expressed in control HeLa cells (Fig. 3A). These two TRIM56 knockdown cell lines were then compared with control HeLa cells for their ability to mount IFN responses. In control HeLa cells, extracellular poly-I:C-induced robust expression of ISG15 and ISG56 proteins (Fig. 3A, compare lanes 4 versus 1). Poly-I:C also moderately up-regulated TRIM56 protein abundance (see also in Fig. 3D, compare lanes 3 versus 1), as we observed previously (26). In contrast, poly-I:C failed to up-regulate ISG15 or ISG56 in T56i-7 and T56i-12 cells (Fig. 3A, compare lanes 5 versus 2 and 6 versus 3, respectively). This was not attributed to defective Jak-Stat signaling downstream of the IFN receptor, as induction of either ISG by IFN-α was normal in T56i-7 and T56i-12 cells compared with control HeLa cells (lanes 7–9). Northern blot analysis revealed that TRIM56 depletion in T56i-7 and T56i-12 cells diminished extracellular poly-I:C-induced up-regulation of ISG56 transcript, in contrast to control HeLa cells which mounted a robust response (Fig. 3B, compare lanes 5 and 6 versus 4 in upper panel). Reporter gene assay showed that activation of the IFN-β promoter by pIC was almost ablated in T56i-7 and T56i-12 cells as compared with control HeLa cells (Fig. 3C), suggesting that the defect in IFN/ISG induction by dsRNA in TRIM56 knockdown cells occurred at promoter level.
FIGURE 3.

Effects of TRIM56 knockdown on IFN induction and ISG expression in HeLa cells following various stimuli. A, control HeLa and HeLa cells with stable knockdown of TRIM56 (T56i-7 and T56i-12) were mock-treated, stimulated with poly-I:C or IFN-α for 12 h. Cells were lysed for immunoblotting of ISG15, ISG56, TRIM56 (two different exposures of the blot are shown) and actin expression. Filled circles denote nonspecific bands. B, control HeLa, HeLa T56i-7 and T56i-12 cells were mock-treated, stimulated with poly-I:C, infected with SeV, or transfected with poly-dA:dT for 12 h. Northern blotting was performed to quantify the expression of ISG56 transcript. The ethidium bromide-stained 18S and 28S rRNAs served as loading controls (upper panel). Western blotting (lower panel) was carried out to determine the expression levels of ISG56 protein (two different exposures of the blot are shown). Actin was blotted to demonstrate equal sample loading. C, IFN-β promoter activities in control HeLa, HeLa T56i-7, and T56i-12 cells mock-stimulated or stimulated by poly-I:C for 8 h. ** indicates statistical difference exists with a p value of <0.01 when compared with HeLa cells stimulated by poly-I:C. D, HeLa cells were transiently transfected with a scrambled control siRNA or TRIM56 siRNA#2. 48 h later, cells were mock-treated or stimulated by poly-I:C for 12 h, followed by cell lysis and immunoblotting of ISG56, ISG15, TRIM56, and actin expression. Note that poly-I:C moderately up-regulated TRIM56 expression in cells transfected with control siRNA (compare lanes 3 versus 1).
We also found that TRIM56 silencing severely compromised the induction of ISG56 transcript and protein by transfected poly-dA:dT (Fig. 3B, compare lanes 11 and 12 versus 10), a dsDNA surrogate, confirming the previous finding that TRIM56 participates in innate immune responses to intracellular dsDNA (27). However, SeV-induced ISG56 expression was not affected by TRIM56 knockdown (compare lanes 8 and 9 versus 7), consistent with our previous observation in MDBK cells that TRIM56 was not essential for RIG-I signaling (26).
To rule out the possibility that the severely compromised response to dsRNA in T56i-7 and T56i-12 cells was due to clonal variation, we performed transient knockdown of TRIM56 in HeLa cells. Compared with negative control siRNA-transfected cells, cells receiving a synthetic TRIM56 siRNA (which targets a distinct region of TRIM56 coding sequence from the shRNA used to establish stable TRIM56 knockdown cells) were substantially less responsive to extracellular dsRNA in up-regulating ISG56 and ISG15 proteins (Fig. 3D). These results were also confirmed by qPCR analysis, which showed poly-I:C-induced expression of ISG56, RANTES and OASL mRNAs was profoundly reduced in TRIM56 siRNA-transfected cells compared with cells transfected with the negative control siRNA (supplemental Fig. S2).
We also considered whether TRIM56 silencing may impair dsRNA response by simply reducing TLR3 expression. We thus conducted transient TRIM56 knockdown in 293-TLR3 cells stably overexpressing Flag-TLR3. In these cells, transfection of either of the two TRIM56 siRNAs, but not of a negative control siRNA, greatly reduced the expression of endogenous TRIM56 protein and mRNA prior to and after poly-I:C stimulation. However, under these conditions the expression of TLR3 protein and mRNAs was unaffected (Fig. 4, A and C). We found that knockdown of TRIM56 in 293-TLR3 cells substantially curtailed poly-I:C-induction of ISG56 protein (Fig. 4A), IFN-β promoter (Fig. 4B) and transcript for IFN-β, ISG56, OASL, and RANTES (Fig. 4C). For all of these known IRF3-target genes, a 50–70% reduction in poly-I:C responsiveness was observed in cells transfected with TRIM56 siRNA compared with cells receiving control siRNA. Taken together, data from the transient and stable RNA interference experiments in HeLa and 293-TLR3 cells reveal an essential physiologic role for TRIM56 in the TLR3 signaling pathway.
FIGURE 4.
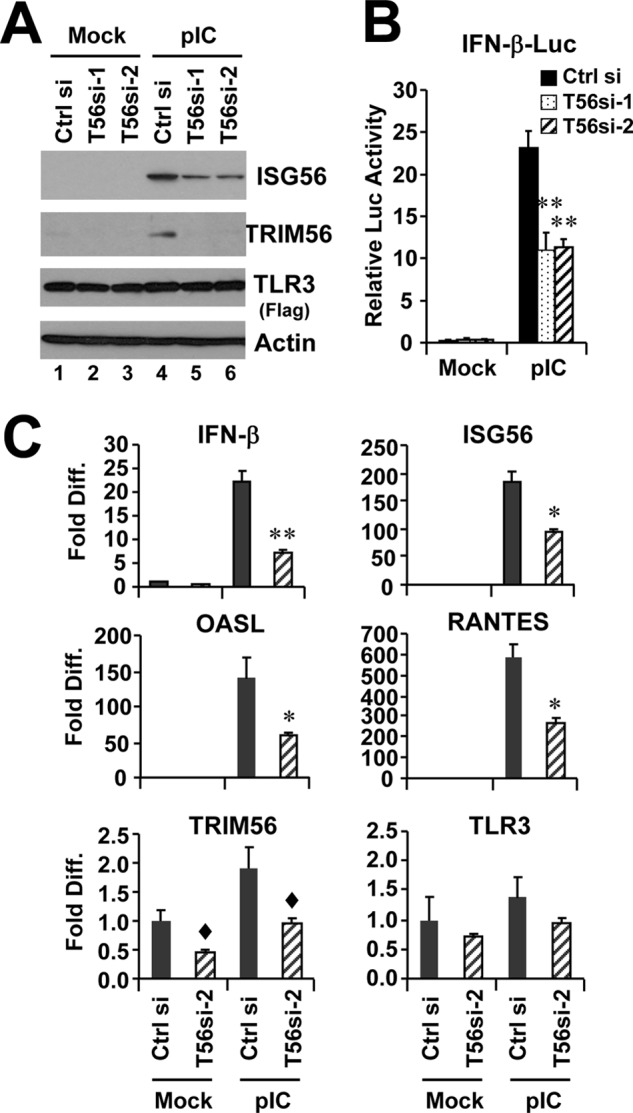
Knockdown of TRIM56 impairs induction of type I IFN response by poly-I:C in HEK293-TLR3 cells. HEK293-TLR3 cells were transiently transfected with control siRNA or different TRIM56 siRNAs for 48 h, followed by mock-stimulation or stimulation by poly-I:C for 12 h (panel A) or 6 h (panel C). Cells were then harvested for immunoblotting of ISG56, TRIM56, TLR3 (using anti-Flag antibody) and actin (panel A) or for qPCR analysis of the abundance of IFN-β, ISG56, OASL, RANTES, TRIM56, and TLR3 mRNAs (panel C). Note that poly-I:C moderately up-regulated TRIM56 expression in cells transfected with control siRNA (compare lanes 4 versus 1 in panel A). In panel C, * and ** indicate statistical differences exist with a p value of <0.05 and <0.01, respectively, when compared with cells transfected with control siRNA and stimulated by poly-I:C. ♦ denotes p = 0.05∼0.06 between cells transfected with control siRNA and TRIM56 siRNA-2. B, poly-I:C (12 h)-induced IFN-β promoter activation in HEK293-TLR3 cells transiently transfected with control siRNA or different TRIM56 siRNAs. ** indicates statistical difference exists with a p value of <0.01 when compared with cells transfected with control siRNA and stimulated by poly-I:C.
Finally, to provide unequivocal evidence that TRIM56 depletion was responsible for the defective response to extracellular dsRNA in HeLa cells with stable knockdown of TRIM56, we conducted rescue experiments in HeLa T56i-7 cells. We generated TRIM56 shRNA (which was used to create HeLa T56i-7 and T56i-12 cells)-insensitive TRIM56-encoding vectors by introducing eight silent mutations in the shRNA targeting sequence (supplemental Fig. S3A). The various shRNA-resistant TRIM56-encoding vectors (WT or deletion mutants) were transfected into HeLa T56i-7 cells and their effects on rescuing poly-I:C induced IFN response were determined. We found that WT or the RING-deleted TRIM56 mutant (mutant A), but not the C-terminal deletion mutants (M or N), conferred HeLa T56i-7 cells the capacity to activate the IFN-β promoter (supplemental Fig. S3B) and to upregulate ISG56 expression (supplemental Fig. S3C) following poly-I:C stimulation. These data corroborate our earlier finding that TRIM56 promotes TLR3 signaling independent of its RING-domain but instead relying on its C-terminal portion (Fig. 2).
Silencing of TRIM56 Expression Undermines TLR3-mediated Antiviral Response
To directly evaluate how TRIM56 regulates TLR3-dependent antiviral response, we studied the effect of TRIM56 silencing on establishment of an antiviral state by extracellular dsRNA. To this end, control HeLa cells, T56i-7 and T56i-12 cells were either mock-treated or stimulated with poly-I:C, followed by infection with a recombinant VSV encoding luciferase (VSV-Luc) (Fig. 5). In mock-treated HeLa, T56i-7, and T56i-12 cells, VSV-Luc replicated with similar efficiency as determined by the VSV-encoded luciferase activity. This is consistent with our previous observation in MDBK cells that TRIM56 expression per se had no effect on VSV propagation (26). Stimulation by poly-I:C produced an antiviral state in control HeLa cells, resulting in a 95% reduction of VSV-Luc replication compared with unstimulated cells. However, poly-I:C was substantially less effective in T56i-7 and T56i-12 cells, inhibiting viral replication by only 37 and 28%, respectively (Fig. 5). These data are consistent with our earlier finding that knockdown of TRIM56 impairs extracellular dsRNA-induced ISG expression (Figs. 3 and 4). More importantly, they provide direct evidence that TRIM56 plays an essential part in TLR3-mediated antiviral responses.
FIGURE 5.
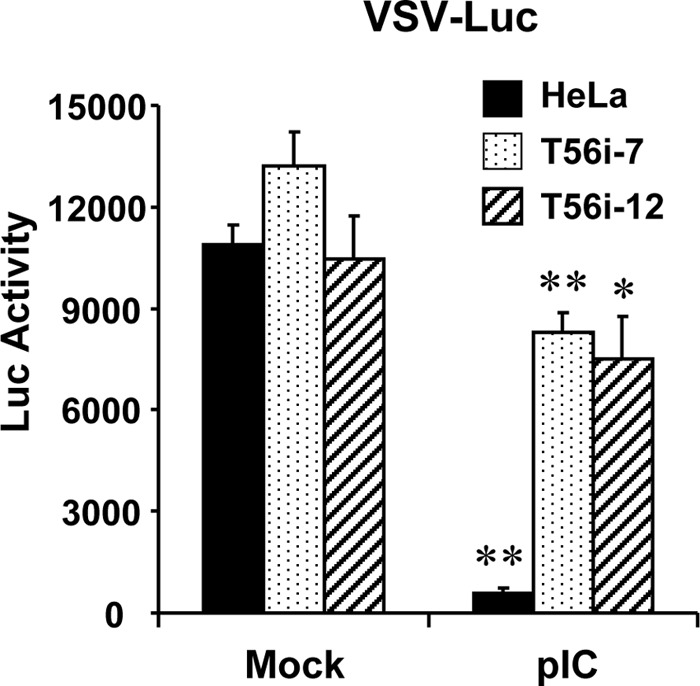
TRIM56 depletion compromises the establishment of an antiviral state by TLR3 ligand. Control HeLa, HeLa T56i-7, and T56i-12 cells were mock-stimulated or stimulated by poly-I:C, followed by infection with VSV-Luc (MOI = 0.1). At 3 h postinfection, cells were lysed for firefly luciferase assay. * and ** indicate statistical differences exist between mock-treated and poly-I:C-stimulated cells with a p value of <0.05 and <0.01, respectively.
Knockdown of TRIM56 Compromises Viral Induction of Chemokines via the TLR3 Signaling Pathway
To corroborate the role of TRIM56 in TLR3 signaling in a viral infection setting, we took advantage of a human hepatoma cell model (Huh7.5 cells reconstituted with TLR3 expression, i.e. 7.5-TLR3 cells) in which infection by hepatitis C virus (HCV), a positive-strand RNA virus, activates chemokine transcription upon TLR3 sensing of dsRNAs produced during HCV replication (36). Because 7.5-TLR3 cells are defective for RIG-I function, this HCV infection model is spared from any confounding effect from RIG-I signaling (29). 7.5-TLR3 cells were transfected with TRIM56 siRNA or negative control siRNA, followed by infection by HCV (MOI = 0.5). 48 h postinfection, cells were lysed for quantification of RANTES, IP-10, and TRIM56 mRNAs and HCV RNAs by qPCR. As shown in Fig. 6, HCV significantly up-regulated the expression of RANTES and IP-10 (by 226- and 11-fold, respectively) in cells transfected with control siRNA. However, these responses were curtailed by 50–60% in TRIM56 knockdown cells. This was not a result of reduced expression of HCV PAMP, as HCV RNA replication was unaffected by TRIM56 silencing (Fig. 6).
FIGURE 6.
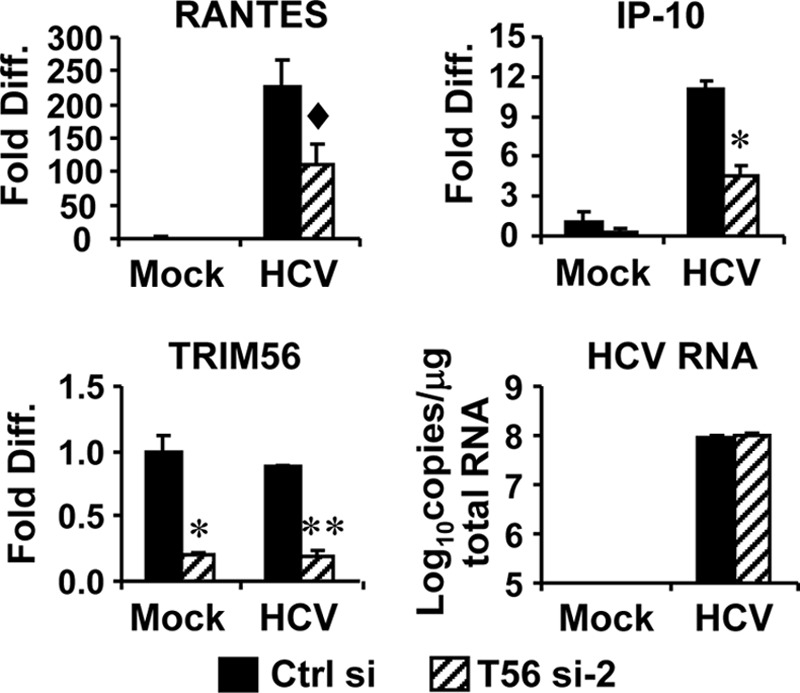
Knockdown of TRIM56 dampens TLR3-mediated chemokine induction by HCV infection in Huh7.5-TLR3 cells. Huh7.5-TLR3 cells were transiently transfected with control siRNA or TRIM56 siRNA#2. 30 h later, cells were mock-infected or infected by HCV (MOI = 0.5). At 48 h postinfection, cells were lysed for total RNA isolation, followed by quantification of RANTES, IP-10, and TRIM56 mRNAs and HCV RNA levels by qPCR. * and ** indicate statistical differences exist with a p value of <0.05 and <0.01, respectively, when compared with cells transfected with control siRNA. ♦ denotes p = 0.06 between cells transfected with control siRNA and TRIM56 siRNA-2.
TRIM56 Is Essential for TLR3-mediated Activation of IRF3
IRF3 is a key transcription factor that controls viral- and dsRNA-induction of the IFN antiviral response (12). Because we observed TRIM56 knockdown severely impaired dsRNA-induced up-regulation of numerous ISGs (ISG56, ISG15, OASL, RANTES, and IFN-β, etc.) that are transcriptionally regulated by IRF3 (35, 39, 40), we investigated whether TRIM56 contributes to IRF3 activation downstream of TLR3. While poly-I:C-induced robust phosphorylation of IRF3 in HeLa cells transfected with control siRNA, it did little in cells in which TRIM56 was transiently silenced (Fig. 7A, compare lanes 3 versus 4 and 5 versus 6). Consistent with this, poly-I:C-induced nuclear translocation of IRF3 was abrogated in HeLa T56i-7 cells with stable TRIM56 depletion, but was intact in control HeLa cells (Fig. 7B, compare lanes 10 versus 9 and 12 versus 11). In aggregate, these data demonstrate that TRIM56 contributes to TLR3-dependent antiviral responses by participating in critical signaling process(es) leading to activation of IRF3.
FIGURE 7.
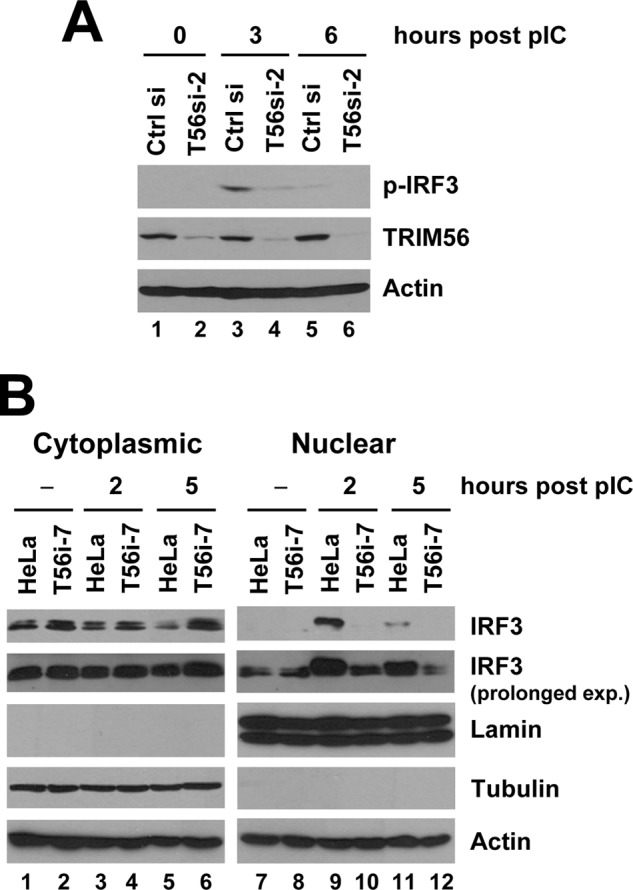
Silencing the expression of TRIM56 diminishes poly-I:C-induced IRF3 phosphorylation and nuclear translocation. A, HeLa cells were transiently transfected with control siRNA or TRIM56 siRNA#2 for 48 h, followed by stimulation by poly-I:C for the indicated time periods. Whole cell lysates were subjected to immunoblot analysis of phosphorylated IRF3 (using anti-IRF3 p396 antibody), TRIM56, and actin. B, control HeLa and HeLa T56i-7 cells were stimulated by poly-I:C for the indicated time periods. Cytoplasmic and nuclear extracts were isolated and subjected to immunoblotting of IRF3, Lamin A/C (a nuclear protein marker), tubulin (a cytoplasmic protein marker), and actin. Comparison of the two different exposures of the IRF3 blot showed that the IRF3 protein present in poly-I:C-stimulated HeLa nuclear extracts was phosphorylated IRF3.
TRIM56 Interacts with TRIF
We next sought to characterize the mechanism by which TRIM56 regulates TLR3-mediated IRF3 activation. To explore where TRIM56 acts in the pathway, we examined the effect of TRIM56 knockdown on activation of IFN response by overexpression of various signaling molecules in the TLR3 pathway. Forced expression of a constitutively active TRIF (TRIFSA) was equally effective in upregulating ISG56 expression in control HeLa cells and cells with stable TRIM56 depletion (Fig. 8A, compare lanes 3 versus 4). Likewise, we did not observe any inhibitory effect of TRIM56 knockdown on activation of IFN-β promoter by overexpression of the downstream signaling molecules, IKKϵ or the constitutively active IRF3–5D (data not shown). These data imply that TRIM56 likely operates at the level of TRIF or above.
FIGURE 8.
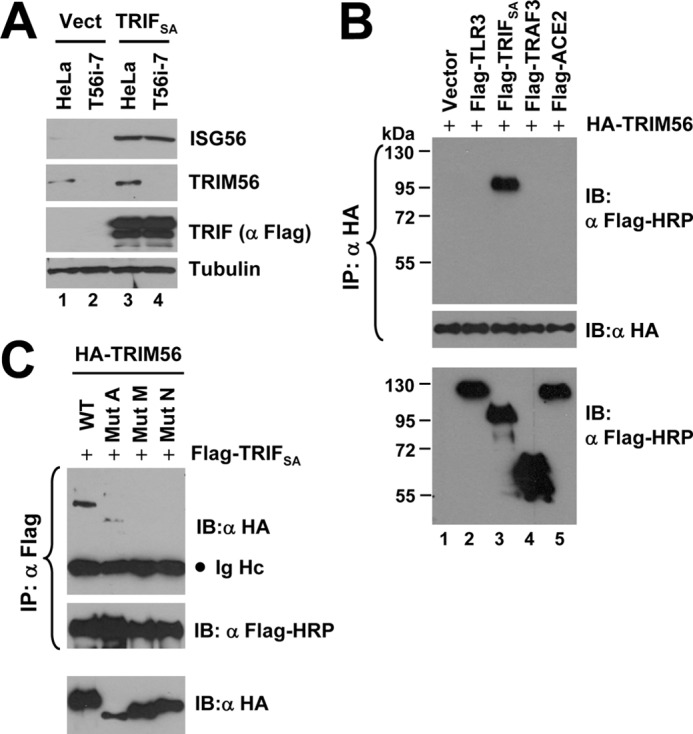
TRIM56 forms a complex with TRIF. A, immunoblot analysis of ISG56, TRIM56, TRIF (using anti-Flag antibody), and actin in control HeLa and HeLa T56i-7 cells transfected with an empty vector (lanes 1–2) or an vector encoding Flag-TRIFSA (lanes 3–4)for 24 h. B, co-IP experiment in HEK293 cells co-expressing HA-TRIM56 (WT) and various Flag-tagged signaling proteins. Cells co-expressing Flag-ACE2 (lane 5) or empty vector (lane 1) served as negative controls. Cell lysates were immunoprecipitated (IP) with anti-HA, followed by immunoblotting (IB) with anti-Flag-HRP or anti-HA. The bottom blot showed expression of the various Flag-tagged proteins in cell lysates. C, co-IP experiment in HEK293 cells co-expressing Flag- TRIFSA and WT HA-TRIM56 or various mutant HA-TRIM56. Cell lysates were immunoprecipitated (IP) with anti-Flag, followed by immunoblotting (IB) with anti-HA or anti-Flag-HRP. The bottom blot showed expression of the various HA-TRIM56 proteins (WT or mutant) in cell lysates. Ig Hc: IgG heavy chain.
Because the E3 ubiquitin ligase-deficient TRIM56 mutant (ΔRING, i.e. mutant A) retained the ability to enhance TLR3 signaling (Fig. 2), the augmentation of TLR3 response by TRIM56 was not due to ubiquitination of signaling proteins. Apart from serving as E3 ubiquitin ligases, the TRIM proteins are proposed to function as a scaffold for assembly of multiple protein complexes (17). We reasoned that TRIM56 (and the ΔRING mutant) may promote TLR3 signaling by interacting with critical components in the pathway to facilitate signaling complex formation. Co-IP experiments showed that TRIM56 formed a complex with TRIF (Fig. 8B, lane 3), but not with TLR3, TRAF3 (lane 2 and 4, respectively) or IRF3 (data not shown), in co-transfected cells. TRIM56 also did not interact with ACE2, a negative control (lane 5). Interestingly, when different TRIM56 mutants were studied, the RING-deleted TRIM56 (mutant A) still associated with TRIF, while the two C-terminal deletion mutants (M and N) were no longer able to do so (Fig. 8C and supplemental Fig. S4). These data demonstrate that the C-terminal portion of TRIM56 is critical for its interaction with TRIF. Given that the C-terminal TRIM56 mutants but not the ΔRING mutant lost the ability to enhance poly-I:C-induced IFN response (Fig. 2), we conclude that the ability to associate with TRIF correlates with the capacity of TRIM56 to promote TLR3 signaling.
DISCUSSION
In this study, we have uncovered a novel role of TRIM56 in innate antiviral immunity by revealing it is a positive regulator of the TLR3 signaling pathway. Overexpression of TRIM56 substantially augmented extracellular dsRNA induced expression of IFN-β and numerous ISGs (Figs. 1 and 2), while knockdown of TRIM56 had the opposite effect (Figs. 3 and 4). Importantly, these findings were not cell type specific and similar results were obtained regardless of the expression/silencing systems used. Furthermore, we demonstrated that TRIM56 depletion greatly compromised the establishment of an antiviral state by TLR3 ligand (Fig. 5) and that silencing of TRIM56 severely impaired TLR3-mediated host response in a viral infection setting (Fig. 6), highlighting the biological significance of these observations.
The heightened TLR3 antiviral response in TRIM56-overexpressing cells was not attributed to increased TLR3 expression, as TRIM56 expression per se did not activate IFN-β transcription, nor did it up-regulate the expression of ISGs, including TLR3 (Fig. 1). Conversely, the impairment of TLR3 signaling upon TRIM56 knockdown was not a result of decreased TLR3 expression (Fig. 4). Rather, our data suggest that TRIM56 participates in critical protein-protein interactions downstream of TLR3 (Fig. 8) that are essential for activating signaling cascades leading to phosphorylation and nuclear translocation of IRF3 (Fig. 7), a pivotal transcription factor that controls type I IFN response. In addition, our data in TRIM56 knockdown cells demonstrate that TRIM56 does not regulate signaling events via the Jak-STAT pathway downstream of the IFN receptor, once type I IFNs are produced (Fig. 3A). Previously, we reported that overexpression of TRIM56 restricted the replication of BVDV in permissive MDBK cells in a way that was not a result of general augmentation of the IFN response (26). We note that this finding was not inconsistent with our current study, because the TLR3 pathway is not active in MDBK cells (these cells do not respond to extracellular poly-I:C) (33) and BVDV does not induce a detectable IFN response because of its encoded N-terminal protease, which efficiently degrades IRF3 via the proteasome (33).
Of the handful of TRIMs reported to positively regulate innate immune signaling, the E3 ligase-dependent ubiquitination of signaling components represents a canonical mechanism, as demonstrated for TRIM25-mediated K63-linked ubiquitination of RIG-I (25) and TRIM23-dependent K27-linked ubiquitination of NEMO (41). However, the mechanism TRIM56 adopts to promote TLR3 signaling revealed in the current study is distinct, obviating the involvement of the entire RING domain (hence the E3 ubiquitin ligase activity) but instead relying on the C-terminal portion of the protein (Fig. 2). This is reminiscent of the TRIM56 determinants that are critical for the interaction between TRIM56 and the N-terminal protease of BVDV (26), and led us to hypothesize that TRIM56 may associate with a TLR3 pathway component(s), serving as a scaffold for assembling signaling protein complexes required for optimal TLR3 signaling. Because we found that TRIM56 was essential for the IFN response activated via the TLR3 pathway but not required for ISG expresson induced via the RLRs, we postulated the molecule(s) which TRIM56 interacts with and which is involved in the enhancement of TLR3 responses is not shared by the TLR3 and RLR pathways. Indeed, we found TRIM56 specifically formed a complex with TRIF, the TLR3 adaptor, but not with the downstream signaling components, TRAF3 and IRF3 (Fig. 8), which are common to both pathways. Recently, it was reported that TRIM56 associates with and promotes K63-linked polyubiquitination of STING, an ER-resident adaptor protein, thereby enhancing cytoplasmic dsDNA-induced IFN expression (27). However, gene knock-out studies have demonstrated that STING is dispensable for TLR3 signaling (42), suggesting that STING is unlikely the protein interaction partner responsible for the effect of TRIM56 on TLR3 signaling.
TRIM56 is diffusely distributed in the cytoplasm (26, 27), as is the case with TRIF in resting cells. We speculate that TRIM56 may serve as a scaffold for assembly of TRIF-associated protein complexes essential for TLR3 signaling. We did not observe an interaction of TRIM56 with TLR3 or TRAF3 in co-IP experiments; it is possible that the association of TRIM56 is a prerequisite for TRIF to recruit (or to be recruited to) an unknown signaling protein in the pathway. Although the exact mechanism by which the TRIM56-TRIF interaction contributes to TLR3-mediated IRF3 activation awaits further investigation, our data that the C-terminal portion of TRIM56 was important both for potentiating TLR3 responses (Fig. 2) and for forming a complex with TRIF (Fig. 8) and that TRIM56 acted at a level at or above TRIF (Fig. 8A) suggest the biological relevance of this interaction for the participation of TRIM56 in TLR3-mediated antiviral responses.
Our data also substantiated the contribution of TRIM56 to cytosolic dsDNA-induced IFN response (Figs. 1, B, E, G, and 3B) (27). However, the role of TRIM56 in RLR signaling is much less certain. Although the interaction with and ubiquitination of STING by TRIM56 were suggested to facilitate RLR signaling (27), STING deficiency had no impact on EMCV replication or IFN induction by transfected poly-I:C (both stimuli trigger MDA5 signaling), and complete loss of STING only partially reduced SeV-induced IFN production (42). Of note, the dampening effect of TRIM56 knockdown on New Castle disease virus-induced IFN-β expression Tsuchida et al. observed was very moderate compared with that on IFN expression stimulated by cytosolic dsDNA (27). In a previous study, we found that stable knockdown of TRIM56 by >80% in MDBK cells had little effect on ISG induction by SeV or transfected poly-I:C (26). In the current study, conditional expression of TRIM56 in HeLa cells did not potentiate the induction of ISG mRNAs following stimulation by SeV or transfected poly-I:C (Fig. 1E), and stable knockdown of TRIM56 by >80% in HeLa cells had little, if any, impact on SeV-induced ISG56 expression (Fig. 3B). The negligible contribution of TRIM56 to RLR-mediated innate immune response contrasted starkly with its essential role in ISG induction by extracellular dsRNA (via TLR3) or cytosolic dsDNA. Surprisingly and not currently understood, tet-regulated expression of TRIM56 reproducibly reduced ISG56 and ISG15 protein levels following SeV infection (Fig. 1C). Future studies in TRIM56 knock-out mice will be required to clarify the physiologic role that TRIM56 may play in RLR signaling.
Acknowledgments
We thank Lawrence Pfeffer (University of Tennessee Health Science Center) for critical review of the manuscript and for the gift of IFN-α. We are grateful to Stephen Taylor (University of Manchester) for sharing the HeLa Flp-IN T-Rex cells, Sean Whealan (Harvard Medical School) for providing the VSV-Luc virus, and Margaret Offermann (Emory University) and Jürg Tschopp (University of Lausanne) for the Flag-TRIF-mRHIM and Flag-TRAF3 constructs, respectively.
This study was supported, in whole or in part, by National Institutes of Health Grant AI069285 (to K. L.).

This article contains supplemental Figs. S1–S4.
- IFN
- interferon
- TRIM56
- tripartite motif protein 56
- TLR3
- Toll-like receptor 3
- IRF
- interferon regulatory factor
- ISG
- interferon-stimulated gene
- RING
- really interesting new gene
- RIG-I
- retinoic inducible gene I
- MDA5
- melanoma differentiation-associated gene 5
- RLR
- RIG-I-like receptors
- MAVS
- mitochondrial antiviral signaling protein, a.k.a., IPS-1/Cardif/VISA
- STING
- stimulator of IFN genes, a.k.a. MITA/ERIS/MPYS
- ER
- endoplasmic reticulum
- TRIF
- Toll-IL1 receptor homology domain containing adaptor inducing interferon-β, a.k.a., TICAM1
- STAT
- signal transducers and activators of transcription
- OASL
- 2′,5′-oligoadenylate synthetase like gene
- RANTES
- regulated upon activation, normal T-cell expressed, and secreted
- SeV
- Sendai virus
- VSV
- vesicular stomatitis virus
- HCV
- hepatitis C virus
- BVDV
- bovine viral diarrhea virus
- TRAF3
- tumor necrosis factor receptor-associated factor 3
- NEMO
- NF-κB essential modulator
- ACE2
- angiotensin-converting enzyme 2.
REFERENCES
- 1. Borden E. C., Sen G. C., Uze G., Silverman R. H., Ransohoff R. M., Foster G. R., Stark G. R. (2007) Interferons at age 50: past, current, and future impact on biomedicine. Nat. Rev. Drug Discov. 6, 975–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sen G. C. (2001) Viruses and interferons. Annu. Rev. Microbiol. 55, 255–281 [DOI] [PubMed] [Google Scholar]
- 3. Kawai T., Akira S. (2008) Toll-like receptor and RIG-I-like receptor signaling. Ann. N.Y. Acad. Sci. 1143, 1–20 [DOI] [PubMed] [Google Scholar]
- 4. Yoneyama M., Fujita T. (2010) Recognition of viral nucleic acids in innate immunity. Rev. Med. Virol. 20, 4–22 [DOI] [PubMed] [Google Scholar]
- 5. Seth R. B., Sun L., Ea C. K., Chen Z. J. (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 122, 669–682 [DOI] [PubMed] [Google Scholar]
- 6. Yamamoto M., Sato S., Mori K., Hoshino K., Takeuchi O., Takeda K., Akira S. (2002) Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-β promoter in the Toll-like receptor signaling. J. Immunol. 169, 6668–6672 [DOI] [PubMed] [Google Scholar]
- 7. Fitzgerald K. A., McWhirter S. M., Faia K. L., Rowe D. C., Latz E., Golenbock D. T., Coyle A. J., Liao S. M., Maniatis T. (2003) IKKϵ and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496 [DOI] [PubMed] [Google Scholar]
- 8. Sharma S., tenOever B. R., Grandvaux N., Zhou G. P., Lin R., Hiscott J. (2003) Triggering the interferon antiviral response through an IKK-related pathway. Science 300, 1148–1151 [DOI] [PubMed] [Google Scholar]
- 9. Häcker H., Redecke V., Blagoev B., Kratchmarova I., Hsu L. C., Wang G. G., Kamps M. P., Raz E., Wagner H., Häcker G., Mann M., Karin M. (2006) Specificity in Toll-like receptor signaling through distinct effector functions of TRAF3 and TRAF6. Nature 439, 204–207 [DOI] [PubMed] [Google Scholar]
- 10. Oganesyan G., Saha S. K., Guo B., He J. Q., Shahangian A., Zarnegar B., Perry A., Cheng G. (2006) Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 439, 208–211 [DOI] [PubMed] [Google Scholar]
- 11. Zhao T., Yang L., Sun Q., Arguello M., Ballard D. W., Hiscott J., Lin R. (2007) The NEMO adaptor bridges the nuclear factor-κB and interferon regulatory factor signaling pathways. Nat. Immunol. 8, 592–600 [DOI] [PubMed] [Google Scholar]
- 12. Hiscott J. (2007) Triggering the innate antiviral response through IRF-3 activation. J. Biol. Chem. 282, 15325–15329 [DOI] [PubMed] [Google Scholar]
- 13. Munir M. (2010) TRIM proteins: another class of viral victims. Sci. Signal 3, jc2. [DOI] [PubMed] [Google Scholar]
- 14. Ozato K., Shin D. M., Chang T. H., Morse H. C., 3rd. (2008) TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 8, 849–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Short K. M., Cox T. C. (2006) Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J. Biol. Chem. 281, 8970–8980 [DOI] [PubMed] [Google Scholar]
- 16. Meroni G., Diez-Roux G. (2005) TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. Bioessays 27, 1147–1157 [DOI] [PubMed] [Google Scholar]
- 17. Reymond A., Meroni G., Fantozzi A., Merla G., Cairo S., Luzi L., Riganelli D., Zanaria E., Messali S., Cainarca S., Guffanti A., Minucci S., Pelicci P. G., Ballabio A. (2001) The tripartite motif family identifies cell compartments. EMBO J. 20, 2140–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nisole S., Stoye J. P., Saïb A. (2005) TRIM family proteins: retroviral restriction and antiviral defence. Nat. Rev. Microbiol. 3, 799–808 [DOI] [PubMed] [Google Scholar]
- 19. McNab F. W., Rajsbaum R., Stoye J. P., O'Garra A. (2011) Tripartite-motif proteins and innate immune regulation. Curr. Opin. Immunol. 23, 46–56 [DOI] [PubMed] [Google Scholar]
- 20. Higgs R., Ní Gabhann J., Ben Larbi N., Breen E. P., Fitzgerald K. A., Jefferies C. A. (2008) The E3 ubiquitin ligase Ro52 negatively regulates IFN-β production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J. Immunol. 181, 1780–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shi M., Deng W., Bi E., Mao K., Ji Y., Lin G., Wu X., Tao Z., Li Z., Cai X., Sun S., Xiang C., Sun B. (2008) TRIM30α negatively regulates TLR-mediated NF-κB activation by targeting TAB2 and TAB3 for degradation. Nat. Immunol. 9, 369–377 [DOI] [PubMed] [Google Scholar]
- 22. Zha J., Han K. J., Xu L. G., He W., Zhou Q., Chen D., Zhai Z., Shu H. B. (2006) The Ret finger protein inhibits signaling mediated by the noncanonical and canonical IkappaB kinase family members. J. Immunol. 176, 1072–1080 [DOI] [PubMed] [Google Scholar]
- 23. Liang Q., Deng H., Li X., Wu X., Tang Q., Chang T. H., Peng H., Rauscher F. J., 3rd, Ozato K., Zhu F. (2011) Tripartite motif-containing protein 28 is a small ubiquitin-related modifier E3 ligase and negative regulator of IFN regulatory factor 7. J. Immunol. 187, 4754–4763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao W., Wang L., Zhang M., Wang P., Yuan C., Qi J., Meng H., Gao C. (2012) Tripartite motif-containing protein 38 negatively regulates TLR3/4- and RIG-I-mediated IFN-β production and antiviral response by targeting NAP1. J. Immunol. 188, 5311–5318 [DOI] [PubMed] [Google Scholar]
- 25. Gack M. U., Shin Y. C., Joo C. H., Urano T., Liang C., Sun L., Takeuchi O., Akira S., Chen Z., Inoue S., Jung J. U. (2007) TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920 [DOI] [PubMed] [Google Scholar]
- 26. Wang J., Liu B., Wang N., Lee Y. M., Liu C., Li K. (2011) TRIM56 is a virus- and interferon-inducible E3 ubiquitin ligase that restricts pestivirus infection. J. Virol. 85, 3733–3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tsuchida T., Zou J., Saitoh T., Kumar H., Abe T., Matsuura Y., Kawai T., Akira S. (2010) The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33, 765–776 [DOI] [PubMed] [Google Scholar]
- 28. Kaiser W. J., Offermann M. K. (2005) Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J. Immunol. 174, 4942–4952 [DOI] [PubMed] [Google Scholar]
- 29. Wang N., Liang Y., Devaraj S., Wang J., Lemon S. M., Li K. (2009) Toll-like receptor 3 mediates establishment of an antiviral state against hepatitis C virus in hepatoma cells. J. Virol. 83, 9824–9834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tighe A., Staples O., Taylor S. (2008) Mps1 kinase activity restrains anaphase during an unperturbed mitosis and targets Mad2 to kinetochores. J. Cell Biol. 181, 893–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cureton D. K., Massol R. H., Saffarian S., Kirchhausen T. L., Whelan S. P. (2009) Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog. 5, e1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wakita T., Pietschmann T., Kato T., Date T., Miyamoto M., Zhao Z., Murthy K., Habermann A., Kräusslich H. G., Mizokami M., Bartenschlager R., Liang T. J. (2005) Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11, 791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen Z., Rijnbrand R., Jangra R. K., Devaraj S. G., Qu L., Ma Y., Lemon S. M., Li K. (2007) Ubiquitination and proteasomal degradation of interferon regulatory factor-3 induced by Npro from a cytopathic bovine viral diarrhea virus. Virology 366, 277–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li K., Chen Z., Kato N., Gale M., Jr., Lemon S. M. (2005) Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-β production in hepatocytes. J. Biol. Chem. 280, 16739–16747 [DOI] [PubMed] [Google Scholar]
- 35. Wang N., Dong Q., Li J., Jangra R. K., Fan M., Brasier A. R., Lemon S. M., Pfeffer L. M., Li K. (2010) Viral induction of the zinc finger antiviral protein is IRF3-dependent but NF-κB-independent. J. Biol. Chem. 285, 6080–6090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li K., Li N. L., Wei D., Pfeffer S. R., Fan M., Pfeffer L. M. (2012) Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on Toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology 55, 666–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiang Z., Zamanian-Daryoush M., Nie H., Silva A. M., Williams B. R., Li X. (2003) Poly(I-C)-induced Toll-like receptor 3 (TLR3)-mediated activation of NFκB and MAP kinase is through an interleukin-1 receptor-associated kinase (IRAK)-independent pathway employing the signaling components TLR3-TRAF6-TAK1-TAB2-PKR. J. Biol. Chem. 278, 16713–16719 [DOI] [PubMed] [Google Scholar]
- 38. Chen Z., Benureau Y., Rijnbrand R., Yi J., Wang T., Warter L., Lanford R. E., Weinman S. A., Lemon S. M., Martin A., Li K. (2007) GB virus B disrupts RIG-I signaling by NS3/4A-mediated cleavage of the adaptor protein MAVS. J. Virol. 81, 964–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Elco C. P., Guenther J. M., Williams B. R., Sen G. C. (2005) Analysis of genes induced by Sendai virus infection of mutant cell lines reveals essential roles of interferon regulatory factor 3, NF-κB, and interferon but not toll-like receptor 3. J. Virol. 79, 3920–3929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Grandvaux N., Servant M. J., tenOever B., Sen G. C., Balachandran S., Barber G. N., Lin R., Hiscott J. (2002) Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J. Virol. 76, 5532–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Arimoto K., Funami K., Saeki Y., Tanaka K., Okawa K., Takeuchi O., Akira S., Murakami Y., Shimotohno K. (2010) Polyubiquitin conjugation to NEMO by triparite motif protein 23 (TRIM23) is critical in antiviral defense. Proc. Natl. Acad. Sci. U.S.A. 107, 15856–15861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ishikawa H., Barber G. N. (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signaling. Nature 455, 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Keskinen P., Nyqvist M., Sareneva T., Pirhonen J., Melen K., Julkunen I. (1999) Impaired antiviral response in human hepatoma cells. Virology 263, 364–375 [DOI] [PubMed] [Google Scholar]