

Background: P-TEFb regulates transcription elongation, cell growth, and proliferation.
Results: BET bromodomain inhibition by JQ1 transiently releases free P-TEFb from the inactive 7SK snRNP, thus activating HEXIM1 and HIV transcription.
Conclusion: JQ1 affects the P-TEFb equilibrium.
Significance: P-TEFb release from and reassembly into 7SK snRNP by JQ1 inhibits tumor cell growth and reactivates latent HIV.
Keywords: Chromatin Regulation, Histone Deacetylase Inhibitors, HIV, Stress, Transcription Elongation Factors, 7SK snRNP, BRD4, HEXIM1, P-TEFb, SEC
Abstract
By phosphorylating elongation factors and the C-terminal domain of RNA polymerase II, the positive transcription elongation factor b (P-TEFb) is the critical kinase for transcription elongation and co-transcriptional processing of eukaryotic genes. It exists in inactive small nuclear ribonucleoprotein (7SK snRNP) and active (free P-TEFb) complexes in cells. The P-TEFb equilibrium determines the state of cellular activation, proliferation, and differentiation. Free P-TEFb, which is required for growth, can be recruited to RNA polymerase II via transcription factors, BRD4, or the super elongation complex (SEC). UV light, various signaling cascades, transcriptional blockade, or compounds such as hexamethylene bisacetamide (HMBA), suberoylanilide hydroxamic acid (SAHA), and other histone deacetylase inhibitors lead to a rapid release of free P-TEFb, followed by its reassembly into the 7SK snRNP. As a consequence, transcription of HEXIM1, a critical 7SK snRNP subunit, and HIV is induced. In this study, we found that a bromodomain and extra-terminal (BET) bromodomain inhibitor, JQ1, which inhibits BRD4 by blocking its association with chromatin, also leads to the rapid release of free P-TEFb from the 7SK snRNP. Indeed, JQ1 transiently increased levels of free P-TEFb and BRD4·P-TEFb and SEC·P-TEFb complexes in cells. As a consequence, the levels of HEXIM1 and HIV proteins rose. Importantly, the knockdown of ELL2, a subunit of the SEC, blocked the ability of JQ1 to increase HIV transcription. Finally, the effects of JQ1 and HMBA or SAHA on the P-TEFb equilibrium were cooperative. We conclude that HMBA, SAHA, and JQ1 affect transcription elongation by a similar and convergent mechanism.
Introduction
Eukaryotic transcription by RNA polymerase II (RNAPII)3 can be divided into distinct regulated steps, from initiation to promoter clearance, pausing, and elongation (1–5). Although in the past, the emphasis had been placed on the recruitment of the transcription complex to promoters, regulation of gene expression at subsequent steps of transcription has been recognized to play an equally important role. Indeed, pausing of RNAPII downstream of many eukaryotic promoters, whether these genes are active or not, allows for rapid transcriptional activation following environmental or developmental cues (1–5). In this scenario, after promoter clearance, negative elongation factors assemble on the elongating RNAPII, blocking its movement. Recruitment of the positive transcription elongation factor b (P-TEFb) and the super elongation complex (SEC) removes this block and promotes transcription elongation. P-TEFb phosphorylates not only the negative elongation factors DSIF (5,6-dichloro-1-β-d-ribofuranosylbenzimidazole sensitivity-inducing factor) and NELF (negative transcription elongation factor), converting DSIF into an elongation factor, but also the C-terminal domain of RNAPII, thereby regulating co-transcriptional processes such as RNA splicing and polyadenylation (1–5).
The activity of P-TEFb, composed of cyclin T1 (CycT1) or T2 and CDK9 (cyclin-dependent kinase 9), is tightly regulated in cells. Besides microRNA-dependent regulation of CycT1 protein expression in resting hematopoietic cells (6), in growing cells, P-TEFb partitions between an active complex, alone or associated in a mutually exclusive fashion with BRD4 (bromodomain-containing protein 4) or the SEC, and an inactive complex with 7SK snRNA, HEXIM1/2 (hexamethylene bisacetamide-inducible protein 1/2), LARP7 (La-related protein 7), and MePCE(methylphosphate capping enzyme), which is known as the 7SK small nuclear ribonucleoprotein (snRNP) (3, 7). As a central component of the 7SK snRNP, HEXIM1 binds to and inhibits the kinase activity of P-TEFb (3, 7, 8). Active P-TEFb can be recruited to its target genes via interaction with various DNA-bound activators such as NF-κB, c-Myc, the class II transactivator, and steroid hormone receptors, which otherwise function poorly when levels of free P-TEFb are low (1, 3). On the other hand, the HIV transcriptional transactivator Tat can utilize P-TEFb directly from the 7SK snRNP (9, 10). BRD4 can also compete with HEXIM1 for binding to P-TEFb (11, 12), and the BRD4·P-TEFb complex is important for the activation of primary response genes (13, 14).
Previously, we studied the effects of hexamethylene bisacetamide (HMBA) and the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) on HIV transcription in transformed and primary cells, with special emphasis on proviral latency and reactivation (15–17). Both compounds released P-TEFb from the 7SK snRNP and potently induced HIV gene expression when the viral LTR was not occluded by transcriptional interference (integration into active genes) (15–17). In this study, the bromodomain and extra-terminal (BET) bromodomain inhibitor JQ1 (18, 19) did the same and acted cooperatively with HMBA and SAHA. It also increased transcription of HEXIM1 and its incorporation into the 7SK snRNP. Moreover, its effects on HIV transcription could be inhibited by depletion of ELL2, a subunit of the SEC (20).
EXPERIMENTAL PROCEDURES
Cell Lines, Antibodies, and Plasmids
JΔK cells and Jurkat cells were grown in RPMI 1640 medium containing 100 IU/ml penicillin, 100 μg/ml streptomycin, and 10% FBS at 37 °C with 5% CO2. HIV release in the supernatant was quantified by p24 capsid ELISA (PerkinElmer Life Sciences). Antibodies used in this study for co-immunoprecipitations and Western blotting were as follows: anti-HEXIM1 (ab25388), anti-BRD4 (ab75898), anti-AFF4 (ab57077), and anti-tubulin (ab6046) from Abcam; anti-CDK9 (sc-484), anti-CycT1 (sc-10750), and anti-RNAPII (sc-899) from Santa Cruz Biotechnology; and anti-ELL2 from Bethyl Laboratories, Inc. Antibodies used in ChIP assays were as follows: normal rabbit IgG (sc-2027), anti-CDK9 (sc-484), and anti-RNAPII (sc-899) from Santa Cruz Biotechnology; anti-BRD4 (ab75898) from Abcam; and anti-ELL2 (21), which was a kind gift of Dr. Christine Milcarek (University of Pittsburgh). The anti-ELL2 shRNA-encoding plasmid (pSuper-shELL2) was a kind gift of Dr. Qiang Zhou (University of California, Berkeley). SAHA was purchased from Selleck Chemicals and HMBA from Sigma, and stock solutions were prepared in dimethyl sulfoxide (DMSO) and water, respectively. JQ1 was a kind gift from Dr. James Bradner (Harvard University), and stock solutions were prepared in DMSO.
Co-immunoprecipitation
Jurkat cells (5 × 106) were lysed on ice (10 min) in buffer A (20 mm HEPES-KOH (pH 7.8), 0.2 mm EDTA, and 0.5% Nonidet P-40) containing medium salt (100 mm KCl) for detection of CycT1/BRD4 and CycT1/SEC interactions or high salt (300 mm KCl) for detection of CycT1/HEXIM1 interaction. The cell lysates were centrifuged (10,000 × g, 2 min, 4 °C), and the supernatants were collected. Supernatants were then precleared with protein A-Sepharose beads (Invitrogen) for 1 h at 4 °C. Precleared lysates were incubated with 3 μg of the appropriate antibodies (indicated) overnight at 4 °C. The lysates were then centrifuged at 10,000 × g for 10 min at 4 °C, and supernatants were incubated with protein A-Sepharose beads for 1 h at 4 °C. Beads were washed five times with 800 μl of lysis buffer, and immunoprecipitated complexes were boiled in SDS sample buffer and analyzed by Western blotting.
RNA Immunoprecipitations
JΔK cells (2 × 106) were untreated or treated with 5 μm JQ1 for 0.5 or 2 h. Cells were lysed in buffer A containing low salt (10 mm KCl) on ice for 10 min. Cell lysates were centrifuged at 5000 × g for 5 min at 4 °C, and supernatants were collected. Supernatants were then precleared with protein A-Sepharose beads and divided into three aliquots. Each aliquot was incubated with 1 μg of normal rabbit IgG, anti-HEXIM1, or anti-CDK9 antibody overnight at 4 °C and then with 20 μl of protein A-Sepharose beads precoated with BSA and yeast tRNA for an additional 2 h at 4 °C. Beads were washed five times with medium-salt buffer A (100 mm KCl). RNA was then extracted by TRIzol (Invitrogen) and analyzed by RT-quantitative PCR (RT-qPCR). Data were normalized to input amounts of 7SK snRNA and calculated as percent values relative to the amount obtained with untreated cells (set to 100%).
Differential Salt Extraction
Differential salt extraction was carried out to determine fractions of free P-TEFb or 7SK snRNP according to Biglione et al. (22) with some modifications. Jurkat cells (5 × 105) were collected and washed twice with cold PBS. Cells were lysed in 80 μl of low-salt buffer (10 mm KCl, 10 mm MgCl2, 10 mm HEPES-KOH (pH 7.5), 1 mm EDTA, 1 mm DTT, 0.5% Nonidet P-40, and proteinase inhibitor mixture) and incubated on ice for 10 min. Lysates were then centrifuged at 5000 × g for 5 min, and supernatants were collected and designated as 7SK snRNP fractions. Pellets were washed once with 200 μl of low-salt buffer and resuspended in 80 μl of high-salt buffer (450 mm NaCl, 1.5 mm MgCl2, 20 mm HEPES (pH 7.5), 0.5 mm EDTA, 1 mm DTT, 0.5% Nonidet P-40, and proteinase inhibitor mixture). Suspensions were mixed by vortexing briefly and incubated on ice for 10 min. Lysates were then centrifuged at 10,000 × g for 5 min, and supernatants were collected and designated as free P-TEFb fractions.
Chromatin Immunoprecipitation
ChIPs were carried out according to Nelson et al. (23) with some modifications. Briefly, JΔK cells (2 × 107) were treated with JQ1 (5 μm) or DMSO for 1 h. Cells were fixed with 1% formaldehyde in PBS for 15 min at room temperature. By adding 125 mm glycine for 5 min at room temperature, cross-linking was stopped. Sonication of chromatin was carried out using a Sonic Dismembrator 100 (Fisher) for 20 cycles of 15 s at setting 4, followed by 30 s on ice. Sheared chromatin was precleared by 50 μl of protein G-Sepharose beads for 1 h at 4 °C. 2 μg of specific antibodies were added to the precleared lysate corresponding to 2 × 106 cells and incubated at 4 °C overnight. Lysates were then centrifuged at 10,000 × g for 10 min. 90% of the supernatant was used for further processing. 30 μl of protein G beads precoated with BSA and salmon sperm DNA were added to each tube and incubated at 4 °C for 1 h. The chromatin-protein-bead complexes were washed six times with ChIP buffer. The DNA was purified with 10% Chelex beads (Bio-Rad). The DNA was used as a template for qPCR.
Transient Transfection and Luciferase Assays
Jurkat cells (2 × 107) growing in log phase were transfected with 10 μg of plasmid DNA by electroporation using the Bio-Rad Gene Pulser II system (270 V, 975 microfarads). After electroporation, cells were allowed to sit in the cuvette for 15 min at room temperature and then transferred to 5 ml of prewarmed RPMI 1640 medium with 10% FBS and kept in 5% CO2 at 37 °C for 48 h. Luciferase activity in the cell lysate was determined using the luciferase assay system (Promega) according to the manufacturer's instructions.
qPCR and RT-qPCR
Total cellular RNA was extracted by TRIzol reagent according to the manufacturer's instruction and reverse-transcribed with random hexamers using SuperScript III reverse transcriptase (Invitrogen). The cDNA was quantified using the Stratagene Mx3004P real-time PCR system and SensiFAST SYBR Green reagents (Bioline) with specific primers. The primer sequences used in this study were as follows: for RNA immunoprecipitation of human 7SK, GAGGGCGATCTGGCTGCGACAT (forward) and ACATGGAGCGGTGAGGGAGGAA (reverse); for HIV expression and ChIP, TGGTTAGACCAGATCTGAGCCTGGGA (forward LTR), TAAGCAGTGGGTTCCCTAGTTAGCCA (reverse LTR), AGCAGGAAGATGGCCAGTAA (forward coding), and AACAGGCGGCCTTAACTGTA (reverse coding); for HEXIM1 mRNA quantification, GACCTGGGAAGAGAAGAAAAAG (forward) and GAGGAACTGCGTGGTGTTATAG (reverse).
Glycerol Gradients
Glycerol gradients (10–30%) were established by pipetting 2 ml of each glycerol fraction (10, 15, 20, 25, and 30%, v/v) in medium-salt buffer A into centrifugation tubes (Beckman 331372). Gradients were formed by standing for 6 h at 4 °C. JΔK cells (2 × 106) were untreated or treated with 5 μm JQ1 for 1 h and lysed in 0.6 ml of medium-salt buffer A for 30 min at 4 °C. Lysates were centrifuged at 5000 × g for 5 min, and supernatants were loaded into tubes with preformed glycerol gradients. Protein complexes were then fractionated by centrifugation in an SW40T1 rotor (Beckman) at 38,000 rpm for 21 h. 10 fractions (1 ml) were collected, precipitated with trichloroacetic acid, and analyzed with the indicated antibodies by Western blotting.
RESULTS
JQ1 Activates HIV Transcription
To investigate the effects of BET bromodomain inhibition on HIV transcription, we administered JQ1 (19, 24) to Jurkat cells that contained one integrated copy of the HIV provirus that lacks both NF-κB-binding sites (JΔK cells) (Fig. 1, A and B) (25). Thus, its transcriptional activation is independent of NF-κB induction. As shown in Fig. 1A and measured by p24 capsid release into the medium, JQ1 activated HIV production by JΔK cells. Titration of the compound indicated that JQ1 elicited a maximum stimulation at a concentration of 5 μm (Fig. 1A, bar 4). Expression of HIV mRNA after JQ1 treatment, as quantified by RT-qPCR, further demonstrated that JQ1 stimulated HIV transcription (Fig. 1B). Similar results were obtained with another BET bromodomain inhibitor, I-BET (26) (data not shown). These results indicate that the BET bromodomain inhibition by JQ1 activates HIV transcription, which is reminiscent of other HIV activators such as SAHA and HMBA (15–17).
FIGURE 1.
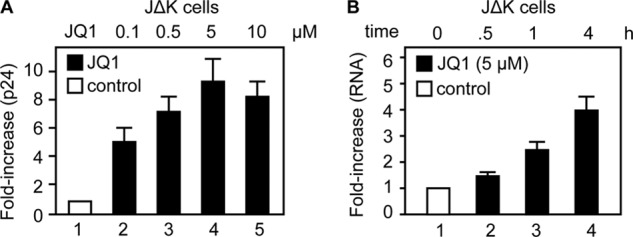
JQ1 activates HIV transcription. A, JΔK cells were treated with increasing concentrations of JQ1 as indicated for 24 h. The production of new viral particles was determined by p24 capsid ELISA and is presented as -fold increase over the DMSO control (bar 1). B, JΔK cells were treated with JQ1 (5 μm) for the indicated times, and HIV-specific mRNA levels were determined by RT-qPCR and normalized to actin mRNA. The results are presented as -fold increase over the DMSO control (bar 1). Error bars represent S.E. of duplicate (A) or triplicate (B) experiments.
JQ1 Transiently Releases P-TEFb from the 7SK snRNP
Treatment of cells with SAHA or HMBA leads to a rapid but transient release of free P-TEFb from the 7SK snRNP, followed by increased expression of P-TEFb-dependent genes, including HEXIM1 and the HIV LTR (16, 17, 27). Increased synthesis of HEXIM1 then leads to the reassembly of the 7SK snRNP and the subsequent growth arrest of treated cells (27). In contrast, HIV transcription continues due to the synthesis of Tat, which competes with HEXIM1 for the binding of P-TEFb to 7SK snRNA and its recruitment to the HIV TAR (transactivation response) element RNA stem-loop (9, 10). Thus, once Tat is made, HIV replication is sustained despite the return of cells to their resting state. To verify that JQ1 also affects this P-TEFb equilibrium, we performed glycerol gradient centrifugation to separate free P-TEFb and the 7SK snRNP in cell lysates under medium-salt (100 mm KCl) conditions, which extract these complexes but not BRD4 from its chromatin-bound state (22, 28). Minute amounts of BRD4 were extracted from chromatin in untreated JΔK cells with the medium-salt buffer (Fig. 2A, upper left panel). In contrast, incubating JΔK cells with JQ1 for 1 h resulted in a massive release of BRD4 from chromatin into low-molecular mass fractions (Fig. 2A, lower left panel, lanes 2–5). At the same time, the levels of free P-TEFb, which was released from the 7SK snRNP, also increased (Fig. 2A, right panels). Quantitative analysis of the 7SK snRNP and free P-TEFb by sequential extractions of the cells with low-salt (10 mm KCl) and high-salt (450 mm NaCl) buffers, respectively, revealed that the levels of free P-TEFb increased after JQ1 treatment, peaked between 30 min and 1 h, and decreased to basal levels by 6 h of incubation (Fig. 2B, lanes 7–12), suggesting that the 7SK snRNP had reassembled by 6 h after JQ1 treatment. The time course of P-TEFb release and reassembly of the 7SK snRNP by JQ1 was similar to that with HMBA and SAHA (16, 17). The release of P-TEFb from the 7SK snRNP was further confirmed by RNA immunoprecipitations. Anti-HEXIM1 and anti-CDK9 immunoprecipitations, followed by RT-qPCR quantification of co-immunoprecipitated 7SK snRNA, indicated that the association of 7SK snRNA with HEXIM1 or CDK9 decreased after JQ1 treatment (Fig. 2C). We conclude that JQ1 not only releases BRD4 from chromatin but also leads to the release of P-TEFb from the 7SK snRNP.
FIGURE 2.
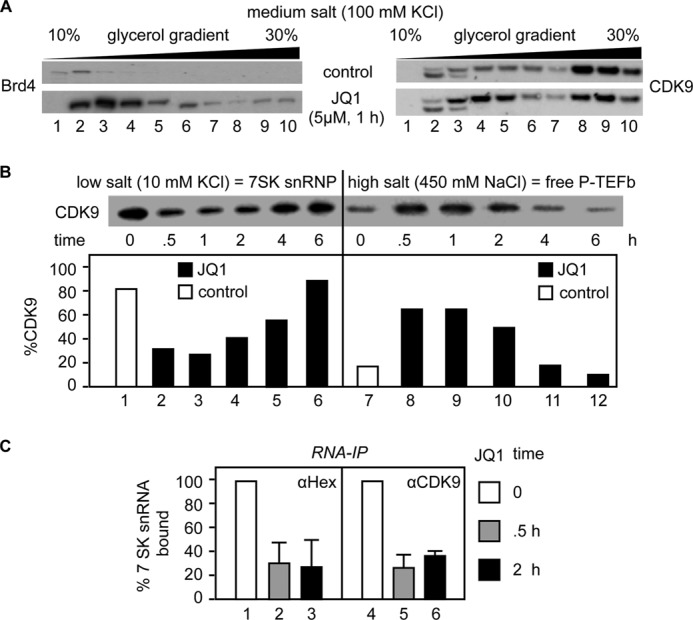
JQ1 directs the disruption and reassembly of the 7SK snRNP. A, cell lysates of JΔK cells treated with DMSO (upper panels) or JQ1 (5 μm for 1 h; lower panels) were subjected to 10–30% glycerol gradient sedimentation. BRD4 (left panels) and CDK9 (right panels) in each fraction were detected by Western blotting. B, Jurkat cells were subjected to differential salt extraction after JQ1 treatment (5 μm) for the indicated times. The levels of CDK9 extracted in low-salt (7SK snRNP; lanes 1–6) and high-salt (free P-TEFb; lanes 7–12) buffers were determined by Western blotting (upper panel) and quantified. The results are presented as relative distributions in the 7SK snRNP and free P-TEFb fractions (lower panels). C, the association between 7SK snRNA and HEXIM1 or CDK9 after JQ1 treatment (5 μm) for the indicated times was determined by RNA immunoprecipitations (RNA-IP) with anti-HEXIM1 (αHex; left panel) and anti-CDK9 (right panel) antibodies, followed by RT-qPCR with primers specific to 7SK snRNA. Data are presented relative to the DMSO control (bars 1 and 4). Error bars represent S.E. of triplicate experiments.
JQ1 Induces HEXIM1 Gene Expression and Promotes 7SK snRNP Reassembly
A known consequence of the release of free P-TEFb from the 7SK snRNP is transcriptional activation of HEXIM1, the inhibitory subunit of the 7SK snRNP, and the subsequent incorporation of P-TEFb into the 7SK snRNP (27). Indeed, previous microarray analyses revealed that HEXIM1 mRNA is increased greatly by JQ1 (19, 24). To confirm this finding, we quantified HEXIM1 mRNA levels in Jurkat cells at different time points after JQ1 treatment. As shown in Fig. 3A, JQ1 activated greatly HEXIM1 gene expression. Additionally, JQ1 increased transcription from a plasmid target carrying the HEXIM1 promoter linked to the luciferase reporter gene (Fig. 3B, Hex.Luc). Concomitantly, JQ1 also increased the production of the HEXIM1 protein (Fig. 3C). Co-immunoprecipitations revealed increased association between the newly synthesized HEXIM1 protein and CycT1 at 6 and 24 h (Fig. 3D), which paralleled the time course of reassembly of the 7SK snRNP (Fig. 2B). These data indicate that like HMBA and SAHA, JQ1 increases the synthesis of the P-TEFb inhibitor HEXIM1, which drives the reassembly of the 7SK snRNP.
FIGURE 3.
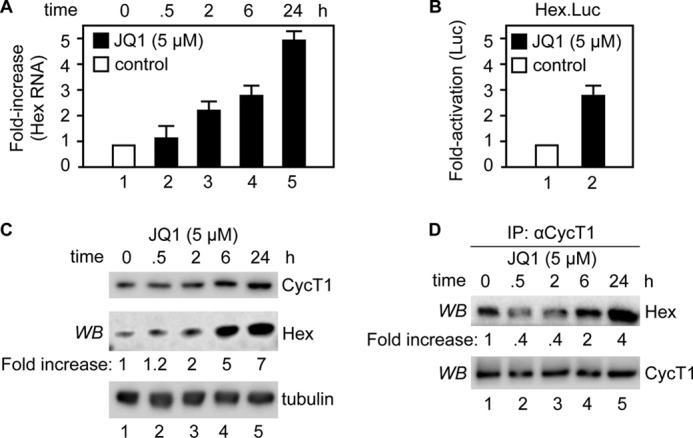
JQ1 increases HEXIM1 levels for reassembly of the 7SK snRNP. A, Jurkat cells were treated with JQ1 (5 μm) for the indicated times. HEXIM1 (Hex) mRNA levels were determined by RT-qPCR and normalized to actin mRNA. The results are presented as -fold increase over the DMSO control (bar 1). Error bars represent S.E. of triplicate experiments. B, Jurkat cells were transfected with the HEXIM1 promoter-luciferase reporter gene plasmid (Hex.Luc) for 24 h and treated with DMSO or JQ1 (5 μm) for an additional 24 h prior to luciferase assays. Luciferase activity is presented as -fold activation over the DMSO control (bar 1). The error bar represents S.E. of triplicate experiments. C, the protein levels of HEXIM1, CycT1, and tubulin in Jurkat cells treated with JQ1 (5 μm) for the indicated times were determined by Western blotting (WB). Ratios between HEXIM1 and tubulin were calculated for each time point and are presented as -fold increase over the DMSO control (lane 1). D, Jurkat cells were treated with JQ1 (5 μm) for the indicated times, and cell lysates were subjected to immunoprecipitations (IP) with anti-CycT1 antibody. The levels of HEXIM1 and CycT1 proteins in the immunoprecipitations were determined by Western blotting. Ratios of immunoprecipitated HEXIM1 to CycT1 were calculated and are presented as -fold increase over the DMSO control (lane 1).
Released P-TEFb Associates with the SEC
Free P-TEFb increases transcription elongation of target genes via many different protein/protein interactions (1, 11, 18, 21, 27, 29–32). Thus, we next examined the association of P-TEFb with other known protein complexes after JQ1 treatment. As shown in Fig. 4A, we observed an increased association between BRD4 and P-TEFb. This association peaked between 30 min and 2 h and was diminished by 6 and 24 h, which paralleled the disruption and reassembly of the 7SK snRNP (Fig. 4A, lanes 2–6; see also Fig. 2B). Another complex in which P-TEFb resides as a transcriptional activator is with the SEC (18, 27, 28, 33). Of several different SECs, P-TEFb associates prominently with those that contain AFF4 as the central scaffold (27). Following JQ1 treatment, we observed increased association between P-TEFb and AFF4, which also paralleled the time course of the disruption and reassembly of the 7SK snRNP (Fig. 4B, lanes 6–10). Indeed, BRD4 and AFF4 were found in distinct and separate complexes with P-TEFb (29), which was confirmed by co-immunoprecipitations that failed to detect any interaction between BRD4 and AFF4 (Fig. 4C, lanes 6–10). Thus, JQ1 led to a transient release of free P-TEFb and its concomitant increased association with BRD4 and the SEC.
FIGURE 4.
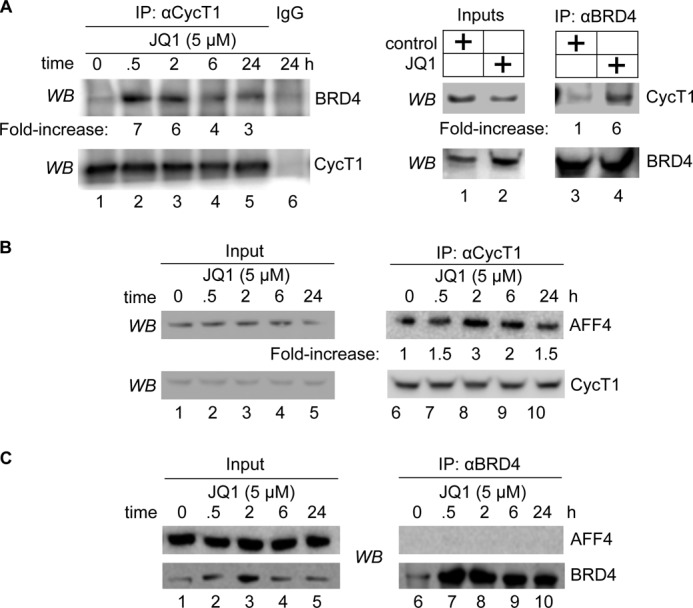
JQ1 increases the association between P-TEFb, BRD4, and AFF4 from the SEC. A, Jurkat cells were treated with JQ1 (5 μm) for the indicated times (left panels) or for 30 min (right panels), and cell lysates were subjected to immunoprecipitations (IP) with anti-CycT1 (left panels) and anti-BRD4 (right panels) antibodies. The levels of the indicated proteins in the 5% input (right panels) and immunoprecipitations (left panels) were determined by Western blotting (WB). Ratios between immunoprecipitated BRD4 and CycT1 (left panels, BRD4/CycT1; right panels, CycT1/BRD4) were calculated and are presented as -fold increase over the DMSO control (lanes 1 and 6). B, Jurkat cells were treated with JQ1 (5 μm) for the indicated times, and cell lysates were subjected to immunoprecipitations with anti-CycT1 antibody. The levels of the indicated proteins in the 5% input (lanes 1–5) and immunoprecipitations (lanes 6–10) were determined by Western blotting. Ratios of immunoprecipitated AFF4 to CycT1 were calculated and are presented as -fold increase over the DMSO control (lane 6). C, Jurkat cells were treated with JQ1 (5 μm) for the indicated times, and cell lysates were subjected to immunoprecipitations with anti-BRD4 antibody. The levels of the indicated proteins in the 5% input (lanes 1–5) and immunoprecipitations (lanes 6–10) were determined by Western blotting.
JQ1 Increases P-TEFb and SEC Levels at the HIV LTR
Because HIV transcription was increased by JQ1 in JΔK cells, we performed ChIPs with the HIV LTR and coding regions (Fig. 5A, upper panel). As shown in Fig. 5A (lower panels), whereas the recruitment of RNAPII, CDK9, and the SEC (AFF4 and ELL2) to the HIV LTR (bars 1–5) and coding regions (bars 6–10) was increased by JQ1, that of BRD4 were decreased. This finding is not surprising, as JQ1 removes BRD4 from chromatin (Fig. 2A) (18, 24), and ELL2 and AFF4 can recruit P-TEFb to the viral genome (27). Indeed, in a previous study, the exogenous coexpression of AFF4 and ELL2 activated the HIV LTR by up to 100-fold (29). Thus, we examined if ELL2 is required for the effects of JQ1 on HIV transcription. To this end, we knocked down ELL2 with specific shRNAs for 48 h in Jurkat cells. A previously validated ELL2 shRNA plasmid effector was co-transfected with the HIV-luciferase plasmid target for 48 h. Subsequently, cells were treated with JQ1 or DMSO for 24 h, and luciferase activity was determined (Fig. 5B, lower panels). Indeed, JQ1 increased the luciferase activity by 4-fold (Fig. 5B, upper panel, bar 2), which was abrogated when ELL2 was knocked down (bar 4). Of note, this JQ1- and ELL2-dependent activation of HIV transcription occurred in the absence of Tat. We conclude and confirm that as with MLL fusion proteins MED26 and the polymerase-associated factor complex (20, 31, 32), the SEC can recruit P-TEFb to transcription units, in this case, to the HIV LTR.
FIGURE 5.

JQ1 increases AFF4 and ELL2 levels at the HIV LTR and coding regions, and inactivation of ELL2 blocks its effects on HIV transcription. A, upper panel, schematic representation of the HIV promoter (LTR) and the Gag-Pol coding regions. Arrows indicate positions of primers for qPCR after ChIP. Lower panels, JΔK cells treated with DMSO (white bars) or JQ1 (5 μm; black bars) for 1 h were subjected to ChIP with the indicated antibodies. The qPCR signals obtained with the LTR primers (bars 1-5) or the coding primers (bars 6–10) were normalized to the input and are presented as -fold enrichment over the signals obtained with the IgG control. Error bars represent S.E. of triplicate experiments. B, upper panel, Jurkat cells were co-transfected with the HIV LTR-luciferase (HIV.Luc) plasmid target and scrambled (SCR; bars 1 and 2) and ELL2 (ELL2, bars 2 and 4) shRNA plasmids for 48 h prior to the addition of DMSO (bars 1 and 3) or JQ1 (5 μm; bars 2 and 4). After an additional 24 h, luciferase activity was determined. Data are presented relative to the DMSO control. ELL2 knockdown was confirmed by Western blotting (WB; middle panel) with tubulin as the loading control (lower panel). Error bars represent S.E. of triplicate experiments.
JQ1, HMBA, and SAHA Have Comparable Effects on HIV Transcription
This and previous studies suggested that HMBA, SAHA, and JQ1 share a common mechanism of transcriptional activation (16, 17, 34). To this end, we treated JΔK cells with HMBA or SAHA alone or in combination with JQ1 for 24 h and measured the production of HIV by p24 capsid ELISA. Of note, JQ1 alone had lesser effects on HIV production than SAHA (Fig. 6A, gray bars versus white bars) or HMBA (Fig. 6B, gray bars versus white bars) at optimal doses (15–17). However, when suboptimal doses of these compounds were used, JQ1 acted cooperatively with SAHA (Fig. 6A, black bars) or HMBA (Fig. 6B, black bars) to activate HIV gene expression. In no instance did we find inhibition or multiplicative effects of these compounds, indicating that they act by a similar mechanism with respect to HIV transcription. Importantly, although we observed respectable increases of new viral particles with JQ1, we did not observe the reactivation of HIV in latently infected primary cells or in those from patients on optimal antiretroviral therapy, in which levels of P-TEFb are vanishingly low (data not shown). This finding is in contrast to HMBA and SAHA, which can reactivate latent viruses in these cells (16, 17).
FIGURE 6.

JQ1, SAHA, and HMBA have similar effects on HIV production. A and B, JΔK cells were treated with the indicated concentrations of JQ1 (gray bars), SAHA (A, white bars), HMBA (B, white bars), or combinations (black bars). After 24 h, the production of HIV was determined by p24 capsid ELISA and is presented as -fold increase over the DMSO control. Error bars represent S.E. of triplicate experiments.
DISCUSSION
In this study, we have described a mechanism of transcriptional activation by the BET bromodomain inhibitor JQ1. Indeed, JQ1 activated HIV and HEXIM1 transcription via the transient release of free P-TEFb from the 7SK snRNP and its concomitant recruitment to the HIV LTR and coding regions by the SEC. Consequently, the increased synthesis of HEXIM1 led to the reassembly of the 7SK snRNP and thus the inactivation of P-TEFb. Importantly, HIV transcription was activated by JQ1 and then transactivated by Tat, which can not only utilize P-TEFb from its active and inactive complexes but also compete with BRD4 for its binding (9, 10, 13).
We also found that HMBA, SAHA, and JQ1 share a common mechanism of transcriptional activation (16, 17, 34). SAHA also affects histone acetylation, and JQ1 affects the “readout” of these marks by BRD4. Interestingly, recent work demonstrated that HMBA can also rapidly de- and reacetylate histones with a concomitant transient release of BRD4 from chromatin. It was proposed that BRD4·P-TEFb complexes can thus be found in at least two different states, one chromatin-bound and the other promoter-associated (28). However, how much P-TEFb is bound to chromatin via BRD4 tethered to acetylated histones versus BRD4 in the Mediator complex remains to be investigated. Of note, BRD4 interacts best with the acetylated CycT1 protein, thus opening the possibility of a competition between acetylated histones and P-TEFb for its binding (11). Nevertheless, all of these compounds, although structurally and functionally distinct, seem to target P-TEFb. One simple unifying hypothesis is that by modifying chromatin, they all stress cells, whose survival depends on the rapid release of free P-TEFb from the 7SK snRNP. This consideration applies to other epigenetic modifiers that affect transcription elongation.
Although the transcription of many P-TEFb-dependent genes decreases during reassembly into the 7SK snRNP, HIV transcription continues because Tat and TAR compete with HEXIM1 and 7SK snRNA for binding to P-TEFb (9, 10). Our results indicate that JQ1 activates HIV transcription from JΔK cells as well as other latently infected transformed cell lines (data not shown). Although JQ1 alone did not reactivate HIV from latently infected primary resting T cells because their levels of P-TEFb are vanishingly low (6, 35), JQ1 could become a candidate for combinatorial anti-latency therapy with SAHA and/or other T cell-activating compounds. To this end, during the preparation of this manuscript, Montano and colleagues (36) reported that JQ1 could reactivate minimally latent HIV in cells isolated from one of three HIV-infected patients on highly active antiretroviral therapy, which supports this hypothesis.
BET bromodomain inhibitors such as JQ1 and I-BET are being investigated as potent anticancer drugs (18, 19, 24, 37) Attention has been focused on multiple myeloma, the growth of which depends on increased expression of c-Myc. Indeed, JQ1 rapidly depletes c-Myc mRNA, which results in G1 cell cycle arrest and apoptosis (24). Of note, the expression of the c-myc gene and its transcriptional effects depend on P-TEFb, which is inactivated by HEXIM1 (38). In this light, it is of interest that HEXIM1 transcription is decreased by estrogen in breast cancer cells, and its exogenous expression inhibits their proliferation (39). Depleting LARP7, which is an essential component of the 7SK snRNP, also increases their metastatic potential and malignant transformation (27). Thus, the effects of JQ1 on multiple myeloma cells could also result from increased sequestration of free P-TEFb into the 7SK snRNP. The lack of response of some c-Myc-dependent tumors to BET bromodomain inhibition could then result from a failed reassembly of the 7SK snRNP because of mutations in its subunits or their inadequate synthesis (37).
This study reveals an interesting new dimension to JQ1. Although primarily thought of as an epigenetic modifier, it also affects the P-TEFb equilibrium in growing cells. These effects could be direct, as BRD4 can remove P-TEFb from the 7SK snRNP (11, 13, 30), or indirect, via stress, as the structure of chromatin changes upon the release of BRD4 (40). Cooperative removal of BRD4 from chromatin and increased reassembly of the 7SK snRNP following the administration of JQ1 could act synergistically to silence the transcription of c-Myc, which results in growth arrest and apoptosis of multiple myeloma cells. Synergistic effects of histone deacetylases and JQ1 could also be exploited for better treatment of these conditions and even for possible future interventions in HIV latency and reactivation.
Acknowledgments
We thank Drs. James Bradner, Christine Milcarek, and Qiang Zhou for reagents.
This work was supported, in whole or in part, by National Institutes of Health CARE Center Grant U19 AI076113. This work was also supported by grants from the University of California San Francisco Center for AIDS Research (to K. B.) and amfAR (to K. F.).
- RNAPII
- RNA polymerase II
- P-TEFb
- positive transcription elongation factor b
- SEC
- super elongation complex
- CycT1
- cyclin T1
- snRNP
- small nuclear ribonucleoprotein
- HMBA
- hexamethylene bisacetamide
- SAHA
- suberoylanilide hydroxamic acid
- BET
- bromodomain and extra-terminal
- DMSO
- dimethyl sulfoxide
- qPCR
- quantitative PCR.
REFERENCES
- 1. Peterlin B. M., Price D. H. (2006) Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 23, 297–305 [DOI] [PubMed] [Google Scholar]
- 2. Saunders A., Core L. J., Lis J. T. (2006) Breaking barriers to transcription elongation. Nat. Rev. Mol. Cell Biol. 7, 557–567 [DOI] [PubMed] [Google Scholar]
- 3. Zhou Q., Li T., Price D. H. (2012) RNA polymerase II elongation control. Annu. Rev. Biochem. 81, 119–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nechaev S., Adelman K. (2011) Pol II waiting in the starting gates: regulating the transition from transcription initiation into productive elongation. Biochim. Biophys. Acta 1809, 34–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li J., Gilmour D. S. (2011) Promoter proximal pausing and the control of gene expression. Curr. Opin. Genet. Dev. 21, 231–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chiang K., Sung T. L., Rice A. P. (2012) Regulation of cyclin T1 and HIV-1 replication by microRNAs in resting CD4+ T lymphocytes. J. Virol. 86, 3244–3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Peterlin B. M., Brogie J. E., Price D. H. (2012) 7SK snRNA: a noncoding RNA that plays a major role in regulating eukaryotic transcription. Wiley Interdiscip. Rev. RNA 3, 92–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Czudnochowski N., Bösken C. A., Geyer M. (2012) Serine 7 but not serine 5 phosphorylation primes RNA polymerase II CTD for P-TEFb recognition. Nat. Commun 3, 842. [DOI] [PubMed] [Google Scholar]
- 9. Sedore S. C., Byers S. A., Biglione S., Price J. P., Maury W. J., Price D. H. (2007) Manipulation of P-TEFb control machinery by HIV: recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res. 35, 4347–4358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barboric M., Yik J. H., Czudnochowski N., Yang Z., Chen R., Contreras X., Geyer M., Matija Peterlin B., Zhou Q. (2007) Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res. 35, 2003–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schröder S., Cho S., Zeng L., Zhang Q., Kaehlcke K., Mak L., Lau J., Bisgrove D., Schnölzer M., Verdin E., Zhou M. M., Ott M. (2012) Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J. Biol. Chem. 287, 1090–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krueger B. J., Varzavand K., Cooper J. J., Price D. H. (2010) The mechanism of release of P-TEFb and HEXIM1 from the 7SK snRNP by viral and cellular activators includes a conformational change in 7SK. PLoS ONE 5, e12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang Z., Yik J. H., Chen R., He N., Jang M. K., Ozato K., Zhou Q. (2005) Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 19, 535–545 [DOI] [PubMed] [Google Scholar]
- 14. Hargreaves D. C., Horng T., Medzhitov R. (2009) Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138, 129–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lenasi T., Contreras X., Peterlin B. M. (2008) Transcriptional interference antagonizes proviral gene expression to promote HIV latency. Cell Host Microbe 4, 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Contreras X., Schweneker M., Chen C. S., McCune J. M., Deeks S. G., Martin J., Peterlin B. M. (2009) Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J. Biol. Chem. 284, 6782–6789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Contreras X., Barboric M., Lenasi T., Peterlin B. M. (2007) HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog. 3, 1459–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W. B., Fedorov O., Morse E. M., Keates T., Hickman T. T., Felletar I., Philpott M., Munro S., McKeown M. R., Wang Y., Christie A. L., West N., Cameron M. J., Schwartz B., Heightman T. D., La Thangue N., French C. A., Wiest O., Kung A. L., Knapp S., Bradner J. E. (2010) Selective inhibition of BET bromodomains. Nature 468, 1067–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Delmore J. E., Issa G. C., Lemieux M. E., Rahl P. B., Shi J., Jacobs H. M., Kastritis E., Gilpatrick T., Paranal R. M., Qi J., Chesi M., Schinzel A. C., McKeown M. R., Heffernan T. P., Vakoc C. R., Bergsagel P. L., Ghobrial I. M., Richardson P. G., Young R. A., Hahn W. C., Anderson K. C., Kung A. L., Bradner J. E., Mitsiades C. S. (2011) BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smith E., Lin C., Shilatifard A. (2011) The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 25, 661–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martincic K., Alkan S. A., Cheatle A., Borghesi L., Milcarek C. (2009) Transcription elongation factor ELL2 directs immunoglobulin secretion in plasma cells by stimulating altered RNA processing. Nat. Immunol. 10, 1102–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Biglione S., Byers S. A., Price J. P., Nguyen V. T., Bensaude O., Price D. H., Maury W. (2007) Inhibition of HIV-1 replication by P-TEFb inhibitors DRB, seliciclib, and flavopiridol correlates with release of free P-TEFb from the large, inactive form of the complex. Retrovirology 4, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nelson J. D., Denisenko O., Bomsztyk K. (2006) Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc. 1, 179–185 [DOI] [PubMed] [Google Scholar]
- 24. Mertz J. A., Conery A. R., Bryant B. M., Sandy P., Balasubramanian S., Mele D. A., Bergeron L., Sims R. J., 3rd (2011) Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. U.S.A. 108, 16669–16674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Antoni B. A., Rabson A. B., Kinter A., Bodkin M., Poli G. (1994) NF-κB-dependent and -independent pathways of HIV activation in a chronically infected T cell line. Virology 202, 684–694 [DOI] [PubMed] [Google Scholar]
- 26. Nicodeme E., Jeffrey K. L., Schaefer U., Beinke S., Dewell S., Chung C. W., Chandwani R., Marazzi I., Wilson P., Coste H., White J., Kirilovsky J., Rice C. M., Lora J. M., Prinjha R. K., Lee K., Tarakhovsky A. (2010) Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. He N., Pezda A. C., Zhou Q. (2006) Modulation of a P-TEFb functional equilibrium for the global control of cell growth and differentiation. Mol. Cell. Biol. 26, 7068–7076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ai N., Hu X., Ding F., Yu B., Wang H., Lu X., Zhang K., Li Y., Han A., Lin W., Liu R., Chen R. (2011) Signal-induced Brd4 release from chromatin is essential for its role transition from chromatin targeting to transcriptional regulation. Nucleic Acids Res. 39, 9592–9604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. He N., Liu M., Hsu J., Xue Y., Chou S., Burlingame A., Krogan N. J., Alber T., Zhou Q. (2010) HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 38, 428–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jang M. K., Mochizuki K., Zhou M., Jeong H. S., Brady J. N., Ozato K. (2005) The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 19, 523–534 [DOI] [PubMed] [Google Scholar]
- 31. Mueller D., García-Cuéllar M. P., Bach C., Buhl S., Maethner E., Slany R. K. (2009) Misguided transcriptional elongation causes mixed lineage leukemia. PLoS Biol. 7, e1000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takahashi H., Parmely T. J., Sato S., Tomomori-Sato C., Banks C. A., Kong S. E., Szutorisz H., Swanson S. K., Martin-Brown S., Washburn M. P., Florens L., Seidel C. W., Lin C., Smith E. R., Shilatifard A., Conaway R. C., Conaway J. W. (2011) Human mediator subunit MED26 functions as a docking site for transcription elongation factors. Cell 146, 92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Luo Z., Lin C., Guest E., Garrett A. S., Mohaghegh N., Swanson S., Marshall S., Florens L., Washburn M. P., Shilatifard A. (2012) The super elongation complex family of RNA polymerase II elongation factors: gene target specificity and transcriptional output. Mol. Cell. Biol. 32, 2608–2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen R., Liu M., Li H., Xue Y., Ramey W. N., He N., Ai N., Luo H., Zhu Y., Zhou N., Zhou Q. (2008) PP2B and PP1α cooperatively disrupt 7SK snRNP to release P-TEFb for transcription in response to Ca2+ signaling. Genes Dev. 22, 1356–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sung T. L., Rice A. P. (2009) miR-198 inhibits HIV-1 gene expression and replication in monocytes, and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 5, e1000263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Banerjee C., Archin N., Michaels D., Belkina A. C., Denis G. V., Bradner J., Sebastiani P., Margolis D. M., Montano M. (2012) BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukoc. Biol. 92, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dawson M. A., Kouzarides T. (2012) Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27 [DOI] [PubMed] [Google Scholar]
- 38. Wittmann B. M., Fujinaga K., Deng H., Ogba N., Montano M. M. (2005) The breast cell growth inhibitor estrogen-down-regulated gene 1 modulates a novel functional interaction between estrogen receptor α and transcriptional elongation factor cyclin T1. Oncogene 24, 5576–5588 [DOI] [PubMed] [Google Scholar]
- 39. Wittmann B. M., Wang N., Montano M. M. (2003) Identification of a novel inhibitor of breast cell growth that is down-regulated by estrogens and decreased in breast tumors. Cancer Res. 63, 5151–5158 [PubMed] [Google Scholar]
- 40. Zhao R., Nakamura T., Fu Y., Lazar Z., Spector D. L. (2011) Gene bookmarking accelerates the kinetics of post-mitotic transcriptional reactivation. Nat. Cell Biol. 13, 1295–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]