

Background: CCR2 is a chemokine receptor up-regulated in breast cancer cells.
Results: Inhibiting the activity of CCR2 and downstream signaling proteins Smad3 and MAPK significantly reduces CCL2-induced survival and motility.
Conclusion: CCR2 regulates CCL2-induced breast cancer cell survival and motility through MAPK- and Smad3-dependent mechanisms.
Significance: Learning how CCR2 functions in breast cancer cells enhances our understanding of how cells survive and migrate.
Keywords: Breast Cancer, Cell Motility, Chemokines, MAP Kinases (MAPKs), Signal Transduction, Smad3, Cell Survival
Abstract
Increased cell motility and survival are important hallmarks of metastatic tumor cells. However, the mechanisms that regulate the interplay between these cellular processes remain poorly understood. In these studies, we demonstrate that CCL2, a chemokine well known for regulating immune cell migration, plays an important role in signaling to breast cancer cells. We report that in a panel of mouse and human breast cancer cell lines CCL2 enhanced cell migration and survival associated with increased phosphorylation of Smad3 and p42/44MAPK proteins. The G protein-coupled receptor CCR2 was found to be elevated in breast cancers, correlating with CCL2 expression. RNA interference of CCR2 expression in breast cancer cells significantly inhibited CCL2-induced migration, survival, and phosphorylation of Smad3 and p42/44MAPK proteins. Disruption of Smad3 expression in mammary carcinoma cells blocked CCL2-induced cell survival and migration and partially reduced p42/44MAPK phosphorylation. Ablation of MAPK phosphorylation in Smad3-deficient cells with the MEK inhibitor U0126 further reduced cell survival but not migration. These data indicate that Smad3 signaling through MEK-p42/44MAPK regulates CCL2-induced cell motility and survival, whereas CCL2 induction of MEK-p42/44MAPK signaling independent of Smad3 functions as an alternative mechanism for cell survival. Furthermore, we show that CCL2-induced Smad3 signaling through MEK-p42/44MAPK regulates expression and activity of Rho GTPase to mediate CCL2-induced breast cancer cell motility and survival. With these studies, we characterize an important role for CCL2/CCR2 chemokine signaling in regulating the intrinsic relationships between breast cancer cell motility and survival with implications on the metastatic process.
Introduction
Chemokines are a family of small soluble proteins that regulate cell migration through the formation of concentration gradients. Chemokines exhibit a high degree of conservation between mice and humans and have long been recognized as critical mediators of immune cell trafficking during embryonic development, wound healing, and infection (1–3). Currently ∼47 chemokine ligands and 23 chemokine receptors have been identified and are subdivided into several categories depending on composition of the cysteine motif present on the ligand (4). CCL22 belongs to the C-C class of chemokines and has been shown to be a critical modulator of inflammation, regulating macrophage recruitment during wound healing, infections, and autoimmune diseases. CCL2 exhibits a particular affinity for the receptor CCR2 (5–7), and signaling through CCR2 leads to activation of downstream signaling pathways including p42/44MAPK, phospholipase C-γ, and PKC through G protein-dependent mechanisms to regulate cellular adhesion and motility in macrophages (8–10).
Alterations in protein and RNA expression levels of CCL2 have recently been implicated in cancer. Elevated CCL2 expression correlates with tumor grade and poor patient prognosis as demonstrated in flow cytometry studies of cell suspensions prepared from tumor biopsies (11), immunohistochemistry studies of breast tumors (12, 13), and analysis of blood serum levels from breast cancer patients (14–17). Further expression analysis studies have revealed significant correlations between CCL2 expression and macrophage levels (13, 18). Functional studies reveal that blockade of CCL2 activity in mammary tumor-bearing mice decreases tumor growth and metastasis accompanied by decreased macrophage recruitment and angiogenesis (18–23). These studies indicate that CCL2 regulates tumor progression through a macrophage-dependent mechanism that is sustained through a positive feedback loop.
Although the importance of CCL2 signaling in macrophages has been established, recent studies in our laboratory indicate that CCL2 regulates tumor progression through additional mechanisms. In previous studies, we had shown that targeting expression of CCL2 in fibroblasts significantly reduced mammary tumor metastasis associated with decreased survival without notably affecting tumor growth or macrophage recruitment (21, 24). Furthermore, neutralizing antibodies to CCL2 blocked mammary carcinoma cell invasion induced by fibroblasts (21, 24). One study showed that CCL2 induced migrational responses in breast cancer cell lines (25). These studies indicate a role for CCL2 signaling in breast epithelial cells during the metastatic process.
Studies have shown that metastatic tumor cells acquire unique properties that enable them to escape the primary tumor including enhanced motility and survival (26, 27). To further understand the mechanisms that drive metastatic tumor cell behavior, we examined the role of CCL2 signaling in breast cancer cells, utilizing a panel of mouse and human breast cancer cell lines. We found that CCL2 increased cell survival and migration associated with increased phosphorylation of Smad3 and p42/44MAPK proteins. Delivery of CCR2 siRNAs inhibited CCL2-induced survival, migration, and phosphorylation of Smad3 and p42/44MAPK proteins. Through studies involving the use of shRNAs and pharmacologic inhibitors, we found that CCL2/CCR2 signaling enhances Smad3 and MEK-p42/44MAPK signaling, which activates Rho GTPase to regulate cell motility and survival. As an alternative mechanism for cell survival, CCL2/CCR2 signaling regulates signaling through p42/44MAPK independently of Smad3.
Previous studies have stressed the importance of CCL2 signaling in macrophages. The present study demonstrates an important role for CCL2/CCR2 signaling in regulating the intrinsic relationship between survival and migration in breast cancer cells.
EXPERIMENTAL PROCEDURES
Cell Culture
The following cell lines were obtained from American Tissue Culture Collection: Raw 264.7, Phoenix, 293A, MCF10A, MCF-7, MDA-MB-231, NMuMG, and 4T1. PyVmT cells were isolated from MMTV-PyVmT transgenic mice (28) using approaches described previously (29). Mammary fibroblasts from control Tgfbr2Flox/Flox and Tgfbr2FspKO mice were spontaneously immortalized and characterized previously (30). All cell lines except MCF10A were cultured on plastic in DMEM containing 10% FBS with 0.1% amphotericin, 1% penicillin-streptomycin (catalog number 30-004-CI, Cellgro). MCF10A cells were cultured in DMEM containing 50% F-12 (catalog number 10-080, Cellgro), 5% horse serum (catalog number 35-030-CV, Cellgro), 500 μg/ml hydrocortisone, 100 ng/ml cholera toxin (catalog number 8052, Sigma), 10 ng/ml insulin (catalog number I6634, Sigma), and 20 ng/ml EGF (catalog number E964, Sigma) with 0.1% amphotericin, 1% penicillin-streptomycin (31).
Generation of Conditioned Medium
500,000 cells were plated in 10-cm dishes in DMEM, 10% FBS for 24 h. The complete medium was aspirated, and cells were then incubated in 6 ml of serum-free medium for 24 h. The conditioned medium was then removed, filtered through a 0.4-μm filtration unit, and used for the experiments indicated.
Transient siRNA Transfection
Negative control siRNAs (catalog number AM4613) were obtained from Ambion. siRNAs targeting CCR2 (catalog number sc-44766), CCR2A (catalog number sc-270220), and Smad3 (catalog number sc-38376) were obtained from Santa Cruz Biotechnology. siRNAs were transfected into cells according to the manufacturer's protocol. Briefly, carcinoma cells were seeded at a density of 400,000 cells in 6-cm dishes and cultured for 24 h. Cells were then washed with PBS and incubated in Opti-MEM (catalog number 11058-021, Invitrogen) with a complex of 240 pmol of Smad3, CCR2 siRNA, or control siRNA and 24 μl of Lipofectamine 2000 transfection reagent (catalog number 11668027, Invitrogen) for 24 h. The medium containing siRNA-transfection reagent complexes was then aspirated and replaced with DMEM, 10% FBS for 24 h prior to stimulation with CCL2.
Retroviral Transduction
Phoenix cells were transfected with 10 μg of empty pBabe retroviral construct, pBabe expressing dominant negative RhoA (kindly provided by Jin Chen, M.D., Ph.D., Vanderbilt University, Nashville, TN), or pRetrosuper retroviral construct carrying a puromycin selection marker and shRNAs targeting two different regions of Smad3 (Smad3-1 and Smad3-6) (32) or GFP as a specificity control (33). The targeting sequences for each shRNA construct are as follows (5′-3′): GFP, GCT GAC GGA GAA CAA CATC; Smad3-1, GGCCATCACCACGCAGAAC; Smad3-6, CCTGAAGATCTTCAACAAT. Medium containing retrovirus was collected after 48 h of transfection and used to transduce 4T1 carcinoma cells seeded at 60% subconfluence in 10-cm dishes in the presence of 5 μg/ml Polybrene (catalog number AL-118, Sigma). 48 h postinfection, cells were placed under puromycin selection (2 μg/ml) (catalog number P9620, Sigma) in DMEM, 10% FBS containing antibiotics. Non-infected 4T1 cells were treated with puromycin as a control.
Adenoviral Infection
Adenovirus was generated using the pAdTrack/pAdEasy system as described (34). Briefly, wild type Smad3 cDNA was subcloned into pAdTrack as described (35). pAdTrack plasmid was cotransfected with pAdEasy into electrocompetent Escherichia coli BJ5183 cells (catalog number 200154, Agilent) to generate recombinant plasmid. 10 μg of recombinants were linearized with PacI restriction enzyme (catalog number R05047, New England Biolabs) and transfected into 293A packaging cells. Supernatant was harvested and concentrated using an Ultracel 50,000 molecular weight filtration unit (catalog number UFC T05008, Millipore). Cells were harvested in PBS and lysed by three freeze-thaw cycles in methanol/dry ice and at 37 °C. Virus from supernatant and cells were combined and measured to determine the number of plaque-forming units (pfu) according to Martin (36). 4T1 cells were infected with vehicle adenovirus or Ad-Sm3 at 107 pfu/ml for 24 h and analyzed as described.
Western Blot
Carcinoma cells were seeded in 6-cm dishes at a density of 400,000 cells, cultured for 24 h, and starved in serum-free medium for 24 h. Cells were then treated with 2 ml of conditioned medium or serum-free medium at 37 °C in the presence or absence of 20 ng/ml CCL2 (catalog number 479-JE-010, R&D Systems), 5 ng/ml TGF-β (catalog number 101-B1–001, R&D System), 10–100 μm Rho kinase inhibitor II (catalog number 555551, Calbiochem), 10 μg/ml goat IgG (Sigma), 10 μg/ml anti-CCL2 (catalog number AB-279-NA, R&D Systems), 5–10 μm SB431542 (catalog number 616461, Calbiochem), 100–300 μm pertussis toxin (catalog number P7208, Sigma), or 1 μm U0126 (catalog number 9903, Cell Signaling Technology). The cells were lysed in radioimmune precipitation assay buffer containing 10 mm Tris-HCl, pH 8.0, 0.1 mm EDTA, 0.1% sodium deoxycholate, 0.1% SDS, and 140 mm NaCl supplemented with a protease inhibitor mixture containing aprotinin, leupeptin, bestatin, and pepstatin A (catalog number P8340, Sigma) and 10 mm phosphatase inhibitor sodium orthovanadate (catalog number S6508, Sigma). 50 μg of protein were resolved by 8–12% SDS-PAGE. The proteins were transferred to nitrocellulose membranes (Fisher) and then probed with antibodies (1:1000) to phospho-p42/44MAPK (Thr-202/Tyr-204) (catalog number 4370, Cell Signaling Technology), p42/44MAPK (catalog number 4695, Cell Signaling Technology), phospho-Smad3 (Ser-423/425) (catalog number 9520, Cell Signaling Technology), Smad3 (catalog number 9523, Cell Signaling Technology), phospho-AKT (Ser-473) (catalog number 4060, Cell Signaling Technology), AKT (catalog number 4685, Cell Signaling Technology), phospho-Src (Tyr-416) (catalog number 6943, Cell Signaling Technology), Src (catalog number 2109, Cell Signaling Technology), phospho-focal adhesion kinase (Tyr-397) (catalog number 3293, Cell Signaling Technology), focal adhesion kinase (C-20, Santa Cruz Biotechnology), RhoA (catalog number 2117, Cell Signaling Technology), CCR2 (M-50, Santa Cruz Biotechnology), CCR2A (H-61, Santa Cruz Biotechnology), or pan-actin (catalog number 8456, Cell Signaling Technology). Specific immunoreaction was detected with goat (sc-2020, Santa Cruz Biotechnology), rabbit (166-2408EDU, Bio-Rad), or mouse (catalog number 172-1011-EDU, Bio-Rad) secondary antibodies conjugated to horseradish peroxidase and Pierce ECL Western blotting substrate (catalog number 32106, Fisher).
Cleaved Caspase-3 Assay
Cells were seeded at a density of 250,000 on glass coverslips in 6-cm dishes. Apoptosis was induced by serum starvation, gentamicin (catalog number G1264, Sigma), or 5-fluorouracil (5-FU; catalog number F6627, Sigma) for 24 h in the presence or absence of 20 ng/ml CCL2, 1 μm U0126, or 10–100 μm Rho kinase inhibitor II. Cells were fixed in 10% neutral formalin buffer and permeabilized with ice-cold methanol for 10 min at −20 °C, blocked in PBS containing 1% goat serum, immunostained for antibodies to cleaved caspase-3 (Asp-175) (Cell Signaling Technology) at 1:200 dilution overnight at 4 °C in blocking buffer, and visualized by secondary rabbit antibodies conjugated to Alexa Fluor 488 (catalog number A11008, Invitrogen) at a 1:500 dilution. Samples were counterstained with DAPI (catalog number D9542, Sigma) at 1:1000 and mounted onto glass slides with ProLong antifade reagent (catalog number P36930, Invitrogen). Images were captured at 20× magnification using a Motic AE 31 microscope with Infinity 2-1c color digital camera. Samples were plated in duplicate, and three fields per sample were captured and quantitated by NIH ImageJ software.
TUNEL Assay
Cells were seeded at a density of 250,000 on glass coverslips in 6-cm dishes. Apoptosis was induced by serum starvation, gentamicin, or 5-FU for 24 h in the presence or absence of CCL2. Samples were fixed in 10% neutral formalin buffer, stained for TUNEL-positive nuclei, counterstained with propidium iodide according to the manufacturer's instructions (catalog number A23210, Invitrogen), and mounted onto glass slides with ProLong antifade reagent. Images were captured at 20× magnification using a Motic AE 31 microscope with Infinity 2-1c color digital camera. Samples were plated in duplicate, and three fields per sample were captured and quantitated by NIH ImageJ software.
Wound Closure
Wound closure assays were conducted as described previously (30). Briefly, carcinoma cell lines (50,000 cells/well) were seeded in 24-well plates, serum-deprived for 24 h, and then stimulated with CCL2 for 8 h. A vertical wound was made to the cells with a sterile pipette tip, and phase-contrast images were taken of each sample at 0 and 24 h at 10× magnification using a Motic AE 31 microscope with Infinity 2-1c color digital camera. Wound closure was assessed using NIH ImageJ software.
Transwell Migration
8-μm-pore Transwell supports (catalog number 3422, Costar) were coated with PBS buffer containing 50 μg/ml fibronectin (catalog number F1141, Sigma) and 0.1% gelatin (catalog number G9136, Sigma). Carcinoma cells were serum-starved for 24 h and then seeded (75,000) on top of the Transwell in the presence or absence of 20 ng/ml CCL2 in the presence or absence of inhibitors at 37 °C for 8 h. The cells were fixed in 10% neutral formalin buffer (VWR) for 10 min and stained with 0.1% crystal violet (catalog number AC21212-0250, Fisher) for 10 min. Tumor cells on the top side of the filter were removed by a cotton swab. Carcinoma cells that migrated to the underside of the filter were micrographed with a Nikon SMZ-800 stereo microscope with a chargecoupled device camera; three fields per sample were captured at 10× magnification. Quantitation of tumor cells was determined by measuring the pixel density of crystal violet-stained cells using NIH ImageJ software (arbitrary units).
RhoA GTPase Assay
Cells were seeded in duplicate in 6-cm dishes (250,000) in DMEM, 10% FBS with antibiotics. Cells were serum-starved for 24 h, stimulated with 20 ng/ml CCL2 in the presence or absence of inhibitors for 8 h, and assayed for RhoA GTPase activity by RhoA G-LISA (catalog number BK124, Cytoskeleton) according to the commercial protocol. Briefly, cell lysates were prepared, and the protein concentration was measured. Protein samples were normalized to concentration prior to incubation in the 96-well plates provided, and RhoA activity was assessed by absorbance reading of colorimetric signals at A490 nm using a BioTek microplate reader.
Immunohistochemistry
CCL2 and CCR2 staining were performed on array slides containing de-identified cores of normal (n = 14) and invasive breast ductal carcinoma tissues (n = 20) sectioned at 5-μm thickness (catalog number 8022, US Biomax). Additional studies were performed on de-identified normal and breast cancer tissues (n = 5 per group) obtained from the Biospecimen Shared Resource, an Institutional Review Board-approved facility at the University of Kansas Medical Center. These tissues were obtained under a human subject exemption policy. For immunostaining, tissue sections (5 μm) were dewaxed, rehydrated, and then subjected to antigen retrieval in sodium citrate buffer, pH 6.0 for 10 min at 100 °C. Samples were blocked in PBS containing 5% rabbit serum and incubated with antibodies (1:100) to CCL2 (catalog number sc-1784, Santa Cruz Biotechnology) and CCR2 (catalog number 25788, Abcam) overnight at 4 °C. Samples were washed in PBS and incubated with secondary goat biotinylated antibodies (1:500) (catalog number BA-5000, Vector Laboratories), conjugated with streptavidin peroxidase (catalog number PK-4000, Vector Laboratories), and incubated with 3,3′-diaminobenzidine substrate (catalog number K346711, Dako). Sections were counterstained with Mayer's hematoxylin for 1 min, dehydrated, and mounted with Cytoseal. For analysis, four fields were captured at 10× magnification using a Motic AE 31 microscope with Infinity 2-1c color digital camera. Total 3,3′-diaminobenzidine staining was quantified by pixel density analysis using NIH ImageJ software.
Flow Cytometry
Cells were cultured in complete medium in 10-cm dishes as described above. To detach cells from the plastic, cells were washed with PBS twice and incubated with 3 mm EDTA at 37 °C for 10–15 min. Cells were washed with 10 ml of complete medium twice, fixed in 10% neutral formalin buffer for 10 min at room temperature, and washed with PBS twice to remove traces of formalin. For CCR2 staining in mouse cells, 4T1 and PyVmT cells (300,000) were incubated with anti-CCR2 conjugated to phycoerythrin (catalog number FB151P, R&D Systems) according to the manufacturer's instructions. Cells were washed with PBS three times and filtered in PBS prior to analysis; cells were compared with unstained control. For CCR2 staining in human cells, MDA-MB-231 and MCF-7 cells (300,000) were incubated with goal polyclonal anti-CCR2 (catalog number 85711, Abcam) in PBS containing 2% BSA overnight at 4 °C at a 1:50 dilution. Cells were washed with PBS three times and incubated with goat secondary antibodies conjugated to Alexa Fluor 488 (Invitrogen) in PBS at a 1:500 dilution at 4 °C while covered in foil. Cells were washed with PBS three times and filtered in PBS prior to analysis; cells were compared with unstained control and secondary antibody-only controls. CCR2 expression was analyzed on an LSRII flow cytometer (BD Biosciences).
ELISA
Conditioned media generated from the indicated cell lines were subjected to CCL2 ELISA specific to murine (catalog number 900-K126, Peprotech) or human protein (catalog number 900-K31 Peprotech) or to TGF-β ELISA compatible with mouse and human protein (catalog number DY240, R&D Systems). Samples were analyzed according to the manufacturer's protocol. Reactions were catalyzed using tetramethylbenzidine substrate (catalog number DY999, R&D Systems) according to the manufacturer's instructions. The reaction was stopped with 1 m HCl, and absorbances were read at A450 nm using a BioTek microplate reader.
Immunocytochemistry
Cells were seeded at a density of 250,000 on glass coverslips in 6-cm dishes. For Smad3 nuclear localization studies, cells were serum-deprived for 24 h and then stimulated with CCL2 for 24 h. For CCR2 and TGF-β receptor I receptor localization studies, cells were serum-starved for 24 h and stimulated with CCL2 or TGF-β for 5 min. Cells were fixed in 10% neutral formalin buffer for 30 min, permeabilized in methanol for 10 min at −20 °C. After blocking in PBS containing 1% goat serum, samples were incubated with primary antibodies to Smad3 at a 1:100 dilution (Cell Signaling Technology), CCR2 at a 1:50 dilution (anti-CCR2-phycoerythrin, R&D Systems), or TGF-β receptor I at a 1:200 dilution (catalog number 06-1086, Millipore) overnight. Samples were washed with PBS three times. Smad3 and TGF-β receptor I were visualized by incubation with rabbit secondary antibodies conjugated to Alexa Fluor 568 (catalog number A1101, Invitrogen) at a 1:500 dilution. Cells were washed in PBS three times, counterstained with DAPI, and mounted on glass slides in ProLong antifade reagent (Invitrogen).
Statistical Analysis
Experiments were performed in triplicate at the minimum. Data are expressed as mean ± S.E. Statistical analysis was performed using two-tailed t test or analysis of variance with Bonferroni's post-test of comparisons using GraphPad software. Significance was determined by p < 0.05.
RESULTS
CCL2 Stimulates Survival and Motility of Mammary Carcinoma Cells
In recent studies, we used the MMTV-PyVmT transgenic mouse model and 4T1 transplant model of breast ductal carcinoma formation to demonstrate that CCL2 derived from fibroblasts regulated breast cancer metastasis independently of macrophage recruitment (21, 24). To determine a role for CCL2 signaling in mammary carcinoma cells, cultured 4T1 and PyVmT cells were treated with recombinant CCL2 and then evaluated for changes in cell proliferation, survival, and migration. CCL2 had no significant effect on breast cancer cell proliferation as determined by BrdU incorporation studies (data not shown). To induce cellular apoptosis, 4T1 and PyVmT cells were deprived of serum and treated with gentamicin or 5-fluoruracil. Gentamicin is an anti-inflammatory compound that was shown to block inflammatory disease including mastitis and arthritis (37–39). Gentamicin effectively induces apoptosis in epithelial and mesenchymal cell types (40–42) through caspase-dependent mechanisms (42–45). 5-FU is a synthetic nucleotide analog commonly used to treat non-invasive and invasive breast cancer (46, 47). 5-FU induces cell cycle checkpoint arrest and apoptosis through caspase-dependent mechanisms (48, 49). As CCL2 signaling is a key regulator of inflammatory processes (50–53) and is overexpressed in breast tumors (see Fig. 3), we determined the possibility that CCL2 would block apoptosis induced by gentamicin and 5-FU. CCL2 significantly reduced apoptosis of 4T1 and PyVmT cells induced by serum deprivation or gentamicin or 5-FU treatment as determined by cleaved caspase-3 expression (Fig. 1A). CCL2 treatment of MDA-MB-231 cells, a human breast cancer cell line, also significantly reduced apoptosis induced by serum deprivation or gentamicin or 5-FU treatment, indicating similar prosurvival effects of CCL2 on human breast cancer cells (Fig. 1A). Because breast tumors have been shown to acquire somatic mutations in apoptosis pathways during cancer progression (54), we determined the effect of CCL2 on MCF-7 breast cancer cells, which exhibit inactivating mutations in caspase-3 (55). Serum deprivation and gentamicin treatment after 24 h still resulted in detectable levels of apoptosis in MCF-7 cells that were significantly reduced with CCL2 treatment as determined by TUNEL analysis (Fig. 1B). These data indicate that CCL2 significantly blocks apoptosis in both mouse and human mammary carcinoma cells. We next determined the effect of CCL2 on the motility of mammary carcinoma cells by Transwell and wound closure assays. Treatment of 4T1 and PyVmT mammary carcinomas with CCL2 resulted in significant cell migration over 8 h as determined by Transwell assay. MCF-7 and MDA-MB-231 cells also showed significant increases in Transwell migration in response to CCL2 (Fig. 2A). In wound closure assays, CCL2 treatment significantly enhanced migration of all four cell lines over a period of 24 h, consistent with the promigratory effects of CCL2 observed in the Transwell assays (Fig. 2B). In summary, these data show that CCL2 significantly enhances survival and motility of epithelial cells in mouse and human cell lines.
FIGURE 3.

CCR2 expression is up-regulated in breast cancer tissues and corresponds to increased CCL2 expression. A, CCL2 and CCR2 staining was performed on array slides containing de-identified cores of normal (n = 14) and invasive breast ductal carcinoma tissues (n = 20) sectioned at 5-μm thickness (catalog number 8022, US Biomax). Additional studies were performed on de-identified normal and breast cancer tissues (n = 5 per group) obtained from the Biospecimen Shared Resource at the University of Kansas Medical Center. Representative images of staining for CCL2 and CCR2 are from the tissue arrays. Arrowheads point to staining in ductal epithelial cells. The magnified inset shows CCL2 staining in the stroma. Tumor scale bar, 50 μm. Levels of CCR2 and CCL2 expression were measured by pixel density using NIH ImageJ software. Arbitrary units are shown. Statistical analysis was performed by two-tailed t test: ***, p < 0.05 compared with normal tissue. B, expression of CCR2 was analyzed in mouse and human mammary carcinoma cells by flow cytometry analysis with the Raw 264.7 murine macrophage cell line as a positive control. C, expression of CCL2 was analyzed in mouse and human mammary carcinoma cells by ELISA of conditioned medium. For B and C, statistical analysis was performed by analysis of variance test with Bonferroni's post-test of comparisons: ***, p < 0.05; ****, p > 0.05. Values are expressed as mean ± S.E. Error bars represent S.E.
FIGURE 1.
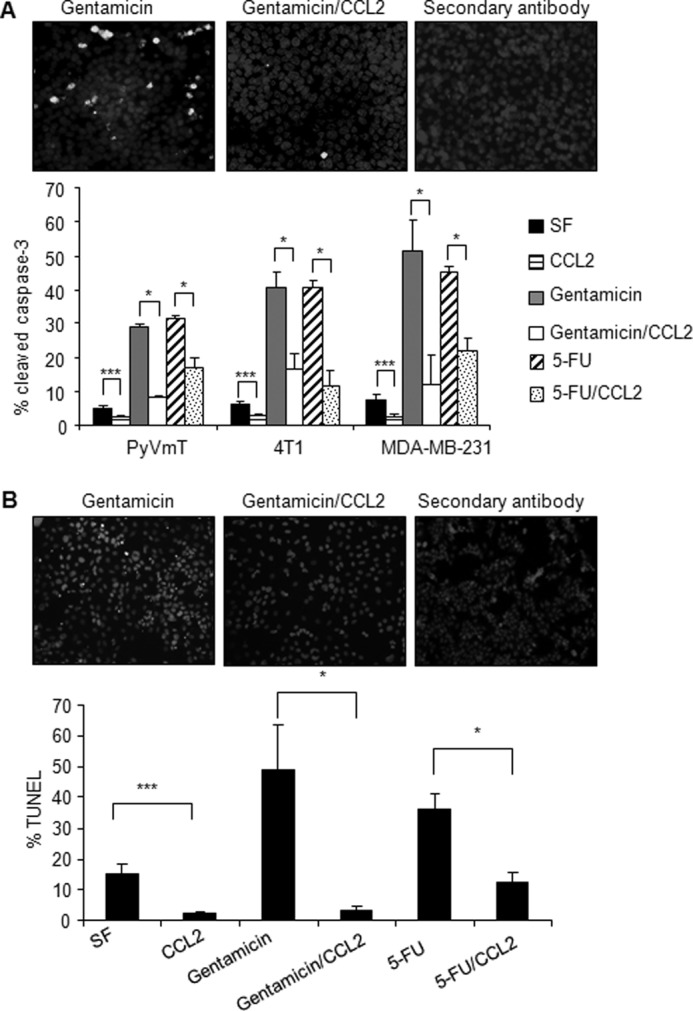
CCL2 blocks serum-deprived and gentamicin-induced apoptosis of mouse and human mammary carcinoma cells. A, mammary carcinoma cells were incubated in serum-free medium in the presence or absence of 20 ng/ml CCL2, 250 μg/ml gentamicin, or 250 μg/ml 5-FU for 24 h and analyzed for apoptosis by immunofluorescence staining for cleaved caspase-3 expression. Representative images of cleaved caspase-3 staining with DAPI overlay are shown for 4T1 cells. B, MCF-7 breast cancer cells were incubated in serum-free medium (SF) in the presence or absence of 250 μg/ml gentamicin and 20 ng/ml CCL2 for 24 h and analyzed for apoptosis by immunofluorescence staining for TUNEL-positive nuclei. Representative images of TUNEL staining with propidium iodide overlay are shown. Statistical analysis was determined by two-tailed t test: *, p < 0.05; ***, p < 0.001. Values are expressed as mean ± S.E. Error bars represent S.E.
FIGURE 2.

CCL2 enhances migration of mammary carcinoma cells. Mouse and human mammary carcinoma cell lines were incubated in serum-free medium (SF) in the presence or absence of 20 ng/ml CCL2 and analyzed for changes in migration by Transwell assay (A) and wound closure assay (B). Representative images of 4T1 mammary carcinoma cells that migrated across a Transwell membrane after 8-h incubation with CCL2 are shown (A). Representative images of wound closure of 4T1 cells after 24-h incubation with CCL2 are shown (B). Statistical analysis was determined by two-tailed t test: *, p < 0.001; **, p < 0.01; ***, p < 0.05. Values are expressed as mean ± S.E. Error bars represent S.E.
CCR2 Expression in Breast Cancer Cells Is Essential for CCL2 Signaling
CCL2 has been shown to exhibit strong binding affinity to the chemokine receptor CCR2 (kd = 0.77 nm) and signal primarily through this receptor in macrophages to regulate migration (5). The relevance of CCR2 in breast cancer cells remained unclear; therefore, we performed immunostaining analysis of CCR2 in invasive breast carcinomas in comparison with normal breast tissues. CCR2 expression was significantly increased in breast tumors compared with normal tissues and was primarily localized to cells of epithelial origin. CCR2 expression in breast tumors corresponded to increased CCL2 expression in breast tumors that was detected in the breast tumor stroma and epithelium (Fig. 3A). The expression patterns of CCR2 in breast epithelial cells were confirmed by flow cytometry analysis of breast epithelial cell lines. Compared with NMuMG cells, non-tumorigenic immortalized mouse mammary epithelial cells (56), 4T1 and PyVmT mammary carcinoma cells expressed significantly higher levels of CCR2, comparable with receptor levels in macrophages (Fig. 3B and supplemental Fig. 1). Similarly, MCF-7 and MDA-MB-231 breast cancer cells expressed higher levels of CCR2 compared with MCF10A cells (Fig. 3B), which are non-tumorigenic immortalized human breast epithelial cells (57). By ELISA analysis, we observed that CCL2 secretion in breast cancer cells varied among cell lines. In murine PyVmT carcinoma cells and NMuMG non-tumorigenic cells, CCL2 was expressed at marginally higher levels than CCL2 expression found in macrophages. In contrast, 4T1 mammary carcinoma cells expressed significantly lower levels of CCL2 compared with NMuMG cells. In human MCF-7 breast carcinoma cells, significantly higher levels of CCL2 were expressed compared with non-tumorigenic MCF10A cells. MDA-MB-231 cells expressed CCL2 at levels lower than those in MCF-7 cells and marginally higher than those in MCF10A cells (Fig. 3C). These data indicate that CCR2, but not CCL2, corresponds to tumorigenicity of breast carcinoma cell lines.
In previous studies, we had shown that TGF-β suppressed CCL2 expression in breast stromal cells to regulate tumor progression (21). As TGF-β negatively regulates CCL2, we hypothesized that CCL2 would suppress TGF-β signaling in breast cancer cells. CCL2 had no significant effect on Smad2 phosphorylation (data not shown) but unexpectedly increased Smad3 phosphorylation over time. We screened for phosphorylation of other candidate proteins showing a functional association with migration and survival including MAPK, focal adhesion kinase, Src, and AKT (58, 59). CCL2 significantly enhanced phosphorylation of p42/44MAPK and Smad3 proteins in multiple breast cancer cell lines over an extended period of time. Expression levels of these proteins were slightly weaker or comparable with TGF-β treatment in their respective cancer cell lines and with MCF10A breast epithelial cells (supplemental Fig. 2A and Fig. 4C). CCL2 treatment also resulted in increased nuclear Smad3 expression as determined by increased Smad3 immunofluorescence staining (supplemental Fig. 2B), consistent with the role of Smad3 as a transcription factor (60). In contrast to Smad3 and MAPK, focal adhesion kinase and Src showed no significant changes in phosphorylation in CCL2-treated cells. In CCL2-treated cells, AKT showed weak transient phosphorylation at 1 h, which then decreased over time (supplemental Fig. 3). These data indicate that MAPK and Smad3 pathways may play important roles in CCL2 signaling in breast cancer cells.
FIGURE 4.

CCR2 is required for CCL2 signaling in mouse and human breast carcinoma cells. 4T1 or MCF-7 parental (Par) cells, control (Con) siRNA-transfected cells, or cells transfected with CCR2 siRNAs were stimulated with 20 ng/ml CCL2 and analyzed for changes in gentamicin-induced apoptosis by cleaved caspase-3 or TUNEL assay as indicated (A), changes in migration by wound closure assay (B), and expression of the indicated proteins by Western blot after 8-h stimulation (C). TGF-β treatment of MCF10A cells is shown as a comparison of phospho-Smad3 expression between cell lines. The level of CCR2 knockdown was determined by densitometry analysis using NIH ImageJ software. CCR2 knockdown was compared with control siRNA samples. Values are normalized to actin. Statistical analysis was determined by two-tailed t test: *, p < 0.001; **, p < 0.01; ***, p < 0.05. Values are expressed as mean ± S.E. Error bars represent S.E.
The functional significance of CCR2 was determined by CCR2 knockdown in 4T1 and MCF-7 mammary carcinoma cells. Compared with parental cells or cells expressing control siRNAs, CCL2 did not significantly affect gentamicin-induced apoptosis in CCR2-deficient cells as determined by cleaved caspase-3 staining (Fig. 4A). CCR2 knockdown in 4T1 and MCF-7 cells also significantly blocked CCL2-induced wound closure, indicating defects in CCL2-induced cell migration (Fig. 4B). CCR2 knockdown significantly reduced CCL2-induced phosphorylation of p42/44MAPK and Smad3 in 4T1 and MCF-7 cells (Fig. 4C). In summary, these data indicate that CCR2 is essential for CCL2 induction of breast cancer cell motility and survival.
Smad3 and MEK Signaling through p42/44MAPK Regulate CCL2-induced Cell Survival and Motility
To determine the functional role of Smad3 and p42/44MAPK signaling pathways in CCL2-induced cell motility and survival, we used a combination of approaches involving RNA interference and pharmacologic inhibitors. 4T1 mammary carcinoma cell lines were generated to stably express shRNAs to Smad3 that targeted two different regions of Smad3 (referred to as Smad3-1 and Smad3-6). Compared with 4T1 parental cells and cells expressing control shRNAs to GFP, Smad3 knockdown significantly decreased Smad3 expression and phosphorylation. Smad3 knockdown resulted in a partial loss of p42/44MAPK phosphorylation, whereas U0126 treatment of control 4T1 mammary carcinoma cells inhibited Smad3 phosphorylation (Fig. 5A). Transient Smad3 knockdown in other mammary carcinoma cell lines including MCF-7 (Fig. 5A), MDA-MB-231, and PyVmT (supplemental Fig. 4) resulted in similar reductions in MAPK phosphorylation. Re-expression of Smad3 in Smad3-deficient mammary carcinoma cells enhanced the levels of phospho-p42/44MAPK protein (supplemental Fig. 5A). These data indicate that CCL2-induced p42/44MAPK activity is partially dependent on Smad3 expression and that Smad3 phosphorylation is also dependent on p42/44MAPK activity in breast cancer cells. The presence of phosphorylated p42/44MAPK protein in Smad3-deficient cells indicated an additional upstream regulator of p42/44MAPK. Previous studies have shown that important upstream regulators of p42/44MAPK phosphorylation are MEK1/2 proteins, which often function in concert to directly phosphorylate p42/44MAPK proteins at threonine and tyrosine residues (61). Therefore, we hypothesized that MEK signaling through p42/44MAPK and Smad3 signaling through p42/44MAPK would mediate CCL2 induction of breast cancer cell motility and survival.
FIGURE 5.

Effect of Smad3 and MAPK inhibition on CCL2-induced cell survival and migration of mammary carcinoma cells. Parental cells (Par), 4T1 cells stably expressing shRNAs to GFP (GFP−) or Smad3 (Sm3-1 and Sm3-6), and MCF-7 cells transiently expressing control siRNAs (Con) or Smad3 siRNAs (Smad3−) were treated with CCL2 (20 ng/ml) in the presence or absence of 1 μm U0126 and analyzed by Western blot for changes in phosphorylation of Smad3 and p42/44MAPK (A). Densitometry analysis was performed on three independent experiments. Values are expressed as mean ± S.E. B, gentamicin (Gent)-induced apoptosis. C, migration by wound closure assay. Statistical analysis was determined by two-tailed t test: **, p < 0.01; ***, p < 0.05; ****, p > 0.05. Values are expressed as mean ± S.E. Error bars represent S.E. SF, serum-free medium.
We first assessed the role of these signaling pathways in gentamicin-induced apoptosis in 4T1 and MCF-7 carcinoma cell lines. Control cells or Smad3-deficient cells were treated with CCL2 in the presence or absence of U0126, an MEK kinase inhibitor (61, 62), which blocked CCL2-induced p42/44MAPK phosphorylation (Fig. 5A). We observed that Smad3 knockdown inhibited CCL2-induced cell survival under gentamicin treatment. Re-expression of wild type Smad3 protein by adenoviral infection of Smad3-deficient 4T1 cells restored the ability of CCL2 to promote cell survival in gentamicin-treated cells to levels similar to those observed in 4T1 parental cells (supplemental Fig. 5, A and B), further supporting a role for Smad3 in CCL2-induced cell survival. U0126 treatment increased apoptosis of CCL2-treated cells, indicating an important role for MEK-MAPK signaling in CCL2-induced cell survival. We then determined whether the MEK-MAPK pathway affected Smad3 signaling to regulate CCL2-induced breast cancer cell survival. We hypothesized that if MEK-MAPK functioned independently of Smad3 to mediate cell survival then Smad3 knockdown combined with U0126 treatment would further inhibit CCL2-induced cell survival compared with Smad3 knockdown or MEK inhibition alone. Supporting this hypothesis, treatment of Smad3-deficient cells combined with U0126 further increased gentamicin-induced apoptosis (Fig. 5B) compared with Smad3 deficiency alone or U0126 treatment alone. These data indicate that MEK signaling through MAPK functions independently of Smad3 to regulate CCL2-induced mammary carcinoma cell survival.
We then assessed the role of these signaling pathways in CCL2-induced cell migration in 4T1 and MCF-7 carcinoma cells. Smad3-deficient cells (Smad3-1 and Smad3-6) showed significant reductions in CCL2-induced wound closure (Fig. 5C). Re-expression of Smad3 in Smad3-deficient 4T1 cells rescued the ability of CCL2 to induce cell migration to levels similar to those observed in 4T1 parental cells, validating an important role for Smad3 in CCL2-induced cell migration (supplemental Fig. 5C). U0126 treatment of 4T1 or MCF-7 control cells significantly decreased CCL2-induced wound closure, indicating an important role for MEK-MAPK signaling in CCL2-induced cell migration (Fig. 5C). To determine whether MEK-MAPK depended on Smad3 signaling to mediate cell migration, Smad3-deficient cells were also treated with U0126 and analyzed for changes in CCL2-induced wound closure. We predicted that if MEK-MAPK functioned independently of Smad3 then Smad3 knockdown combined with U0126 treatment would further inhibit CCL2-induced migration compared with Smad3 knockdown or MEK inhibition alone. However, Smad3 knockdown combined with U0126 treatment did not further inhibit CCL2-induced wound closure (Fig. 5C), indicating that MEK-MAPK signaling depended on Smad3 to regulate CCL2-induced breast cancer cell migration.
Rho GTPase Functions Downstream of p42/44MAPK to Regulate CCL2-induced Motility and Survival
We further characterized the molecular mechanisms through which Smad3 and MEK signaling through MAPK would regulate CCL2-induced cell survival and motility. As a small GTPase, RhoA expression has recently been shown to regulate CCL2-induced migration of PC-3 prostate cancer cells (63). However, the functional significance of RhoA in CCL2 signaling in breast cancer cells had not been addressed. In these studies, 4T1 cells were treated with CCL2 in the presence or absence of U0126 treatment or Smad3 shRNA expression and analyzed for RhoA expression. Although Smad3-deficient cells showed higher basal levels of RhoA expression compared with control cells, there was no significant difference in Rho expression with CCL2 treatment. U0126 treatment did not significantly affect CCL2 induction of RhoA in control cells. Smad3-deficient cells treated with U0126 did not show any significant differences in RhoA expression compared with Smad3-deficient alone or U0126 treatment alone (Fig. 6A). These data indicate that RhoA expression in both 4T1 cells depends on normal levels of Smad3 expression, and targeting of Smad3 deregulates CCL2 induction of RhoA expression.
FIGURE 6.
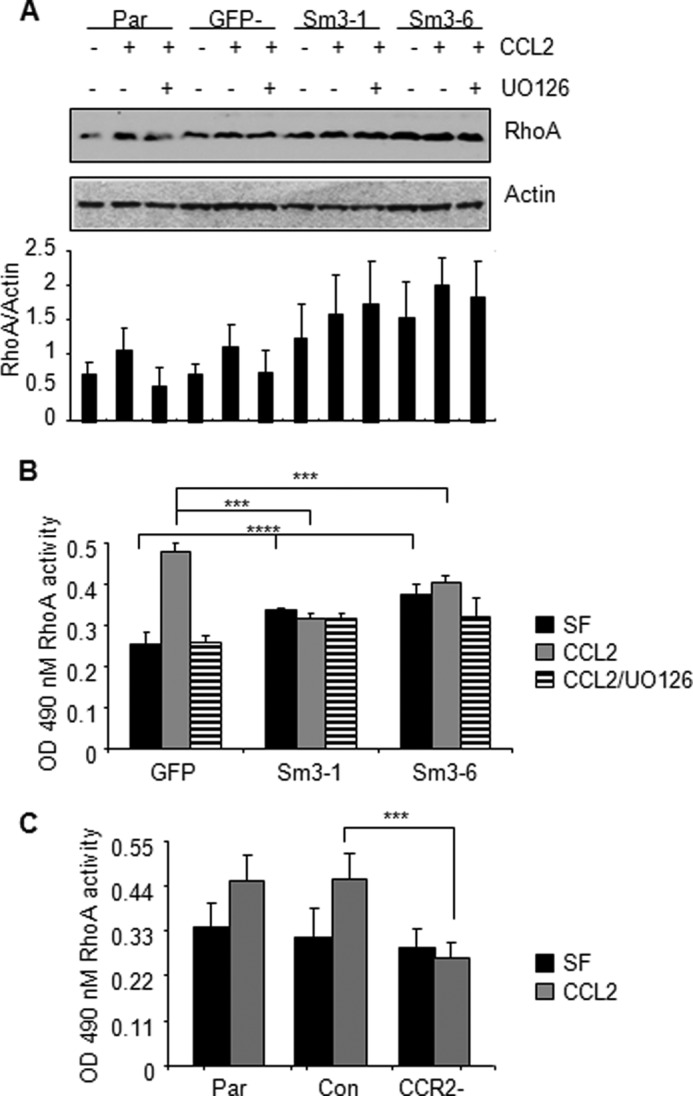
CCL2/CCR2 signaling mediates RhoA activity through Smad3- and MAPK-dependent mechanisms. Parental (Par) 4T1 cells and 4T1 cells stably expressing control shRNAs (GFP−) or Smad3 shRNAs (Sm3-1 and Sm3-6) were treated with 20 ng/ml CCL2 in the presence or absence of 1 μm U0126. A, samples were analyzed for expression of RhoA by Western blot analysis after 4 h of treatment. RhoA expression was determined by densitometry analysis of three independent Western blot experiments using NIH ImageJ software. Values are expressed as mean ± S.E. B, RhoA GTPase activity was analyzed by G-LISA after 8-h incubation. Statistical analysis was determined by analysis of variance with Bonferroni's post-test of comparisons. C, parental (Par) 4T1 or cells expressing control siRNA (Con) or CCR2 siRNAs were analyzed for RhoA activity by G-LISA. Statistical analysis was determined by two-tailed t test: ***, p < 0.05; ****, p > 0.05. Values are expressed as mean ± S.E. Error bars represent S.E. SF, serum-free medium.
We next determined whether CCL2/CCR2-induced RhoA activity was regulated by Smad3 and MEK signaling through p42/44MAPK in 4T1 mammary carcinoma cells. These studies involved commercially available ELISA-based assays in which the levels of activated RhoA protein were measured by the amount of RhoA bound to rhotekin substrate on 96-well plates. Smad3-deficient cells showed a higher basal level of Rho GTPase activity, which was not observed to be statistically significant and was not significantly affected by CCL2 treatment (Fig. 6B). U0126 treatment significantly inhibited CCL2-induced Rho GTPase activity, indicating an important role for MEK-MAPK in regulating RhoA activity. However, U0126 treatment of Smad3-deficient cells did not further affect CCL2-induced RhoA activity. These data indicate that CCL2-induced RhoA activity is regulated by both Smad3 and MEK-MAPK signaling. Furthermore, we found that transient CCR2 knockdown inhibited CCL2-induced RhoA activity in 4T1 mammary carcinoma (Fig. 6C), indicating a requirement for CCR2 expression in CCL2 induction of RhoA GTPase activity in mammary carcinoma cells. In summary, these data indicate that CCL2/CCR2 signaling mediates RhoA activity through Smad3 and MAPK pathways in mammary carcinoma cells.
The expression of RhoA, a small GTPase, has been shown to regulate cell migration of epithelial cell types by regulating the formation of actin stress fibers (64); however, its role in epithelial cell survival has remained poorly understood. To determine the functional role of RhoA in CCL2-induced cell survival and motility, 4T1 cells were transduced with retrovirus expressing dominant negative RhoA, which contained a Thr to Asn point mutation in codon 19 that inactivated GTPase activity (65). Compared with vehicle retrovirus control, expression of dominant negative RhoA significantly reduced RhoA GTPase activity and significantly inhibited CCL2-induced survival and migration (Fig. 7, A–D). To further confirm the role of RhoA signaling in CCL2-induced survival and migration, 4T1 mammary carcinoma cells were treated with CCL2 in the presence or absence of Rho kinase inhibitor II, a selective inhibitor of Rho-associated protein kinase, an effector immediately downstream of RhoA (66). Increasing doses of Rho kinase inhibitor II significantly reduced CCL2-induced survival and migration of 4T1 cells similarly to expression of dominant negative RhoA (Fig. 7, C and D). In summary, these data indicate that RhoA functions downstream of Smad3 and MEK to regulate CCL2-induced mammary carcinoma cell survival and motility.
FIGURE 7.

CCL2 mediation of RhoA function is important for mammary carcinoma cell survival and migration. 4T1 mammary carcinoma cells were infected with control vehicle retrovirus (veh) or retrovirus overexpressing dominant negative RhoA (Rho.DN) or treated with Rho kinase inhibitor II (Rocki) and analyzed for changes. A, expression of RhoA by immunoblot analysis. The protein band shown is at the predicted molecular mass for RhoA (25 kDa). B, RhoA activity by G-LISA. C, gentamicin-induced apoptosis. D, migration by wound closure assay. Statistical analysis was determined by two-tailed t test: *, p < 0.001; **, p < 0.01; ***, p < 0.05; ****, p > 0.05. Values are expressed as mean ± S.E. Error bars represent S.E. Par, parental; SF, serum-free medium.
CCL2 Signaling Occurs Independently of TGF-β Expression
The signaling pathways regulated by CCL2/CCR2 are also regulated by a number of mechanisms in breast tumors including TGF-β signaling. Given the importance of autocrine TGF-β signaling in breast cancer cells (67, 68), we evaluated the possibility that CCL2 signaling was dependent on TGF-β expression. 4T1 mammary carcinoma cells were treated with neutralizing antibodies to TGF-β in the presence or absence of CCL2. Anti-TGF-β reduced basal Smad3 phosphorylation in serum-free controls and modestly reduced CCL2-induced Smad3 phosphorylation but did not inhibit the ability of CCL2 to stimulate detectable levels of phospho-Smad3 (Fig. 8A). Anti-TGF-β did not significantly affect CCL2-induced Rho GTPase activity (Fig. 8B). As the cells were serum-starved for 24 h prior to stimulation, these data indicate that mammary carcinoma cells still express endogenous TGF-β expression that maintains low level Smad3 phosphorylation but does not significantly affect CCL2/CCR2 signaling. To determine whether CCL2/CCR2 signaling pathways were mediated by induction of TGF-β expression, CCR2 was knocked down in 4T1 mammary carcinoma cells and analyzed for TGF-β expression by ELISA. There were no significant changes in TGF-β expression in CCR2 knockdown cells compared with controls (Fig. 8C). Smad3-deficient 4T1 cells also did not exhibit any changes in TGF-β expression (Fig. 8D). These data indicate that CCL2/CCR2 signaling occurs independently of TGF-β expression.
FIGURE 8.

CCL2/CCR2 signaling is not significantly dependent on TGF-β expression. A, 4T1 cells were treated with CCL2 (20 ng/ml) or TGF-β as a positive control (5 ng/ml) with or without 10 μg/ml neutralizing antibodies to TGF-β and analyzed for changes in Smad3 phosphorylation by immunoblot analysis. B, 4T1 cells were treated with CCL2 with or without 10 μg/ml neutralizing antibodies to TGF-β and analyzed for changes in RhoA activity. C, CCR2 was transiently knocked down (CCR2kd) in 4T1 cells, and cells were analyzed for expression of TGF-β by ELISA. D, Smad3-deficient 4T1 cells were treated with CCL2 and analyzed for changes in TGF-β expression by ELISA. Statistical analysis was determined by two-tailed t test: ****, p > 0.05. Values are expressed as mean ± S.E. Error bars represent S.E. Con, control; Par, parental; SF, serum-free medium.
DISCUSSION
Recent studies have shown that CCR2 is overexpressed in prostate tumors (69, 70) and that CCL2 signaling promotes human PC-3 prostate cancer cell proliferation and enhances cancer cell survival by autophagic cell death (71). Here, we show that CCL2 has no significant effect on mammary carcinoma cell proliferation or autophagy (data not shown), indicating tissue-specific functions for CCL2 signaling. Based on these current studies, we propose a working model for the role of CCL2/CCR2 signaling in epithelial cells during breast cancer progression. In breast cancer cells, CCL2 binds to CCR2 to stimulate Smad3 and p42/44MAPK pathways. In one pathway, CCL2 induces MEK signaling through p42/44MAPK independently of Smad3 to promote cell survival. In addition, CCL2 signaling activates Smad3, which cooperates with the MEK-p42/44MAPK pathway to regulate cell motility and survival through RhoA-dependent mechanisms (Fig. 9). In combination, these cellular processes function to promote invasion and metastasis during breast cancer progression.
FIGURE 9.
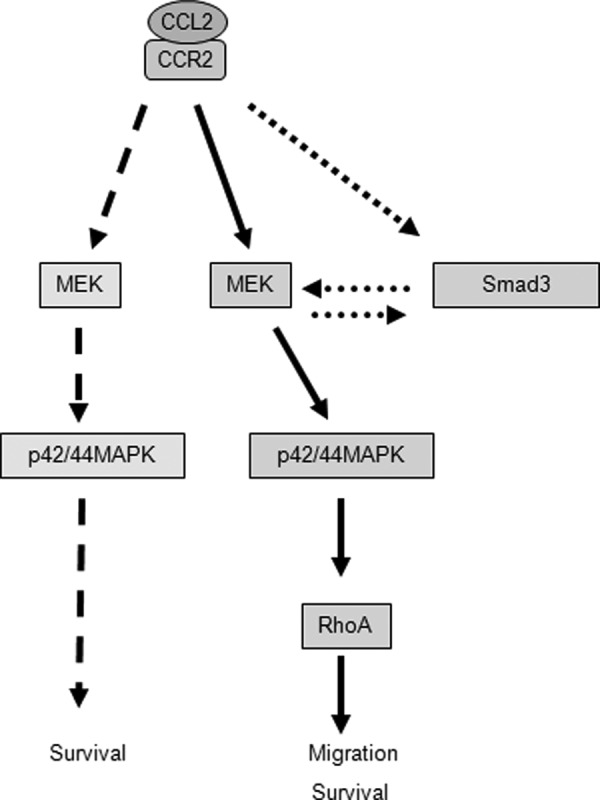
Proposed model for how CCL2 signaling through CCR2 signaling regulates survival and motility of breast cancer cells. CCL2 binds to CCR2 to stimulate Smad3 and p42/44MAPK pathways. In one pathway, CCL2 induces MEK signaling through p42/44MAPK independently of Smad3 to promote cell survival. In addition, CCL2 activates Smad3, which cooperates with the MEK-p42/44MAPK pathway to regulate cell motility and survival through RhoA-dependent mechanisms.
Although CCL2 and CCR2 proteins were detected in normal breast ductal epithelial cells, the significance of CCL2 and CCR2 expression in normal cells remains unclear as CCL2 treatment of MCF10A cells does not significantly affect survival or migration (supplemental Fig. 6). Studies of CCR2 knock-out mice are underway to determine whether CCR2 expression in ductal epithelial cells is necessary for mammary gland development and homeostasis.
We observed that CCL2 expression did not correspond to tumorigenicity of breast cancer cell lines. Studies indicate that the stroma may be an important source of CCL2 expression and that a paracrine mechanism may be important for CCL2 signaling in breast cancer cells. We observed positive CCL2 expression in the breast tumor stroma, consistent with previous studies that show that CCL2 expression in the breast tumor stroma, but not in tumor epithelium, positively correlates with tumor grade and poor patient prognosis (18). CCL2 is highly expressed in macrophages (18), which have been shown to enhance breast cancer cell invasion and metastasis in mouse models (26, 72, 73). Fibroblasts represent another potential source of CCL2 expression. In previous studies, we showed that CCL2 derived from Tgfbr2-deficient fibroblasts (Tgfbr2FspKO) enhanced mammary carcinoma invasion and metastasis, which were blocked by CCL2-neutralizing antibodies (21, 24). In the current studies, we show that CCL2-neutralizing antibodies inhibit phospho-p42/44MAPK and phospho-Smad3 induced by Tgfbr2FspKO fibroblasts but do not affect phospho-p42/44MAPK or phospho-Smad3 regulated by autocrine CCL2 signaling in breast cancer cells (supplemental Fig. 8A). Compared with mammary carcinoma cells, Tgfbr2FspKO fibroblasts express significantly higher levels of CCL2 (supplemental Fig. 8B), further indicating that a paracrine mechanism may be important for CCL2 signaling in breast cancer. We are currently conducting studies of CCL2 expression in carcinoma-associated fibroblasts to further understand the relevance of TGF-β signaling and stromal CCL2 expression in breast tumors. If autocrine CCL2 signaling is important for regulating breast cancer cell migration and survival, we anticipate that CCR2 knockdown alone would inhibit cell survival or migration. We observed that CCR2 knockdown alone has no significant effect on cell migration or survival of 4T1 and MCF-7 breast cancer cells (Fig. 4). We cannot completely rule out a role for autocrine CCL2 signaling in breast cancer cells. The model systems used in this report and previous studies suggest that in the context of the tumor microenvironment CCL2 derived from stromal cells regulates signaling in breast cancer cells to promote survival and metastasis.
For the first time, we also show that CCL2 regulates Smad3 phosphorylation in concert with MAPK signaling to regulate survival and motility in breast epithelial cells. Previous studies have shown that the TGF-β receptors are important regulators of Smad protein function. TGF-β ligand binding to the type II receptor leads to interactions with the TGF-β type I receptor, which acts as a kinase to phosphorylate Smad3 and the related protein Smad2 at the C terminus (74, 75). In TGF-β-treated cells, phosphorylation of Smad3 and -2 proteins leads to homodimeric or heterodimeric protein complexes that translocate to the nucleus to regulate expression of genes related to proliferation, survival, and motility (76, 77). In contrast, CCL2 stimulated Smad3 phosphorylation in multiple mammary carcinoma cell lines without affecting Smad2 phosphorylation (data not shown), indicating a specific function for Smad3 in the CCL2/CCR2 signaling axis.
The role of CCL2-mediated phospho-Smad3 in cell survival and motility is consistent with functional studies on Smad3 knock-out mice. Smad3-deficient mice exhibit fewer metastases (78), indicating an important role for Smad3 in regulating late stage breast cancer progression. These functional studies are consistent with prognostic studies of phospho-Smad3 expression. Although studies have reported the absence of Smad3 mutations in breast cancers (79), phospho-Smad3 expression is associated with late stage invasive breast carcinomas (80), consistent with the increased CCL2 and CCR2 expression in invasive breast tumors observed in our studies.
We would also anticipate that TGF-β expression positively correlates with phospho-Smad3 expression and breast tumor grade. However, there are conflicting reports regarding the prognostic significance of TGF-β expression in breast tumors (81–83). TGF-β expression in breast tumors may be dependent on several factors including the degree of stromal reactivity and clinical subtype (84). Another possibility is that other factors such as CCL2 may regulate Smad3 activity in breast tumors. Future studies would need to be performed to analyze expression of CCL2, TGF-β, and phospho-Smad3 to understand their prognostic significance in breast tumors while accounting for these variables.
Studies indicate that CCL2/CCR2 signaling is not dependent on TGF-β expression in 4T1 mammary carcinoma cells (Fig. 8). We also evaluated the possibility that CCL2 signaling in breast cancer cells was dependent on TGF-β receptor signaling. CCL2 did not stimulate receptor clustering between CCR2 and TGF-β receptor I or affect receptor localization as determined in confocal imaging studies. Treatment with a TGF-β type I receptor inhibitor (SB431542) did not affect localization or clustering of CCR2 and TGF-β receptors (supplemental Fig. 7A) and did not significantly affect CCL2-induced phosphorylation of Smad3 and p42/44MAPK proteins (supplemental Fig. 7B). These data indicate that CCL2 signaling is not dependent on TGF-β receptor I function. However, pertussis toxin did inhibit CCL2-induced p42/44MAPK and Smad3 signaling, indicating that these signaling pathways are dependent on G protein mechanisms (supplemental Fig. 7C). It is possible that CCL2 signaling operates independently of TGF-β signaling to regulate breast cancer cell survival and motility.
Multiple factors elevated in breast tumors including insulin-like growth factor and EGF have been shown to activate the p42/44MAPK pathway (85–87). Our studies indicate that CCL2/CCR2 signaling in breast cancer cells is also an important regulator of p42/44MAPK. With elevated CCL2 and CCR2 expression in breast tumors, we would expect increased phospho-42/44MAPK expression in breast tumors. Studies show that increased expression of phospho-p42/44MAPK correlates with tumor grade, poor patient survival, and reduced responses to chemotherapy (85). Currently we are conducting studies to determine how CCL2/CCR2 signaling coordinates breast cancer signaling with other cytokines and growth factors including EGF. These studies will yield important insight into how breast cancer cell signaling is regulated during tumor progression.
There are several possible mechanisms through which CCL2/CCR2 signaling would regulate Smad3 phosphorylation in mammary carcinoma cells. It is possible that CCR2 blocks the activity of phosphatases, leading to Smad3 phosphorylation. G protein-coupled receptors including bradykinin receptor BR2 and somatostatin receptor SST2 have been shown to regulate SHP-1 and SHP-2 tyrosine phosphatases to mediate cell proliferation (88, 89), and PPM1A/PP2Ca has been identified as a phosphatase for Smad proteins (90). Another possible mechanism is that CCR2 regulates a protein kinase responsible for Smad3 phosphorylation as cyclic GMP/protein kinase G has been shown to regulate Smad3 phosphorylation in cardiac fibroblasts during fibrogenesis (91).
Although recent studies have uncovered multiple roles for p42/44MAPK signaling, it is best known for its role in regulating cell proliferation in part by regulating transcription activity (92–94). Our studies indicate that CCL2 induction of p42/44MAPK functions to promote survival and migration rather than cell proliferation. The decreased RhoA expression and activity in mammary carcinoma cells treated with U0126 indicate that CCL2 induction of p42/44MAPK signaling functions to regulate the expression of genes associated with survival and migration. Studies are currently being performed to more thoroughly investigate the effects of CCL2-induced p42/44MAPK signaling on gene expression in mammary carcinoma cells.
Although the role of MEK as an upstream regulator of p42/44MAPK is well established (95–97), the mechanisms through which Smad3 affects the MEK-MAPK signaling pathway are less clear. TGF-β receptor kinase inhibitors did not significantly affect CCL2 induction of Smad3 phosphorylation, indicating a TGF-β receptor-independent mechanism. Smad3 knockdown cells exhibited decreased phosphorylation of p42/44MAPK but did not affect expression of MAPK. Therefore, it is unlikely that Smad3 regulates MAPK phosphorylation by modulating p42/44MAPK transcription. Previous studies have demonstrated that MAPK proteins function upstream of Smad3 in human mesangial cells (98), human alveolar basal epithelial cells, and smooth muscle cells (99). MEK inhibitors had no effect on Smad3 phosphorylation in the non-tumorigenic mammary epithelial cell line NMuMG (98). However, in our studies, MEK inhibition of 4T1 mammary carcinoma cells decreased Smad3 phosphorylation, whereas Smad3 knockdown inhibited MAPK phosphorylation. These data indicate that Smad3 functions cooperatively with MEK-p42/44MAPK in mammary carcinoma cells. Based on published studies, it is likely that the effects between Smad3 and MEK-p42/44MAPK are cell type-dependent. There are possible mechanisms through which Smad3 may regulate phosphorylation of MAPK. One possibility is that regulation of MEK-p42/44MAPK by Smad3 may occur through Smad3-regulated transcription of kinases including Src, Ras, Raf, MEK (100), protein kinase C, and trafficking protein particle complex TrappC4 (101) that directly or indirectly interact with p42/44MAPK to regulate phosphorylation. Another possibility is that Smad3 suppresses transcription of phosphatases such as PP2A, a phosphatase demonstrated to dephosphorylate MEK and p42/44MAPK in neuronal cells (102). These possibilities will be explored in future studies to more fully understand the mechanisms through which Smad3 cooperates with the MEK-MAPK pathway.
Our studies indicate that CCL2 enhances expression and activity of RhoA through Smad3 and MEK signaling through p42/44MAPK signaling to regulate breast cancer cell motility and survival. Interestingly, we observed that increased RhoA protein levels do not always correspond to the same increases in RhoA activity. CCL2 induction of RhoA expression corresponds to increased RhoA activity and to increased migration and survival. However, the increased RhoA expression in Smad3 knockdown cells does not correspond to RhoA activity, which was only slightly elevated, and does not correspond to the decreased migration and survival observed in Smad3 knockdown cells. These data indicate that CCL2-mediated migration and survival may be a function of RhoA activity rather than RhoA expression levels. The functional consequences of CCL2 induction of RhoA expression currently remain unclear. Our data indicate that RhoA activity is dependent on CCL2 induction of Smad3 and MAPK activity. However, RhoA activity is also regulated by GDP/GTP exchange. It is possible that the amount of GTP/GTP or guanine nucleotide exchange factor protein available limits the amount of RhoA protein activation. Thus, the increased RhoA expression would not necessarily result in increased RhoA activity.
RhoA expression has been found to be increased in many types of cancers (103, 104) and has been shown to be an important regulator of CCL2-induced cell migration of mesenchymal cell types (63). Currently, the molecular mechanisms through which RhoA functions to simultaneously regulate cell migration and survival remain poorly understood. Our studies indicate that RhoA participates in CCL2-induced cell migration and cell survival by functioning downstream in signaling pathways regulated independently by Smad3 and MEK signaling. It is possible that RhoA would signal downstream to regulate gene expression to regulate cell survival. Studies have shown that RhoA signals to p53 transcription factors to regulate expression of Bax, a protein involved in mediating Bcl-2 activity and subsequent cytochrome c release from the mitochondria during apoptosis (105). Further studies to understand the molecular mechanisms through which RhoA functions to regulate cell survival would provide further understanding of the intrinsic relationships between cell survival and migration. In summary, these studies demonstrate that one functional consequence of CCL2 signaling is increased RhoA activity, which mediates breast cancer cell survival and migration.
These studies have clarified important functional roles for CCL2/CCR2 signaling in breast cancer cells in regulating cell survival and motility, cellular processes that are important to the metastatic spread (26, 106). Recent studies have shown that therapeutic targeting of specific oncogenic proteins in preclinical mouse models of cancer is more effective than actual treatment of cancer patients at a clinical level (107), underscoring a need to identify the signaling pathways that are relevant to cancer progression in humans. Our studies show similarities in CCL2 induction of cell motility and survival between mouse and human breast cancer cells, indicating that targeting the CCL2 pathway would be potentially effective in treating metastatic disease, the effects of which may be more easily predicted in preclinical models of cancer. As invasive breast cancer cells are often drug-resistant (108) and CCL2 regulates both cell motility and survival, targeting the CCL2 signaling pathway may affect multiple mechanisms of breast cancer progression, thus representing an attractive target in therapeutics. By further understanding the molecular and cellular mechanisms that regulate the behavior of invasive cancer cells, we may be able to design new strategies to more effectively diagnose and treat metastatic cancer.
Acknowledgments
We thank Jay Vivian, Ph.D. (University of Kansas Medical Center, Kansas City, KS) and Rebecca Cook, Ph.D. (Vanderbilt University, Nashville, TN) for critical reading of this manuscript. Flow cytometry studies were conducted through the Kansas University Medical Center Flow Cytometry Core. Statistical analysis was conducted in consultation with the Kansas University Medical Center Biostatistics Core.
This work was supported, in whole or in part, by National Institutes of Health Grant R00 CA127357-03 from the NCI and United States Department of Health and Human Services (to N. C.). This work was also supported by funds from the Kansas Bioscience Authority (to N. C.).

This article contains supplemental Figs. 1–8.
- CCL2
- chemokine (C-C motif) ligand 2
- CCR2
- chemokine (C-C motif) receptor 2
- MMTV
- murine mammary tumor virus
- PyVmT
- polyoma virus middle T antigen
- 5-FU
- 5-fluoruracil
- NMuMG
- normal murine mammary gland.
REFERENCES
- 1. Gillitzer R., Goebeler M. (2001) Chemokines in cutaneous wound healing. J. Leukoc. Biol. 69, 513–521 [PubMed] [Google Scholar]
- 2. Laing K. J., Secombes C. J. (2004) Chemokines. Dev. Comp. Immunol. 28, 443–460 [DOI] [PubMed] [Google Scholar]
- 3. Lentsch A. B. (2002) The Duffy antigen/receptor for chemokines (DARC) and prostate cancer. A role as clear as black and white? FASEB J. 16, 1093–1095 [DOI] [PubMed] [Google Scholar]
- 4. Lazennec G., Richmond A. (2010) Chemokines and chemokine receptors: new insights into cancer-related inflammation. Trends Mol. Med. 16, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ernst C. A., Zhang Y. J., Hancock P. R., Rutledge B. J., Corless C. L., Rollins B. J. (1994) Biochemical and biologic characterization of murine monocyte chemoattractant protein-1. Identification of two functional domains. J. Immunol. 152, 3541–3549 [PubMed] [Google Scholar]
- 6. Huang D. R., Wang J., Kivisakk P., Rollins B. J., Ransohoff R. M. (2001) Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J. Exp. Med. 193, 713–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fife B. T., Huffnagle G. B., Kuziel W. A., Karpus W. J. (2000) CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J. Exp. Med. 192, 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Johnson Z., Power C. A., Weiss C., Rintelen F., Ji H., Ruckle T., Camps M., Wells T. N., Schwarz M. K., Proudfoot A. E., Rommel C. (2004) Chemokine inhibition—why, when, where, which and how? Biochem. Soc. Trans. 32, 366–377 [DOI] [PubMed] [Google Scholar]
- 9. Mellado M., Rodríguez-Frade J. M., Aragay A., del Real G., Martín A. M., Vila-Coro A. J., Serrano A., Mayor F., Jr., Martínez-A. C. (1998) The chemokine monocyte chemotactic protein 1 triggers Janus kinase 2 activation and tyrosine phosphorylation of the CCR2B receptor. J. Immunol. 161, 805–813 [PubMed] [Google Scholar]
- 10. Jiménez-Sainz M. C., Fast B., Mayor F., Jr., Aragay A. M. (2003) Signaling pathways for monocyte chemoattractant protein 1-mediated extracellular signal-regulated kinase activation. Mol. Pharmacol. 64, 773–782 [DOI] [PubMed] [Google Scholar]
- 11. Chavey C., Bibeau F., Gourgou-Bourgade S., Burlinchon S., Boissière F., Laune D., Roques S., Lazennec G. (2007) Oestrogen receptor negative breast cancers exhibit high cytokine content. Breast Cancer Res. 9, R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ueno T., Toi M., Saji H., Muta M., Bando H., Kuroi K., Koike M., Inadera H., Matsushima K. (2000) Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin. Cancer Res. 6, 3282–3289 [PubMed] [Google Scholar]
- 13. Saji H., Koike M., Yamori T., Saji S., Seiki M., Matsushima K., Toi M. (2001) Significant correlation of monocyte chemoattractant protein-1 expression with neovascularization and progression of breast carcinoma. Cancer 92, 1085–1091 [DOI] [PubMed] [Google Scholar]
- 14. Lebrecht A., Grimm C., Lantzsch T., Ludwig E., Hefler L., Ulbrich E., Koelbl H. (2004) Monocyte chemoattractant protein-1 serum levels in patients with breast cancer. Tumour Biol. 25, 14–17 [DOI] [PubMed] [Google Scholar]
- 15. Qualls J. E., Murray P. J. (2011) Tumor macrophages protective and pathogenic roles in cancer development. Curr. Top. Dev. Biol. 94, 309–328 [DOI] [PubMed] [Google Scholar]
- 16. Talmadge J. E. (2011) Immune cell infiltration of primary and metastatic lesions: mechanisms and clinical impact. Semin. Cancer Biol. 21, 131–138 [DOI] [PubMed] [Google Scholar]
- 17. Pagès F., Galon J., Dieu-Nosjean M. C., Tartour E., Sautès-Fridman C., Fridman W. H. (2010) Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene 29, 1093–1102 [DOI] [PubMed] [Google Scholar]
- 18. Fujimoto H., Sangai T., Ishii G., Ikehara A., Nagashima T., Miyazaki M., Ochiai A. (2009) Stromal MCP-1 in mammary tumors induces tumor-associated macrophage infiltration and contributes to tumor progression. Int. J. Cancer 125, 1276–1284 [DOI] [PubMed] [Google Scholar]
- 19. Lu X., Kang Y. (2009) Chemokine (C-C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J. Biol. Chem. 284, 29087–29096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qian B. Z., Li J., Zhang H., Kitamura T., Zhang J., Campion L. R., Kaiser E. A., Snyder L. A., Pollard J. W. (2011) CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475, 222–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hembruff S. L., Jokar I., Yang L., Cheng N. (2010) Loss of transforming growth factor-β signaling in mammary fibroblasts enhances CCL2 secretion to promote mammary tumor progression through macrophage-dependent and -independent mechanisms. Neoplasia 12, 425–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yamashiro S., Takeya M., Nishi T., Kuratsu J., Yoshimura T., Ushio Y., Takahashi K. (1994) Tumor-derived monocyte chemoattractant protein-1 induces intratumoral infiltration of monocyte-derived macrophage subpopulation in transplanted rat tumors. Am. J. Pathol. 145, 856–867 [PMC free article] [PubMed] [Google Scholar]
- 23. Hoshino Y., Hatake K., Kasahara T., Takahashi Y., Ikeda M., Tomizuka H., Ohtsuki T., Uwai M., Mukaida N., Matsushima K. (1995) Monocyte chemoattractant protein-1 stimulates tumor necrosis and recruitment of macrophages into tumors in tumor-bearing nude mice: increased granulocyte and macrophage progenitors in murine bone marrow. Exp. Hematol. 23, 1035–1039 [PubMed] [Google Scholar]
- 24. Fang W. B., Jokar I., Chytil A., Moses H. L., Abel T., Cheng N. (2011) Loss of one Tgfbr2 allele in fibroblasts promotes metastasis in MMTV: polyoma middle T transgenic and transplant mouse models of mammary tumor progression. Clin. Exp. Metastasis 28, 351–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Youngs S. J., Ali S. A., Taub D. D., Rees R. C. (1997) Chemokines induce migrational responses in human breast carcinoma cell lines. Int. J. Cancer 71, 257–266 [DOI] [PubMed] [Google Scholar]
- 26. Joyce J. A., Pollard J. W. (2009) Microenvironmental regulation of metastasis. Nat. Rev. Cancer 9, 239–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bogenrieder T., Herlyn M. (2003) Axis of evil: molecular mechanisms of cancer metastasis. Oncogene 22, 6524–6536 [DOI] [PubMed] [Google Scholar]
- 28. Maglione J. E., Moghanaki D., Young L. J., Manner C. K., Ellies L. G., Joseph S. O., Nicholson B., Cardiff R. D., MacLeod C. L. (2001) Transgenic polyoma middle-T mice model premalignant mammary disease. Cancer Res. 61, 8298–8305 [PubMed] [Google Scholar]
- 29. Medina D., Kittrell F. (2000) in Methods in Mammary Gland Biology and Breast Cancer Research (Ip M. M., Asch B. B., eds) pp. 137–147, Kluwer Academic/ Plenum Publishers, New York [Google Scholar]
- 30. Cheng N., Bhowmick N. A., Chytil A., Gorksa A. E., Brown K. A., Muraoka R., Arteaga C. L., Neilson E. G., Hayward S. W., Moses H. L. (2005) Loss of TGF-β type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF-α-, MSP- and HGF-mediated signaling networks. Oncogene 24, 5053–5068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moon A., Kim M. S., Kim T. G., Kim S. H., Kim H. E., Chen Y. Q., Kim H. R. (2000) H-ras, but not N-ras, induces an invasive phenotype in human breast epithelial cells: a role for MMP-2 in the H-ras-induced invasive phenotype. Int. J. Cancer 85, 176–181 [DOI] [PubMed] [Google Scholar]
- 32. Brown K. A., Pietenpol J. A., Moses H. L. (2007) A tale of two proteins: differential roles and regulation of Smad2 and Smad3 in TGF-β signaling. J. Cell. Biochem. 101, 9–33 [DOI] [PubMed] [Google Scholar]
- 33. Cheng N., Chytil A., Shyr Y., Joly A., Moses H. L. (2007) Enhanced hepatocyte growth factor signaling by type II transforming growth factor-β receptor knockout fibroblasts promotes mammary tumorigenesis. Cancer Res. 67, 4869–4877 [DOI] [PubMed] [Google Scholar]
- 34. He T. C., Zhou S., da Costa L. T., Yu J., Kinzler K. W., Vogelstein B. (1998) A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. U.S.A. 95, 2509–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Frederick J. P., Liberati N. T., Waddell D. S., Shi Y., Wang X. F. (2004) Transforming growth factor β-mediated transcriptional repression of c-myc is dependent on direct binding of Smad3 to a novel repressive Smad binding element. Mol. Cell. Biol. 24, 2546–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martin S. (1978) Biochemistry of Viruses, pp. 21–23, Cambridge University Press, Cambridge, UK [Google Scholar]
- 37. Tylaska L. A., Boring L., Weng W., Aiello R., Charo I. F., Rollins B. J., Gladue R. P. (2002) Ccr2 regulates the level of MCP-1/CCL2 in vitro and at inflammatory sites and controls T cell activation in response to alloantigen. Cytokine 18, 184–190 [DOI] [PubMed] [Google Scholar]
- 38. Dixon R. A. (1981) Curative effects of tobramycin or gentamicin therapy on mouse arthritis caused by Mycoplasma pulmonis. Antimicrob. Agents Chemother. 20, 321–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Erskine R. J., Wilson R. C., Riddell M. G., Jr., Tyler J. W., Spears H. J., Davis B. S. (1992) Intramammary administration of gentamicin as treatment for experimentally induced Escherichia coli mastitis in cows. Am. J. Vet. Res. 53, 375–381 [PubMed] [Google Scholar]
- 40. Kahn T., Stein R. M. (1972) Gentamicin and renal failure. Lancet 1, 498. [DOI] [PubMed] [Google Scholar]
- 41. Paape M. J., Nickerson S. C., Ziv G. (1990) In vivo effects of chloramphenicol, tetracycline, and gentamicin on bovine neutrophil function and morphologic features. Am. J. Vet. Res. 51, 1055–1061 [PubMed] [Google Scholar]
- 42. El Mouedden M., Laurent G., Mingeot-Leclercq M. P., Tulkens P. M. (2000) Gentamicin-induced apoptosis in renal cell lines and embryonic rat fibroblasts. Toxicol. Sci. 56, 229–239 [DOI] [PubMed] [Google Scholar]
- 43. Choi K. H., Kim T. I., Chong D. L., Lee H. Y., Han D. S. (2000) Gentamicin induced apoptosis of renal tubular epithelial (LLC-PK1) cells. Korean J. Int. Med. 15, 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lang H., Liu C. (1997) Apoptosis and hair cell degeneration in the vestibular sensory epithelia of the guinea pig following a gentamicin insult. Hear. Res. 111, 177–184 [DOI] [PubMed] [Google Scholar]
- 45. Juan S. H., Chen C. H., Hsu Y. H., Hou C. C., Chen T. H., Lin H., Chu Y. L., Sue Y. M. (2007) Tetramethylpyrazine protects rat renal tubular cell apoptosis induced by gentamicin. Nephrol. Dial. Transplant. 22, 732–739 [DOI] [PubMed] [Google Scholar]
- 46. Cameron D. A., Gabra H., Leonard R. C. (1994) Continuous 5-fluorouracil in the treatment of breast cancer. Br. J. Cancer 70, 120–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Iwata H., Sato N., Masuda N., Nakamura S., Yamamoto N., Kuroi K., Kurosumi M., Tsuda H., Akiyama F., Ohashi Y., Toi M. (2011) Docetaxel followed by fluorouracil/epirubicin/cyclophosphamide as neoadjuvant chemotherapy for patients with primary breast cancer. Jpn. J. Clin. Oncol. 41, 867–875 [DOI] [PubMed] [Google Scholar]
- 48. Zoli W., Ulivi P., Tesei A., Fabbri F., Rosetti M., Maltoni R., Giunchi D. C., Ricotti L., Brigliadori G., Vannini I., Amadori D. (2005) Addition of 5-fluorouracil to doxorubicin-paclitaxel sequence increases caspase-dependent apoptosis in breast cancer cell lines. Breast Cancer Res. 7, R681–R689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Okamoto S., Sakai M., Uchida J., Saito H. (1996) 5-Fluorouracil induces apoptotic cell death with G2 phase arrest in human breast cancer grafted in nude mice. Anticancer Res. 16, 2699–2704 [PubMed] [Google Scholar]
- 50. Serbina N. V., Jia T., Hohl T. M., Pamer E. G. (2008) Monocyte-mediated defense against microbial pathogens. Annu. Rev. Immunol. 26, 421–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gangur V., Birmingham N. P., Thanesvorakul S. (2002) Chemokines in health and disease. Vet. Immunol. Immunopathol. 86, 127–136 [DOI] [PubMed] [Google Scholar]
- 52. Dawson J., Miltz W., Mir A. K., Wiessner C. (2003) Targeting monocyte chemoattractant protein-1 signalling in disease. Expert Opin. Ther. Targets 7, 35–48 [DOI] [PubMed] [Google Scholar]
- 53. Xia M., Sui Z. (2009) Recent developments in CCR2 antagonists. Expert Opin. Ther. Pat. 19, 295–303 [DOI] [PubMed] [Google Scholar]
- 54. Soung Y. H., Jeong E. G., Ahn C. H., Kim S. S., Song S. Y., Yoo N. J., Lee S. H. (2008) Mutational analysis of caspase 1, 4, and 5 genes in common human cancers. Hum. Pathol. 39, 895–900 [DOI] [PubMed] [Google Scholar]
- 55. Blanc C., Deveraux Q. L., Krajewski S., Jänicke R. U., Porter A. G., Reed J. C., Jaggi R., Marti A. (2000) Caspase-3 is essential for procaspase-9 processing and cisplatin-induced apoptosis of MCF-7 breast cancer cells. Cancer Res. 60, 4386–4390 [PubMed] [Google Scholar]
- 56. Mazzoni E., Adam A., Bal de Kier Joffe E., Aguirre-Ghiso J. A. (2003) Immortalized mammary epithelial cells overexpressing protein kinase Cγ acquire a malignant phenotype and become tumorigenic in vivo. Mol. Cancer Res. 1, 776–787 [PubMed] [Google Scholar]
- 57. Pilat M. J., Schwab E. D., Yao K. L., Pienta K. J. (1998) Examination of the DNA methylation properties in nontumorigenic and tumorigenic breast epithelial cell lines. Anticancer Res. 18, 2575–2582 [PubMed] [Google Scholar]
- 58. Arefieva T. I., Kukhtina N. B., Antonova O. A., Krasnikova T. L. (2005) MCP-1-stimulated chemotaxis of monocytic and endothelial cells is dependent on activation of different signaling cascades. Cytokine 31, 439–446 [DOI] [PubMed] [Google Scholar]
- 59. Yanagisawa K., Osada H., Masuda A., Kondo M., Saito T., Yatabe Y., Takagi K., Takahashi T. (1998) Induction of apoptosis by Smad3 and down-regulation of Smad3 expression in response to TGF-β in human normal lung epithelial cells. Oncogene 17, 1743–1747 [DOI] [PubMed] [Google Scholar]
- 60. Weinstein M., Yang X., Deng C. (2000) Functions of mammalian Smad genes as revealed by targeted gene disruption in mice. Cytokine Growth Factor Rev. 11, 49–58 [DOI] [PubMed] [Google Scholar]
- 61. Crews C. M., Alessandrini A., Erikson R. L. (1992) The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science 258, 478–480 [DOI] [PubMed] [Google Scholar]
- 62. Favata M. F., Horiuchi K. Y., Manos E. J., Daulerio A. J., Stradley D. A., Feeser W. S., Van Dyk D. E., Pitts W. J., Earl R. A., Hobbs F., Copeland R. A., Magolda R. L., Scherle P. A., Trzaskos J. M. (1998) Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 273, 18623–18632 [DOI] [PubMed] [Google Scholar]
- 63. Loberg R. D., Tantivejkul K., Craig M., Neeley C. K., Pienta K. J. (2007) PAR1-mediated RhoA activation facilitates CCL2-induced chemotaxis in PC-3 cells. J. Cell. Biochem. 101, 1292–1300 [DOI] [PubMed] [Google Scholar]
- 64. Hotta K., Tanaka K., Mino A., Kohno H., Takai Y. (1996) Interaction of the Rho family small G proteins with kinectin, an anchoring protein of kinesin motor. Biochem. Biophys. Res. Commun. 225, 69–74 [DOI] [PubMed] [Google Scholar]
- 65. Qiu R. G., Chen J., McCormick F., Symons M. (1995) A role for Rho in Ras transformation. Proc. Natl. Acad. Sci. U.S.A. 92, 11781–11785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fujisawa K., Fujita A., Ishizaki T., Saito Y., Narumiya S. (1996) Identification of the Rho-binding domain of p160ROCK, a Rho-associated coiled-coil containing protein kinase. J. Biol. Chem. 271, 23022–23028 [DOI] [PubMed] [Google Scholar]
- 67. Moses H., Barcellos-Hoff M. H. (2011) TGF-β biology in mammary development and breast cancer. Cold Spring Harb. Perspect. Biol. 3, a003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Moore-Smith L., Pasche B. (2011) TGFBR1 signaling and breast cancer. J. Mammary Gland Biol. Neoplasia 16, 89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lu Y., Cai Z., Xiao G., Liu Y., Keller E. T., Yao Z., Zhang J. (2007) CCR2 expression correlates with prostate cancer progression. J. Cell. Biochem. 101, 676–685 [DOI] [PubMed] [Google Scholar]
- 70. Lu Y., Chen Q., Corey E., Xie W., Fan J., Mizokami A., Zhang J. (2009) Activation of MCP-1/CCR2 axis promotes prostate cancer growth in bone. Clin. Exp. Metastasis 26, 161–169 [DOI] [PubMed] [Google Scholar]
- 71. Roca H., Varsos Z. S., Pienta K. J. (2009) CCL2 is a negative regulator of AMP-activated protein kinase to sustain mTOR complex-1 activation, survivin expression, and cell survival in human prostate cancer PC3 cells. Neoplasia 11, 1309–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mantovani A., Germano G., Marchesi F., Locatelli M., Biswas S. K. (2011) Cancer-promoting tumor-associated macrophages: new vistas and open questions. Eur. J. Immunol. 41, 2522–2525 [DOI] [PubMed] [Google Scholar]
- 73. Balkwill F. R., Mantovani A. (2012) Cancer-related inflammation: common themes and therapeutic opportunities. Semin. Cancer Biol. 22, 33–40 [DOI] [PubMed] [Google Scholar]
- 74. Zhang Y., Feng X., We R., Derynck R. (1996) Receptor-associated Mad homologues synergize as effectors of the TGF-β response. Nature 383, 168–172 [DOI] [PubMed] [Google Scholar]
- 75. Qin B. Y., Lam S. S., Correia J. J., Lin K. (2002) Smad3 allostery links TGF-β receptor kinase activation to transcriptional control. Genes Dev. 16, 1950–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Samanta D., Datta P. K. (2012) Alterations in the Smad pathway in human cancers. Front. Biosci. 17, 1281–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ikushima H., Miyazono K. (2012) TGF-β signal transduction spreading to a wider field: a broad variety of mechanisms for context-dependent effects of TGF-β. Cell Tissue Res. 347, 37–49 [DOI] [PubMed] [Google Scholar]
- 78. Tian F., DaCosta Byfield S., Parks W. T., Yoo S., Felici A., Tang B., Piek E., Wakefield L. M., Roberts A. B. (2003) Reduction in Smad2/3 signaling enhances tumorigenesis but suppresses metastasis of breast cancer cell lines. Cancer Res. 63, 8284–8292 [PubMed] [Google Scholar]
- 79. Riggins G. J., Kinzler K. W., Vogelstein B., Thiagalingam S. (1997) Frequency of Smad gene mutations in human cancers. Cancer Res. 57, 2578–2580 [PubMed] [Google Scholar]
- 80. Araki S., Eitel J. A., Batuello C. N., Bijangi-Vishehsaraei K., Xie X. J., Danielpour D., Pollok K. E., Boothman D. A., Mayo L. D. (2010) TGF-β1-induced expression of human Mdm2 correlates with late-stage metastatic breast cancer. J. Clin. Investig. 120, 290–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ghellal A., Li C., Hayes M., Byrne G., Bundred N., Kumar S. (2000) Prognostic significance of TGFβ1 and TGFβ3 in human breast carcinoma. Anticancer Res. 20, 4413–4418 [PubMed] [Google Scholar]
- 82. Koumoundourou D., Kassimatis T., Zolota V., Tzorakoeleftherakis E., Ravazoula P., Vassiliou V., Kardamakis D., Varakis J. (2007) Prognostic significance of TGFβ-1 and pSmad2/3 in breast cancer patients with T1–2,N0 tumours. Anticancer Res. 27, 2613–2620 [PubMed] [Google Scholar]
- 83. Richardsen E., Uglehus R. D., Johnsen S. H., Busund L. T. (2012) Immunohistochemical expression of epithelial and stromal immunomodulatory signalling molecules is a prognostic indicator in breast cancer. BMC Res. Notes 5, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Herrera A. C., Panis C., Victorino V. J., Campos F. C., Colado-Simão A. N., Cecchini A. L., R. C. (2012) Cancer Immunol. Immunother., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Guo L., Abraham J., Flynn D. C., Castranova V., Shi X., Qian Y. (2007) Individualized survival and treatment response predictions for breast cancers using phospho-EGFR, phospho-ER, phospho-HER2/neu, phospho-IGF-IR/In, phospho-MAPK, and phospho-p70S6K proteins. Int. J. Biol. Markers 22, 1–11 [DOI] [PubMed] [Google Scholar]
- 86. Mizukami Y., Nonomura A., Noguchi M., Taniya T., Koyasaki N., Saito Y., Hashimoto T., Matsubara F., Yanaihara N. (1991) Immunohistochemical study of oncogene product ras p21, c-myc and growth factor EGF in breast carcinomas. Anticancer Res. 11, 1485–1494 [PubMed] [Google Scholar]
- 87. Mizukami Y., Nonomura A., Yamada T., Kurumaya H., Hayashi M., Koyasaki N., Taniya T., Noguchi M., Nakamura S., Matsubara F. (1990) Immunohistochemical demonstration of growth factors, TGF-α, TGF-β, IGF-I and neu oncogene product in benign and malignant human breast tissues. Anticancer Res. 10, 1115–1126 [PubMed] [Google Scholar]
- 88. Duchene J., Schanstra J. P., Pecher C., Pizard A., Susini C., Esteve J. P., Bascands J. L., Girolami J. P. (2002) A novel protein-protein interaction between a G protein-coupled receptor and the phosphatase SHP-2 is involved in bradykinin-induced inhibition of cell proliferation. J. Biol. Chem. 277, 40375–40383 [DOI] [PubMed] [Google Scholar]
- 89. Florio T., Thellung S., Arena S., Corsaro A., Bajetto A., Schettini G., Stork P. J. (2000) Somatostatin receptor 1 (SSTR1)-mediated inhibition of cell proliferation correlates with the activation of the MAP kinase cascade: role of the phosphotyrosine phosphatase SHP-2. J. Physiol. Paris 94, 239–250 [DOI] [PubMed] [Google Scholar]
- 90. Lin X., Duan X., Liang Y. Y., Su Y., Wrighton K. H., Long J., Hu M., Davis C. M., Wang J., Brunicardi F. C., Shi Y., Chen Y. G., Meng A., Feng X. H. (2006) PPM1A functions as a Smad phosphatase to terminate TGFβ signaling. Cell 125, 915–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Buxton I. L., Duan D. (2008) Cyclic GMP/protein kinase G phosphorylation of Smad3 blocks transforming growth factor-β-induced nuclear Smad translocation: a key antifibrogenic mechanism of atrial natriuretic peptide. Circ. Res. 102, 151–153 [DOI] [PubMed] [Google Scholar]
- 92. Rubinfeld H., Seger R. (2005) The ERK cascade: a prototype of MAPK signaling. Mol. Biotechnol. 31, 151–174 [DOI] [PubMed] [Google Scholar]
- 93. Lawrence M. C., Jivan A., Shao C., Duan L., Goad D., Zaganjor E., Osborne J., McGlynn K., Stippec S., Earnest S., Chen W., Cobb M. H. (2008) The roles of MAPKs in disease. Cell Res. 18, 436–442 [DOI] [PubMed] [Google Scholar]
- 94. Kondoh K., Torii S., Nishida E. (2005) Control of MAP kinase signaling to the nucleus. Chromosoma 114, 86–91 [DOI] [PubMed] [Google Scholar]
- 95. Seger R., Krebs E. G. (1995) The MAPK signaling cascade. FASEB J. 9, 726–735 [PubMed] [Google Scholar]
- 96. McKay M. M., Morrison D. K. (2007) Integrating signals from RTKs to ERK/MAPK. Oncogene 26, 3113–3121 [DOI] [PubMed] [Google Scholar]
- 97. Sugden P. H., Clerk A. (1997) Regulation of the ERK subgroup of MAP kinase cascades through G protein-coupled receptors. Cell. Signal. 9, 337–351 [DOI] [PubMed] [Google Scholar]
- 98. Hayashida T., Decaestecker M., Schnaper H. W. (2003) Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-β-dependent responses in human mesangial cells. FASEB J. 17, 1576–1578 [DOI] [PubMed] [Google Scholar]
- 99. Ross K. R., Corey D. A., Dunn J. M., Kelley T. J. (2007) SMAD3 expression is regulated by mitogen-activated protein kinase kinase-1 in epithelial and smooth muscle cells. Cell. Signal. 19, 923–931 [DOI] [PubMed] [Google Scholar]
- 100. Roberts P. J., Der C. J. (2007) Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26, 3291–3310 [DOI] [PubMed] [Google Scholar]
- 101. Zhao S. L., Hong J., Xie Z. Q., Tang J. T., Su W. Y., Du W., Chen Y. X., Lu R., Sun D. F., Fang J. Y. (2011) TRAPPC4-ERK2 interaction activates ERK1/2, modulates its nuclear localization and regulates proliferation and apoptosis of colorectal cancer cells. PLoS One 6, e23262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mao L., Yang L., Arora A., Choe E. S., Zhang G., Liu Z., Fibuch E. E., Wang J. Q. (2005) Role of protein phosphatase 2A in mGluR5-regulated MEK/ERK phosphorylation in neurons. J. Biol. Chem. 280, 12602–12610 [DOI] [PubMed] [Google Scholar]
- 103. Tsukamoto H., Mishima Y., Hayashibe K., Sasase A. (1992) α-Smooth muscle actin expression in tumor and stromal cells of benign and malignant human pigment cell tumors. J. Invest. Dermatol. 98, 116–120 [DOI] [PubMed] [Google Scholar]
- 104. Tamiolakis D., Papadopoulos N., Cheva A., Lambropoulou M., Kotini A., Jivannakis T., Simopoulos C. (2002) Immunohistochemical expression of α-smooth muscle actin in infiltrating ductal carcinoma of the breast with productive fibrosis. Eur. J. Gynaecol Oncol. 23, 469–471 [PubMed] [Google Scholar]
- 105. Del Re D. P., Miyamoto S., Brown J. H. (2007) RhoA/Rho kinase up-regulate Bax to activate a mitochondrial death pathway and induce cardiomyocyte apoptosis. J. Biol. Chem. 282, 8069–8078 [DOI] [PubMed] [Google Scholar]
- 106. Kopfstein L., Christofori G. (2006) Metastasis: cell-autonomous mechanisms versus contributions by the tumor microenvironment. Cell. Mol. Life Sci. 63, 449–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Schuh J. C. (2004) Trials, tribulations, and trends in tumor modeling in mice. Toxicol. Pathol. 32, Suppl. 1, 53–66 [DOI] [PubMed] [Google Scholar]
- 108. Amar S., Roy V., Perez E. A. (2009) Treatment of metastatic breast cancer: looking towards the future. Breast Cancer Res. Treat. 114, 413–422 [DOI] [PubMed] [Google Scholar]