

Background: To investigate the involvement of HEXIM1 in the p53 signaling pathway, we examine the functional connection between HEXIM1 and p53.
Results: HEXIM1 directly interacts with p53 and enhances the protein stability of p53.
Conclusion: HEXIM1 is a novel regulator of p53.
Significance: Our results reveal a new function of HEXIM1 in regulating the protein stability and induction of p53.
Keywords: Cancer, p53, RNA Polymerase II, Transcription Elongation Factors, Ubiquitination, HDM2, HEXIM1, P-TEFb
Abstract
Hexamethylene bisacetamide-inducible protein 1 (HEXIM1) is best known as the inhibitor of positive transcription elongation factor b (P-TEFb), which regulates the transcription elongation of RNA polymerase II and controls 60–70% of mRNA synthesis. Our previous studies show that HEXIM1 interacts with two key p53 regulators, nucleophosmin and human double minute-2 protein (HDM2), implying a possible connection between HEXIM1 and the p53 signaling pathway. Here we report the interaction between p53 and HEXIM1 in breast cancer, acute myeloid leukemia, and colorectal carcinoma cells. The C-terminal regions of p53 and HEXIM1 are required for the protein-protein interaction. Overexpression of HEXIM1 prevents the ubiquitination of p53 by HDM2 and enhances the protein stability of p53, resulting in up-regulation of p53 target genes, such as Puma and p21. Induction of p53 can be achieved by several means, such as UV radiation and treatment with anti-cancer agents (including doxorubicin, etoposide, roscovitine, flavopiridol, and nutlin-3). Under all the conditions examined, elevated protein levels of p53 are found to associate with the increased p53-HEXIM1 interaction. In addition, knockdown of HEXIM1 significantly inhibits the induction of p53 and releases the cell cycle arrest caused by p53. Finally, the transcription of the p53 target genes is regulated by HEXIM1 in a p53-dependent fashion. Our results not only identify HEXIM1 as a positive regulator of p53, but also propose a novel molecular mechanism of p53 activation caused by the anti-cancer drugs and compounds.
Introduction
Positive transcription elongation factor b (P-TEFb)2 plays an essential role in the regulation of RNA polymerase II (Pol II) transcription elongation and human immunodeficiency virus (HIV) Tat transactivation (1–3). Treatment with P-TEFb inhibitors, such as flavopiridol and 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB), blocks 60–70% of total RNA Pol II transcription and effectively inhibits HIV replication, clearly indicating the importance of P-TEFb in regulation of cellular and viral transcription (1, 2, 4, 5). Recent genome-wide analysis of Drosophila and human embryonic stem cells re-confirms the significance of transcription elongation in gene regulation (6–8).
In cells, P-TEFb is found in two forms of protein complexes. The free, active complex is composed of cyclin-dependent kinase 9 (CDK9) and cyclin T1, while the larger, inactive form consists of CDK9, cyclin T1, hexamethylene bisacetamide-inducible protein 1 (HEXIM1), and a small nuclear RNA (snRNA), 7SK (1, 9–14). HEXIM1 functions as a P-TEFb inhibitor in a 7SK snRNA-dependent manner. Association of 7SK snRNAs with HEXIM1 homodimers causes a conformational change in the proteins and consequently exposes the C-terminal domain of HEXIM1 for CDK9/cyclin T1 binding, resulting in inhibition of the kinase activity of CDK9 (1, 12, 15). HEXIM1, also known as estrogen down-regulated gene 1 (EDG1), was identified as an estrogen receptor α (ERα)-binding protein, and its involvement in breast cancers had been reported (16–18). Other studies suggest that HEXIM1 plays an important role in acquired immunodeficiency syndrome, cancers, cardiac hypertrophy, and inflammation through the inhibition of P-TEFb (15, 19–21).
The tumor suppressor p53 is a stress-inducible transcription factor which regulates cellular genes required for cell cycle arrest, DNA repair, apoptosis, differentiation, and prevention of angiogenesis (22). About 50% of adult cancer contains mutated or deleted p53 gene while another 50% is caused by the suppression of p53 functions (23). Therefore, the p53 signaling pathway represents a major target for development of novel cancer therapeutics. For example, our recent study identified p53 as a potential drug target for the selective treatment of acute myeloid leukemia (AML) (24).
We previously identified two novel HEXIM1-binding proteins, nucleophosmin (NPM) and human double minute-2 protein (HDM2), two key regulators of the p53 signaling pathway. We found that both NPM and HDM2 regulate P-TEFb activity through the modulation of HEXIM1 (25, 26). Overexpression of NPM results in proteasome-mediated degradation of HEXIM1 (25). NPMc+, the cytoplasmic-misallocated mutant form of NPM, was found to interact and sequester a portion of HEXIM1 in the cytoplasm of the NPMc+ AML cell line, leading to increases in P-TEFb-dependent transcription (25). Since 35% of AML patients carry the NPMc+ mutant, our findings suggest the potential involvement of HEXIM1 in tumorigenesis of AML (27). We also demonstrated that HDM2 ubiquitinates HEXIM1; however, the HDM2-ubiquitinated HEXIM1 does not lead to proteasome-mediated protein degradation (26). Furthermore, fusion of ubiquitin to HEXIM1 exhibits stronger inhibition on P-TEFb-dependent transcription, indicating a role for HDM2 in regulation of P-TEFb activity (26). These findings show functional interactions between HEXIM1 and p53 regulators, and indicate a possible involvement of HEXIM1 in the p53 signaling pathway. In this report, we identify HEXIM1 as a p53-binding protein and our results suggest a novel role for HEXIM1 in the regulation of p53 induction.
EXPERIMENTAL PROCEDURES
Cells, Plasmids, and Short Hairpin RNAs (shRNAs)
HEK293 and MCF7 cells were obtained from American Type Culture Collection, and AML2 cells were purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen. Primary human foreskin fibroblasts were obtained from Dr. Mark Stinski (28). HCT116 p53+/+ and p53−/− cells were kindly provided by Dr. Bert Vogelstein (29). Yellow fluorescent protein (YFP)-tagged HEXIM1 (YFP-HEXIM1), YFP-Basic Region (YFP-BR), FLAG-HEXIM1 wild-type (WT), FLAG-HEXIM1(120–359), FLAG-HEXIM1(180–359), FLAG-HEXIM1(1–180), FLAG-HEXIM1(ΔBR), and FLAG-HEXIM1-ILAA mutant expression plasmids were kindly provided by Dr. B. Matija Peterlin and Dr. Olivier Bensaude (12, 30, 31). The untagged HEXIM1-ILAA mutant vector was generated by polymerase chain reaction (PCR) using FLAG-HEXIM1-ILAA plasmid as the template. The amplified PCR fragment was subcloned into pcDNA6 (Invitrogen). To generate the hemagglutinin (HA) tagged full-length and deletions of p53 DNA constructs, including HA-p53 WT, HA-p53(1–369), HA-p53(1–324), HA-p53(45–393), and HA-p53(293–393) vectors, the full-length p53 cDNA (kindly provided by Dr. David P. Lane) was used as the template for PCR and the amplified DNA fragments were subcloned into the pCMV-HA vector (Clontech). The luciferase (Luc) reporters, Puma-Luc (Addgene plasmid #16591), p21-Luc (also known as WWP-Luc, Addgene plasmid #16451), and p53-Luc (also known as PG13-Luc, Addgene plasmid #16442) were gifts from Dr. Bert Vogelstein (32–34). The HDM2 and histidine-tagged ubiquitin (His-Ub) expression plasmids were described previously (26). A nonspecific shRNA and four shRNAs specifically against HEXIM1 were purchased from Origene. All shRNA plasmids contain the green fluorescent protein (GFP) coding sequence driven by a CMV promoter. Expression of GFP can be used to sort the transfected cells by fluorescence-activated cell sorting (FACS).
Transfection and Luciferase Assays
Transient transfections of DNA and shRNA plasmids were performed using FuGENE 6 (Roche) according to the manufacturer's instructions. Firefly luciferase activities were measured 48 h post-transfection using the Bright-Glo assay system (Promega), and the activities were determined using an Infinite 200 multiplate reader (Tecan).
Immunoprecipitation (IP) and Western Blotting
IP was performed using an immunoprecipitation kit according to manufacturer's manual (Roche). HEK293 cells were used for HEXIM1 and p53 domain studies. Western blotting was carried out according to the standard protocols. The horseradish peroxidase-conjugated secondary antibodies were purchased from Pierce. After the antibody incubation, the blots were washed and incubated with SuperSignal West Pico Substrate (Pierce), and the chemiluminescent signal was detected using an x-ray film (Roche). The film was then scanned, and the protein bands were quantified by the GS-800 densitometer (Bio-Rad). The primary antibodies utilized in this study include the HEXIM1 antiserum (25), antibodies against HEXIM1, p53, p53 phospho-Ser33, p53 phospho-Ser315, p53 phospho-Ser392, GFP, p21, HDM2, His tag, CDK9 (Santa Cruz Biotechnology), Puma, p27 (Abcam), FLAG tag, HA tag (Sigma), and β-actin (Millipore).
UV Radiation and Drug Treatment
MCF7 cells were seeded into tissue culture plates and grown for 24 h to reach confluence. The culture medium was aspirated and cells were washed twice with PBS. The cells then were exposed to 50J/m2 of UV irradiation using a crosslinker UV chamber (UV Stratalinker 1800, Stratagene). Fresh medium was added to the UV-treated cells, and the cell lysates were prepared 6 h after UV irradiation. Non UV-treated cells were used as a control. For drug/compound treatment, cells were incubated with an indicated drug or compound, including flavopiridol, DRB, doxorubicin, nutlin-3, etoposide, roscovitine or actinomycin D (Sigma). Cells treated with dimethyl sulfoxide (DMSO) were used as a control since all compounds were dissolved in DMSO. After 3-h incubation, cell lysates were prepared and analyzed by IP, followed by Western blotting.
p53 Ubiquitination and Stability Assays
MCF7 cells were transiently co-transfected with the indicated His-Ub, HDM2, and HEXIM1 expression plasmids. The transfected cells were harvested 2 days after transfection. 16 h before harvest, cells were treated with 10 μm proteasome inhibitor MG132 (Calbiochem). The ubiquitination assay was carried out as described previously (26). For stability assays, cycloheximide (100 μg/ml; Sigma) was added into the culture 0–3 h before harvesting.
Quantitative Reverse Transcriptase-Polymerase Chain Reaction (QRT-PCR)
RNAs of the transfected cells were isolated using RNeasy kit (Qiagen). Improm II Reverse Transcription System (Promega) and SYBR Green PCR Master Mix (Applied Biosystems) were utilized to carry out reverse transcription and real time PCR, respectively. The primer pairs used in this study include: p53 (5′-TCAACAAGATGTTTTGCCAACTG-3′ and 5′-ATGTGCTGTGACTGCTTGTAGATG-3′), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (5′-AACAGCCTCAAGATCATCAGC-3′ and 5′-GGATGATGTTCTGGAGGACC-3′). GAPDH was used as a control to normalize the cDNA inputs. Amplification and detection of specific mRNAs were performed using ABI Prism 7000 Thermal-Cycler (Applied Biosystems).
FACS
HCT116 cells were transiently transfected with an empty (i.e. pcDNA6) or a p53 expression vectors along with an indicated shRNA (i.e. shControl or shHEXIM1). Cells were harvested 48–60 h post-transfection. Cells were further processed using the CycleTestTM plus DNA reagent kit (Becton Dickinson) according to the manufacturer's protocol. The transfected cells with higher fluorescence intensity of GFP were then gated and analyzed. Data were collected using LSR II flow cytometer (BD Biosciences) and analyzed with FlowJo (TreeStar) to determine the cell cycle status.
RESULTS
HEXIM1 Interacts with p53
Interactions between HEXIM1, NPM, and HDM2 indicate a possible connection between HEXIM1 and the p53 signaling pathway (25, 26). We first examined the association between p53 and HEXIM1 in MCF7 cells by IP using an anti-HEXIM1 antibody. Interaction between the endogenous HEXIM1 and p53 proteins was detected (Fig. 1A). It was noted that the HEXIM1-p53 interaction significantly increased after cells were exposed to UV radiation, which enhanced the protein levels of p53 but showed little effects on HEXIM1 (Fig. 1A). Association between HEXIM1 and CDK9 was significantly inhibited by UV treatment as previously reported (12). Reciprocal IP was carried out using an anti-p53 antibody and HEXIM1 was detected in the immunoprecipitated complexes, confirming the interaction between p53 and HEXIM1 (Fig. 1B). However, no interaction between p53 and CDK9 was detected (Fig. 1B). We next performed immunofluorescence to examine the subcellular localization of HEXIM1 and p53 under UV radiation. Without UV treatment, little HEXIM1 was found to co-localize with p53 (supplemental Fig. S1). Increased co-localization of HEXIM1 and p53 was observed 1, 3, and 6 h after UV treatment (supplemental Fig. S1). The p53-HEXIM1 interaction in primary cells was also examined. The association between endogenous p53 and HEXIM1 proteins was detected in primary human foreskin fibroblasts by IP (supplemental Fig. S2). We further performed in vitro binding assays using purified p53 and HEXIM1 proteins and demonstrated the direct protein-protein interaction between p53 and HEXIM1 (supplemental Fig. S3).
FIGURE 1.
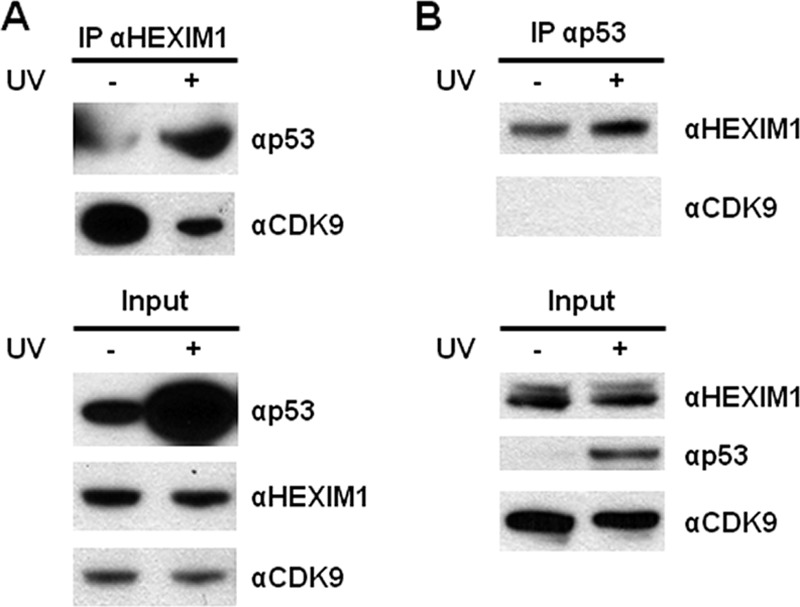
HEXIM1 interacts with p53. MCF7 cells were treated with or without UV radiation. Lysates of the cells were analyzed by IPs using the antibodies against HEXIM1 (A) and p53 (B), followed by Western blotting to examine the interactions between endogenous HEXIM1 and p53. Associations between endogenous HEXIM1/CDK9 (A) and p53/CDK9 (B) proteins were also examined.
A series of HEXIM1 truncations tagged with FLAG or YFP were utilized for the domain study of HEXIM1 (Fig. 2A). HEK293 cells were transfected with an individual HEXIM1 truncation plasmid followed by IP using an anti-p53 antibody. To confirm the specific binding between p53 and the full-length HEXIM1, a normal IgG was used as a negative control for the IP (Fig. 2B). Besides the full-length HEXIM1, only HEXIM1(120–359) was found to interact with p53 (Fig. 2B). It was noted that neither N-terminal (amino acids (a.a.) 1–180) nor C-terminal (a.a. 180–359) regions of HEXIM1 associated with p53 (Fig. 2B). In addition, deletion of the BR (a.a. 150–180) abolished the interaction between HEXIM1 and p53 (Fig. 2B) and a weak interaction between BR and p53 was observed (Fig. 2C). These results suggested that the BR was required to cooperate with the C terminus of HEXIM1 for sufficient p53 binding. We next assessed the requirement of 7SK snRNA for the p53-HEXIM1 interaction. The 7SK snRNA binding motif, KHRR, is located within the BR (a.a. 152–156). The HEXIM1 ILAA mutant, in which KHRR residues are replaced with ILAA, is unable to associate with 7SK snRNA (30). As shown in Fig. 2D, the HEXIM1 ILAA mutant did not exhibit significant changes in p53 binding, indicating that 7SK snRNA was not involved in the p53-HEXIM1 interaction.
FIGURE 2.

Domain study. A, diagram of HEXIM1 truncations. The HEXIM1 constructs were FLAG- or YFP-tagged, while the HEXIM1-ILAA mutant was not tagged. The p53 binding data are summarized on the right. ID, inhibitory domain; BR, basic region; AR, acidic region; DD, dimerization domain. B, HEK293 cells were transiently transfected with the indicated FLAG-tagged HEXIM1 plasmids. Lysates of the transfected cells were immunoprecipitated with an anti-p53 antibody or a normal IgG (as a negative control). The precipitated protein complexes were analyzed by immunoblotting using an anti-FLAG antibody. C, HEK293 cells were transfected with YFP, BR-YFP, or HEXIM1-YFP plasmids. IPs were performed using the lysates of the transfected cells and an anti-p53 antibody. The immunoprecipitated complexes were analyzed by Western blotting using an anti-GFP antibody, which recognized YFP as well. HC, the heavy chain of anti-p53 antibody. D, cell lysates prepared from the HEXIM1 WT or HEXIM1 ILAA-overexpressing HEK293 cells were utilized for IP analyses. IPs were carried out using an anti-HEXIM1 antibody. The immunoprecipitated protein complexes were analyzed by Western blotting using an anti-p53 antibody. E, diagram of p53 deletions. The HEXIM1 binding data are summarized on the right. TA, transactivation domain; PR, proline-rich region; DBD, DNA binding domain; OLI, oligomerization domain; NEG, negative regulation domain. F, HEK293 cells were transiently transfected with an indicated p53 plasmid. Lysates of the transfected cells were analyzed by IPs with an anti-HEXIM1 antibody or a normal IgG, followed by immunoblotting using an anti-p53 antibody.
The domain of p53 required for HEXIM1 binding was investigated next. HEK293 cells were transfected with the indicated p53 deletion plasmids followed by IP and Western blotting (Fig. 2E). The IP with a normal IgG was utilized as a negative control (Fig. 2F). Both p53(1–369) and p53(1–324) did not interact with HEXIM1, suggesting that the oligomerization (OLI; a.a. 324–368) and the negative regulation (NEG; a.a. 369–393) domains were required for binding (Fig. 2F). However, the OLI-NEG regions (i.e. a.a. 293–393) failed to interact with HEXIM1 (Fig. 2F). Only the transactivation (TA) domain-less p53 truncation (a.a. 45–393) was found to bind to HEXIM1 (Fig. 2F). We conclude that the NEG region is essential for HEXIM1 binding and the NEG region needs to coordinate with other domains, including OLI, proline-rich (PR), and DNA binding domains (DBD), to ensure efficient association with HEXIM1.
Overexpression of HEXIM1 Increases p53 Protein Levels and Stimulates p53 Activation
To determine the functional importance of the p53-HEXIM1 interaction, effects of HEXIM1 overexpression were investigated. A significant increase in the p53 level was seen in the HEXIM1-transfected MCF7 cells (Fig. 3A, lanes 1 and 3). After UV treatment, ectopic expression of HEXIM1 did not show significant effects on p53 (Fig. 3A, lanes 2 and 4). HEXIM1 is known as a transcription inhibitor and a key regulator of P-TEFb. We thus examined if HEXIM1 regulated the expression of p53 at transcriptional level. RNAs isolated from the HEXIM1-transfected cells were analyzed by QRT-PCR, showing that overexpression of HEXIM1 decreased the mRNA levels of p53 (Fig. 3B). On the other hand, IP analysis showed the increased interaction between HEXIM1 and p53 in the HEXIM1-overexpressing cells (Fig. 3C, lanes 1 and 3). Taken together, these results suggest that HEXIM1 may regulate p53 at the protein level and enhance p53 stability through protein-protein interaction.
FIGURE 3.

Overexpression of HEXIM1 stabilizes and activates p53. A, HEXIM1-transfected MCF7 cells were treated with or without UV. Cells transfected with an empty vector were used as the negative control. Expression of p53 and HEXIM1 proteins was analyzed by immunoblotting. Actin was used as the loading control. B, mRNA levels of p53 in the HEXIM1-transfected cells were measured by QRT-PCR. The cells transfected with an empty plasmid were used as a control. C, cell lysates prepared from the mock- and HEXIM1-transfected MCF7 cells were analyzed by IP. MG132 was added into the culture before harvesting. IP with a normal IgG was used as a negative control to exhibit the nonspecific background bindings. D, effects of HEXIM1 overexpression on the phosphorylation of p53 and the expression of p53 downstream targets were analyzed by Western blotting. Actin and p27 were used as controls.
The impact of HEXIM1 overexpression on p53 was further investigated. P-TEFb has been shown to phosphorylate p53 at Ser-33, Ser-315, and Ser-392 (35, 36). Phosphorylation of Ser-33, Ser-315, and Ser-392 enhances the DNA binding ability and induces transactivation of p53 (37–39). Overexpression of HEXIM1 maintained the phosphorylation of p53 at Ser-33 and Ser-392, but failed to do so on the phosphorylation of Ser-315, as the protein level of p53 increased (Fig. 3D). As expected, overexpression of HEXIM1 resulted in up-regulation of p53 downstream targets, p21 and Puma (Fig. 3D). No effects on p27, another tumor suppressor protein which is not regulated by p53 (40), were detected (Fig. 3D). These findings demonstrate that HEXIM1 specifically regulates p53 and its signaling pathway.
HEXIM1 Decreases the Ubiquitination of p53 by Disrupting the Interaction between p53 and HDM2
The domain study demonstrated an essential role of p53 NEG region in HEXIM1 binding (Fig. 2, E and F). The NEG domain is important for p53 degradation since the lysine residues required for the ubiquitination of p53 are located within this region (41). Therefore, we hypothesized that the HEXIM1 binding might interfere with the ubiquitination of p53 by HDM2. We co-transfected MCF7 cells with the indicated expression vectors, including His-Ub, HDM2, and HEXIM1. His-tagged ubiquitinated proteins were captured by Ni-NTA and analyzed by Western blotting (Fig. 4A). Significant decreases in p53 ubiquitination were observed in the cells with high expressing levels of HEXIM1 (Fig. 4A; compare the ubiquitinated p53 in lane 2 to lanes 4/5, and lane 3 to lanes 6/7). A decrease in the endogenous ubiquitination of p53 was also detected when HEXIM1 was overexpressed (supplemental Fig. S4). To ensure the counter effect of HEXIM1 on the HDM2-mediated p53 protein degradation, we co-transfected cells with HDM2, HEXIM1, and GFP expression vectors in the indicated combinations. The GFP plasmid was included in all transfections as an internal control to normalize transfection efficiency (Fig. 4B). A significant decrease in the p53 level was detected in the HDM2-expressing cells (Fig. 4B; compare lane 1 to lane 3). When HEXIM1 was co-expressed, HDM2-mediated p53 degradation was alleviated as the expression levels of HEXIM1 increased (Fig. 4B; compare lane 3 to lanes 4, 5, and 6). Influence of HEXIM1 overexpression on the protein stability of p53 was also investigated. The mock- and HEXIM1-transfected cells were treated with cycloheximide, and the p53 protein levels were monitored 1–3 h after the compound treatment. The results indicated that the half-life of endogenous p53 in the HEXIM1-transfected cells (i.e. 2.04 h) was much longer than that in the mock-transfected cells (i.e. 0.88 h) (Fig. 4C and supplemental Fig. S5).
FIGURE 4.

Overexpression of HEXIM1 inhibits p53 ubiquitination and disrupts the interaction between p53 and HDM2. A, MCF7 cells were co-transfected with the His-Ub, HDM2, and HEXIM1 expression vectors as indicated. To prevent the degradation of ubiquitinated p53, MG132 was added into the culture before harvesting. The cell lysates prepared from the transfected cells were incubated with Ni-NTA-agarose to capture His-Ub-conjugated proteins. Elutions from the Ni resin were analyzed by Western blot using an anti-p53 antibody. The expression of His-Ub, HDM2, HEXIM1, p53, and actin was examined. B, MCF7 cells were transfected with HDM2, HEXIM1, and GFP expression vectors in the indicated combinations. The GFP plasmid was included in all transfections to normalize the transfection efficiency. The quantitation of p53 protein is presented in percentage after normalizing with GFP proteins. C, MCF7 cells were transfected with an empty vector or a HEXIM1 expression plasmid. Mock- and HEXIM1-transfected cells were incubated with 100 μg/ml cycloheximide (CHX) for 0, 1, 2, and 3 h. The quantitation of p53 is presented in percentage after normalizing with actin. D, cell lysates of the mock- and HEXIM1-transfected MCF7 cells were used to analyze the association between p53 and HDM2 by IPs. MG132 was included in the culture before harvesting. The amounts of p53 and HDM2 proteins immunoprecipitated by an anti-HDM2 antibody were quantified as described under “Experimental Procedures.” The HDM2 protein precipitated in the IP against HDM2 was used as the input to normalize the amount of p53 protein detected in the IP. Reverse IP using an anti-p53 antibody was carried out and the same quantitative methods were utilized to examine the changes of p53-HDM2 interaction caused by HEXIM1 overexpression. The experiments were performed at least twice and similar results were obtained.
Effect of HEXIM1 overexpression on the p53-HDM2 interaction was examined next. The lysates prepared from the HEXIM1-transfected cells were divided into two halves and analyzed by IPs with anti-HDM2 and anti-p53 antibodies, respectively. In the IP assay using an anti-HDM2 antibody, the amounts of p53 and HDM2 proteins in the anti-HDM2 immunoprecipitated complexes were quantified. To determine the changes in the p53-HDM2 interaction, the immunoprecipitated HDM2 proteins by an anti-HDM2 antibody were used to normalize the amounts of precipitated p53 proteins in the same IP (Fig. 4D). A 36% decrease in the interaction between p53 and HDM2 was found when HEXIM1 was overexpressed (Fig. 4D). Reciprocal IP was performed using an anti-p53 antibody. Amounts of the precipitated p53 and HEXIM1 proteins in the anti-p53 IPs were quantified and normalized as described earlier. A similar result was obtained showing a 35% decrease in p53-HDM2 interaction (Fig. 4D). Collectively, these results suggest that HEXIM1 enhances p53 stability by blocking the HDM2-mediated p53 protein degradation.
We next investigated the effect of HEXIM1 on p53 in a different cancer cell line. Using a colorectal carcinoma cell line, HCT116, we detected an increased p53-HEXIM1 interaction and decreased endogenous ubiquitination of p53 in the HEXIM1-overexpressing cells (supplemental Fig. S6A). In addition, overexpression of HEXIM1 also significantly disrupted the interaction between p53 and HDM2 in HCT116 cells (supplemental Fig. S6B). Collectively, our results indicate that the effects of HEXIM1 on p53 protein stability may not be cell-specific.
Elevated Protein Levels of p53 Are Accompanied with an Increased p53-HEXIM1 Interaction
Besides UV and DNA damage compounds (doxorubicin and etoposide), induction of p53 can also be achieved by the treatment of CDK compounds (flavopiridol, DRB, roscovitine), transcription inhibitors (actinomycin D), and HDM2 antagonists (nutlin-3) (42–47). Changes in the p53-HEXIM1 interaction during p53 induction were examined using MCF7 cells. Higher p53 levels were observed in the treated cells as expected (Fig. 5A). Treatment of UV and DRB is known to disrupt the formation of large P-TEFb complexes and release free HEXIM1 from the association with CDK9/cyclin T1 (11, 48). Besides UV and DRB, incubation with flavopiridol, roscovitine, and actinomycin D also disrupted the association between CDK9/cyclin T1 and HEXIM1 (Fig. 5A). Most importantly, the p53-HEXIM1 interaction was significantly increased under all of these treatments (Fig. 5A). We further examined the impact of doxorubicin, etoposide, and nutlin-3 on the p53-HEXIM1 interaction. Increases in p53 protein levels were detected, while the association between HEXIM1 and CDK9 remained unchanged (Fig. 5B). Again, incubations of doxorubicin, etoposide, and nutlin-3 all enhanced the binding between p53 and HEXIM1 (Fig. 5B), reconfirming a potential role for HEXIM1 to enhance the stability of p53.
FIGURE 5.

Functional connection between p53 induction and p53-HEXIM1 interaction. A, MCF7 cells were treated with UV, flavopiridol (i.e. Flavo, 1 μm), DRB (100 μm), roscovitine (i.e. Ros, 20 μm), or actinomycin D (i.e. ActD, 30 nm). The DMSO-treated cells were used as controls since all compounds were dissolved in DMSO. The HEXIM1-p53 and HEXIM1-CDK9 associations were determined by IP using an anti-HEXIM1 antibody (i.e. αH1), followed by Western analysis. The normal IgG was used as the control for IP experiments. The protein levels of p53, HEXIM1, and CDK9 in cells were also examined. B, after incubated with doxorubicin (i.e. Dox, 1 μm), etoposide (i.e. Etop, 10 μm), or nutlin-3 (i.e. Nut, 10 μm), the cell lysates prepared from the treated MCF7 cells were analyzed by IP, followed by Western blotting. C, AML2 cells were incubated with flavopiridol (i.e. Flavo, 1 μm) or nutlin-3 (i.e. Nut, 30 μm) and the interaction between p53 and HEXIM1 was analyzed by IP. The protein levels of p53 and HEXIM1 in the cell lysates were also examined. Flavo, DRB, and Ros are CDK-inhibiting compounds; ActD, transcription inhibiting compound; Dox and Etop, DNA damage compounds; Nut, HDM2 antagonist.
A similar set of experiments was carried out using an acute myeloid leukemia cell line, AML2, and the interaction between endogenous p53 and HEXIM1 was detected (Fig. 5C). In addition, cells were incubated with flavopiridol and nutlin-3 to induce p53. We again observed increases in p53 levels and p53-HEXIM1 interactions in the treated cells (Fig. 5C). A lower HEXIM1 protein level was detected in the flavopiridol-treated AML2 cells; however, it did not affect the increases in the p53-HEXIM1 interaction and p53 protein level (Fig. 5C). Taken together, these results demonstrate the positive correlation between p53 protein stability and the p53-HEXIM1 association in two different cancer cell lines.
Knockdown of HEXIM1 Inhibits Induction of p53
HEXIM1 knockdown assay was performed to examine the impact of HEXIM1 on p53. We obtained four different shRNAs targeting HEXIM1 and examined the effectiveness of the shRNAs by Western blot. Only one HEXIM1 shRNA showing consistent and efficient knockdown of HEXIM1 protein was chosen for the following knockdown experiments (supplemental Fig. S7, #1 shRNA). MCF7 cells were transfected with a nonspecific control shRNA or the selected HEXIM1 shRNA. In agreement with the results of HEXIM1 overexpression assays (Fig. 3D), knockdown of HEXIM1 decreased the protein levels of p53 as well as its downstream target, p21, while no effects were found in p27 (Fig. 6A). A parallel set of experiments was performed with MG132 treatment. Incubation of MG132 recovered the decreased p53 protein level caused by HEXIM1 knockdown, and furthermore, the increased protein level of p53 was much higher than the level found in the control (Fig. 6A, compare lane 4 to lane 1). These data suggest that HEXIM1 may be one of the cellular factors involved in the regulation of p53 protein turnover.
FIGURE 6.

Knockdown of HEXIM1 inhibits the induction of p53. A, MCF7 cells were transiently transfected with a control (i.e. shCon) or a HEXIM1 (i.e. shH1) shRNA in the absence or presence of MG132. Expression of the indicated proteins was analyzed by Western blotting. Actin and p27 were used as negative controls. B, MCF7 cells were transiently transfected by an indicated shRNA plasmid. Two days after transfection, cells were incubated with 1 μm of doxorubicin (Dox) or flavopiridol (Flavo) for 3 h before harvesting. The DMSO-treated cells were used as controls since both Dox and Flavo were dissolved in DMSO. Expression of p53, HEXIM1, Puma, p21, p27, and actin proteins was analyzed by immunoblotting. Actin and p27 were used as controls.
To examine the effects of HEXIM1 knockdown on the action of p53-inducing compounds, MCF7 cells were first transfected with the selected HEXIM1 shRNA, followed by doxorubicin or flavopiridol treatment. Compared with the control (i.e. cells transfected with a nonspecific shRNA), significant lower protein levels of p53 and its downstream targets, Puma and p21, were detected in the HEXIM1 knockdown cells under the treatment of doxorubicin and flavopiridol (Fig. 6B). No changes in the p27 protein levels were observed (Fig. 6B). These results further confirm the function of HEXIM1 as a positive regulator of p53.
HEXIM1 Regulates the p53-dependent Cell Cycle Arrest and Activation of Caspases 3/7
The impact of HEXIM1 knockdown on the p53-mediated cell arrest was studied next. HCT116 p53+/+ cells were transfected with the indicated combinations of vectors to overexpress p53 or knock down HEXIM1 (Fig. 7). Effectiveness of HEXIM1 knockdown and p53 overexpression in the transfected cells was examined and confirmed by immunoblotting (supplemental Fig. S8). Little effects on cell cycle were observed when HEXIM1 was knocked down by the HEXIM1 shRNA (Fig. 7; compare pcDNA6 + shHEXIM1 to pcDNA6 + shControl). Ectopic expression of p53 increased cell cycle arrest in S and G2/M phases (Fig. 7; compare p53 + shControl to pcDNA6 + shControl). Such p53-induced arrestation of cells were significantly reversed when HEXIM1 was knocked down in the p53-overexpressing cells (Fig. 7; compare p53 + shHEXIM1 to p53 + shControl), suggesting the functional importance of the p53-HEXIM1 interaction in the regulation of the p53-dependent cell cycle arrest.
FIGURE 7.

Knockdown of HEXIM1 releases the cell cycle arrest induced by p53. HCT116 cells were transiently transfected with the indicated expression (i.e. p53 or an empty vector, pcDNA6) and shRNA (shControl or shHEXIM1) plasmids. The transfected GFP+ cells were gated and analyzed for cell cycle status by FACS as described under “Experimental Procedures.” Similar observations were obtained in at least two independent experiments.
Caspases 3 and 7 are the key regulators of p53-mediated apoptosis (49). To assess the involvement of HEXIM1 in p53-dependent apoptosis, HCT116 p53+/+ and p53−/− cells were transfected with a HEXIM1 expression vector. Up to a 60% increase in the caspase activity was detected in HCT116 p53+/+ cells when HEXIM1 was overexpressed (supplemental Fig. S9). However, no changes in caspase activities were observed in the HEXIM1-overexpressing HCT116 p53−/− cells (supplemental Fig. S9). These results suggest that overexpression of HEXIM1 can induce p53-mediated apoptosis.
HEXIM1 Transcriptionally Regulates the Expression of p53 Target Genes in a p53-dependent Manner
Our data suggested that HEXIM1 up-regulated the downstream targets of p53, such as Puma and p21, by enhancing the protein stability of p53. To test our hypothesis, we examined the impact of HEXIM1 on the expression of the p53 target genes in a p53-null background. HCT116 p53−/− cells were co-transfected with a Luc reporter (i.e. Puma-Luc, p21-Luc, or p53-Luc) and an indicated expression vector (i.e. HEXIM1 or an empty vector). Expression of Luc in the Puma-Luc and p21-Luc reporter is driven by the Puma and p21 promoters, respectively (32, 33). The reporter p53-Luc, also known as PG13-Luc, contains 13 copies of the p53-binding consensus sequence (34). A parallel experiment was carried out using HCT116 p53+/+ cells. Overexpression of HEXIM1 resulted in 50–80% increases in Luc expression of the reporter plasmids in the p53+/+ background (Fig. 8A). On the contrary, no activation of the p53-dependent transcription was detected in the HEXIM1-transfected HCT116 p53−/− cells (Fig. 8A). A similar set of experiments were carried out to examine the effects of HEXIM1 knockdown on the p53 target genes. In the HCT116 p53+/+ cells, 30–55% decreases in Luc expression of the reporter plasmids were caused by HEXIM1 knockdown (Fig. 8B). However, knockdown of HEXIM1 failed to inhibit the p53-dependent transcription in the p53−/− background (Fig. 8B). Taken together, we conclude that HEXIM1 regulates the expression of p53 target genes at transcriptional level and in a p53-dependent fashion.
FIGURE 8.

HEXIM1 regulates the transcription of p53 target genes in a p53-dependent fashion. HCT116 p53+/+ and p53−/− cells were transfected with a Luc reporter and an indicated expression vector for the overexpression experiments (A) or an indicated shRNA plasmid for the knockdown assays (B). The total amounts of plasmids transfected were kept constant by adjusting the mock vector. Luciferase activity was determined 2 days post-transfected as described under “Experimental Procedures.”
DISCUSSION
Besides binding to CDK9/cyclin T1, HEXIM1 has been shown to associate with ERα, MyoD, histone deacetylases, importin α, HDM2, NPM, NF-κB, and glucocorticoid receptors (17, 21, 25, 26, 50–52). Here, we identify p53 as a novel HEXIM1 binding protein through direct interaction with HEXIM1 (Fig. 1 and supplemental Fig. S3). No interaction between CDK9 and p53 is detected (Fig. 1B). Our results contradict to a previous report demonstrating that p53 binds to CDK9, but not HEXIM1 (53). In the report, Napolitano et al. transiently transfected H1299 (a non-small cell lung cancer cell line) and U2OS (an osteosarcoma cell line) cells with p53 and cyclin T1 plasmids to overexpress both proteins, followed by IP analyses (53). Instead, our study aims to examine the interaction between the endogenous p53 and HEXIM1 proteins in MCF7 (a breast cancer cell line) cells (Fig. 1). The p53-HEXIM1 interaction is also seen in the domain studies using HEK293 (an embryonic kidney cell line) cells (Fig. 2). Furthermore, the association between endogenous p53 and HEXIM1 proteins is also detected in AML2 (an acute myeloid leukemia cell line) and HCT116 (a colorectal carcinoma cell line) cells (Fig. 5C and supplemental Fig. S6A), as well as primary human foreskin fibroblasts (supplemental Fig. S2). Still, since different cell lines were used in these studies, we could not rule out the possibility that the observed p53-HEXIM1 interaction might be cell-specific.
HEXIM1, also known as EDG1, was identified from a cDNA library of MCF7 cells by yeast two-hybrid screenings as an ERα-binding protein and found to play an important role in ERα-induced gene expression (16, 17). Involvement of HEXIM1 in breast cancers has been reported. Lower expression levels of HEXIM1 were detected in breast tumor tissues when compared with the adjacent normal breast tissues in all tumor grades (16). Furthermore, a recent study demonstrates HEXIM1 as a critical factor for cancer recurrence in breast cancer patients treated with tamoxifen, a selective ER modulator used for treatment of advanced stage breast cancers (18). Our results presented here further emphasize the importance of HEXIM1 in breast cancers. Furthermore, the interaction between p53 and HEXIM1 was also detected in an acute myeloid leukemia cell line (AML2; Fig. 5C) and a colorectal carcinoma cell line (HCT116; supplemental Fig. S6A), suggesting a potential role for HEXIM1 in other cancers.
The IP analysis showed that p53 interacted with the “free” HEXIM1, instead of the “CDK9/cyclin T1-bound” HEXIM1 (Fig. 1B). Domain study indicated that the BR, acidic region (AR), and dimerization domain (DD) of HEXIM1 were essential for the p53 interaction (Fig. 2, A and B). Furthermore, the HEXIM1 ILAA mutant, which fails to associate with 7SK snRNA and to form protein complex with CDK9/cyclin T1, is still able to interact with p53 (Fig. 2D). These results demonstrate that HEXIM1 interacts with p53 in a 7SK snRNA-independent manner and not in the large P-TEFb form. The AR of HEXIM1 has been shown to interact with the adjacent BR in the absence of 7SK snRNA (31). Since the P-TEFb binding motif, PYNT (a.a. 202–205), is located between the BR (a.a. 150–180) and AR (a.a. 210–250), it has been proposed that the BR-AR interaction establishes a “close” conformation and prevents the association with CDK9/cyclin T1 (30, 31). The DD of HEXIM1 is responsible for the formation of HEXIM1 homodimers (54). It remains to be determined if the association between HEXIM1 and p53 may interfere with the homodimerization of HEXIM1. Collectively, our data suggest that the 7SK snRNA-free HEXIM1 binds to p53 in the monomeric or homodimeric “close” forms.
The PR, DBD, OLI, and NEG regions of p53 are required for the stable interaction with HEXIM1 (Fig. 2, E and F). Notably, no interaction between p53 and HEXIM1 was detected when the NEG region (a.a. 370–393) was deleted (Fig. 2, E and F), demonstrating an essential role of the NEG domain for the interaction. The NEG domain contains six lysine residues, including Lys-370, -372, -373, -381, -382, and -386, which are ubiquitinated by HDM2 (41). Therefore, through the interaction with the NEG domain, HEXIM1 could block the ubiquitination by HDM2 and enhance the protein stability of p53. This hypothesis is supported by the data showing that overexpression of HEXIM1 increased the protein levels of p53 and decreased the ubiquitination of p53 (Figs. 3A and 4A). Recently, Poyurovsky et al. showed that the C-terminal region of p53 (367–393 a.a.) was required for the interaction with HDM2. The p53-HDM2 interaction was significantly decreased when the C-terminal 30 a.a. of p53 was deleted (55). In this study, we unraveled that overexpression of HEXIM1 disrupted the p53-HDM2 interaction (Fig. 4D and supplemental Fig. S6B). Taken together, we conclude that HEXIM1 may compete with HDM2 in binding to the NEG domain of p53 and protect p53 from being ubiquitinated by HDM2.
It has been reported that P-TEFb phosphorylates p53 on Ser-33, -315, and -392 (35, 36). Phosphorylation of p53 at these three serine residues is known to enhance the DNA binding and transactivating abilities of p53 (37–39, 56, 57). We investigated the effects of HEXIM1 on the phosphorylation of Ser-33, -315, and -392. In the HEXIM1-overexpressing cells, phosphorylation of p53 at Ser-33 and Ser-392 increased proportionally with the protein level of p53 (Fig. 3D). In contrast, overexpression of HEXIM1 failed to maintain the phosphorylation of p53 at Ser-315 as the protein level of p53 increased (Fig. 3D). Increasing in the p53 protein level and maintaining the p53 phosphorylation at Ser-33 and Ser-392 help to explain the activation of the p53 target genes, such as p21 and Puma, by HEXIM1 overexpression (Fig. 3D). In addition, we performed the cell-based reporter assays using HCT116 p53 wild-type and knock-out cells, and demonstrated that HEXIM1 regulated the transcription of p21 and Puma in a p53-dependent manner (Fig. 8). We did not detect the association between p53 and P-TEFb by IP (Fig. 1B). However, this is expected since the binding between the kinase (i.e. CDK9), and its substrate (i.e. p53) could be very weak and temporary. Therefore, IP analysis may not be the ideal approach for this kind of interaction. The p53-P-TEFb and p53-HEXIM1 interactions may be two independent events. It is possible that P-TEFb may phosphorylate p53 first. After dissociating with P-TEFb, the phosphorylated p53 binds to HEXIM1, which further stabilizes the phosphorylation of p53 at Ser-33 and Ser-392.
Treatment with UV radiation, CDK inhibiting (flavopiridol, DRB, roscovitine), transcription inhibiting (actinomycin D), and p53 inducing compounds (doxorubicin, etoposide, and nutlin-3) results in the induction of p53 in cells (42–47, 58). Among the compounds, doxorubicin and etoposide are anti-cancer drugs, while flavopiridol, roscovitine, and nutlin-3 are currently being evaluated as potential anti-cancer therapeutics (49, 59). We examined the p53-HEXIM1 interaction during these processes and found that all these treatments enhanced the association between p53 and HEXIM1 in MCF7 cells (Fig. 5, A and B). This finding was confirmed in another cancer cell line, AML2 (Fig. 5C). Furthermore, the induction of p53 by doxorubicin and flavopiridol was significantly inhibited when HEXIM1 was knocked down (Fig. 6B). Our findings suggest an important role of HEXIM1 in p53 induction through the interaction with p53. Besides increasing the p53 protein level, treatments of UV, flavopiridol, DRB, roscovitine, and actinomycin D disrupt the formation of large P-TEFb complexes (Fig. 5A, IPαHEXIM1, CDK9 immunoblotting), resulting in releasing more HEXIM1 from the large complexes. Such treatments should increase the pool of “free” HEXIM1 in cells and may contribute to the increase in the p53-HEXIM1 interaction. However, this hypothesis was ruled out by the results obtained from the treatment with the p53-inducing compounds. Incubation of doxorubicin, etoposide, and nutlin-3 was found to enhance the p53-HEXIM1 association without disturbing the equilibrium between large and small P-TEFb complexes (Fig. 5B, IPαHEXIM1, CDK9 immunoblotting). Previous studies showed significant amounts of HEXIM1 in cells did not associate with CDK9/cyclin T1, indicating that cells could have plenty of the “free” HEXIM1 to interact with p53 upon the stimulation (12, 48).
The role of HEXIM1 as a positive regulator of p53 is further supported by the HEXIM1 knockdown assays (Figs. 6A and 8B). Knockdown of the cellular HEXIM1 proteins by the HEXIM1 shRNA decreases the expression of p53 and its downstream targets (Figs. 6A and 8B), suggesting the possible involvement of HEXIM1 in p53-dependent cell cycle arrest. This hypothesis is confirmed by FACS analysis presented in Fig. 7, showing that the cell cycle arrest induced by p53 overexpression is alleviated when HEXIM1 is knocked down. We further demonstrated the involvement of HEXIM1 in the p53-dependent apoptosis using HCT116 p53+/+ and p53−/− cells (supplemental Fig. S9). The results of cell cycle analysis further strengthen the significance of the p53-HEXIM1 interaction in regulating the function of p53.
In our previous study, we showed that HDM2 bound and ubiquitinated HEXIM1 (26). Based on the previous data and the results presented here, we propose two mechanisms utilized by HEXIM1 to stabilize p53 protein. The proposed model is summarized in Fig. 9. First, HEXIM1 directly interacts with p53 by binding to the C-terminal region of p53 and protects p53 from being associated with and ubiquitinated by HDM2 (Figs. 1, 2, and supplemental Fig. S3). Secondly, HEXIM1 competes with p53 binding to HDM2 and is subsequently ubiquitinated by HDM2. This HEXIM1-HDM2 interaction protects a portion of p53 from being associated with and ubiquitinated by HDM2, and indirectly contributes to the stability of p53 (Fig. 4D and supplemental Fig. S6B) (26).
FIGURE 9.

Proposed model for the effect of p53-HEXIM1 interaction on the induction of p53. Our results suggest that two regulatory mechanisms are utilized by HEXIM1 to enhance the protein stability of p53: 1) HEXIM1 directly binds to p53 and protects p53 from being ubiquitinated by HDM2. The p53-HEXIM1 interaction can be induced by UV, CDK inhibitors, transcription inhibitors, and p53 inducers. 2) HEXIM1 binds to HDM2 and is subsequently ubiquitinated by HDM2. Therefore, this HEXIM1-HDM2 interaction indirectly protects a portion of p53 from being ubiquitinated by HDM2. Flavo, flavopiridol; Ros, roscovitine; ActD, actinomycin D; Dox, doxorubicin; Etop, etoposide; Nut, nutlin-3.
In summary, our results reveal a novel role of HEXIM1 in regulation of p53 and suggest a molecular mechanism of p53 activation induced by the anti-cancer drugs/compounds through the interaction with HEXIM1. This new discovery should help us better in understanding on the molecular actions of the p53-inducing agents and may lead to the development of new strategies for cancer therapy. Future studies are required to further investigate the functional interactions between HEXIM1 and other p53 regulators, as well as the involvement of HEXIM1 in breast and other cancers.
Acknowledgments
We thank Dr. B. Matija Peterlin, Dr. Olivier Bensaude, and Dr. David P. Lane for providing plasmids, Dr. Bert Vogelstein for supplying HCT116 cell lines and luciferase reporter plasmids, Dr. Mark Stinski for providing primary human foreskin fibroblasts, Dr. Zhiwei Song for critical review of the manuscript, and Chuan Hao Tan and Vikneswari Rajasegaran for expert technical assistance.
This work is supported by the Agency for Science, Technology, and Research (A*STAR), Singapore.

This article contains supplemental Figs. S1–S9.
- P-TEFb
- positive transcription elongation factor b
- HEXIM1
- hexamethylene bisacetamide-inducible protein 1
- HDM2
- human double minute-2 protein
- RNA Pol II
- RNA polymerase II
- DRB
- 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole
- CDK9
- cyclin-dependent kinase 9
- snRNA
- small nuclear RNA
- EDG1
- estrogen down-regulated gene 1
- ERα
- estrogen receptor α
- AML
- acute myeloid leukemia
- NPM
- nucleophosmin
- shRNA
- short hairpin RNA
- YFP
- yellow fluorescent protein
- BR
- basic region
- Luc
- luciferase
- His-Ub
- histidine-tagged ubiquitin
- FACS
- fluorescence-activated cell sorting
- IP
- immunoprecipitation
- DMSO
- dimethyl sulfoxide
- QRT-PCR
- quantitative reverse transcriptase-polymerase chain reaction
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- OLI
- oligomerization
- NEG
- negative regulation
- TA
- transactivation
- PR
- proline-rich
- DBD
- DNA binding domains
- AR
- acidic region
- DD
- dimerization domain.
REFERENCES
- 1. Peterlin B. M., Price D. H. (2006) Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 23, 297–305 [DOI] [PubMed] [Google Scholar]
- 2. Pumfery A., de la Fuente C., Berro R., Nekhai S., Kashanchi F., Chao S. H. (2006) Potential use of pharmacological cyclin-dependent kinase inhibitors as anti-HIV therapeutics. Curr. Pharm. Des. 12, 1949–1961 [DOI] [PubMed] [Google Scholar]
- 3. Zhu Y., Pe'ery T., Peng J., Ramanathan Y., Marshall N., Marshall T., Amendt B., Mathews M. B., Price D. H. (1997) Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 11, 2622–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chao S. H., Fujinaga K., Marion J. E., Taube R., Sausville E. A., Senderowicz A. M., Peterlin B. M., Price D. H. (2000) Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J. Biol. Chem. 275, 28345–28348 [DOI] [PubMed] [Google Scholar]
- 5. Chao S. H., Price D. H. (2001) Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J. Biol. Chem. 276, 31793–31799 [DOI] [PubMed] [Google Scholar]
- 6. Guenther M. G., Levine S. S., Boyer L. A., Jaenisch R., Young R. A. (2007) A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130, 77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zeitlinger J., Stark A., Kellis M., Hong J. W., Nechaev S., Adelman K., Levine M., Young R. A. (2007) RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat. Genet. 39, 1512–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Muse G. W., Gilchrist D. A., Nechaev S., Shah R., Parker J. S., Grissom S. F., Zeitlinger J., Adelman K. (2007) RNA polymerase is poised for activation across the genome. Nat. Genet. 39, 1507–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Peng J., Zhu Y., Milton J. T., Price D. H. (1998) Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 12, 755–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Graña X., De Luca A., Sang N., Fu Y., Claudio P. P., Rosenblatt J., Morgan D. O., Giordano A. (1994) PITALRE, a nuclear CDC2-related protein kinase that phosphorylates the retinoblastoma protein in vitro. Proc. Natl. Acad. Sci. U.S.A. 91, 3834–3838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nguyen V. T., Kiss T., Michels A. A., Bensaude O. (2001) 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 414, 322–325 [DOI] [PubMed] [Google Scholar]
- 12. Michels A. A., Nguyen V. T., Fraldi A., Labas V., Edwards M., Bonnet F., Lania L., Bensaude O. (2003) MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Mol. Cell Biol. 23, 4859–4869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang Z., Zhu Q., Luo K., Zhou Q. (2001) The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 414, 317–322 [DOI] [PubMed] [Google Scholar]
- 14. Yik J. H., Chen R., Nishimura R., Jennings J. L., Link A. J., Zhou Q. (2003) Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell 12, 971–982 [DOI] [PubMed] [Google Scholar]
- 15. Dey A., Chao S. H., Lane D. P. (2007) HEXIM1 and the control of transcription elongation: from cancer and inflammation to AIDS and cardiac hypertrophy. Cell Cycle 6, 1856–1863 [DOI] [PubMed] [Google Scholar]
- 16. Wittmann B. M., Wang N., Montano M. M. (2003) Identification of a novel inhibitor of breast cell growth that is down-regulated by estrogens and decreased in breast tumors. Cancer Res. 63, 5151–5158 [PubMed] [Google Scholar]
- 17. Wittmann B. M., Fujinaga K., Deng H., Ogba N., Montano M. M. (2005) The breast cell growth inhibitor, estrogen down regulated gene 1, modulates a novel functional interaction between estrogen receptor α and transcriptional elongation factor cyclin T1. Oncogene 24, 5576–5588 [DOI] [PubMed] [Google Scholar]
- 18. Ketchart W., Ogba N., Kresak A., Albert J. M., Pink J. J., Montano M. M. (2011) HEXIM1 is a critical determinant of the response to tamoxifen. Oncogene 30, 3563–3569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fraldi A., Varrone F., Napolitano G., Michels A. A., Majello B., Bensaude O., Lania L. (2005) Inhibition of Tat activity by the HEXIM1 protein. Retrovirology 2, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sano M., Abdellatif M., Oh H., Xie M., Bagella L., Giordano A., Michael L. H., DeMayo F. J., Schneider M. D. (2002) Activation and function of cyclin T-Cdk9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat. Med. 8, 1310–1317 [DOI] [PubMed] [Google Scholar]
- 21. Ouchida R., Kusuhara M., Shimizu N., Hisada T., Makino Y., Morimoto C., Handa H., Ohsuzu F., Tanaka H. (2003) Suppression of NF-κB-dependent gene expression by a hexamethylene bisacetamide-inducible protein HEXIM1 in human vascular smooth muscle cells. Genes Cells 8, 95–107 [DOI] [PubMed] [Google Scholar]
- 22. Vogelstein B., Lane D., Levine A. J. (2000) Surfing the p53 network. Nature 408, 307–310 [DOI] [PubMed] [Google Scholar]
- 23. Brown C. J., Lain S., Verma C. S., Fersht A. R., Lane D. P. (2009) Awakening guardian angels: drugging the p53 pathway. Nat. Rev. Cancer 9, 862–873 [DOI] [PubMed] [Google Scholar]
- 24. Lew Q. J., Tan C. H., Gurumurthy M., Chu K. L., Cheong N., Lane D. P., Chao S. H. (2011) NPMc(+) AML cell line shows differential protein expression and lower sensitivity to DNA-damaging and p53-inducing anticancer compounds. Cell Cycle 10, 1978–1987 [DOI] [PubMed] [Google Scholar]
- 25. Gurumurthy M., Tan C. H., Ng R., Zeiger L., Lau J., Lee J., Dey A., Philp R., Li Q., Lim T. M., Price D. H., Lane D. P., Chao S. H. (2008) Nucleophosmin interacts with HEXIM1 and regulates RNA polymerase II transcription. J. Mol. Biol. 378, 302–317 [DOI] [PubMed] [Google Scholar]
- 26. Lau J., Lew Q. J., Diribarne G., Michels A. A., Dey A., Bensaude O., Lane D. P., Chao S. H. (2009) Ubiquitination of HEXIM1 by HDM2. Cell Cycle 8, 2247–2254 [DOI] [PubMed] [Google Scholar]
- 27. Falini B., Mecucci C., Tiacci E., Alcalay M., Rosati R., Pasqualucci L., La Starza R., Diverio D., Colombo E., Santucci A., Bigerna B., Pacini R., Pucciarini A., Liso A., Vignetti M., Fazi P., Meani N., Pettirossi V., Saglio G., Mandelli F., Lo-Coco F., Pelicci P. G., Martelli M. F. (2005) Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 352, 254–266 [DOI] [PubMed] [Google Scholar]
- 28. Stinski M. F. (1976) Human cytomegalovirus: glycoproteins associated with virions and dense bodies. J. Virol. 19, 594–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bunz F., Dutriaux A., Lengauer C., Waldman T., Zhou S., Brown J. P., Sedivy J. M., Kinzler K. W., Vogelstein B. (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497–1501 [DOI] [PubMed] [Google Scholar]
- 30. Michels A. A., Fraldi A., Li Q., Adamson T. E., Bonnet F., Nguyen V. T., Sedore S. C., Price J. P., Price D. H., Lania L., Bensaude O. (2004) Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J. 23, 2608–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barboric M., Kohoutek J., Price J. P., Blazek D., Price D. H., Peterlin B. M. (2005) Interplay between 7SK snRNA and oppositely charged regions in HEXIM1 direct the inhibition of P-TEFb. EMBO J. 24, 4291–4303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu J., Zhang L., Hwang P. M., Kinzler K. W., Vogelstein B. (2001) PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 7, 673–682 [DOI] [PubMed] [Google Scholar]
- 33. el-Deiry W. S., Tokino T., Velculescu V. E., Levy D. B., Parsons R., Trent J. M., Lin D., Mercer W. E., Kinzler K. W., Vogelstein B. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825 [DOI] [PubMed] [Google Scholar]
- 34. el-Deiry W. S., Kern S. E., Pietenpol J. A., Kinzler K. W., Vogelstein B. (1992) Definition of a consensus binding site for p53. Nat. Genet. 1, 45–49 [DOI] [PubMed] [Google Scholar]
- 35. Radhakrishnan S. K., Gartel A. L. (2006) CDK9 phosphorylates p53 on serine residues 33, 315, and 392. Cell Cycle 5, 519–521 [DOI] [PubMed] [Google Scholar]
- 36. Claudio P. P., Cui J., Ghafouri M., Mariano C., White M. K., Safak M., Sheffield J. B., Giordano A., Khalili K., Amini S., Sawaya B. E. (2006) Cdk9 phosphorylates p53 on serine 392 independently of CKII. J. Cell Physiol. 208, 602–612 [DOI] [PubMed] [Google Scholar]
- 37. Zheng H., You H., Zhou X. Z., Murray S. A., Uchida T., Wulf G., Gu L., Tang X., Lu K. P., Xiao Z. X. (2002) The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 419, 849–853 [DOI] [PubMed] [Google Scholar]
- 38. Zacchi P., Gostissa M., Uchida T., Salvagno C., Avolio F., Volinia S., Ronai Z., Blandino G., Schneider C., Del Sal G. (2002) The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature 419, 853–857 [DOI] [PubMed] [Google Scholar]
- 39. Hupp T. R., Meek D. W., Midgley C. A., Lane D. P. (1992) Regulation of the specific DNA binding function of p53. Cell 71, 875–886 [DOI] [PubMed] [Google Scholar]
- 40. Slingerland J., Pagano M. (2000) Regulation of the cdk inhibitor p27 and its deregulation in cancer. J. Cell Physiol. 183, 10–17 [DOI] [PubMed] [Google Scholar]
- 41. Rodriguez M. S., Desterro J. M., Lain S., Lane D. P., Hay R. T. (2000) Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol. Cell Biol. 20, 8458–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kwok T. T., Mok C. H., Menton-Brennan L. (1994) Up-regulation of a mutant form of p53 by doxorubicin in human squamous carcinoma cells. Cancer Res. 54, 2834–2836 [PubMed] [Google Scholar]
- 43. Fritsche M., Haessler C., Brandner G. (1993) Induction of nuclear accumulation of the tumor-suppressor protein p53 by DNA-damaging agents. Oncogene 8, 307–318 [PubMed] [Google Scholar]
- 44. Vassilev L. T., Vu B. T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C., Fotouhi N., Liu E. A. (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 [DOI] [PubMed] [Google Scholar]
- 45. Demidenko Z. N., Blagosklonny M. V. (2004) Flavopiridol induces p53 via initial inhibition of Mdm2 and p21 and, independently of p53, sensitizes apoptosis-reluctant cells to tumor necrosis factor. Cancer Res. 64, 3653–3660 [DOI] [PubMed] [Google Scholar]
- 46. Ljungman M., Zhang F., Chen F., Rainbow A. J., McKay B. C. (1999) Inhibition of RNA polymerase II as a trigger for the p53 response. Oncogene 18, 583–592 [DOI] [PubMed] [Google Scholar]
- 47. Ljungman M., Paulsen M. T. (2001) The cyclin-dependent kinase inhibitor roscovitine inhibits RNA synthesis and triggers nuclear accumulation of p53 that is unmodified at Ser-15 and Lys-382. Mol. Pharmacol. 60, 785–789 [PubMed] [Google Scholar]
- 48. Byers S. A., Price J. P., Cooper J. J., Li Q., Price D. H. (2005) HEXIM2, a HEXIM1-related protein, regulates positive transcription elongation factor b through association with 7SK. J. Biol. Chem. 280, 16360–16367 [DOI] [PubMed] [Google Scholar]
- 49. Shen H., Maki C. G. (2011) Pharmacologic activation of p53 by small-molecule MDM2 antagonists. Curr. Pharm. Des. 17, 560–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Galatioto J., Mascareno E., Siddiqui M. A. (2010) CLP-1 associates with MyoD and HDAC to restore skeletal muscle cell regeneration. J. Cell Sci. 123, 3789–3795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Czudnochowski N., Vollmuth F., Baumann S., Vogel-Bachmayr K., Geyer M. (2010) Specificity of Hexim1 and Hexim2 complex formation with cyclin T1/T2, importin α and 7SK snRNA. J. Mol. Biol. 395, 28–41 [DOI] [PubMed] [Google Scholar]
- 52. Shimizu N., Ouchida R., Yoshikawa N., Hisada T., Watanabe H., Okamoto K., Kusuhara M., Handa H., Morimoto C., Tanaka H. (2005) HEXIM1 forms a transcriptionally abortive complex with glucocorticoid receptor without involving 7SK RNA and positive transcription elongation factor b. Proc. Natl. Acad. Sci. U.S.A. 102, 8555–8560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Napolitano G., Varrone F., Majello B., Lania L. (2007) Activation of P-TEFb induces p21 leading to cell cycle arrest. Cell Cycle 6, 1126–1129 [DOI] [PubMed] [Google Scholar]
- 54. Li Q., Price J. P., Byers S. A., Cheng D., Peng J., Price D. H. (2005) Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. J. Biol. Chem. 280, 28819–28826 [DOI] [PubMed] [Google Scholar]
- 55. Poyurovsky M. V., Katz C., Laptenko O., Beckerman R., Lokshin M., Ahn J., Byeon I. J., Gabizon R., Mattia M., Zupnick A., Brown L. M., Friedler A., Prives C. (2010) The C terminus of p53 binds the N-terminal domain of MDM2. Nat. Struct. Mol. Biol. 17, 982–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bulavin D. V., Saito S., Hollander M. C., Sakaguchi K., Anderson C. W., Appella E., Fornace A. J., Jr. (1999) Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 18, 6845–6854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Blaydes J. P., Luciani M. G., Pospisilova S., Ball H. M., Vojtesek B., Hupp T. R. (2001) Stoichiometric phosphorylation of human p53 at Ser-315 stimulates p53-dependent transcription. J. Biol. Chem. 276, 4699–4708 [DOI] [PubMed] [Google Scholar]
- 58. Maltzman W., Czyzyk L. (1984) UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol. Cell Biol. 4, 1689–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Krystof V., Uldrijan S. (2010) Cyclin-dependent kinase inhibitors as anticancer drugs. Curr. Drug Targets 11, 291–302 [DOI] [PubMed] [Google Scholar]