

Abstract
Primary cultures of rat and murine hippocampal neurons are widely used to reveal cellular mechanisms in neurobiology. By isolating and growing individual neurons, researchers are able to analyze properties related to cellular trafficking, cellular structure and individual protein localization using a variety of biochemical techniques. Results from such experiments are critical for testing theories addressing the neural basis of memory and learning. However, unambiguous results from these forms of experiments are predicated on the ability to grow neuronal cultures with minimum contamination by other brain cell types. In this protocol, we use specific media designed for neuron growth and careful dissection of embryonic hippocampal tissue to optimize growth of healthy neurons while minimizing contaminating cell types (i.e. astrocytes). Embryonic mouse hippocampal tissue can be more difficult to isolate than similar rodent tissue due to the size of the sample for dissection. We show detailed dissection techniques of hippocampus from embryonic day 19 (E19) mouse pups. Once hippocampal tissue is isolated, gentle dissociation of neuronal cells is achieved with a dilute concentration of trypsin and mechanical disruption designed to separate cells from connective tissue while providing minimum damage to individual cells. A detailed description of how to prepare pipettes to be used in the disruption is included. Optimal plating densities are provided for immuno-fluorescence protocols to maximize successful cell culture. The protocol provides a fast (approximately 2 hr) and efficient technique for the culture of neuronal cells from mouse hippocampal tissue.
Keywords: Neuroscience, Issue 65, Physiology, Medicine, Brain, Cell Culture, Hippocampal Neurons
Protocol
1. Set-up Prior to Harvest
To generate prenatal pups for neuron harvest, schedule breeding between adult mice 19 days prior to the day of neuron isolation. (C57BL/6 mice ages 2-8 months were used in matings for the purposes of developing this protocol). Successful mating can be confirmed by detection of a vaginal plug in the female, palpitation or visual confirmation of pregnancy.
- The day prior to neuron isolation:
- For immunofluorescence applications, coat glass coverslips in a 24-well plate with a light coating of 3:1 Collagen 1, Rat Tail:poly-D-Lysine solution.
- For cell culture applications, coat appropriate size tissue culture plastic ware with a light coating of 3:1 Collagen 1, Rat Tail:poly-D-Lysine solution.
Rest the plates uncovered in a tissue culture hood under a UV light overnight.
Wash the plates with sterile Hank's Balance Salt Solution (HBSS) prior to use. Coated plates can be filled with HBSS and stored up to one week at 4 °C in the dark.
2. Tissue Harvest
Sterility is always a factor when growing primary cell cultures and as such, the greatest caution should be exercised to ensure the most sterile environment possible. With careful attention to sterile technique, initial dissection and harvest of neural tissue for this protocol can be completed outside of a laminar flow hood with minimal risk of contamination. After initial harvest, all subsequent steps should be conducted under maximum sterile conditions within a hood rated for cell culture.
Euthanize a pregnant mouse at approximately 19 days post-fertilization by decapitation. Use of anesthesia to euthanize the pregnant female is not recommended as anesthesia is known to cause brain cell death (Stratmann, et al., 2010). Using sterile dissecting scissors and forceps, create an opening in the mid-ventral side of the mouse to completely reveal body cavity. Instruments can be sterilized using alcohol and an open flame.
Prenatal pups will be located towards the posterior of the mouse's body cavity and should be easily visible in the uterus. With autoclaved sterile forceps, open the uterus and remove pups. Decapitate pups with fresh sterile scissors and place removed head on sterile gauze under a dissecting microscope. Sterile, autoclaved instruments can be flame cleaned using alcohol and an open flame prior to and during use.
Using sterile scissors or scalpel, open cranium of pup from back of the neck to the nose. This procedure can normally be completed by inserting one tip of the scissors into the vertebral foramen and then proceeding anteriorly. Carefully remove the entire brain with forceps. Place the brain on sterile gauze. Using a sterile scalpel, remove the cerebellum and incise down the midline of the brain to separate it into two hemispheres (Figure 1).
Grasp a small section of meninges surrounding the hippocampus with sterile forceps and pull it gently away. Although it is not strictly necessary to remove the meninges before isolating the hippocampus, the presence of the meninges can make the dissection of the hippocampus more difficult due to the toughness of the membrane. In either case, the hippocampus will be more clearly visible after the meninges have been removed. The hippocampus is a curved structure that starts in the distal part of the hemisphere and bends ventrally (Figure 2). As the inner, concave, side (caudal) is facing a ventricle, it is already free. Therefore, to isolate the hippocampus, one needs to cut along the convex outer side. After the dissection, gently lift each hippocampi with sterile tissue forceps and transfer into a small tissue culture dish with warmed (37 °C) HBSS under a cell culture hood. Brain tissue can be combined from multiple pups.
3. Tissue Dissociation
Using a sterile scalpel, gently mince brain tissue in 3 ml of sterile HBSS in a 100 mm Tissue Culture dish.
Transfer the minced tissue and HBSS to a 15 ml conical tube. Add 1.5 ml of HBSS and 0.5 ml of 0.25% Trypsin solution to a total volume of 5 ml.
Cap and gently invert the tube 4-5 times to mix. Try to avoid producing bubbles as DNA released from the digested tissue will adhere to the bubbles and cause the minced tissue to float instead of settling to the bottom of the tube (Figure 3).
Incubate hippocampal tissue at 37 °C for 15 minutes, inverting tube as above every 5 minutes.
Allow the tissue to settle to the bottom of the tube.
Carefully remove excess solution using a sterile pipette, leaving tissue undisturbed at the bottom of the tube.
Wash tissue pellet with 5 ml of HBSS at 37 °C for 5 minutes. Repeat a total of 3 times. Allow tissue to completely settle to the bottom of the tube each time before proceeding to the next wash step.
Remove the final wash from the tissue pellet and replace with 2 ml of fresh HBSS.
4. Neuron Trituration
Before beginning trituration steps, you will need to prepare fire polished Pasteur pipettes. Using a Bunson burner, hold a sterile 9 inch Pasteur pipette tip (Figure 4a) in the flame until diameter of the pipette opening is approximately 0.5 mm in size and edges of the pipette opening have been rounded slightly (Figure 4b). Allow pipette to completely cool before beginning the trituration process.
Using a normal sterile 9 inch Pasteur pipette, gently triturate the tissue a total of 7 times. Larger tissue pieces are normal at this point and should be allowed to settle to the bottom of the tube prior to moving to the next step.
Transfer the supernatant to a fresh sterile 50 ml conical tube.
To the remaining tissue, add 2 ml of sterile HBSS and triturate a total of 5 times using the fire polished Pasteur pipette.
Allow all remaining larger tissue pieces to settle to the bottom of the tube and combine supernatant with the previous supernatant for a total of 4 ml dissociated neuronal cells.
5. Cell Plating
Count the dissociated cells using a hemocytometer.
As a general rule, once cell numbers have been determined, subtract 20% from that final number to account for any cell death that may occur after plating.
Cells can be plated using the recommendations below: For coverslips in a 24 well plate - 6 x 104 cells/well in 0.5 ml For 60 mm Tissue Culture plates - 4 x 105 cells/plate in 3 ml For 100 mm Tissue Culture plates - 6 x 106 cells/plate in 6 ml
Mix appropriate cell numbers with indicated volume of Neurobasal Plating Media (Neurobasal Media containing B27 Supplement [1 ml / 50 ml], 0.5 mM Glutamine Solution, 25 μM Glutamate (Mr 147.13 g / mol), Penicillin (10,000 units / ml)/Streptomycin (10,000 μg / ml) [250 μl / 50 ml], 1mM HEPES (Mr 238.3 g / mol), 10% Heat Inactivated Donor Horse Serum) and add cells to plates. Swirl plates gently to distribute cells evenly. HI-Donor Horse Serum is added to the Plating Media to enrich the cells during the first 24 hours of growth. The cells are subsequently weaned from the serum and returned to a Serum-Free environment by serial reduction of the serum at each media replacement. It should also be noted that while glutamate at higher concentrations is toxic to neuronal cell cultures, at the lower concentrations added here, it will inhibit the attachment of non-neuronal cells11. However, it should only be added to the plating media for the first 24 hours in culture and subsequently left out of any Feeding Media to prevent neurotoxicity of the cells.
Place neurons in a 37 °C, 5% CO2 incubator overnight.
Remove half the volume of media from the cells and replace with same volume of Neurobasal Feeding Media (Neurobasal Media containing B27 Supplement [1 ml / 50 ml], 0.5 mM Glutamine Solution, Penicillin (10,000 units / ml) / Streptomycin (10,000 μg / ml) [250 μl / 50 ml], 1 mM HEPES (Mr 238.3 g / mol).
Neurons should be fed every 4 days by removing half of the old media and replacing it with the same volume of fresh Neurobasal Feeding media. Neuronal processes should begin to be visible on Day 1 (Figure 5a) and become prevalent by Day 10 (Figure 5b).
6. Representative Results
The ability to grow and culture primary neuronal cells has become an indispensible part of neuroscience. Primary cultures allow the researcher to analyze specific cellular pathways, chemical modification and treatment, target localization and growth patterns in a controlled environment. Many of these procedures utilize sophisticated methodology to visualize specific changes in cell responses. In this case, hippocampal neurons are used to study specific neuronal pathways that would prove difficult, if not impossible to analyze in the intact brain. Preparation of near homogeneous populations of neurons from specific areas of the brain is critical for studying brain function. Molecular effects in individual neurons can be instrumental in delineating higher order pathways such as memory or learning. As this protocol yields relatively pure cultures of hippocampal neurons, without the need of a feeder layer of glial cells, these neurons are easily utilized for immunofluorescence studies. However, as with all primary culture from organs containing multiple cell types, some contamination by less desired cells can occur. In isolation of neuronal cells, contamination by glial cells can be a common problem. Glial cells can be easily detected upon microscopic visualization of the culture as their morphology differs significantly from the target neurons (Figure 6). The impact of glial cell contamination will depend on the planned use of the cultures. If cells are being used for immuno-fluorescence examination, glial contamination can be nothing more than an inconvenience when trying to photograph individual neurons. However, if the neuronal cultures are to be used for biochemical analysis, any significant contamination by glial cells could cause major changes in the results. Ways to address glial cell contamination are outlined further in the Discussion.
Once neurons have been successfully isolated and grown in culture, one typical application is to examine cellular processes immuno-fluorescence techniques. As illustrated in Figure 7, organelles, such as the mitochondria, can be stained using vital dyes added to the culture media prior to fixation. Endogenous cellular proteins can be visualized from fixed cells using standard immuno-fluorescence techniques (Figure 8). Once neuronal cells are fixed, specific antibodies for proteins of interest can be introduced to the cell and these proteins can be visualized using a fluorescence microscope. Cultured neurons also provide the researcher with the means to examine individual protein effects on neuronal functions. Using a variety of techniques including DNA transfections, electroporation or viral transduction, proteins can be overexpressed in neuronal cells (Figure 9). How neural cells respond to the effects of over-expressed proteins can have direct inferences on how the brain may respond and offers the possibility of identifying cellular targets for drug treatments. The details of these types of experiments go beyond the scope of this paper but they do illustrate that cultures prepared by this technique are suitable for a wide array of down-stream applications. However, the overall simplicity of this protocol, as well as, the short time period required to prepare these neuronal cultures make this an ideal method for use in today's neuroscience laboratory.
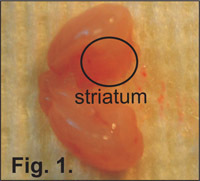 Figure 1. Dissection of the prenatal mouse brain. The first incision is down the midline of the brain separating it into two hemispheres.
Figure 1. Dissection of the prenatal mouse brain. The first incision is down the midline of the brain separating it into two hemispheres.
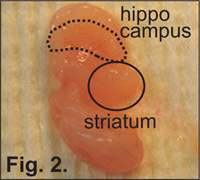 Figure 2. Location of the hippocampus in the prenatal mouse brain. The striatum is moved aside to visualize the hippocampus and is noted by the curved "kidney bean" type structure in the distal region of each hemisphere.
Figure 2. Location of the hippocampus in the prenatal mouse brain. The striatum is moved aside to visualize the hippocampus and is noted by the curved "kidney bean" type structure in the distal region of each hemisphere.
 Figure 3. Dissociation of hippocampal tissue in trypsin solution.
Figure 3. Dissociation of hippocampal tissue in trypsin solution.
 Figure 4. Pasteur pipette tips used in trituration of hippocampal tissue. (a) Normal Pasteur pipette tip. (b) Fire-polished Pasteur pipette tip. Take note of the rounded edges and the approximate 50% decrease in pipette opening size.
Figure 4. Pasteur pipette tips used in trituration of hippocampal tissue. (a) Normal Pasteur pipette tip. (b) Fire-polished Pasteur pipette tip. Take note of the rounded edges and the approximate 50% decrease in pipette opening size.
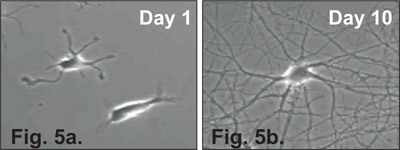 Figure 5. Hippocampal neurons isolated using this procedure and plated in NB Media. (a) Cell growth 1 day post-plating. Neuronal processes begin to be visible during Day 1. (b) Cell growth 10 day post-plating, neurites are branched and overlapping.
Figure 5. Hippocampal neurons isolated using this procedure and plated in NB Media. (a) Cell growth 1 day post-plating. Neuronal processes begin to be visible during Day 1. (b) Cell growth 10 day post-plating, neurites are branched and overlapping.
 Figure 6. Hippocampal neurons contaminated with glial cells grown for 7 days and stained with the organelle marker MitoTracker Red CM-H2XRos (Invitrogen #M7515) and transfected with GFP-LC3 using Lipofectamine 2000 (Invitrogen #11668019). Mitochondria are visible in all cells however only a single neuron was successfully transfected with the fluorescent construct. Contamination with glial cells makes analysis of GFP-LC3 expression in the neuronal processes difficult to visualize.
Figure 6. Hippocampal neurons contaminated with glial cells grown for 7 days and stained with the organelle marker MitoTracker Red CM-H2XRos (Invitrogen #M7515) and transfected with GFP-LC3 using Lipofectamine 2000 (Invitrogen #11668019). Mitochondria are visible in all cells however only a single neuron was successfully transfected with the fluorescent construct. Contamination with glial cells makes analysis of GFP-LC3 expression in the neuronal processes difficult to visualize.
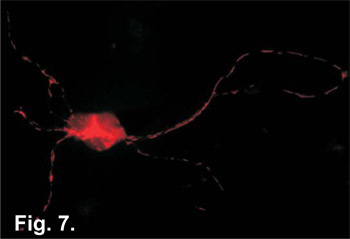 Figure 7. Hippocampal neurons grown for 7 days and stained with the organelle marker MitoTracker Red CM-H2 XRos (Invitrogen #M7513). This vital dye is used to stain active mitochondria in tissue culture cells. The cells were fixed in 4% paraformaldehyde/PBS and visualized by fluorescent microscopy. The dye itself is non-fluorescent until oxidized in the mitochondria. Active mitochondria can be seen throughout the neuronal processes.
Figure 7. Hippocampal neurons grown for 7 days and stained with the organelle marker MitoTracker Red CM-H2 XRos (Invitrogen #M7513). This vital dye is used to stain active mitochondria in tissue culture cells. The cells were fixed in 4% paraformaldehyde/PBS and visualized by fluorescent microscopy. The dye itself is non-fluorescent until oxidized in the mitochondria. Active mitochondria can be seen throughout the neuronal processes.
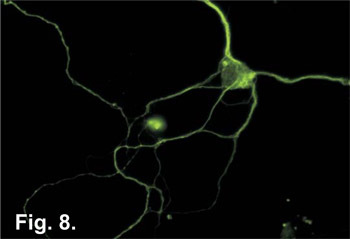 Figure 8. Hippocampal neurons grown for 7 days, fixed with 4% paraformaldehyde/PBS and immuno-stained with monoclonal anti-tubulin β antibody (Sigma #T0198). Following primary antibody, Oregon Green labeled goat-anti-mouse secondary antibody (Invitrogen #O11033) was added and fluorescence visualized by microscopy.
Figure 8. Hippocampal neurons grown for 7 days, fixed with 4% paraformaldehyde/PBS and immuno-stained with monoclonal anti-tubulin β antibody (Sigma #T0198). Following primary antibody, Oregon Green labeled goat-anti-mouse secondary antibody (Invitrogen #O11033) was added and fluorescence visualized by microscopy.
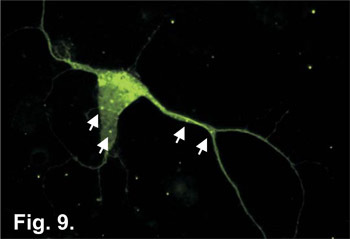 Figure 9. Hippocampal neuronal cultures were grown for 5 days and transfected with GFP-LC3β DNA construct using Lipofectamine 2000 (Invitrogen #11668019). At Day 7, cells were fixed using 4% paraformaldehyde/PBS and aggresomes with GFP tagged LC3β incorporated into their outer membrane were visualized using fluorescent microscopy. Aggresomes are located throughout the cell body and neurites and are denoted with arrows.
Figure 9. Hippocampal neuronal cultures were grown for 5 days and transfected with GFP-LC3β DNA construct using Lipofectamine 2000 (Invitrogen #11668019). At Day 7, cells were fixed using 4% paraformaldehyde/PBS and aggresomes with GFP tagged LC3β incorporated into their outer membrane were visualized using fluorescent microscopy. Aggresomes are located throughout the cell body and neurites and are denoted with arrows.
Discussion
Hippocampal cultures have been used in molecular biology for more than 20 years. While in principle, neuronal cultures can be made from any part of the brain, hippocampal cultures have proven to be the most popular due to the relatively simple architecture of the nerve cell population in the hippocampus7. Hippocampal cultures are typically made from late-stage embryonic tissue. This tissue is easier to dissociate and contains fewer glial cells than does mature brain tissue1. Isolation of hippocampal neurons from embryonic tissue also decreases shearing damage to axons and dendrites due to fewer adhesion contacts3. While hippocampal cultures are most often generated from rats due to the relatively easier isolation of the hippocampus, mice can also be used with the same protocols if appropriate care is taken during tissue isolation. Once neurons are cultured, the ability to use advanced molecular techniques to analyze subcellular localization and trafficking can be employed. This can be especially advantageous when analyzing embryonic lethal transgenic mice as it provides the ability to study protein interactions that would result in the death of the embryo.
The hippocampus has been implicated in both spatial and contextual learning5 and memory6. Growth of primary cultures from the hippocampus can allow a correlation between subcellular biological events and their effects on the brain's ability to learn and remember.
As with all neural cells, neurons grown from hippocampal cultures require critical growth factors, hormones and amino acids. In the brain, these factors are provided by glial cells. This symbiotic relationship can also be carried into a culture environment by growing a "feeder" layer of glial cells along with the cultured neurons. However, glial cells will also produce cytotoxic factors during their lifespan11 which can be toxic to cultured neurons. To circumvent this, neurons have been grown in serum-free media such as Neurobasal medium supplemented with B27. The B27 supplement is optimized for survival of hippocampal neurons but will support growth of other neuronal cultures as well4. L-glutamine is an essential amino acid for energy production and protein synthesis in cell culture. However, glutamine can be labile over time, degrading into ammonia and carboxylic acid byproducts once added to culture media. Glutamax, a cell culture supplement from Invitrogen, can be used as a direct substitute for L-glutamine if desired. Glutamax is more stable in media but slightly more expensive. Growth of neurons in serum-free media allows the study of effects of growth factors and hormones on neuronal growth and differentiation.
We submit a protocol for the rapid isolation of hippocampal neurons from mouse prenatal embryos using Neurobasal media and the B27 supplement for growth of neurons in a serum free environment without the use of feeder cells. As with all cultured primary cell protocols, it is advantageous to minimize the growth of non-desired cell types (i.e. glial cells). This is most readily accomplished by careful dissection of the hippocampus from surrounding regions of the brain. Prenatal cells also contain a comparatively small number of glial cells aiding in this goal. However, glial cell contamination can still occur. It is possible to reduce glial cell contamination by treatment with cytosine arabinoside (AraC) at early time points in the culture8,9. AraC can be used as an anti-mitotic agent to reduce the population of non-neuronal cells capable of DNA synthesis. However, to avoid possible toxic effects of treatment on neurons, it should be used at its lowest effective does (5 μM) and not added after 3-4 days of culture11. Another option would be the use of 5-fluoro-2'-deoxyuridine (FUdR) treatment to decrease the proportion of fibroblastic-reactive microglial cells10.
Once harvested, hippocampal tissue can be treated with a dilute trypsin solution to dissociate/disaggregate adherent cells. However, prolonged exposure to higher concentrations of trypsin can be detrimental to cell subculture so time in trypsin solution is most often limited to between 3-5 minutes. The diluted concentration of trypsin used in this protocol does allow longer times of enzyme incubation to increase individual cellular disassociation but care should be taken to strictly adhere to the time points provided. Papain can be used as an alternative enzyme and has been proven to be more effective and less destructive with certain tissues such as retinal neurons 10.
Cell dissociation is followed by trituration of the tissue. This has proven to be the most important step in consistent neuronal culture and is highlighted by two important points. First, two sterile Pasteur pipettes are used during this process. The first is used to grossly disrupt tissue/cellular association. The opening size of this first pipette should be between 1.0-1.5 mm. Careful selection of pipettes directly from the vendor package can usually result in appropriate pipettes for this first trituration. The second Pasteur pipette used is "fire-polished" over a Bunsen burner to reduce the size of the opening to approximately 0.5 mm in diameter. This also results in "polishing" the glass ring of the opening to a smoother surface reducing mechanical damage to cells as they pass through. Secondly, the speed/force of trituration through the pipette is of major importance. As the cells dissociate from the whole tissue, they are of course more fragile. Thus, "rough" treatment as this point would be detrimental to survival of the neuronal cells. Trypsinized tissue should be passaged through the pipette at a consistent but firm flow rate. A common mistake by the researcher is the idea of having to completely disassociate the tissue until there is no discernible structure remaining. In most cases, this will result in massive neuronal death as the cells will be too damaged by the repeated mechanical manipulation. The indicated number of times to pass the tissue through the pipette will result in sufficient numbers of neurons without resulting in gross damage to the population.
Once the cells have been dissociated and pooled, a Trypan Blue stain of an aliquot of cells will provide a ratio of live to dead cells. Typically, 75-80% of the cells should survive the harvest process and can be used for plating. Trypan Blue staining can also be used to obtain an accurate cell count when done on a hemocytometer. In practice, once a total cell count is achieved, add 20% to the total and use that cell number for plating to account for the approximate amount of cell death. Numbers provided in the above protocol are a good starting point to achieve good densities of neurons. If neuron density is too low at plating, cell growth will likely not be sufficient as plating density is an important variable in propagation of the cell population. Cells should be fed every four days with B27/Neurobasal Feed media. Neuronal growth under these conditions can easily be carried out to 10-14 days in culture and has been used to propagate neurons2 out to 30 days in culture when seeded at 80 cells / mm2.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank Dr. Michael Wooten for his help in preparing the manuscript. This work was supported by NIH 2RO1NS033661 (MWW).
References
- Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–442. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Serum-free B27/neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum, and dentate gyrus. J. Neurosci. Res. 1995;42:674–683. doi: 10.1002/jnr.490420510. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Isolation and culture of adult rat hippocampal neurons. J. Neurosci. Methods. 1997;71:143–155. doi: 10.1016/s0165-0270(96)00136-7. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J. Neurosci. Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Burwell RD, Saddoris MP, Bucci DJ, Wiig KA. Corticohippocampal contributions to spatial and contextual learning. J. Neurosci. 2004;24:3826–3836. doi: 10.1523/JNEUROSCI.0410-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluck MA, Myers C, Meeter M. Cortico-hippocampal interaction and adaptive stimulus representation: a neurocomputational theory of associative learning and memory. Neural Netw. 2005;18:1265–1279. doi: 10.1016/j.neunet.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat. Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Mao L, Wang JQ. Gliogenesis in the striatum of the adult rat: alteration in neural progenitor population after psychostimulant exposure. Brain Res. Dev. Brain Res. 2001;130:41–51. doi: 10.1016/s0165-3806(01)00195-x. [DOI] [PubMed] [Google Scholar]
- Mao L, Wang JQ. Upregulation of preprodynorphin and preproenkephalin mRNA expression by selective activation of group I metabotropic glutamate receptors in characterized primary cultures of rat striatal neurons. Brain Res. Mol. Brain Res. 2001;86:125–137. doi: 10.1016/s0169-328x(00)00276-x. [DOI] [PubMed] [Google Scholar]
- Oorschot DE. Effect of fluorodeoxyuridine on neurons and non-neuronal cells in cerebral explants. Exp. Brain Res. 1989;78:132–138. doi: 10.1007/BF00230692. [DOI] [PubMed] [Google Scholar]
- Price PJ, Brewer GJ. Serum -free media for neural cell cultures. In: Federoff S, Richardson A, editors. Protocols for Neural Cell Culture. Humana Press; 2001. [Google Scholar]
- Shen J, Watanabe S, Kaneko A. Cell dissociation with papain reduces the density of cGMP-activated channels of the retinal rod. Jpn. J. Physiol. 1995;45:151–164. doi: 10.2170/jjphysiol.45.151. [DOI] [PubMed] [Google Scholar]
- Stratmann G, Sall JW, May LD, Loepke AW, Lee MT. Beyond anesthetic properties: The effects of isoflurane on brain cell death, neurogenesis and long-term neurocognitive function. Anesthesia and Analgesia. 2009;110:431–437. doi: 10.1213/ANE.0b013e3181af8015. [DOI] [PubMed] [Google Scholar]
- Wallace TL, Johnson EM. Cytosine arabinoside kills postmitotic neurons: evidence that deoxycytidine may have a role in neuronal survival that is independent of DNA synthesis. J. Neurosci. 1989;9:115–124. doi: 10.1523/JNEUROSCI.09-01-00115.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]