

Background: Flt3 is an important regulator of hematopoiesis and is often found mutated and constitutively active in patients with acute myeloid leukemia.
Results: SOCS6 is up-regulated by Flt3 activation and binds to phosphorylated Flt3.
Conclusion: SOCS6 is a negative regulator of Flt3 signaling.
Significance: Our results provide a role for SOCS6 in Flt3 signaling. The absence of SOCS6 promotes transformation of cells by Flt3 ITD.
Keywords: Phosphorylation, Protein Degradation, Receptor Endocytosis, Receptor Tyrosine Kinase, Ubiquitination, Flt3, SOCS6
Abstract
The receptor tyrosine kinase Flt3 is an important growth factor receptor in hematopoiesis, and gain-of-function mutations of the receptor contribute to the transformation of acute myeloid leukemia. SOCS6 (suppressor of cytokine signaling 6) is a member of the SOCS family of E3 ubiquitin ligases that can regulate receptor tyrosine kinase signal transduction. In this study, we analyzed the role of SOCS6 in Flt3 signal transduction. The results show that ligand stimulation of Flt3 can induce association of SOCS6 and Flt3 and tyrosine phosphorylation of SOCS6. Phosphopeptide fishing indicated that SOCS6 binds directly to phosphotyrosines 591 and 919 of Flt3. By using stably transfected Ba/F3 cells with Flt3 and/or SOCS6, we show that the presence of SOCS6 can enhance ubiquitination of Flt3, as well as internalization and degradation of the receptor. The presence of SOCS6 also induces weaker activation of Erk1/2, but not Akt, in transfected Ba/F3 and UT-7 cells and in OCI-AML-5 cells. The absence of SOCS6 promotes Ba/F3 and UT-7 cell proliferation induced by oncogenic internal tandem duplications of Flt3. Taken together, these results suggest that SOCS6 negatively regulates Flt3 activation, the downstream Erk signaling pathway, and cell proliferation.
Introduction
Flt3 is a type III receptor tyrosine kinase (RTK)3 that belongs to the same family as the receptors for PDGF, stem cell factor (SCF) and macrophagecolony-stimulating factor (M-CSF) (1). Under normal conditions, Flt3 is of great importance for the proliferation and differentiation of hematopoietic stem cells and progenitor cells. Ligand binding leads to dimerization of receptors, activation of their intrinsic tyrosine kinase activity, and phosphorylation of tyrosine residues within the receptor intracellular domain, as well as of downstream signal transduction molecules. The phosphorylated tyrosine residues constitute high affinity binding sites for signal transduction molecules that contain a conserved domain of ∼100 amino acids, the so-called Src homology 2 (SH2) domain (2). Binding to the phosphorylated tyrosine residues is specified by the three to six amino acids immediately C-terminal to the phosphorylated tyrosine residues, thus defining the specificity of individual SH2 domains. To understand the way growth factor receptors signal, it is of utmost importance to define the individual tyrosine residues that are phosphorylated in response to ligand stimulation.
A number of gain-of-function mutations of Flt3 have been found in leukemia patients and human leukemia-derived cell lines. The internal tandem duplication (ITD) mutations were the first mutation to be identified (3). The ITD mutations are characterized by the duplication of a segment of the juxtamembrane region of Flt3, which leads to ligand-independent constitutive activation of Flt3. The ITD mutations range in size from 3 to >400 bp and always occur in multiple of three with the reading frame maintained. The size of the ITD is negatively correlated with 5-year overall survival of acute myeloid leukemia (AML) patients (4). Furthermore, the allelic ratio ITD/WT Flt3 is a significant and independent prognostic factor for relapse in pediatric AML (5). Apart from the ITD mutations, point mutations in the kinase domain of Flt3 that can induce constitutive kinase activity have been described in both pediatric and adult AML (6). Among point mutations, the most commonly occurring is mutation at Asp-835 of Flt3. Both the ITD mutations and point mutations of Flt3 are primarily found in AML, and a considerable body of evidence is now accumulating regarding their incidence and clinical impact.
The signaling mediated by growth factor-induced activation of Flt3 must be tightly regulated. This occurs mainly through the action of specific protein-tyrosine phosphatases and ubiquitin ligases. Protein-tyrosine phosphatases can terminate the signal transduction by dephosphorylating target proteins. Ubiquitin ligases covalently attach ubiquitin moieties to target proteins, which serve as tags for degradation either in proteasomes or, in the case of RTKs, in lysosomes. Oncogenic mutations in cancer can render the RTKs less ubiquitinated, either due to mutations of the binding sites for ubiquitin ligases on the receptors themselves or through inactivating mutations in the ubiquitin ligases (7). Cbl is the most studied ubiquitin ligase, and loss-of-function mutations in Cbl have been described in AML and shown to contribute to oncogenic transformation (8).
SOCS (suppressors of cytokine signaling) is another class of regulator for downstream signal transduction of RTKs. The SOCS family comprises CIS1 and SOCS1–7 (9). Members of this family are characterized by an SH2 substrate recognition domain and a C-terminal SOCS box, which mediates assembly into elongin B/C-cullin ubiquitin ligase complexes. CIS1 and SOCS1–3 are well characterized as negative feedback regulators of cytokine receptor signal transduction via the JAK/STAT pathway, but the function of SOCS4–7 is less known. Although originally implicated as regulators of cytokine signaling, they are also involved in regulation of growth factor receptor signal transduction. It has been shown that both SOCS1 and SOCS6 can regulate signal transduction downstream of the SCF receptor/c-Kit (10, 11). SOCS6 was shown to bind the juxtamembrane region of c-Kit, and the binding caused a 40% reduction in SCF-induced cell proliferation and a similar reduction in the activation of Erk and p38 in Ba/F3-Kit cells (11). SOCS1 and SOCS6 can also interact with the insulin receptor and inhibit insulin-dependent activation of Erk and protein kinase B (12).
In this study, we present evidence that Flt3 can bind SOCS6 in response to ligand stimulation. SOCS6 binds directly to phosphotyrosines 591 and 919 of Flt3. Flt3 ligand (FL) stimulation also up-regulates SOCS6 mRNA expression in Ba/F3 cells. The binding of SOCS6 to Flt3 increases receptor ubiquitination, internalization, and degradation. Correspondingly, the presence of SOCS6 inhibits Flt3-induced Erk activation and cell proliferation.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
The transfection reagent jetPEI was from Polyplus-transfection SA, and Lipofectamine 2000 was from Invitrogen. Cycloheximide was from Sigma. Rabbit polyclonal anti-SOCS6 serum was raised and purified as described (13). Anti-phosphotyrosine antibody 4G10 was from Millipore, and anti-ubiquitin antibody was from Covance Research Products. Anti-Flt3 antibody was described previously (14). Anti-Shc, anti-phospho-p38, and anti-p38 antibodies were from BD Transduction Laboratories. Anti-phospho-Akt antibody was from Epitomics. Polyclonal antibodies against Akt, phospho-Erk, and Erk were purchased from Santa Cruz Biotechnology. Phycoerythrin-labeled anti-Flt3 antibody was from BD Biosciences. Horseradish peroxidase-coupled secondary anti-mouse and anti-rabbit antibodies were from Invitrogen.
Cell Culture
Ba/F3 cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10 ng/ml recombinant murine IL-3. OCI-AML-5 and UT-7 cells were cultured in MEM-alpha supplemented with 20% heat-inactivated FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10 ng/ml recombinant human IL-3. COS-1 and EcoPack cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin.
Expression Constructs
pcDNA3-Flt3-WT, pcDNA3-Flt3-ITD, pMSCV-Flt3-WT and pMSCV-Flt3-ITD constructs were described previously (14). p-FLAG-SOCS6-WT, p-FLAG-SOCS6-R409E and p-FLAG-SOCS6-C504F constructs have been described (15). pMSCV-SOCS6-WT was constructed by subcloning the full-length open reading frame of murine SOCS6 into the pMSCV vector. pcDNA3-Flt3-Y591F/Y919F, pMSCV-Flt3-Y591F/Y919F, and pMSCV-Flt3-ITD-Y591F/Y919F constructs were generated by site-directed mutagenesis using the QuikChange mutagenesis XL kit (Stratagene, La Jolla, CA). All plasmids were verified by sequencing.
Real-time Quantitative PCR (RT-qPCR)
Total RNA was isolated from Ba/F3-Flt3-WT cells using the RNeasy mini kit (Qiagen) according to the manufacturer's directions. cDNAs were synthesized with oligo(dT) and random hexamer primers using a RevertAid premium first-strand cDNA synthesis kit (Fermentas) according to the manufacturer's directions. Gene expression was assessed by RT-qPCR using an Applied Biosystems 7900HT fast sequence detection system and gene-specific RT-qPCR primer assays (SABiosciences). β-Actin was used as an endogenous control to normalize expression data. Thermal cycling conditions included 95 °C for 10 min and 40 cycles at 95 °C for 15 s and 60 °C for 1 min according to the RT-qPCR primer assays protocol. Each sample was analyzed in quadruplicate. The comparative Ct (threshold cycle) method was used to calculate the relative changes in gene expression.
Transient and Stable Transfection
COS-1 cells were transiently transfected using JetPEI according to the manufacturer's directions. Cells were serum-starved overnight 24 h after transfection and then stimulated at 37 °C for the indicated times with 100 ng/ml FL (ProSpec-Tany). For transient transfection of OCI-AML-5 and UT-7 cell lines, the 4D-Nucleofector system (Lonza) was used. To establish Ba/F3 and UT-7 cells stably expressing wild-type or mutant Flt3, EcoPack packaging cells were transfected with the corresponding Flt3 construct in pMSCV-puro, and virus-containing supernatants were collected 72 h after transfection. Retroviral infection of Ba/F3 cells was followed by a 2-week selection in 1.2 μg/ml puromycin. Expression of Flt3 was confirmed by flow cytometry. Flt3-transfected Ba/F3 cells were then further transfected with the SOCS6 construct in pMSCV-neo. Cells were selected with 0.8 mg/ml G-418 for 2 weeks, and SOCS6 expression was verified by Western blotting. Ba/F3 cells were serum-starved for 4 h in RPMI 1640 medium without serum and cytokines and then stimulated at 37 °C for the indicated times with 100 ng/ml FL.
Immunoprecipitation and Western Blotting
After stimulation, cells were washed once with ice-cold PBS, lysed, and processed for immunoprecipitation and Western blotting as described previously (16). Immunodetection was performed by enhanced chemiluminescence using Immobilon Western chemiluminescent horseradish peroxidase substrate (Millipore) and a CCD camera (LAS-3000, Fujifilm, Tokyo, Japan). Signal intensity was quantified by Multi-Gauge software (Fujifilm).
Affinity Fishing of SOCS6 with Immobilized Peptides
Peptides corresponding to the tyrosine motifs of the Flt3 intracellular domain either phosphorylated or not were synthesized (Tyr-589, CGSSDNEYFYVDFREY; phospho-Tyr-566, CHKpYKKNFRYESQLQM; phospho-Tyr-572, CYKKQFRpYESQLQMV; phospho-Tyr-589, CGSSDNEpYFYVDFREY; phospho-Tyr-591, CGSSDNEYFpYVDFREY; phospho-Tyr-599, CYVDFREYEpYDLKWEF; phospho-Tyr-726, CEHNFSFpYPTFQSH; phospho-Tyr-768, CSEDEIEpYENQKRLEE; phospho-Tyr-793, CDLLSFApYQVAKGMEF; phospho-Tyr-842, CIMSDSNpYVVRGNAR; phospho-Tyr-919, CATEEIpYIIMQS; phospho-Tyr-955, CDAEEAMpYQNVDGRVS; phospho-Tyr-969, CSESPHTpYQNRRPFSR; and phospho-Tyr-589/phospho-Tyr-919, CGSSDNEpYFpYVDFREY) and immobilized on UltraLink beads (Thermo Scientific) according to the manufacturer's instructions. Immobilized peptide slurry (50 μl) was incubated at 4 °C for 2 h with SOCS6-transfected COS-1 cell lysates. Peptide-bound proteins were then processed for Western blotting.
Cell Proliferation and Survival Assay
Ba/F3 cells were washed three times with PBS and seeded in 24-well plates (60,000 cells/well). Cells were then incubated with or without 100 ng/ml FL or 10 ng/ml IL-3 for 48 h. For cell proliferation, viable cells were counted using trypan blue exclusion. For PrestoBlue cell viability assays (Molecular Probes), 10,000 cells were seeded per well in 96-well plates. After 70 h of incubation, 10 μl of PrestoBlue was added to each well, followed by 2 h of incubation. Absorbance was measured using a 96-well plate reader according to the manufacturer's protocol. Cell survival was measured using an annexin V/7-aminoactinomycin D kit (Pharmingen). Double-negative (annexin V/7-aminoactinomycin D) cells represent viable cells.
Receptor Ubiquitination, Internalization, and Degradation
To determine receptor ubiquitination, cells were starved for 4 h, followed by 30 min of incubation with the proteasome inhibitor MG132 and the lysosome inhibitor chloroquine diphosphate. Cells were then stimulated with FL for the indicated times in the presence of inhibitors and processed for lysis. Internalization of Flt3 was determined by flow cytometry using phycoerythrin-labeled anti-Flt3 antibody after FL stimulation for the indicated times at 37 °C. For protein degradation, cells were incubated with 100 μg/ml cycloheximide for 4 h at 37 °C in RPMI 1640 medium without serum and cytokines. Cells were then incubated with or without 100 ng/ml FL for 30 min, followed by lysis and immunoprecipitation with anti-Flt3 antibody. Samples were assessed by SDS-PAGE and Western blotting.
Analysis of SOCS6 Expression in Human Samples
The Gene Expression Omnibus (GEO) Database was used for expression analysis. Microarray expression data of three individual sets of patient samples for acute promyelocytic leukemia and AML and the corresponding matched cells (GSE2550, GSE9476, and GSE2191) were downloaded and used for analysis.
RESULTS
SOCS6 Protein Interacts with Flt3 upon Ligand Stimulation
It has been reported that SOCS6 can associate with c-Kit through its SH2 domain upon SCF stimulation (11). Flt3 belongs to the same RTK family as c-Kit and contains a similar region as c-Kit where SOCS6 was shown to bind. To investigate whether SOCS6 can also interact with Flt3, SOCS6 and Flt3 were transiently expressed in COS-1 cells. FL stimulation induced a strong association of SOCS6 with Flt3 (Fig. 1A). SOCS6 was found to be associated with the oncogenic Flt3 ITD mutant in COS-1 cells (Fig. 1B) and also in Ba/F3 cells (Fig. 1D) as well. Furthermore, endogenous Flt3-SOCS6 interaction was detected in OCI-AML-5 cells using specific antibodies (Fig. 1C). The kinetics of Flt3-SOCS6 interaction was also studied. Similar to Flt3 activation, the Flt3-SOCS6 interaction was rapid, reached the maximum after ligand stimulation for 5 min, and was stable for at least for 1 h (Fig. 1E). Although we observed SOCS6 tyrosine phosphorylation in COS-1 cells in response to FL (Fig. 1E), we were unable to detect tyrosine phosphorylation in either Ba/F3 or OCI-AML-5 cells (data not shown). To determine whether the Flt3-SOCS6 interaction is mediated through the SOCS6 SH2 domain, we used SOCS6 SH2 domain and SOCS box mutants. Whereas the SOCS box mutant was able to interact with Flt3, the SOCS6 SH2 domain mutant did not show any interaction with Flt3 (Fig. 1F).
FIGURE 1.

SOCS6 interacts with Flt3 in response to FL-stimulation. A, COS-1 cells were cotransfected with SOCS6 and the WT Flt3 expression vector. Cells were serum-starved overnight and stimulated by FL or not for 5 min before lysis. Cell lysates were subjected to immunoprecipitation (IP) using either anti-Flt3 or anti-SOCS6 antibody and analyzed by Western blotting. B, COS-1 cells were cotransfected with SOCS6 and Flt3 ITD. Cells were then processed as described for A. C, OCI-AML-5 cells were starved overnight and stimulated with FL for 5 min. Cells were lysed and processed for immunoprecipitation and Western blotting. D, Ba/F3 cells transfected with Flt3 ITD and SOCS6 were starved for 4 h, followed by 5 min of FL stimulation. Cells were lysed and processed for immunoprecipitation and Western blotting. E, transfected COS-1 cells were serum-starved overnight and then treated with 100 ng/ml FL for the indicated times before lysis and immunoprecipitated with anti-SOCS6 antibody. F, COS-1 cells were transfected with Flt3 and SOCS6 mutants. After overnight starvation, cells were treated with 100 ng/ml FL for 5 min before lysis and immunoprecipitated with anti-SOCS6 antibody. TCL, total cell lysate.
SOCS6 Binds Directly to Phosphotyrosines 591 and 919 of Flt3
The Flt3 intracellular domain contains at least 12 tyrosine residues that are known to be phosphorylated upon activation. To determine the SOCS6-binding sites, phospho- and non-phosphopeptides corresponding to all known Flt3 tyrosine phosphorylation residues were synthesized. Peptides were immobilized on UltraLink and subjected to pulled down proteins from SOCS6-overexpressing cell lysates. Western blotting with anti-SOCS6 antibody showed that phospho-Tyr-591 and phospho-Tyr-919 are SOCS6-binding sites (Fig. 2A). Interestingly, concomitant phosphorylation of the adjacent Tyr-589 greatly enhanced the binding of SOCS6 to Tyr-591 (Fig. 2B). To determine whether Tyr-591 and Tyr-919 are also SOCS6-binding sites in vivo, we generated pcDNA3-Flt3-Y591F/Y919F. COS-1 cells were cotransfected with either wild-type or double-mutant Flt3 and SOCS6 upon FL stimulation. The interaction of SOCS6 with Flt3 was significantly reduced when Tyr-591 and Tyr-919 were mutated (Fig. 2C), which indicates that Tyr-591 and Tyr-919 of Flt3 are SOCS6-binding sites in vivo.
FIGURE 2.

Identification of Tyr-591 and Tyr-919 as SOCS6-binding sites. A, phosphopeptides corresponding to 12 known tyrosine phosphorylation sites in Flt3 were synthesized and immobilized on UltraLink. Peptide-bound slurry was incubated with SOCS6-transfected COS-1 cell lysates, and pulled down proteins were then processed for Western blotting using anti-SOCS6 antibody to screen the SOCS6-binding sites. The c-Kit Tyr-568 peptide was used as a positive control. pY, phospho-Tyr; IB, immunoblot. B, immobilized phospho- and non-phosphopeptides were used to pull down SOCS6 proteins and processed as described for A to determine whether the binding was dependent on tyrosine phosphorylation. C, COS-1 cells were cotransfected with SOCS6 and WT Flt3 or Flt3(Y591F/Y919F). Cells were serum-starved and stimulated with FL for 5 min before lysis. Lysates were immunoprecipitated (IP) with anti-Flt3 or anti-SOCS6 antibody and analyzed by Western blotting to show that Tyr-591 and Tyr-919 are the SOCS6-binding sites in vivo.
Activation of Flt3 Induces mRNA Transcription of SOCS6
To assess the influence of the Flt3-SOCS6 interaction in hematopoietic cells, we generated Ba/F3-Flt3-WT, Ba/F3-Flt3-WT/SOCS6-WT, Ba/F3-Flt3-ITD, and Ba/F3-Flt3-ITD/SOCS6-WT cell lines. The Flt3 expression levels in these cell lines were verified by flow cytometry (Fig. 3A), and SOCS6 expression levels were checked by Western blotting (Fig. 3B). It has been shown that SCF stimulation can induce mRNA transcription of SOCS6 (11). We tested whether FL stimulation can do the same. Ba/F3 cells stably transfected with Flt3 were treated with 100 ng/ml FL for different times. Total RNAs were extracted and subjected to qPCR analysis. SOCS6 transcription was shown to be linearly increased with the time in response to FL stimulation (Fig. 4A). Furthermore, meta-analysis of published microarray data from patient samples revealed that SOCS6 expression significantly increased in acute promyelocytic leukemia patients expressing the Flt3 ITD (Fig. 4B). SOCS6 expression also increased in AML patients (Fig. 4, C and D).
FIGURE 3.

Expression of Flt3 and SOCS6 in Ba/F3 cells. A, stably transfected Ba/F3 cells were labeled with an isotype control or phycoerythrin-conjugated anti-Flt3 antibody and analyzed by flow cytometry. The gray line indicates cells labeled with the isotype control, and the black line indicates cells labeled with anti-Flt3 antibody. EV, empty vector. B, lysates of stably transfected Ba/F3-Flt3 cells with or without SOCS6 were immunoprecipitated (IP) with anti-SOCS6 antibody and analyzed by Western blotting. IB, immunoblot. C, endogenous SOCS6 and Flt3 expression was detected using specific antibodies in different AML cell lines.
FIGURE 4.

FL induces SOCS6 gene expression in Ba/F3-Flt3 cells. A, Ba/F3-Flt3 cells were serum-starved and stimulated with FL for the indicated times. Cells were lysed, and total RNAs were isolated. Expression of SOCS6 mRNA was analyzed by qPCR as described under “Experimental Procedures.” B, SOCS6 expression in acute promyelocytic leukemia patients carrying either WT Flt3 or the Flt3 ITD mutation. C, SOCS6 expression in an AML patient and in corresponding normal cells. D, SOCS6 expression in AML patients with complete remission or relapse and in corresponding normal cells. ns, not significant. *, p < 0.05; ***, p < 0.001.
SOCS6 Is Involved in Ubiquitination, Internalization, and Degradation of the Flt3 Receptor
It was recently shown that SOCS6 promotes ligand-dependent ubiquitination of the c-Kit receptor (15). To test the ubiquitin ligase activity of SOCS6 on the Flt3 receptor, we stimulated transfected Ba/F3 cells with FL for different times. The results clearly demonstrate an increased amount of ubiquitinated Flt3 in the presence of SOCS6 (Fig. 5, A and B). Internalization of Flt3 following FL stimulation was determined by flow cytometry for the indicated times. Internalization of the Flt3 receptor was significantly increased in SOCS6-expressing cells (Fig. 6A). SOCS6 also promoted ligand-induced degradation of the Flt3 receptor (Fig. 6B).
FIGURE 5.
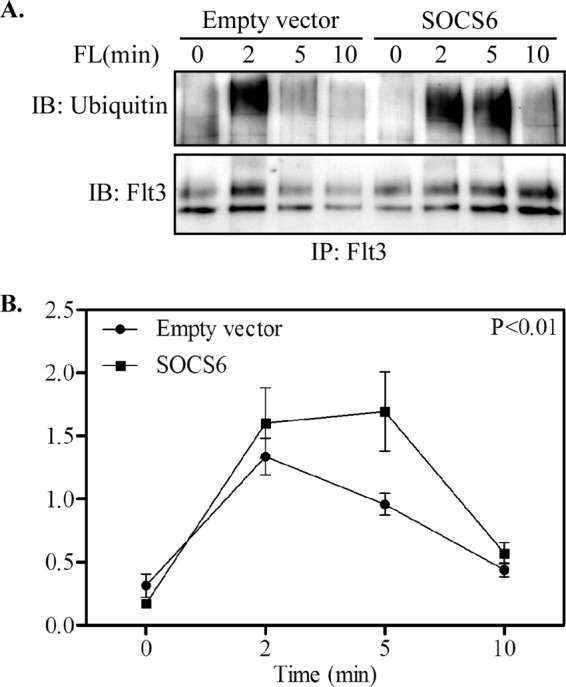
SOCS6 expression increases ubiquitination of the Flt3 receptor. A, Ba/F3-Flt3 cells with or without SOCS6 were serum-starved and stimulated with 100 ng/ml FL for the indicated times. Cells were lysed, and lysates were immunoprecipitated (IP) with anti-Flt3 antibody, followed by Western blotting. IB, immunoblot. B, signal intensities were quantified using Multi-Gauge software to calculate the receptor ubiquitination.
FIGURE 6.

SOCS6 expression increases Flt3 receptor internalization and degradation. A, Ba/F3-Flt3 cells with or without SOCS6 were serum-starved and stimulated with 100 ng/ml FL for the indicated times. Cells were transferred to ice, immediately followed by incubation with phycoerythrin-conjugated anti-Flt3 antibody. The Flt3 surface expression level was analyzed by flow cytometry. Internalization of Flt3 was quantified compared with unstimulated cells. B, Ba/F3-Flt3 cells with or without SOCS6 were serum-starved and preincubated with cycloheximide for 4 h. Cells were then stimulated with 100 ng/ml FL for 30 min before lysis. Cell lysates were immunoprecipitated with anti-Flt3 antibody and analyzed by Western blotting. Signal intensities were quantified using Multi-Gauge software to calculate the receptor degradation. **, p < 0.01.
SOCS6 Negatively Regulates Erk Signaling
Activation of Flt3 is known to regulate several signaling pathways, including the Ras/Erk, p38, and PI3K/Akt pathways (1). To investigate how SOCS6 affects Flt3-mediated signal transduction, activation of Akt, Erk, and p38 was examined by Western blotting using phospho-specific antibodies. The presence of SOCS6 in Ba/F3-Flt3 cells significantly inhibited FL-mediated Erk activation (Fig. 7A), whereas p38 phosphorylation was reduced by ∼20% (not statistically significant), and Akt activation remained mostly unchanged (Fig. 7A). In addition, the selective depletion of SOCS6 using siRNA in OCI-AML-5 cells expressing endogenous Flt3 and SOCS6 (Fig. 3C) significantly increased FL-induced Erk phosphorylation (Fig. 7B). As we observed that the SOCS6(R409E) mutant was unable to associate with Flt3 (Fig. 1F), we compared FL-induced Erk activation in Ba/F3-Flt3 cells expressing WT SOCS6 and SOCS6(R409E). In accordance with interaction data, SOCS6(R409E) did not block FL-induced Erk phosphorylation (Fig. 7C). In Fig. 2C, we showed that mutation of Tyr-591 and Tyr-919 reduced SOCS6 interaction with Flt3. Thus, we transfected UT-7 cells expressing endogenous SOCS6 (Fig. 3C) with WT Flt3 and Flt3(Y591F/Y919F) constructs. The results show that UT-7 cells expressing the Flt3(Y591F/Y919F) mutant exhibited increased Erk phosphorylation in response to FL stimulation compared with WT Flt3 (Fig. 7D). Shc is a signaling molecule that is upstream of the Ras/Erk pathway, similar to Erk activation. A 50% reduction in Shc phosphorylation in SOCS6-expressing Ba/F3-Flt3 cells in response to FL stimulation was observed (Fig. 7, C and D).
FIGURE 7.
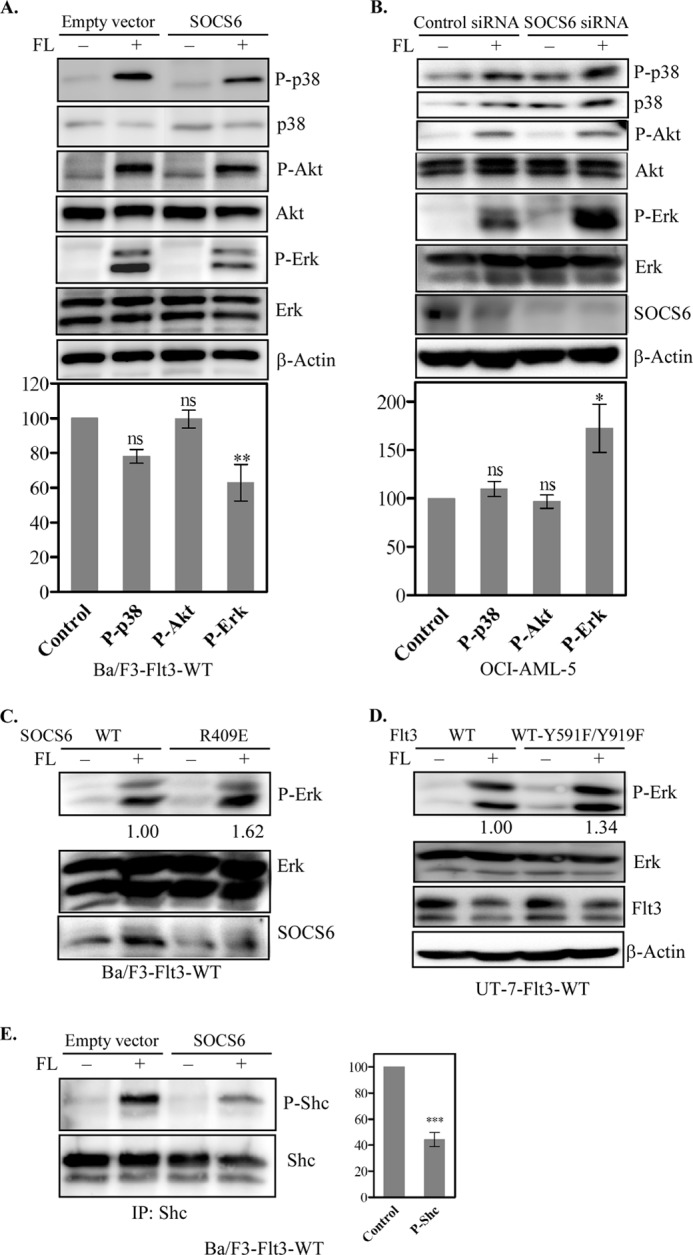
Effects of SOCS6 expression on FL-induced activation of Erk, p38, Akt, and Shc. A, Ba/F3-Flt3 cells with or without SOCS6 were treated with 100 ng/ml FL or not for 5 min before lysis. Total cell lysates were separated by SDS-PAGE, and membranes were probed with phospho-specific antibodies. Membranes were then stripped and reprobed with the respective antibodies to show loading. Signal intensities from three independent experiments were quantified using Multi-Gauge software to calculate the inhibition. ns, not significant. **, p < 0.01. B, OCI-AML-5 cells were transfected with control or SOCS6 siRNA using electroporation. One day after transfection, cells were starved overnight and stimulated with FL, followed by Western blot analysis. *, p < 0.05. C, Ba/F3-Flt3 cells transfected with WT SOCS6 or the SOCS6(R409E) mutant were starved for 4 h before a 5-min FL stimulation. Cells were then processed for Western blotting. D, UT-7 cells transfected with WT Flt3 or Flt3(Y591F/Y919F) were starved overnight before a 5-min FL stimulation. Cells were then processed for Western blotting. E, cell lysates were immunoprecipitated (IP) with anti-Shc antibody and analyzed by Western blotting. Signal intensities were quantified using Multi-Gauge software to calculate the inhibition. ***, p < 0.001.
SOCS6 Negatively Regulates FL-induced Cell Proliferation, but Not Cell Survival
Cell proliferation induced by activated RTKs can be suppressed by SOCS family proteins (10, 11). Several SOCS proteins also exhibit tumor suppressor function. For example, SOCS6 is down-regulated in a variety of cancers and ectopic expression of this protein suppresses cell growth and colony formation of gastric cancer cell lines (17). To explore the biological outcome of the Flt3-SOCS6 interaction, we studied the effect of SOCS6 expression on Flt3-dependent cell proliferation. SOCS6 expression led to a 25% decrease in Ba/F3-Flt3-ITD cell proliferation in response to FL stimulation (Fig. 8A). The effect of SOCS6 expression on cell survival was also examined. By staining the cells with annexin V and 7-aminoactinomycin D, we showed that SOCS6 expression could not alter survival of Ba/F3-Flt3-ITD cells (Fig. 8B). Using PrestoBlue cell viability assays, we demonstrated that WT SOCS6, but not the SOCS6(R409E) mutant, was able to reduce Flt3 ITD-mediated cell proliferation (Fig. 8, C and D). Furthermore, Flt3 ITD(Y591F/Y919F)-induced cell proliferation was not blocked by endogenous SOCS6 in UT-7 cells (Fig. 8E).
FIGURE 8.

Presence of SOCS6 down-regulates cell proliferation of Ba/F3-Flt3-ITD cells. A, Ba/F3-Flt3-ITD cells with or without SOCS6 (Empty vector) were grown for 48 h in the presence or absence of ligand. Viable cells were counted by the trypan blue exclusion method. ns, not significant. B, cells were labeled with annexin V and 7-aminoactinomycin D, and living cells were measured by flow cytometry. IL-3 was used as a positive control. Ba/F3-Flt3-ITD/empty vector cells or Ba/F3-Flt3-ITD/SOCS6-WT cells (C), Ba/F3-Flt3-ITD/SOCS6-WT cells or Ba/F3-Flt3-ITD/SOCS6-R409E (SOCS6-RE) cells (D), and UT-7-Flt3-ITD cells or UT-7-Flt3-ITD-Y591F/Y919F (Flt3-ITD-YYFF) cells (E) were washed to remove IL-3 and seeded in 96-well plates. Cells were then treated with either ligand or not for 48 h, followed by PrestoBlue cell viability assays. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
DISCUSSION
It is widely accepted that deregulated activation of Flt3 has important functions in AML pathogenesis. Flt3 is highly expressed in AML. To understand the mechanism that controls Flt3 signaling, we analyzed the role of SOCS6 in Flt3 regulation. We identified SOCS6 as a new interaction partner of Flt3. This interaction negatively regulates Flt3-dependent signaling pathways by promoting ubiquitination, internalization, and degradation of the Flt3 receptor.
The finding that SOCS6 is capable of interacting with Flt3 and c-Kit (11) indicates that SOCS6 may have a role in RTK signaling. In both cases, the interaction was dependent on ligand stimulation. The SH2 domain of SOCS6, but not the SOCS box, is involved in this association, suggesting that SOCS6 specifically interacts with phosphotyrosine residues (15, 18). Using Flt3 phosphopeptides, we mapped SOCS6 the interaction sites as phospho-Tyr-591 and phospho-Tyr-919. A previous analysis of binding specificity for phosphopeptide showed that the SOCS6 SH2 domain bound preferentially to motifs containing valine at phospho-Tyr position +1 and hydrophobic residues at phospho-Tyr positions +2 and +3 (18). The sequence surrounding Tyr-591 in Flt3 fulfills the consensus requirement (YVDF) for binding of SOCS6 and also shows strong similarity to the region of c-Kit where SOCS6 is shown to bind (11). Interestingly, the Flt3 phospho-Tyr-589 peptide did not associate with SOCS6, but phosphorylation of Tyr-589 increased binding affinity dramatically when introduced with the phospho-Tyr-591 peptide.
The mechanism by which Tyr-589 phosphorylation increases binding affinity for the Flt3 phospho-Tyr-591 peptide is not clear. One possible explanation is that negative charge at phospho-Tyr position −2 may increase affinity for phospho-Tyr-591. Tyr-591 is located in the Flt3 juxtamembrane domain. This domain is of the utmost regulatory importance for many growth factor receptors. Tyr-589, Tyr-591, and Tyr-599 in this domain are suggested to be Src-binding sites (19). Tyr-589 and Tyr-591 have shown to be conserved in the related tyrosine kinases c-Kit (20), PDGF receptor-α (21), and PDGF receptor-β (22). These tyrosine residues are involved in interaction with various signaling proteins, including Src family kinases (SFKs), SHP1, and SHP2. Thus, the Tyr-589/Tyr-591 motif appears to be a major docking site for the protein complexes following type III RTK activation. Another SOCS6-binding site in Flt3 (Tyr-919) is located in the kinase domain, which has not been studied in depth. One report suggests that, together with other phosphotyrosine residues, Tyr-919 is involved in activation of Flt3 kinase domain mutants (23).
The expression profiles of Flt3 and SOCS6 overlap. For example, SOCS6 mRNA is expressed in most hematopoietic progenitors and bone marrow cells (18). The Flt3-positive AML cell line OCI-AML-5 expresses SOCS6 mRNA. In addition, we observed that transcription of SOCS6 mRNA was induced by FL in the Ba/F3-Flt3 cell line. This observation suggests that Flt3 signaling might be regulated by SOCS6. Like SOCS1 and SOCS3, SOCS6 associates with elongins B and C in a SOCS box-dependent manner (18, 24). Furthermore, SOCS6 promotes ligand-dependent ubiquitination of the c-Kit receptor (15) and p56lck kinase (25). We also observed an increased ligand-dependent ubiquitination of the Flt3 receptor in SOCS6-expressing cells. These findings provide further evidence that SOCS6 acts as an E3 ubiquitin ligase in biological systems. Another E3 ligase, Cbl, has also been reported to be involved in ubiquitination and degradation of Flt3 (26). The observation that SOCS6-expressing cells showed accelerated internalization and degradation of the Flt3 receptor can also be explained by the increased ubiquitination effect of the receptor in the same system.
Loss of SOCS function promotes tumor formation, which can occur by several mechanisms, including gene deletion, mutation, or silencing due to hypermethylation (27). Several SOCS proteins, including CIS, SOCS2, SOCS3, and SOCS6, have been implicated in the negative regulation of growth factor signaling. Our data also provide evidence that SOCS6-mediated regulation of Flt3 is biologically important. SOCS6 partially blocked Flt3 ITD-mediated cell proliferation. In addition, SOCS6 partially inhibited ligand-dependent proliferation of Ba/F3-Kit cells, but not Ba/F3-EGF receptor cells (11). Taken together, these observations suggest that SOCS6 controls growth of hematopoietic cells by modulating distinct signaling pathways.
Flt3 physically associates and phosphorylates several signaling proteins, including Ras, phospholipase Cγ, Grb2, SHP2, and SFKs, resulting in further activation of downstream PI3K and MAPK pathways (1, 28). Activation of MAPK pathways results in phosphorylation of Erk and p38 kinases, whereas the PI3K signaling pathway regulates Akt phosphorylation. We observed that activation of Erk, but not Akt, was significantly inhibited by SOCS6. Bayle et al. (11) also observed a very similar inhibition in SOCS6-expressing Ba/F3-Kit cells. These individual observations suggest that SOCS6 interrupts MAPK signaling pathways, but not the PI3K pathway. MAPK signaling pathways can be activated by Flt3 in multiple ways. For example, interaction of Grb2 with Flt3 results in Erk phosphorylation (29), and mutation of Src-binding sites of Flt3 reduces Erk phosphorylation (19). The protein-tyrosine phosphatase SHP2 has also been shown to interact with Flt3, modulating Erk phosphorylation (19). Thus, we suggest that interaction of SOCS6 with Flt3 may disrupt the binding of signaling proteins with Flt3, resulting in reduced phosphorylation of Erk and p38. Activation of MAPK pathways through EGF, phorbol 12-myristate 13-acetate, or anisomycin stimulation is not inhibited by SOCS6 (11), which also provides further evidence that SOCS6 inhibition takes place in the initial stages. SFKs phosphorylate Shc protein upon activation of RTKs, which is an early event in MAPK signaling. One SOCS6 interaction site in Flt3 (Tyr-591) is also a binding site for SFKs (1). We observed that SOCS6 expression inhibited FL-induced phosphorylation of Shc protein. Interaction of SOCS6 with Tyr-591 may partially block SFK activation, followed by reduced Shc phosphorylation. Thus, we conclude that the Flt3-SOCS6 interaction partially blocks MAPK signaling pathways, resulting in reduced cell proliferation in hematopoietic cells.
Acknowledgments
We thank Susanne Bengtsson and Elena Razumovskaya for help in different experiments.
This work was supported in part by the Swedish Cancer Society, the Swedish Children's Cancer Organization, and the Swedish Research Council.
- RTK
- receptor tyrosine kinase
- SCF
- stem cell factor
- SH2
- Src homology 2
- ITD
- internal tandem duplication
- AML
- acute myeloid leukemia
- FL
- Flt3 ligand
- RT-qPCR
- real-time quantitative PCR
- SFK
- Src family kinase.
REFERENCES
- 1. Masson K., Rönnstrand L. (2009) Oncogenic signaling from the hematopoietic growth factor receptors c-Kit and Flt3. Cell. Signal. 21, 1717–1726 [DOI] [PubMed] [Google Scholar]
- 2. Pawson T. (2004) Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 116, 191–203 [DOI] [PubMed] [Google Scholar]
- 3. Levis M., Small D. (2003) FLT3: ITDoes matter in leukemia. Leukemia 17, 1738–1752 [DOI] [PubMed] [Google Scholar]
- 4. Stirewalt D. L., Kopecky K. J., Meshinchi S., Engel J. H., Pogosova-Agadjanyan E. L., Linsley J., Slovak M. L., Willman C. L., Radich J. P. (2006) Size of FLT3 internal tandem duplication has prognostic significance in patients with acute myeloid leukemia. Blood 107, 3724–3726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meshinchi S., Alonzo T. A., Stirewalt D. L., Zwaan M., Zimmerman M., Reinhardt D., Kaspers G. J., Heerema N. A., Gerbing R., Lange B. J., Radich J. P. (2006) Clinical implications of FLT3 mutations in pediatric AML. Blood 108, 3654–3661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamamoto Y., Kiyoi H., Nakano Y., Suzuki R., Kodera Y., Miyawaki S., Asou N., Kuriyama K., Yagasaki F., Shimazaki C., Akiyama H., Saito K., Nishimura M., Motoji T., Shinagawa K., Takeshita A., Saito H., Ueda R., Ohno R., Naoe T. (2001) Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 97, 2434–2439 [DOI] [PubMed] [Google Scholar]
- 7. Peschard P., Park M. (2003) Escape from Cbl-mediated down-regulation: a recurrent theme for oncogenic deregulation of receptor tyrosine kinases. Cancer Cell 3, 519–523 [DOI] [PubMed] [Google Scholar]
- 8. Reindl C., Quentmeier H., Petropoulos K., Greif P. A., Benthaus T., Argiropoulos B., Mellert G., Vempati S., Duyster J., Buske C., Bohlander S. K., Humphries K. R., Hiddemann W., Spiekermann K. (2009) CBL exon 8/9 mutants activate the FLT3 pathway and cluster in core binding factor/11q deletion acute myeloid leukemia/myelodysplastic syndrome subtypes. Clin. Cancer Res. 15, 2238–2247 [DOI] [PubMed] [Google Scholar]
- 9. Yoshimura A., Naka T., Kubo M. (2007) SOCS proteins, cytokine signaling, and immune regulation. Nat. Rev. Immunol. 7, 454–465 [DOI] [PubMed] [Google Scholar]
- 10. De Sepulveda P., Okkenhaug K., Rose J. L., Hawley R. G., Dubreuil P., Rottapel R. (1999) Socs1 binds to multiple signaling proteins and suppresses steel factor-dependent proliferation. EMBO J. 18, 904–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bayle J., Letard S., Frank R., Dubreuil P., De Sepulveda P. (2004) Suppressor of cytokine signaling 6 associates with KIT and regulates KIT receptor signaling. J. Biol. Chem. 279, 12249–12259 [DOI] [PubMed] [Google Scholar]
- 12. Mooney R. A., Senn J., Cameron S., Inamdar N., Boivin L. M., Shang Y., Furlanetto R. W. (2001) Suppressors of cytokine signaling 1 and 6 associate with and inhibit the insulin receptor. A potential mechanism for cytokine-mediated insulin resistance. J. Biol. Chem. 276, 25889–25893 [DOI] [PubMed] [Google Scholar]
- 13. Blume-Jensen P., Siegbahn A., Stabel S., Heldin C. H., Rönnstrand L. (1993) Increased Kit/SCF receptor-induced mitogenicity but abolished cell motility after inhibition of protein kinase C. EMBO J. 12, 4199–4209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Razumovskaya E., Masson K., Khan R., Bengtsson S., Rönnstrand L. (2009) Oncogenic Flt3 receptors display different specificity and kinetics of autophosphorylation. Exp. Hematol. 37, 979–989 [DOI] [PubMed] [Google Scholar]
- 15. Zadjali F., Pike A. C., Vesterlund M., Sun J., Wu C., Li S. S., Rönnstrand L., Knapp S., Bullock A. N., Flores-Morales A. (2011) Structural basis for c-KIT inhibition by the suppressor of cytokine signaling 6 (SOCS6) ubiquitin ligase. J. Biol. Chem. 286, 480–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Voytyuk O., Lennartsson J., Mogi A., Caruana G., Courtneidge S., Ashman L. K., Rönnstrand L. (2003) Src family kinases are involved in the differential signaling from two splice forms of c-Kit. J. Biol. Chem. 278, 9159–9166 [DOI] [PubMed] [Google Scholar]
- 17. Lai R. H., Hsiao Y. W., Wang M. J., Lin H. Y., Wu C. W., Chi C. W., Li A. F., Jou Y. S., Chen J. Y. (2010) SOCS6, down-regulated in gastric cancer, inhibits cell proliferation and colony formation. Cancer Lett. 288, 75–85 [DOI] [PubMed] [Google Scholar]
- 18. Krebs D. L., Uren R. T., Metcalf D., Rakar S., Zhang J. G., Starr R., De Souza D. P., Hanzinikolas K., Eyles J., Connolly L. M., Simpson R. J., Nicola N. A., Nicholson S. E., Baca M., Hilton D. J., Alexander W. S. (2002) SOCS6 binds to insulin receptor substrate 4, and mice lacking the Socs6 gene exhibit mild growth retardation. Mol. Cell. Biol. 22, 4567–4578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heiss E., Masson K., Sundberg C., Pedersen M., Sun J., Bengtsson S., Rönnstrand L. (2006) Identification of Y589 and Y599 in the juxtamembrane domain of Flt3 as ligand-induced autophosphorylation sites involved in binding of Src family kinases and the protein-tyrosine phosphatase SHP2. Blood 108, 1542–1550 [DOI] [PubMed] [Google Scholar]
- 20. Price D. J., Rivnay B., Avraham H. (1999) CHK down-regulates SCF/KL-activated Lyn kinase activity in Mo7e megakaryocytic cells. Biochem. Biophys. Res. Commun. 259, 611–616 [DOI] [PubMed] [Google Scholar]
- 21. Gelderloos J. A., Rosenkranz S., Bazenet C., Kazlauskas A. (1998) A role for Src in signal relay by the platelet-derived growth factor α receptor. J. Biol. Chem. 273, 5908–5915 [DOI] [PubMed] [Google Scholar]
- 22. Valgeirsdóttir S., Paukku K., Silvennoinen O., Heldin C. H., Claesson-Welsh L. (1998) Activation of Stat5 by platelet-derived growth factor (PDGF) is dependent on phosphorylation sites in PDGF β-receptor juxtamembrane and kinase insert domains. Oncogene 16, 505–515 [DOI] [PubMed] [Google Scholar]
- 23. Ishiko J., Mizuki M., Matsumura I., Shibayama H., Sugahara H., Scholz G., Serve H., Kanakura Y. (2005) Roles of tyrosine residues 845, 892, and 922 in constitutive activation of murine FLT3 kinase domain mutant. Oncogene 24, 8144–8153 [DOI] [PubMed] [Google Scholar]
- 24. Zhang J. G., Farley A., Nicholson S. E., Willson T. A., Zugaro L. M., Simpson R. J., Moritz R. L., Cary D., Richardson R., Hausmann G., Kile B. J., Kent S. B., Alexander W. S., Metcalf D., Hilton D. J., Nicola N. A., Baca M. (1999) The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc. Natl. Acad. Sci. U.S.A. 96, 2071–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Choi Y. B., Son M., Park M., Shin J., Yun Y. (2010) SOCS6 negatively regulates T cell activation through targeting p56lck to proteasomal degradation. J. Biol. Chem. 285, 7271–7280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sargin B., Choudhary C., Crosetto N., Schmidt M. H., Grundler R., Rensinghoff M., Thiessen C., Tickenbrock L., Schwäble J., Brandts C., August B., Koschmieder S., Bandi S. R., Duyster J., Berdel W. E., Müller-Tidow C., Dikic I., Serve H. (2007) Flt3-dependent transformation by inactivating c-Cbl mutations in AML. Blood 110, 1004–1012 [DOI] [PubMed] [Google Scholar]
- 27. Elliott J., Hookham M. B., Johnston J. A. (2008) The suppressors of cytokine signaling E3 ligases behave as tumor suppressors. Biochem. Soc. Trans. 36, 464–468 [DOI] [PubMed] [Google Scholar]
- 28. Takahashi S. (2011) Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: biology and therapeutic implications. J. Hematol. Oncol. 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Masson K., Liu T., Khan R., Sun J., Rönnstrand L. (2009) A role of Gab2 association in Flt3 ITD-mediated Stat5 phosphorylation and cell survival. Br. J. Haematol. 146, 193–202 [DOI] [PubMed] [Google Scholar]