

Background: Activation of [FeFe]-hydrogenases requires coordinated interactions between the maturases HydE, HydF, and HydG.
Results: We characterized the interactions of HydF with HydE, HydG, and the hydrogenase that occur independently of the HydF GTPase activity, related to the dissociation step.
Conclusion: Additional insights into the [FeFe]-hydrogenases maturation process are provided.
Significance: This study contributes to a more detailed picture of how [FeFe]-hydrogenases are assembled.
Keywords: GTPase, Hydrogenase, Iron-Sulfur Protein, Metalloproteins, Protein-Protein Interactions
Abstract
[FeFe]-hydrogenases are iron-sulfur proteins characterized by a complex active site, the H-cluster, whose assembly requires three conserved maturases. HydE and HydG are radical S-adenosylmethionine enzymes that chemically modify a H-cluster precursor on HydF, a GTPase with a dual role of scaffold on which this precursor is synthesized, and carrier to transfer it to the hydrogenase. Coordinate structural and functional relationships between HydF and the two other maturases are crucial for the H-cluster assembly. However, to date only qualitative analysis of this protein network have been provided. In this work we showed that the interactions of HydE and HydG with HydF are distinct events, likely occurring in a precise functional order driven by different kinetic properties, independently of the HydF GTPase activity, which is instead involved in the dissociation of the maturases from the scaffold. We also found that HydF is able to interact with the hydrogenase only when co-expressed with the two other maturases, indicating that under these conditions it harbors per se all the structural elements needed to transfer the H-cluster precursor, thus completing the maturation process. These results open new working perspectives aimed at improving the knowledge of how these complex metalloenzymes are biosynthesized.
Introduction
Iron-sulfur clusters are essential in several major biochemical processes in prokaryotic and eukaryotic organisms, including catalysis, electron transfer, determining of protein structure, and regulation of gene expression (1–3). Different FeS centers may exist in nature, ranging from the simplest [2Fe-2S] and [4Fe-4S] units, found in plant and bacterial ferredoxins, as well as in respiratory complexes I–III of bacteria and mitochondria, to more complex polymetallic clusters characterized in metalloproteins such as nitrogenase and hydrogenase (4), involved in nitrogen fixation and hydrogen metabolism, respectively. The biosynthesis of FeS clusters is a highly complex and strictly coordinated process driven by different, phylogenetically unrelated molecular systems, all sharing common biosynthetic principles (2, 5, 6). These pathways are key events in the overall cellular physiology, and according to their crucial role an increasing number of diseases are related to an impaired biogenesis of iron-sulfur proteins (2).
The paradigm of this complexity is represented by the biosynthesis and maturation machinery of the [FeFe]-hydrogenases, in which the active site (the so-called H-cluster) is composed by a [4Fe-4S] unit connected by a cysteinyl residue to a [2Fe-2S] center coordinated by three CO, two CN−, and a bridging dithiolate (see Ref. 7 for a comprehensive review on this topic). This unique organometallic cluster serves as a catalytic site for the reversible reduction of protons to molecular hydrogen, which is central for both hydrogen metabolism in several prokaryotic and eukaryotic microorganisms and potential bioenergy applications exploiting this class of metalloenzymes. Because of the complexity of the H-cluster and the presence of species that are toxic in their free form, such as iron, CO and CN−, a highly controlled and coordinated process is needed for its assembly (see Refs. 5, 6, and 8 for the most recent reviews on this topic). Three conserved proteins drive this maturation pathway, i.e., HydE and HydG, two radical S-adenosylmethionine (SAM)3 FeS enzymes (5, 9–13), and HydF, a GTPase containing the Walker A P-loop and Walker B Mg2+-binding motifs, as well as a FeS cluster-binding sequence (14, 15). Site-specific mutagenesis analysis revealed that the conserved radical SAM and GTPase consensus motifs are all essential for the [FeFe]-hydrogenase maturation and activation (16). According to several recent in vitro studies, this process occurs in a multistep pathway, in which the [4Fe-4S] unit is synthesized by the Isc/Suf FeS general cell machinery, whereas the biosynthesis and insertion of the 2Fe subcluster, together with its ligands, is driven by HydE, HydG, and HydF maturases (5–8, 17). Current data indicate that the addition of the CO/CN−/dithiolate ligands to the 2Fe subcluster is accomplished through the action of the HydE/HydG radical SAM chemistry coupled to the presence of HydF, which would have a double involvement as scaffold and carrier to build and insert the modified 2Fe subcluster into a hydrogenase containing a preformed [4Fe-4S] unit (17–21).
Because of the multistep nature of the molecular pathway leading to the [FeFe]-hydrogenase maturation, a network of protein interactions between the players of this process must be established to accomplish and coordinate the H-cluster assembly. The dynamic behavior of HydF as scaffold and carrier assigns to this protein a key role along the entire maturation process and indicates its capability to interact with both HydE and HydG in the first step, when the 2Fe subcluster is processed and modified, and finally with the hydrogenase, when the complete 2Fe unit is ready to be transferred to the latter. The interactions of HydF with the other accessory proteins have been previously inferred from the co-purification of HydE and HydG with HydF (19), and recent data suggest that the GTP binding and/or hydrolysis could be associated with the interactions between the maturases, because both HydE and HydG increase by 50% the rate of GTP hydrolysis catalyzed by HydF (15). This led the authors to suggest that GTP binding and/or hydrolysis may induce structural changes in HydF, which would in turn influence the interactions between the three maturases. However, the molecular details of HydF GTPase activity during [FeFe]-hydrogenase maturation and its precise role in this process are still unknown.
We recently solved the crystal structure of a recombinant HydF from Thermotoga neapolitana (22). HydF is organized in three distinct domains, i.e., (i) domain I, which carries all the conserved amino acids considered important for GTP binding and hydrolysis; (ii) domain II, responsible for HydF dimerization; and (iii) domain III, the FeS cluster-binding domain, which may be in principle involved in the interprotein interactions of this maturase with its potential partners.
In this work we address and characterize the protein-protein interactions of HydF with the two other maturases and the hydrogenase, which are expected to be pivotal in all steps of [FeFe]-hydrogenase maturation pathway, with the aim of gaining further insights into this complex molecular process.
EXPERIMENTAL PROCEDURES
All of the chemicals were of the highest purity commercially available.
Heterologous Expression of Hyd Maturation and Structural Proteins from Clostridium acetobutylicum
The C. acetobutylicum hydE, hydF, hydG, and hydA1 coding sequences were cloned in the pCDFDuet-1, pACYCDuet-1, pRSFDuet-1, and pETDuet-1 vectors (Novagen®) suitable for T7 driven (co)expression in Escherichia coli, either as such or in frame with a tag sequence (6His or StrepII tag, depending on the experiment; see “Results” and “Discussion”), thus obtaining the recombinant plasmids listed in Table 1. The pCDFDuet-1/hydF-StrepII, pETDuet-1/hydA1-StrepII, pETDuet-1/hydA1-StrepII/hydE, and pRSFDuet-1/hydF/hydG plasmids were kindly provided by Dr. Matthew C. Posewitz (Department of Chemistry and Geochemistry, Colorado School of Mine, Golden, CO) and obtained as described previously (16). Some of these vectors were used as templates for PCR amplification with specific oligonucleotides designed with 5′ and 3′ end restriction sites for directional subcloning into the dual multiple cloning site (MCS 1 and MCS 2) of plasmids pACYCDuet-1 (hydE), pCDFDuet-1 (hydF), or pRSFDuet-1 (hydG). When required, the restriction sites were selected to clone the gene of interest in frame with a 6His tag coding sequence localized immediately downstream the BamHI restriction site of MCS 1. hydE and hydG were cloned either in MCS 1 between the BamHI and NotI restriction sites (forming the pACYCDuet-1/hydE-6His and pRSFDuet-1/hydG-6His plasmid, respectively) or in MCS 2 between the NdeI and BglII restriction sites (forming the pACYCDuet-1/hydE and pRSFDuet-1/hydG plasmid, respectively). hydF was cloned in MCS 1 between the BamHI and NotI restriction sites (forming the pCDFDuet-1/hydF-6His plasmid). The PCRs were performed using the high fidelity Phusion DNA polymerase (Finnzymes). The sequence and reading frame of each gene were confirmed by DNA sequencing (BMR Genomics, University of Padova). E. coli BL21(DE3) cells were transformed with the recombinant plasmid(s), and positive clones were selected by antibiotic resistance. The protein(s), either wild type or mutant (see below), were expressed as described previously (16) by adding 1 mm isopropyl β-thiogalactopyranoside, in aerobiosis or anaerobiosis depending on the experiment, and purified.
TABLE 1.
Plasmid constructs for T7 promoter driven expression of [FeFe]-hydrogenase structural and maturation genes in E. coli
| Plasmid | |
|---|---|
| pCDFDuet-1/hydF-StrepII | Expression of wild type and mutant HydF proteins with a N-terminal StrepII tag or 6His tag |
| pCDFDuet-1/hydF-StrepII_G24A/K25A | |
| pCDFDuet-1/hydF-StrepII_D67A | |
| pCDFDuet-1/hydF-6His_C304S | |
| pCDFDuet-1/hydF-6His_G24A/K25A | |
| pCDFDuet-1/hydF-6His | |
| pACYCDuet-1/hydE | Expression of untagged HydE protein |
| pACYCDuet-1/hydE-6His | Expression of a HydE protein with a N-terminal 6His tag |
| pRSFDuet-1/hydG | Expression of untagged HydG protein |
| pRSFDuet-1/hydG-6His | Expression of a HydG protein with a N-terminal 6His tag |
| pETDuet-1/hydA1-StrepII | Expression of a HydA1 protein with a C-terminal StrepII tag |
| pETDuet-1/hydA1-StrepII/hydE | Co-expression of untagged HydE and a HydA1 proteins with a C-terminal StrepII tag |
| pRSFDuet-1/hydF/hydG | Co-expression of untagged HydF and HydG proteins |
Co-purification of HydF with Potential Interaction Partners
To evaluate the interactions of HydF-StrepII with HydE-6His and HydG-6His and of HydF-6His with HydA1-StrepII, E. coli cells (100 ml of culture) co-expressing the proteins of interest were collected by centrifugation at 4,000 × g for 10 min at 4 °C. The cell pellet was resuspended in lysis buffer (100 mm Tris-HCl, pH 8, 150 mm NaCl, 2 mm DTT, 2 mm Na2S, 2 mm (NH4)2Fe(SO4)2·6H2O, and protease inhibitors 1 μg/ml pepstatin A, 1 μg/ml leupeptin, 1 μg/ml antipain, 1 mm PMSF) and broken in a French press (at 1.35 kbar; One Shot Constant System Cell Disrupter, from Constant Systems Ltd). A clarified crude extract was then obtained by centrifugation and incubated 1 h at 4 °C under mild shaking either with 200 μl of a StrepTactin-Sepharose suspension (IBA, Göttingen, Germany) or with 200 μl of a nickel affinity gel (HIS-Select® nickel affinity gel; Sigma-Aldrich), both pre-equilibrated with lysis buffer. At the end of this incubation, the mix was transferred into a chromatography column. The column was then washed with 5 volumes of lysis buffer, and the tagged proteins were eluted with 5 volumes of lysis buffer containing 2.5 mm desthiobiotin or 200 mm imidazole. The elution fractions were pooled together, analyzed by 12% SDS-PAGE, and electroblotted onto a nitrocellulose membrane. For immunoblotting analysis, the membrane was probed with a monoclonal anti-6His tag (Sigma-Aldrich) or anti-StrepII tag (IBA) antibody and with a horseradish peroxidase-conjugated goat anti-mouse IgG (Kirkegaard & Perry Laboratories). Labeled proteins were then visualized with an ECL Western blotting detection kit (Thermo Scientific Pierce Protein Research).
Analysis of the Stoichiometry of HydF-StrepII·HydE-6His and HydF-StrepII·HydG-6His Interactions
Recombinant HydF-StrepII (also as mutant HydF-StrepII_G24A/K25A and HydF-StrepII_D67A proteins) and HydE-6His or HydG-6His were co-expressed in E. coli as described previously, and the complexes between these proteins purified by a double affinity chromatography approach, by exploiting first the HydF StrepII epitope and in a second step the HydE or HydG 6His tag. Briefly, in both cases the Strep-Tactin elution fractions containing HydF-StrepII and HydE-6His or HydG-6His were pooled and subjected to a nickel-nitrilotriacetic acid affinity chromatography to retain HydE-6His or HydG-6His. The imidazole eluted fractions were pooled together and resolved on 12% SDS-PAGE with known amounts of BSA, ranging from 0.5 to 2 μg. The proteins were visualized with Coomassie Brilliant Blue stain, and their amount was estimated by densitometry on a Image Station 4000 MM PRO instrument (Kodak). The data were analyzed with Carestream molecular imaging software.
Purification of HydE-6His and HydG-6His Proteins to Homogeneity for Biacore Analysis
HydE-6His and HydG-6His were purified to homogeneity by subjecting affinity-purified proteins to gel filtration chromatography performed with a Superdex 200 HR 10/30 (GE Healthcare), equilibrated in 25 mm Tris-HCl, pH 8, 200 mm NaCl elution buffer. Each run was performed by injecting the appropriate sample volume at a flow rate of 0.75 ml/min and monitoring the UV absorbance at 280 nm, by a fixed wavelength detector. To estimate the molecular weight of the analyzed samples, the column was equilibrated in the same buffer and calibrated with the standards thyroglobulin (669,000 Da), ferritin (440,000 Da), catalase (232,000 Da), aldolase (158,000 Da), bovine serum albumin (67,000 Da), ovalbumin (43,000 Da), and ribonuclease (13,700 Da). Purified proteins were quantified by using a Micro BCA Protein Assay kit (Thermo Scientific Pierce Protein Research). The presence of monomeric HydE-6His and HydG-6His proteins in the selected peaks was confirmed by Western blotting analysis using a monoclonal anti-6His tag antibody.
Site-directed Mutagenesis of hydF-StrepII Coding Sequence
Site-directed mutagenesis of the hydF-StrepII was performed with QuikChange® II site-directed mutagenesis kit (Stratagene) using as template the pCDFDuet-1/hydF-StrepII or the pCDFDuet-1/hydF-6His plasmids. Oligonucleotides were designed according to the manufacturer's guidelines, and the mutant constructs were analyzed by DNA sequencing. The oligonucleotide sequences (with the modified bases underlined) were: mutWalkerAfor, 5′-GGAAAAACTAATGTTGCAGCATCCAGTGTAATAAATG-3′; mutWalkerArev, 5′-CATTTATTACACTGGATGCTGCAACATTAGTTTTTCC-3′; mutWalkerBfor, 5′-ACCAGTTATGCTTATAGCTACTGCTGGTCTTGATC-3′; mutWalkerBrev, 5′-GATCAAGACCAGCAGTAGCTATAAGCATAACTGGT-3′; mutCys304for, 5′-TTAATAGCAGAAGCCAGCACCCACCACCGTC-3′; and mutCys304rev, 5′-GACGGTGGTGGGTGCTGGCTTCTGCTATTAA-3′.
Surface Plasmon Resonance Analysis
For the surface plasmon resonance analysis, a BIAcoreTM T100 system (GE Healthcare) was used. HydF-StrepII and HydF-StrepII_G24A/K25A proteins were covalently coupled to a CM5 (series S) sensor chip (carboxymethylated dextran surface) by amine-coupling chemistry to a final density of 6000 resonance units, as described (23); a 10 mm acetate pH 5.0 buffer was used for the immobilization. A flow cell with no immobilized protein was used as a control. Binding analysis was carried out in a running buffer consisting of 10 mm Hepes, pH 7.6, 150 mm NaCl, 2 mm MgCl2, applying a flow rate of 30 μl/min. The absence of mass transport limitation was assessed by checking that signals observed at different flow rates (10–30 μl/min) were superimposable. Each sensorgram (time course of the surface plasmon resonance signal) was corrected for the response obtained in the control flow cell and normalized to baseline. After each injection, the surface was regenerated by a double injection of 2 m MgCl2 for 1 min; this treatment restored the base line to the initial resonance unit value. For kinetics experiments, a Biacore method program was used that included a series of three start-up injections (running buffer), zero control (running buffer), and six different concentrations of the analytes (HydE-6His or HydG-6His), one of which was duplicated. Serial dilutions of the analytes were performed in running buffer from a 2 μm top concentration. High performance injection parameters were used; the contact time was of 120 s followed by a 120-s dissociation phase. The kinetic data were analyzed using the 2.0.3 BIAevaluation software (GE Healthcare). The curves (both association and dissociation phases) were fitted with the classical Langmuir 1:1 model or with a two-state binding model; the quality of the fits was assessed by visual inspection of the fitted data and their residual, and by chi-squared values. Although the KD values calculated with the two models were very similar, better fits were generated by the two-state reaction model (as an example see Fig. 1, which reports the fits referring to the complex HydE-6His·HydF-StrepII), according to which 1:1 binding is followed by a conformational change that stabilizes the complex (24). Two independent experiments were performed.
FIGURE 1.

Comparison between the 1:1 Langmuir and the two-state reaction fitting models for the binding HydE-6His·HydF-StrepII analyzed by means of SPR signal detection. The kinetics shown are the same as those in Fig. 3A. For more details, see the legend of Fig. 3. The color curves are real sensorgrams, and black curves correspond to the fitting.
GTP Hydrolysis Assay
Under aerobic conditions, HydF-StrepII, HydF-StrepII_G24A/K25A, and HydF-StrepII_D67A proteins were assayed for their ability to hydrolyze GTP using the protocol optimized by Shepard et al. (15), with slight modifications. Briefly, 40 μm of affinity-purified proteins were incubated at 30 °C with 2 mm GTP and 2 mm MgCl2 in 20 mm Tris-HCl buffer, pH 8.0, containing 200 mm KCl. At time intervals, aliquots of the reaction mixture were collected and assayed for production of GDP. Assay aliquots were incubated at 95 °C for 3 min and centrifuged at 14,000 rpm at 4 °C in a benchtop microcentrifuge, and the supernatants were analyzed by reverse phase HPLC on a Synergi MAX-RP 80A (150 × 4.6 mm, 4 μm; Phenomenex). The samples were eluted with an isocratic mobile phase of 50 mm sodium phosphate buffer, pH 7.0, 10 mm tetrabutylammonium bromide, 10% CH3CN. The guanosine nucleotides were detected by their absorbance at 254 nm. Under these conditions, GDP and GTP eluted after ∼8.1 and ∼18.6 min, respectively. Integration of peak areas (using the software Agilent Chemstation) of the samples taken at identical time points allowed the quantification of the μmoles of GDP produced liter−1 min−1, from which the ratio between the kcat were finally determined.
Hydrogen Evolution Assay
Hydrogenase activity of whole extracts obtained from cells co-expressing HydA1-StrepII with HydE, HydG, and HydF-StrepII or HydF-6His (also as HydF-StrepII_G24A/K25A, HydF-StrepII_D67A, and HydF-6His_C304S mutant proteins) were measured in vitro, as described previously (16). Briefly, 1 ml of 2× enzyme reaction buffer was added to 1 ml of E. coli cell cultures giving exactly the same absorbance at 600 nm, and the evolution of H2 gas from reduced methyl viologen was measured using nitrogen-flushed 13.5-ml sealed serum vials and a gas chromatograph PerkinElmer Clarus GC500, fitted with a Restek 5 Å Molecular Sieve 80/100 6′ 1/8″ column and a thermal conductivity detector. All of the steps were performed in an anaerobic chamber (MBRAUN).
UV-visible Absorption
The UV-visible absorption spectra of HydF-6His and HydF-6His_C304S proteins were acquired as described previously (22) using a Lambda Bio 40 UV-visible spectrometer (PerkinElmer Life Sciences).
RESULTS AND DISCUSSION
To gain new biochemical insights into the dynamic roles of HydF, we analyzed the interactions of this protein with both HydE and HydG, as well as with the hydrogenase (HydA1). These interactions are central for the entire maturation process and are supposed to be associated with its scaffold and carrier activities, respectively.
Biochemical Analysis of Protein-Protein Interactions of HydF Scaffold with HydE and HydG Maturases
It has been previously shown that the recombinant HydE, HydF, and HydG proteins co-elute from an affinity chromatography column when co-expressed in E. coli (19), thus suggesting an interaction between the three maturases. The setup of a coordinated and regulated network of protein interactions between HydE, HydF, and HydG is the first crucial step in the HydA maturation pathway. According to all recent literature data concerning the H-cluster assembly, this complex multistep process requires the ability of the HydF scaffold to interact with both HydG and HydE, but the molecular and biochemical details of this key event are still not completely understood. Based on this, we first evaluated whether HydE and HydG are both able to directly and individually interact with HydF using recombinant proteins from C. acetobutylicum fused to different tags to be exploited for affinity chromatography purification and Western blotting analysis. To this end, we co-transformed the E. coli strain BL21(DE3) either with the plasmids pCDFDuet/hydF-StrepII and pACYCDuet/HydE-6His or with the plasmids pCDFDuet/hydF-StrepII and pRSFDuet/hydG-6His, which allowed the isopropyl β-thiogalactopyranoside-inducible T7 co-expression of the corresponding recombinant proteins (Fig. 2, A–D, lanes 1). The HydF-StrepII tag protein was then purified from the soluble fraction of the two cultures by Strep-Tactin affinity chromatography (Fig. 2, A and B, lanes 3), as described under “Experimental Procedures,” and the presence of HydE-6His or HydG-6His in the eluted fractions was verified by Western blotting analysis using an anti-6His tag monoclonal antibody. Fig. 2 (C and D, lanes 3) clearly shows that both HydE-6His and HydG-6His co-purify with HydF-StrepII, indicating that the interactions between HydF scaffold and the two other maturases can be envisaged as distinct, independent, and possibly unrelated events (i.e., HydFΔEG may interact both with HydEΔG and HydGΔE).
FIGURE 2.

Binary interactions of HydF-StrepII scaffold with the HydE-6His and HydG-6His maturases. Western blotting analysis shows the StrepTactin purification of HydF-StrepII co-expressed with HydG-6His (A and C) or with HydE-6His (B and D). Lanes 1, total E. coli cell extract; lanes 2, soluble fraction of cell extract; lanes 3, total pool of desthiobiotin eluted fractions. 25 μl of each sample were loaded on a 12% gel for SDS-PAGE. A and B, Western blotting with anti-StrepII tag monoclonal antibody; C and D, Western blotting with anti-6His tag monoclonal antibody.
To quantify these protein interactions, we determined the stoichiometry of the HydF-StrepII·HydE-6His and HydF-StrepII·HydG-6His heterocomplexes. To exclude from this analysis the free HydF-StrepII protein (i.e., the purified HydF-StrepII, which did not interact with the maturation partner), we isolated the HydF-StrepII·HydE-6His and the HydF-StrepII·HydG-6His complexes by a double affinity chromatography, exploiting first the HydF StrepII epitope and in a second step the HydE-6His or HydG-6His tag, as described under “Experimental Procedures.” The Strep-Tactin elution fractions, containing HydF-StrepII and HydE-6His or HydG-6His, were pooled and subjected to a nickel-nitrilotriacetic acid affinity chromatography to retain HydE-6His or HydG-6His, still associated with HydF-StrepII. In both cases, the imidazole eluted fractions were pooled together and subjected to SDS-PAGE. The gel was then stained with Coomassie Brilliant Blue, and the amount of maturases estimated by densitometry as described under “Experimental Procedures.” Based on this analysis, we found a stoichiometric ratio of roughly 1:4 for the HydE-6His·HydF-StrepII complex and of 1:1 for the HydG-6His·HydF-StrepII complex. The observed stoichiometries could be due to the presence of multiple oligomeric species of HydF-StrepII protein (dimers and tetramers) (22), as well as to the amount of HydE-6His, which is invariably lower when compared with HydG-6His in co-expression experiments (not shown).
To date, only qualitative evidence for protein-protein interaction between Hyd maturases has been reported (19). To obtain further quantitative data for the binding properties of the HydF scaffold protein and to provide the kinetic constants of the HydF·HydE and HydF·HydG interactions, we performed a surface plasmon resonance (SPR) analysis by means of a Biacore T100 instrument. An affinity-purified HydF-StrepII tag protein was covalently immobilized on a chip surface, and solutions at different concentrations of HydG-6His or HydE-6His, previously purified to homogeneity by a combination of affinity and gel filtration chromatography, were individually passed over the chip, as described in detailed under “Experimental Procedures.” As shown in Fig. 3, which reports the curves corresponding to a two-state reaction fitting (see “Experimental Procedures” and Fig. 1), both HydE-6His (Fig. 3A) and HydG-6His (Fig. 3B) give a SPR signal, which is concentration-dependent and clearly indicates the expected binding to HydF-StrepII. However, as immediately evident from the figure, HydE-6His produces a much higher signal when compared with HydG-6His. We performed a quantitative analysis for the kinetics constants with the BIAevaluation software, and the values, reported in Table 2, show that the KD of HydE-6His is 1 order of magnitude lower than that of HydG-6His, indicating a higher affinity for the interaction HydF-StrepII·HydE-6His.
FIGURE 3.

Kinetics of the HydE-6His·HydF-StrepII and HydG-6His·HydF-StrepII interactions by means of SPR signal detection. HydE-6His (panel A) or HydG-6His (panel B) solutions (analytes) at the concentrations (μm) indicated in the boxes were injected over a sensor chip where HydF-StrepII (ligand) was previously covalently immobilized in a Biacore T100 instrument (see the “Experimental Procedures” for details). SPR signal is shown as sensorgram, and the time course of the surface plasmon resonance response reported in resonance units (RU). Each sensorgram has been subtracted of the corresponding signal produced on a control surface and normalized to baseline. One solution of each analyte was injected twice at the same concentration (0.25 μm HydE-6His and 1 μm HydG-6His), as further control. 0 concentrations corresponded to dilution buffer.
TABLE 2.
Kinetics values of the HydE-6His/HydF-StrepII and HydG-6His/HydF-StrepII interactions calculated from Biacore experiments
The constants ± S.E. are calculated from the kinetics shown in Fig. 3, with BIAevaluation software 2.0.3. Only two decimal digits are shown. A two-state reaction model was applied (see “Experimental Procedures” and Fig. 1).
| kon,1 | koff,1 | kon,2 | koff,2 | KD | |
|---|---|---|---|---|---|
| m−1 × s−1 × 104 | s−1 × 10−2 | s−1 × 10−2 | s−1 × 10−3 | m | |
| HydF-StrepII | |||||
| HydE-6His | 7.27 ± 0.04 | 5.29 ± 0.05 | 1.83 ± 0.01 | 2.65 ± 0.02 | 9.19 × 10−8 |
| HydG-6His | 0.65 ± 0.002 | 7.34 ± 0.25 | 1.90 ± 0.03 | 2.52 ± 0.06 | 1.31 × 10−6 |
| HydF-StrepII_G24A/K25A | |||||
| HydE-6His | 9.96 ± 0.06 | 5.92 ± 0.05 | 1.72 ± 0.01 | 2.75 ± 0.02 | 8.20 × 10−8 |
| HydG-6His | 1.02 ± 0.02 | 8.07 ± 0.23 | 1.88 ± 0.02 | 2.58 ± 0.05 | 9.47 × 10−7 |
Because HydE and HydG act on the same [2Fe2S]-cluster prior to its transfer from the scaffold to the [FeFe]-hydrogenase, they likely share the same interaction site within HydF domain III, which, based on the HydF three-dimensional structure recently solved, harbors the cluster-binding pocket (22). To strengthen the Biacore analysis results and to confirm the existence of a possible stepwise mechanism in which the HydF scaffold interacts only with a maturation partner at a time, we first injected the HydE-6His protein near the saturation level on the chip containing the immobilized HydF-StrepII, and in a second step we applied in the same chip a HydG-6His solution; the result was then compared with the one obtained with a similar protocol, in which in the first step the same volume of buffer has been added instead of HydE-6His. Fig. 4A shows that HydG-6His, which as expected interacts with a free HydF-StrepII (line b), is unable to produce any significant signal when injected after HydE-6His (line a), indicating that the occupancy of the HydE-6His sites on HydF-StrepII prevents the binding of HydG-6His. This result also suggests that HydG-6His does not interact with HydE-6His. Moreover, we can also conclude that HydG-6His is not able to displace HydE-6His already bound to the scaffold. Unfortunately, because of the low affinity of HydG-6His for HydF-StrepII, we were not able to perform the opposite protocol (i.e., presaturation of HydF-StrepII with HydG-6His, followed by injection of HydE-6His); thus, we cannot assess whether the presence of HydG-6His prevents the binding of HydE-6His as well, using this Biacore approach. To better address this issue, we co-expressed in E. coli the recombinant HydF-StrepII and HydG-6His proteins and isolated the HydF-StrepII·HydG-6His complex by the same double affinity chromatography approach described above. Fig. 4 shows that the nickel-nitrilotriacetic acid elution fractions contain both HydG-6His (Fig. 4C, lane 2) and HydF-StrepII (Fig. 4B, lane 2). The complex was then incubated for 30 min with the soluble fraction of a cell extract obtained by a HydE-6His overexpressing E. coli culture, and the mixture was subjected to a Strep-Tactin affinity chromatography. The presence of HydE-6His and HydG-6His, together with HydF-StrepII, in the eluted fractions was finally evaluated by Western blotting analysis. Fig. 4 shows that HydG-6His still co-elutes with HydF-StrepII in this third chromatographic step (lanes 3 of C and B, respectively), whereas HydE-6His is exclusively present in the flow-through fraction (C, lane 3, and FT), indicating that a HydF-StrepII·HydG-6His complex is not able to interact with a second maturation partner, exactly as previously observed in the Biacore experiments with the HydF-StrepII·HydE-6His complex.
FIGURE 4.

Analysis of the interactions between HydG-6His and the HydF-StrepII·HydE-6His complex and between HydE-6His and the HydF-StrepII·HydG-6His complex. A, Biacore analysis. Injection of 2 μm HydE-6His solution (sensorgram a) or running buffer (sensorgram b) was performed in a Biacore T100 system over a sensorchip with immobilized HydF-StrepII, at a flow rate of 30 μl/min for 5 min. After further 2 min of buffer flowing (dissociation phase) and washing, a 2 μm HydG-6His solution was injected (1 min of association, 1 min of dissociation). The shown sensorgrams are subtracted from the signal in control flow cell and normalized to a base-line value of 0. B and C, Western blotting analysis. Lane 1, pool of desthiobiotin eluted fractions from the first affinity chromatography; lane 2, pool of imidazole eluted fractions from the second affinity chromatography; lane 3, pool of desthiobiotin eluted fractions from the third affinity chromatography. FT, flow-through of the third affinity chromatography. 25 μl of each sample were loaded on a 12% gel for SDS-PAGE. B, Western blotting with anti-StrepII tag monoclonal antibody. C, Western blotting with anti-6His tag monoclonal antibody.
The sequence of events leading to the synthesis of the complete [2Fe-2S]-cluster on the HydF scaffold is still undefined. Based on several independent compelling studies, a model has been recently proposed (5, 6) in which HydE synthesizes the dithiolate ligand of the H-cluster, thus generating an intermediate that would be then further modified by the addition of CO and CN− catalyzed by HydG (11, 12, 25), leading to a complete H-cluster precursor, to be finally transferred to the [FeFe]-hydrogenase. The chemical rationale of this order is based on the hypothesis that HydE probably modifies the [2Fe-2S]-cluster via a BioB (biotin synthase) type sulfur insertion chemistry (6), even if substrates and products of HydE catalysis are still unidentified. Thus, the reaction catalyzed by HydE would be expected to take place first to protect the sulfide groups and move the reactivity toward the iron ions of the FeS cluster, making them susceptible to the addition of CO and CN− in a second step (21). In this scenario, if HydE and HydG are simultaneously co-expressed in vivo, the higher affinity of HydE for the HydF scaffold, when compared with HydG, would allow the [2Fe-2S]-cluster modification sequence described above to occur. Because HydG is not able to interact with the complex HydF·HydE, nor to displace HydE (Fig. 4), a further step is required to allow the interaction of HydG with the scaffold to complete the FeS cluster chemical modification (see below). Structural analysis of the HydF-StrepII·HydE-6His and HydF-StrepII·HydG-6His heterocomplexes are currently underway in our laboratory to explore the modifications introduced in HydF by the other two maturases and to map the regions lying at the interface between the interacting proteins.
Investigating the Potential Involvement of GTP Binding/Hydrolysis in the Interactions of HydF with HydE and HydG
NTPases are commonly involved in the assembly of metal co-factors of FeS proteins and mediate either the metal delivery to the active site or the cluster transfer to the target protein. Experimental evidences against a role of HydF GTPase activity in FeS cluster precursor transfer to the [FeFe]-hydrogenase have been previously provided (15), and the role of GTP binding/hydrolysis in H-cluster assembly is elusive. As assessed in the Introduction, it has been shown that the HydF-dependent GTP hydrolysis in vitro increases in the presence of HydE or HydG (15), suggesting the existence of a HydF GTPase domain function/structure relationship driving the interactions of this scaffold with the two other maturases. To address this point and to test the requirement of intact HydF GTP hydrolysis properties for the protein-protein interactions described above, we generated two new recombinant HydF-StrepII proteins, carrying (i) two point-mutations in the Walker A P-loop sequence ((G/A)XXXXGK(S/T)), localized at residues 19–26 in HydF from C. acetobutylicum (GKTNVGKS) and responsible for the proper position of the triphosphate moiety of the bound nucleotide, and (ii) a single point-mutation in the Walker B-loop sequence (DXXG), localized at residues 67–70 in HydF from C. acetobutylicum (DTAG) and involved in the interaction with the nucleotide γ-phosphate and Mg2+. We expressed in E. coli the mutant HydF-StrepII_G24A/K25A and HydF-StrepII_D67A proteins (i) alone, to measure their GTPase activity in vitro, (ii) in combination with HydE, HydG, and HydA1-StrepII, to evaluate their capability to activate the [FeFe]-hydrogenase, and (iii) in combination with HydE-6His and HydG-6His, to test their ability to interact with the two other maturases. As expected, the introduced mutations impair the HydF-StrepII GTP hydrolysis and completely abolished the capability of HydF-StrepII to activate HydA1-StrepII (Table 3), confirming the crucial role of the HydF GTPase activity in the maturation process. On the other hand, both mutant proteins retain the ability to interact with HydE and HydG, as assessed by co-purification experiments performed exactly as described above (Fig. 5, A and B, lanes 2), suggesting that the HydF GTP hydrolysis does not introduce in the scaffold structural changes affecting its interactions with the two other accessory proteins. Moreover, the complexes between the mutant HydF-StrepII proteins and the two other 6His-tagged maturases have been purified by double affinity chromatography, exactly as described above, and the stoichiometry of these interactions has been determined by densitometry. We obtained the same ratios estimated with the HydF-StrepII protein, thus further proving that the point mutations introduced in the Walker A and Walker B sequences do not affect the capability of HydF-StrepII to interact with both HydE-6His and HydG-6His. This was also independently confirmed by a Biacore analysis, which showed that the kinetic constants of both proteins for HydF-StrepII_G24A/K25A mutant are similar to those calculated for the interaction with the wild type protein (Table 2).
TABLE 3.
Effects of point mutations of Walker A P-loop and Walker B conserved sequences on purified HydF-StrepII GTPase activity and HydA1-StrepIIEFG hydrogen evolution in whole cell extracts
The values reported for both GTPase and hydrogen evolution activities are the means of three independent experiments ± S.E.
| Protein | GTPase, kcat | Hydrogenase |
|---|---|---|
| min−1 | nmol H2·ml−1·min−1 | |
| HydF-StrepII | 4.84 ± 0.46 | 72.33 ± 3.83 |
| HydF-StrepII_G24A/K25A | 0.35 ± 0.05 | 0.58 ± 0.11 |
| HydF-StrepII_D67A | 0.08 ± 0.02 | 0.87 ± 0.12 |
FIGURE 5.

Involvement of HydF GTPase domain in the interactions with HydE and HydG maturation partners. Western blotting analysis showing the StrepTactin purification of HydF-StrepII_G24A/K25A (A) and HydF-StrepII_D67A (B) co-expressed either with HydE-6His or HydG-6His. Lanes 1, soluble fraction of E. coli cell extract; lanes 2, pool of desthiobiotin eluted fractions. 25 μl of each sample were loaded on a 12% gel for SDS-PAGE. Western blotting with anti-StrepII tag and anti-6His tag monoclonal antibodies is shown.
We also addressed the question whether the nucleotide binding to HydF may influence per se the interaction of the scaffold with HydE and HydG, independently of the GTP hydrolysis. To this end, exactly as described for the experiment reported in Fig. 3, HydE-6His and HydG-6His were individually passed over the BIAcore chip carrying a HydF-StrepII in the presence of the nonhydrolyzable analog GTPγC at concentrations ranging from 0.1 to 5 mm. The results were very similar to those obtained in the absence of GTPγC (see Fig. 6, which refers, as an example, to the experiment with 2 mm GTPγC). The quantitative analysis of the experiment performed with 2 mm GTPγC, reported in Table 4, shows that the KD for HydF-StrepII·HydE-6His and HydF-StrepII·HydG-6His interactions are not significantly different from those obtained in the absence of GTPγC (Table 2), ruling out an effect of the nucleotide binding on the interactions between the maturases. These data indicate that neither the GTP binding to HydF nor the nucleotide hydrolysis are directly involved in the protein interactions between the scaffold and the two other maturases, both in whole cell and in in vitro assays with purified proteins.
FIGURE 6.

Kinetics of the HydE-6His·HydF-StrepII and HydG-6His·HydF-StrepII interactions in the absence or in the presence of GTPγC 2 mm. For more details, see the legend of Fig. 3.
TABLE 4.
Kinetics values of the HydE-6His/HydF-StrepII and HydG-6His/HydF-Strep II interactions in the presence of 2 mm GTPγC, calculated from Biacore experiments
The constants ± S.E. are calculated from Biacore kinetics (not shown), with BIAevaluation software 2.0.3. Only two decimal digits are shown. A two-state reaction model was applied.
| kon,1 | koff,1 | kon,2 | koff,2 | KD | |
|---|---|---|---|---|---|
| m−1 × s−1 × 104 | s−1 × 10−2 | s−1 × 10−2 | s−1 × 10−3 | m | |
| HydF-StrepII | |||||
| HydE-6His | 20.02 ± 0.17 | 4.30 ± 0.06 | 1.10 ± 0.01 | 1.29 ± 0.06 | 2.26 × 10−8 |
| HydG-6His | 2.70 ± 0.007 | 9.21 ± 0.26 | 1.00 ± 0.01 | 1.42 ± 0.09 | 4.21 × 10−7 |
As reported in the Introduction, we recently solved the three-dimensional crystal structure of a nucleotide-free HydF protein (22) and showed that the GTP-binding domain includes a flexible loop that is expected to undergo a structural rearrangement upon nucleotide binding and/or hydrolysis that could in turn influence the interaction of the scaffold with the maturation partners. This prompted us to further investigate the role of this domain in the functional and structural network of the [FeFe]-hydrogenase maturation proteins. Interestingly, as shown in Fig. 7, in Biacore experiments when GTP is injected over HydE-6His (Fig. 7A) or HydG-6His (Fig. 7B) during their dissociation phase from HydF-StrepII, a concentration-dependent change of the curve slope can be observed, indicating an increased dissociation rate. Similar results have been obtained using the nonhydrolyzable analog GTPγC. This suggests that the binding of GTP to HydF can be related to the mechanism by which the displacement of an interaction partner from the scaffold occurs, allowing subsequent association of a different protein.
FIGURE 7.

GTP-induced dissociation of HydE-6His and HydG-6His from the HydF-StrepII scaffold as assessed by Biacore analysis. 2 μm HydE-6His (A) or HydG-6His (B) was injected for 2 min over the immobilized HydF-StrepII, and then, during the dissociation phase, after 4 min of buffer flowing, a second injection was applied of 0.5 mm GTP (sensorgram d), 2 mm GTP (sensorgrams e), or buffer (sensorgrams b and c). The flow rate was 30 μl/min. The effect of 2 mm GTP directly injected, without previous binding of HydE or HydG, is shown by sensorgram a. The sensorgrams are shown for the second injection, after subtraction of the signal in control flow cell and normalization (value of 0) to the baseline at the moment of the second injection (GTP or buffer).
Analysis of Protein Interaction of HydF Carrier with the [FeFe]-Hydrogenase
The role of HydF as a carrier to transfer a complete [2Fe-2S]-subcluster to the [FeFe]-hydrogenase involves the interaction of these two proteins. Interestingly, it has been clearly shown that only a HydF co-expressed with HydE and HydG (i.e., HydFEG) is able to activate a hydrogenase produced in a genetic background completely devoid of maturases (i.e., HydAΔEFG) (15, 18, 19). Instead, the hydrogenase activity was not observed when the three accessory proteins were expressed separately or in varying combinations and added in vitro to HydAΔEFG (18). Based on this, we investigated the protein-protein interactions of HydA1 with both functional HydFEG and nonfunctional HydF proteins produced in different backgrounds. To this end, we co-expressed in E. coli a recombinant HydA1-StrepII protein in combination with (i) HydF-6His; (ii) HydF-6His, HydE, and HydG; (iii) HydF-6His and HydE; and (iv) HydF-6His and HydG. HydE and HydG were expressed without tags, to visualize only HydF-6His in the Western blotting analysis following the purification step. The HydA1-StrepII protein was purified by Strep-Tactin affinity chromatography (Fig. 8, A–D, lanes 2), and the presence of HydFΔEG-6His, HydFEG-6His, HydFE-6His, and HydFG-6His in the eluted fractions was evaluated by Western blotting. Interestingly, Fig. 8 shows that the StrepTactin elution fractions are completely devoid of HydFΔEG-6His (Fig. 8E, lane 2) and that HydF-6His co-purifies with HydA1-StrepII not only when co-expressed with both HydE and HydG (Fig. 8F, lane 2) but also in combination either with only HydE (Fig. 8G, lane 2) or with only HydG (Fig. 8H, lane 2). Taken together, these results indicate that HydE and HydG could independently introduce in HydF structural changes or modulate the HydF scaffold properties, allowing its interaction with HydA. On the other hand, only HydFEG harbors a complete 2Fe subcluster carrying the CO, CN−, and dithiolate ligands, separately added by HydE and HydG, and is able to activate the [FeFe]-hydrogenase.
FIGURE 8.

Co-purification of HydA1-StrepII and HydFEG-6His, HydFE-6His and HydFG-6His. Western blotting analysis shows the StrepTactin purification of HydA1-StrepII expressed in the presence of HydF-6His without HydE-6His and HydG-6His (A and E), with HydG-6His and HydE-6His (B and F), with HydE-6His (C and G), and with HydG-6His (D and H). Lanes 1, soluble fraction of E. coli cell extract; lanes 2, pool of desthiobiotin eluted fractions. 25 μl of each sample were loaded on a 12% gel for SDS-PAGE. Western blotting with anti-6His tag monoclonal and anti-StrepII tag antibodies is shown.
We also investigated whether the presence of a FeS cluster precursor on the HydFEG scaffold is required for its interaction with the hydrogenase. To this end, we obtained a new recombinant HydF-6His protein in which one of the three highly conserved cysteine residues belonging to the FeS cluster-binding consensus sequence (i.e., Cys-304 of the motif CXHX45HCXXC of HydF from C. acetobutylicum) has been mutated. We expressed in E. coli the HydF-6His_C304S protein (i) alone, to evaluate its capability to bind a FeS cluster and (ii) in combination with HydE, HydG and HydA1-StrepII to test both its maturation activity and the capability to interact with the hydrogenase. As reported in Fig. 9A, the UV-visible absorption spectra in the 320–550-nm range of the HydF-6His_C304S mutant protein shows a limited capability to bind iron (gray line) when compared with the HydF-6His protein (black line), confirming the key role of this residue in the binding of the FeS cluster precursor to the scaffold. Accordingly, the same mutation results in a severe impairment of the [FeFe]-hydrogenase maturation (Fig. 9A, inset). On the other hand, the HydFEG-6His_C304S mutant protein co-elutes with HydA1-StrepII (Fig. 9B, lanes 3), as assessed by co-purification experiments performed as described in the previous paragraphs, indicating that the interaction between these proteins occurs independently of the presence of the FeS cluster on the scaffold. Also in this case, the ongoing structural analysis of the HydF-StrepII·HydE-6His and HydF-StrepII·HydG-6His heterocomplexes would add new molecular insights useful to define the protein environment driving the process of the 2Fe subcluster assembly/transfer.
FIGURE 9.
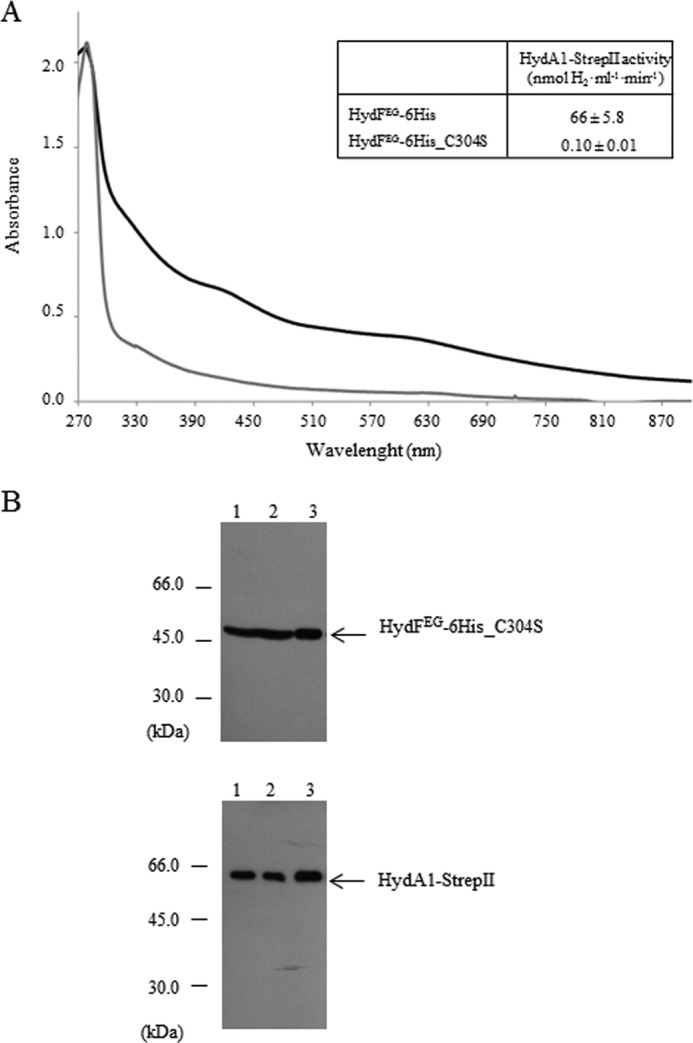
Co-purification of HydA1-StrepII and HydFEG-6His_C304S. A, UV-visible spectroscopy of HydF-6His and HydF-6His_C304S proteins. Black line, HydF-6His; gray line, mutant HydF-6His_C304S protein. The same amount of affinity-purified protein (150 μm) was analyzed for each sample. Inset, H2 evolution activity of HydA1-StrepII anaerobically co-expressed in E. coli with HydE, HydG and HydF-6His, or HydF-6His_C304S. The reported values are the means of three independent experiments ± S.E. B, Western blotting analysis showing the StrepTactin purification of HydA1-StrepII and HydFEG-6His_C304S. Lanes 1, total E. coli cell extract; lanes 2, soluble fraction of cell extract; lanes 3, pool of desthiobiotin eluted fractions. 25 μl of each sample were loaded on a 12% gel for SDS-PAGE. Western blotting with anti-StrepII tag and anti-6His tag monoclonal antibodies is shown.
It was previously shown that the GTPase activity of HydFEG is unrelated to its capability to activate an nonfunctional HydAΔEFG (15), suggesting that the nucleotide binding and/or hydrolysis are not essential for the transfer of the cluster precursor from the scaffold to the hydrogenase. To further address this point, we evaluated whether the HydF GTPase properties are involved in its interaction with the [FeFe]-hydrogenase. To this end, we co-expressed in E. coli HydA1-StrepII in combination with HydE, HydG, and the HydF-6His_G24A/K25A protein. The purification profile of the Strep-Tactin affinity chromatography, performed exactly as described above, clearly indicates that the mutant HydF-6His_G24A/K25A protein retains the capability to interact with the hydrogenase (Fig. 10), ruling out a role of GTP hydrolysis in the binding of the [FeFe]-hydrogenase to the scaffold. Taken together, these data indicate that the [2Fe-2S]-subcluster biosynthesis/modification and delivery represent two functionally related but distinct steps, both involving dynamic HydF scaffold/carrier interactions with the two other accessory proteins as well as with the hydrogenase.
FIGURE 10.
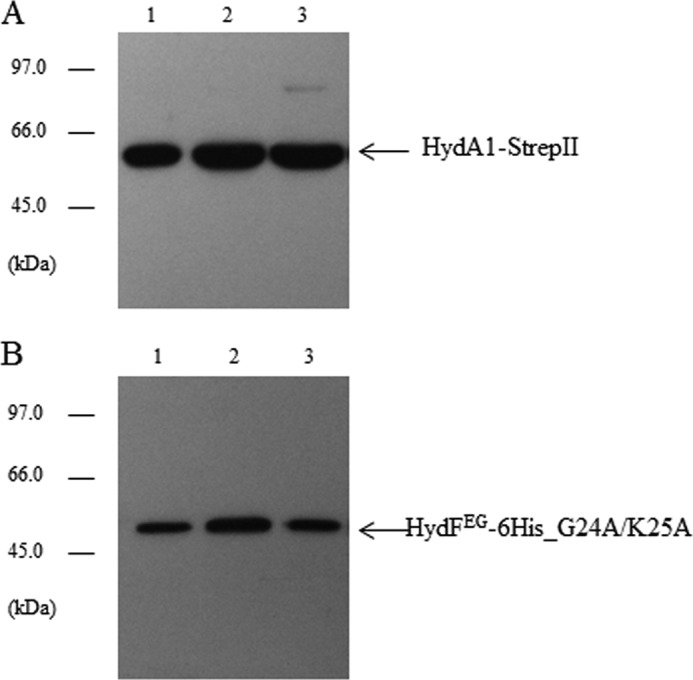
Analysis of the interaction between the mutant HydFEG-6His_G24A/K25A and HydA1-StrepII. Western blotting analysis showing the StrepTactin purification of HydA1-StrepII (A) and HydFEG-6His_G24A/K25A (B). Lanes 1, total E. coli cell extract; lanes 2, soluble fraction of cell extract; lane 3, pool of desthiobiotin eluted fractions. 25 μl of each sample were loaded on a 12% gel for SDS-PAGE. Western blotting with anti-StrepII tag and anti-6His tag monoclonal antibodies is shown.
Conclusions
During recent years remarkable advances have been made in the knowledge of the molecular mechanisms driving the [FeFe]-hydrogenases maturation pathway. Nevertheless, significant gaps remain in the understanding of how this process occurs, and the precise contribution of some of the players involved has still to be assigned. In this work, the interactions between the Hyd structural and functional proteins have been investigated in detail, and a quantitative analysis of these binding events has been provided for the first time. Our kinetic data suggest that the HydE and HydG radical-SAM proteins separately participate in modifying the H-cluster precursor on HydF. We also showed that HydF is able to interact with the two other maturases, as well as with the hydrogenase independently of its GTPase properties, which are otherwise involved in the dissociation of the HydE and HydG maturases from the scaffold. This would allow the coordinate stepwise process needed for the synthesis and chemical modification of the H-cluster precursor. Finally, our data suggest that HydE and HydG separately introduce in the HydF scaffold structural changes enabling its interaction with the hydrogenase, which nevertheless results in activation only when a complete 2Fe subcluster is transferred. The structural features of these intermediates of the hydrogenase maturation process are currently under investigation in our laboratory. These results provide new insights that may improve our understanding of the highly complex molecular pathway leading to the activation of the [FeFe]-hydrogenases, which requires further studies to be completely defined.
This work was supported by Ministero delle Politiche Agricole e Forestali Grant (to G. M. G.).
- SAM
- S-adenosylmethionine
- HydA1
- [FeFe]-hydrogenase 1
- HydFΔEG
- HydF expressed in the absence of HydE and HydG
- HydFEG
- HydF co-expressed with HydE and HydG
- HydFE
- HydF co-expressed with HydE
- HydFG
- HydF co-expressed with HydG
- HydAΔEFG
- [FeFe]-hydrogenase expressed in the absence of HydE, HydF, and HydG
- MCS
- multiple cloning site
- SPR
- surface plasmon resonance.
REFERENCES
- 1. Beinert H., Holm R. H., Münck E. (1997) Iron-sulfur cluster. Nature's modular, multipurpose structure. Science 277, 653–659 [DOI] [PubMed] [Google Scholar]
- 2. Lill R. (2009) Function and biogenesis of iron-sulphur proteins. Nature 460, 831–838 [DOI] [PubMed] [Google Scholar]
- 3. Meyer J. (2008) Iron-sulfur protein folds, iron-sulfur chemistry, and evolution. J. Biol. Inorg. Chem. 13, 157–170 [DOI] [PubMed] [Google Scholar]
- 4. Fontecilla-Camps J. C., Amara P., Cavazza C., Nicolet Y., Volbeda A. (2009) Structure-function relationships of anaerobic gas-processing metalloenzymes. Nature 460, 814–822 [DOI] [PubMed] [Google Scholar]
- 5. Duffus B. R., Hamilton T. L., Shepard E. M., Boyd E. S., Peters J. W., Broderick J. B. (2012) Radical AdoMet enzymes in complex metal cluster biosynthesis. Biochim. Biophys. Acta. 1824, 1254–1263 [DOI] [PubMed] [Google Scholar]
- 6. Peters J. W., Broderick J. B. (2012) Emerging paradigms for complex iron-sulfur cofactor assembly and insertion. Annu. Rev. Biochem. 81, 429–450 [DOI] [PubMed] [Google Scholar]
- 7. Mulder D. W., Shepard E. M., Meuser J. E., Joshi N., King P. W., Posewitz M. C., Broderick J. B., Peters J. W. (2011) Insights into [FeFe]-hydrogenase structure, mechanism, and maturation. Structure 19, 1038–1052 [DOI] [PubMed] [Google Scholar]
- 8. Nicolet Y., Fontecilla-Camps J. C. (2012) Structure-function relationships in [FeFe]-hydrogenase active site maturation. J. Biol. Chem. 287, 13532–13540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Posewitz M. C., King P. W., Smolinski S. L., Zhang L., Seibert M., Ghirardi M. L. (2004) Discovery of two novel radical S-adenosylmethionine proteins required for the assembly of an active [Fe] hydrogenase. J. Biol. Chem. 279, 25711–25720 [DOI] [PubMed] [Google Scholar]
- 10. Rubach J. K., Brazzolotto X., Gaillard J., Fontecave M. (2005) Biochemical characterization of the HydE and HydG iron-only hydrogenase maturation enzymes from Thermotoga maritima. FEBS Lett. 579, 5055–5060 [DOI] [PubMed] [Google Scholar]
- 11. Pilet E., Nicolet Y., Mathevon C., Douki T., Fontecilla-Camps J. C., Fontecave M. (2009) The role of the maturase HydG in [FeFe]-hydrogenase active site synthesis and assembly. FEBS Lett. 583, 506–511 [DOI] [PubMed] [Google Scholar]
- 12. Shepard E. M., Duffus B. R., George S. J., McGlynn S. E., Challand M. R., Swanson K. D., Roach P. L., Cramer S. P., Peters J. W., Broderick J. B. (2010) [FeFe]-hydrogenase maturation. HydG-catalyzed synthesis of carbon monoxide. J. Am. Chem. Soc. 132, 9247–9249 [DOI] [PubMed] [Google Scholar]
- 13. Nicolet Y., Rubach J. K., Posewitz M. C., Amara P., Mathevon C., Atta M., Fontecave M., Fontecilla-Camps J. C. (2008) X-ray structure of the [FeFe]-hydrogenase maturase HydE from Thermotoga maritima. J. Biol. Chem. 283, 18861–18872 [DOI] [PubMed] [Google Scholar]
- 14. Brazzolotto X., Rubach J. K., Gaillard J., Gambarelli S., Atta M., Fontecave M. (2006) The [FeFe]-hydrogenase maturation protein HydF from Thermotoga maritima is a GTPase with an iron-sulfur cluster. J. Biol. Chem. 281, 769–774 [DOI] [PubMed] [Google Scholar]
- 15. Shepard E. M., McGlynn S. E., Bueling A. L., Grady-Smith C. S., George S. J., Winslow M. A., Cramer S. P., Peters J. W., Broderick J. B. (2010) Synthesis of the 2Fe subcluster of the [FeFe]-hydrogenase H cluster on the HydF scaffold. Proc. Natl. Acad. Sci. U.S.A. 107, 10448–10453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. King P. W., Posewitz M. C., Ghirardi M. L., Seibert M. (2006) Functional studies of [FeFe]-hydrogenase maturation in an Escherichia coli biosynthetic system. J. Bacteriol. 188, 2163–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mulder D. W., Boyd E. S., Sarma R., Lange R. K., Endrizzi J. A., Broderick J. B., Peters J. W. (2010) Stepwise [FeFe]-hydrogenase H-cluster assembly revealed in the structure of HydA(ΔEFG). Nature 465, 248–251 [DOI] [PubMed] [Google Scholar]
- 18. McGlynn S. E., Ruebush S. S., Naumov A., Nagy L. E., Dubini A., King P. W., Broderick J. B., Posewitz M. C., Peters J. W. (2007) In vitro activation of [FeFe]hydrogenase. New insights into hydrogenase maturation. J. Biol. Inorg. Chem. 12, 443–447 [DOI] [PubMed] [Google Scholar]
- 19. McGlynn S. E., Shepard E. M., Winslow M. A., Naumov A. V., Duschene K. S., Posewitz M. C., Broderick W. E., Broderick J. B., Peters J. W. (2008) HydF as a scaffold protein in [FeFe] hydrogenase H-cluster biosynthesis. FEBS Lett. 582, 2183–2187 [DOI] [PubMed] [Google Scholar]
- 20. Mulder D. W., Ortillo D. O., Gardenghi D. J., Naumov A. V., Ruebush S. S., Szilagyi R. K., Huynh B., Broderick J. B., Peters J. W. (2009) Activation of HydA(ΔEFG) requires a preformed [4Fe-4S] cluster. Biochemistry 48, 6240–6248 [DOI] [PubMed] [Google Scholar]
- 21. Peters J. W., Szilagyi R. K., Naumov A., Douglas T. (2006) A radical solution for the biosynthesis of the H-cluster of hydrogenase. FEBS Lett. 580, 363–367 [DOI] [PubMed] [Google Scholar]
- 22. Cendron L., Berto P., D'Adamo S., Vallese F., Govoni C., Posewitz M. C., Giacometti G. M., Costantini P., Zanotti G. (2011) Crystal structure of HydF scaffold protein provides insights into [FeFe]-hydrogenase maturation. J. Biol. Chem. 286, 43944–43950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ruzzene M., Brunati A. M., Sarno S., Donella-Deana A., Pinna L. A. (1999) Hematopoietic lineage cell specific protein 1 associates with and down-regulates protein kinase CK2. FEBS Lett. 461, 32–36 [DOI] [PubMed] [Google Scholar]
- 24. Karlsson R., Fält A. (1997) Experimental design for kinetic analysis of protein-protein interactions with surface plasmon resonance biosensor. J. Immunol. Methods 200, 121–133 [DOI] [PubMed] [Google Scholar]
- 25. Driesener R. C., Challand M. R., McGlynn S. E., Shepard E. M., Boyd E. S., Broderick J. B., Peters J. W., Roach P. L. (2010) [FeFe]-hydrogenase cyanide ligands derived from S-adenosylmethionine-dependent cleavage of tyrosine. Angew. Chem. Int. Ed. Engl. 49, 1687–1690 [DOI] [PubMed] [Google Scholar]