

Abstract
Status epilepticus is a clinical emergency defined as continuous seizure activity or rapid, recurrent seizures without regaining consciousness and can lead to the development of acquired epilepsy, characterized by spontaneous, recurrent seizures. Understanding epileptogenesis – the transformation of healthy brain tissue into hyperexcitable neuronal networks – is an important challenge and the elucidation of molecular mechanisms can lend insight into new therapeutic targets to halt this progression. It has been demonstrated that intracellular calcium increases during status epilepticus and that these elevations are maintained past the duration of the injury (Ca2+ plateau). As an important second messenger, Ca2+ elevations can lead to changes in gene expression, neurotransmitter release and plasticity. Thus, characterization of the post-injury Ca2+ plateau may be important in eventually understanding the pathophysiology of epileptogenesis and preventing the progression to chronic epilepsy after brain injury.
Keywords: acquired epilepsy, calcium homeostasis, epileptogenesis, hippocampal neuronal culture, intracellular calcium, Sprague-Dawley rat, status epilepticus
Status epilepticus & acquired epilepsy
Acquired epilepsy (AE) is a neurological condition characterized by the development of spontaneous, recurrent seizures following a CNS injury, such as status epilepticus (SE), stroke or traumatic brain injury (TBI) [1,2]. The process by which healthy brain tissue is transformed into a hyperexcitable circuit of neurons giving rise to seizures is called epileptogenesis [3,4]. In humans, it has proven challenging to determine whether SE is a common etiology behind AE owing to the simultaneous presentation of SE with injuries such as stroke or TBI. Furthermore, in a clinical setting, it is difficult to determine whether SE is the first presentation of epilepsy or the inciting injury that leads to epileptogenesis. Despite these difficulties in delineating one primary cause of epileptogenesis, it is evident that AE is a common occurrence following SE, with 20–40% of all cases of SE developing this chronic condition [4,5]. Thus, experimental models of SE-induced AE remain the most widely studied and best characterized examples of epileptogenesis. Ultimately, the AE produced in these models closely resembles the pathology observed in humans and has provided tremendous insight into the molecular mechanisms of epileptogenesis. Understanding the pathophysiological mechanisms through which SE causes the development of AE would lend insight into mechanisms of neuronal plasticity and also aid in the elucidation of new therapeutic targets to prevent epileptogenesis. This review focuses on the role of SE in causing AE and the associated mechanisms underlying this process.
Status epilepticus had been defined as a medical emergency constituting convulsive or nonconvulsive seizures lasting for at least 30 min, or multiple seizures without regaining consciousness lasting the same duration of time [6,7]. This particular definition was reached following reports in experimental animal models that correlated seizure duration with degree of permanent brain injury; after 30 min, animal data indicated significant declines in brain oxygenation and cardiovascular distress. However, this definition has been revisited because, from a treatment standpoint, after 5 min of ongoing seizures there is a high likelihood that the seizure will continue for at least 30 min and become increasingly difficult to treat [8]. In the USA, the frequency of SE is approximately 100,000–150,000 cases annually, with almost 55,000 deaths per year [9]. Various processes can cause SE including metabolic disorders, drug toxicity, head trauma, hypoxia and stroke. Often chronic conditions such as pre-existing epilepsy, alcohol abuse or CNS tumors can also precipitate SE [9-12]. Therefore, experimental models are needed to study SE and AE following SE to better understand mechanisms and develop treatments.
Models of SE-induced AE provide important tools to study epileptogenesis
There are several different models utilized to study SE-induced AE. The development of these models has made SE-induced AE more accessible to study than AE caused by other brain injuries, such as stroke and TBI. Moreover, models of AE following SE have provided insight into general mechanisms of neuronal injury [13]. Chemoconvulsants, such as pilocarpine and kainic acid, as well as prolonged hippocampal stimulation leading to the spontaneous development of epileptiform activity, are most commonly used to induce SE in animals [14,15]. The animal models that utilize chemicals such as pilocarpine follow a progression that correlates closely with the clinical presentation of SE in humans. Following SE-induced brain injury, there is a latency period characterized by a lack of seizure activity [1,16,17]. This period is then followed by the development of spontaneous, recurrent seizures. The progression from SE to AE is, therefore, often divided into three stages: induction or injury (the episode of SE), a latent period of epileptogenesis, and finally the development and chronic occurrence of AE [16,17]. The animal models also share several other characteristics with the human condition. In the pilocarpine model, similarities including hippocampal neuronal death, astrocyte proliferation and microglial activation are observed [15,18]. Although the in vivo models have the advantage of representing the clinical presentation in an intact animal, they also provide challenges in studying the mechanisms of epileptogenesis, since it is difficult to control many of the variables needed to conduct molecular biological studies in these complex, multidimensional systems. Thus, the development of an in vitro model of epileptogenesis has proven to be a useful addition to understanding this clinical condition.
Our laboratory has developed an in vitro cell culture model of SE and SE-induced AE [19]. This model offers a powerful tool to evaluate potential molecular mechanisms underlying epileptogenesis, but has the disadvantage of not representing clinical epilepsy in an intact brain. The in vitro model utilizes a buffer solution with no added magnesium (Mg2+), referred to as ‘low Mg2+’ to cause in vitro SE in cultured hippocampal neurons plated on glial beds. These cultures are treated with low Mg2+ for 3 h during which time they manifest continuous epileptiform discharges with a frequency greater than 3 Hz, consistent with the frequency and characteristics of seizure activity observed in clinical, electrographic SE. When the neurons are returned to normal, Mg2+-containing maintenance media and patched using whole-cell current clamp electrophysiology, they no longer manifest SE-like continuous epileptiform discharges and, instead, show only occasional depolarization and short epileptiform discharges. By 12 h post-low Mg2+, the neurons consistently exhibit spontaneous, recurrent epileptiform discharges (SREDs), characterized by bursts of action potentials over-riding paroxysmal depolarization shifts. At this and all latter time points, between six and nine distinct SRED episodes are observed per hour. These SREDs meet the electrophysiological characteristics of seizure discharges observed in patients exhibiting seizure activity. The SREDs persist for the life of the neurons in culture and are synchronous events between networked neurons. Thus, this model utilizes an inciting injury that resembles SE to cause the subsequent development of SREDs, an in vitro correlate to AE. The model exhibits several features consistent with the development of AE in both in vivo models and the clinical condition, including pharmacoresistance to anticonvulsants, cell death, homeostatic changes in Ca2+ and spike frequency [19,20]. Moreover, the cell injury observed in the in vitro and in vivo models correspond to that seen in clinical studies documenting the relationship between SE and neuronal injury [21-23].
The low Mg2+ in vitro model and the in vivo models of SE-induced AE are useful tools to study potential molecular mechanisms underlying the induction of AE; however, it is important to note that there are several limitations in these models. Clear differences between the experimental models and the human condition include: duration of latency between SE and the development of epilepsy, percentage of SE cases that develop epilepsy, and comorbidities associated with the pathology [24]. Yet, despite these differences, these models have provided much insight into the molecular mechanisms of epileptogenesis and have been useful tools in the elucidation of new therapeutic modalities.
Ca2+ dynamics in SE-induced AE
Both in vivo and in vitro models of SE-induced AE have provided tremendous insights into the molecular and cellular changes that occur during and following SE. This research is important since the characterization of mechanisms that cause AE after brain injuries has significant implications on the development of new treatments to prevent the progression to AE. Recently, several studies have highlighted the novel observation that intracellular Ca2+ dynamics change during an injury such as SE, stroke or TBI [25-27]. The importance of altered Ca2+ dynamics is largely based on Ca2+ being an important divalent, cationic charge carrier that serves as a second messenger in cell signaling [28]. Under standard conditions, extracellular Ca2+ concentration is significantly higher than intracellular Ca2+ concentration ([Ca2+]i) at approximately 1–2 mM and 100 nM, respectively. Regulators of intracellular Ca2+ and Ca2+ efflux pathways are primarily responsible for maintaining such low concentrations inside the cell [29].
The large concentration gradient between extracellular and intracellular spaces allows for a high signal-to-noise ratio and, thus, enables Ca2+ to serve as a second messenger even upon the smallest of changes in its intracellular concentration. Transient increases in [Ca2+]i are required for higher functions such as learning and memory consolidation through the cellular phenomenon of long-term potentiation [30]. In addition, such ephemeral elevations are necessary for basic neuronal processes and thereby play a significant role in synaptic activity and gene expression. Moderate-to-severe injuries can lead to more sustained elevations in [Ca2+]i, resulting in prolonged and often permanent alterations in cellular function; such increases can overwhelm the cell and regulators of cellular homeostasis leading to the initiation of proapoptotic pathways and, ultimately, to cell death [1]. While it is well known that severe elevations in neuronal [Ca2+]i following injury can lead to both acute and delayed neuronal death [31], studies from our laboratory have demonstrated that the sustained elevations in [Ca2+]i, observed in surviving neurons lead to altered neuronal function and induce neuronal plasticity [1].
The elucidation of the post-injury Ca2+ plateau represents a new discovery. The Ca2+ plateau refers to the prolonged elevation in intracellular Ca2+ following injuries including SE [32,33], stroke [34] and TBI [26]. The neurons that survive these injuries exhibit a marked alteration in their ability to handle Ca2+, as indicated by the presence of this Ca2+ plateau. While SE may be indirectly selecting a population of neurons that can withstand increases in [Ca2+]i without triggering apoptotic pathways, the effect that this persistent elevation has on the phenotype of the surviving cells through second messenger signaling remains an interesting question. These altered neurons represent the substrate for post-traumatic or post-SE epileptogenesis and appear to contribute ultimately to AE, as well as certain cognitive deficits. Evidence from several studies indicates that the Ca2+ plateau profoundly affects the fate of the neuron and leads to changes in long-term neuronal plasticity, as well as the development of AE [32,35].
Through second messenger effects, Ca2+ can alter gene expression, neurotransmitter release, basic cellular functions and plasticity. Thus, the focus of this review is to examine the tremendous implications of altered Ca2+ dynamics on the development and maintenance of AE in light of the recent discoveries in this evolving field. We will emphasize the development of the Ca2+ plateau after SE in contributing to both epileptogenesis and the maintenance of AE. Understanding the mechanisms responsible for the Ca2+ plateau may lead to therapeutic approaches to block this long-term elevation in Ca2+ after injuries such as SE, therefore leading to the prevention of AE. Thus, understanding the mechanisms of altered Ca2+ dynamics after brain injury has potentially important implications for both basic scientists and clinicians. The following presents a brief overview of evidence documenting the development of altered Ca2+ dynamics and how the Ca2+ plateau following SE contributes to epileptogenesis and AE.
SE acutely causes increased [Ca2+]I
In vitro studies
In the hippocampal neuronal culture model of epileptogenesis, it was demonstrated that in vitro SE significantly affected [Ca2+]i [36,37]. When cultures were treated for 3 h with a low Mg2+ solution, the cells exhibited high frequency, continuous epileptiform activity consistent with SE, unlike control neurons, which showed only occasional action potentials (Figure 1). Fluorescent ratio values were obtained using the Ca2+ indicator, fura-2. Ratios were generated using alternating excitation wavelengths of 340/380 nm and the resulting 340/380 ratios correspond directly to [Ca2+]i. Moreover, pseudocolor images were generated wherein individual pixels from the captured image are assigned a color based on the associated grayscale values, which correspond to Ca2+ levels. Red is associated with a higher grayscale value and, therefore, more Ca2+; whereas, at the other end of the colorimetric spectrum, blue is associated with lower grayscale values and less Ca2+. Pseudocolor allows for better visualization of changes in [Ca2+]i. A sevenfold increase from control baseline [Ca2+]i was observed while the neurons were in the low Mg2+ solution, during in vitro SE. The fura-2 340/380 ratios increased from 0.20 ± 0.04 in control neurons to 1.4 ± 0.17 in neurons after 3 h of low Mg2+ (Figure 1). Pseudocolor images confirmed these changes in fura-2 ratios with increased ratios in neurons post-low Mg2+ (Figure 1) but not vehicle treatment (Figure 1). Previous studies have shown with high speed simultaneous electrophysiological and Ca2+ imaging that each epileptiform spike was associated with increases in [Ca2+]i.
Figure 1. Alterations in intracellular Ca2+ concentration immediately following 3 h of low Mg2+-induced in vitro SE.
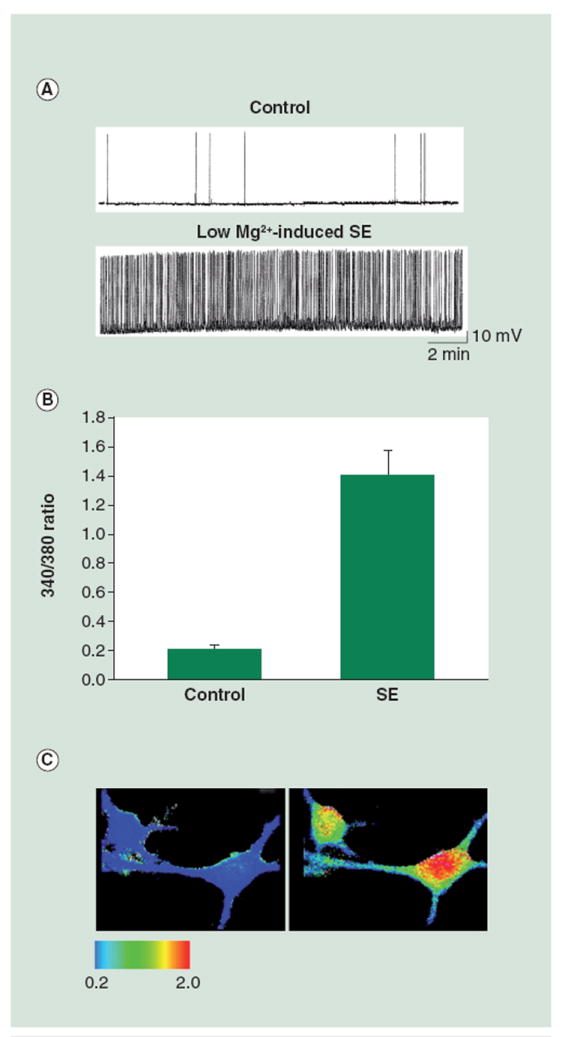
(A) Representative whole-cell current-clamp electrophysiological recording obtained from a control neuron and a neuron in low Mg2+ or in vitro SE. The neuron in low Mg2+ shows epileptiform discharges at a frequency greater than 3 Hz. By contrast, the control neuron shows action potentials intermittently, consistent with baseline firing activity. (B) Bar graph comparing fura-2 340/380 ratios in control neurons versus neurons immediately following 3 h of low Mg2+. The low Mg2+-treated cells exhibit ratio values of 1.4 ± 0.17 as compared with the control neurons with values of 0.20 ± 0.04 (data expressed as mean ± standard error of the mean). (C) Representative pseudocolor ratio images obtained from control (left panel) and low Mg2+-treated (right panel) neurons. Neurons from the low Mg2+ group show an increased 340/380 ratio immediately following treatment. SEM: Status epilepticus.
(A) reproduced with permission from [83].
In vitro analysis of in vivo model of SE-induced AE
These studies were also initiated to evaluate the effects of SE on [Ca2+]i. Similar results to the in vitro studies were observed in the rat pilocarpine model of SE-induced AE [32]. Intraperitoneal injection of the muscarinic agonist pilocarpine induced SE in the rats. The SE lasted for 1 h before being terminated by diazepam, a benzodiazepine. EEG was then used to study the electrical activity in the brain, with pilocarpine-treated animals showing epileptiform activity consistent with clinical SE, unlike vehicle-treated animals, who did not show this high frequency activity (Figure 2). Using this model it was possible to evaluate hippocampal neuronal Ca2+ levels during and after SE. Thus, during and immediately following SE, the rats were sacrificed and hippocampal tissue was acutely isolated, various regions were dissected out, and neurons were dissociated. [Ca2+]i levels were evaluated in these neurons using Ca2+ indicator dyes. Significant elevations in [Ca2+]i were observed in hippocampal neurons from the CA1 region following pilocarpine-induced SE but not in those isolated from vehicle control animals (Figure 2), 850 ± 59 and 90 ± 22 nM, respectively. This represented a 9.4-fold increase in [Ca2+]i between post-SE neurons and control neurons. These studies, in conjunction with the low Mg2+ studies, suggest that [Ca2+]i, increases during SE.
Figure 2. SE causes increased [Ca2+]i acutely in the in vivo pilocarpine model.
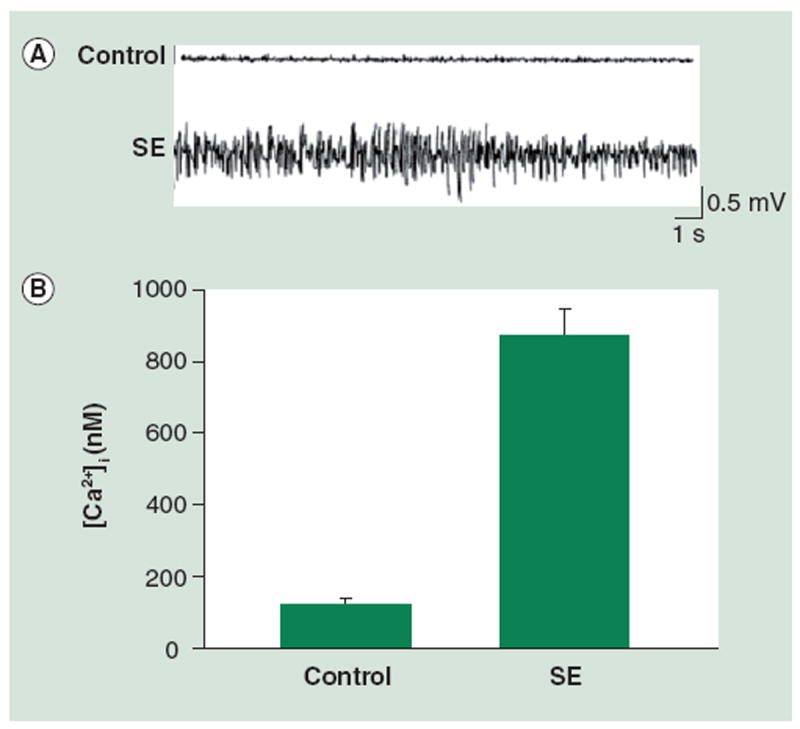
(A) Representative EEG recording from control (vehicle) rats and rats in pilocarpine-induced SE. (B) Bar graph demonstrating that [Ca2+]i is higher in neurons from animals immediately following SE when compared with those from animals in the control group: 850 ± 59 nM and 90 ± 22 nM, respectively (data expressed as mean ± standard error of the mean).
[Ca2+]i: Intracellular Ca2+ concentration.
(A) reproduced with permission from [90] and (B) adapted with permission from [32].
The Ca2+ plateau following SE: long-term elevations in [Ca2+]i
The Ca2+ plateau is a recent novel discovery that was characterized in the rat pilocarpine model of SE-induced AE [32] and in vitro in the culture model of SE [33]. In the rat pilocarpine model, [Ca2+]i was evaluated at 1, 2, 6, 10 and 30 days following SE in acutely isolated CA1 hippocampal neurons (Figure 3). Neurons from animals in the SE group exhibited markedly elevated [Ca2+]i at each of these time points. [Ca2+]i does decrease over time from the levels observed in neurons isolated from animals immediately after SE. By 6 days post-SE, the [Ca2+]i has leveled out but, interestingly, is still significantly higher than concentrations recorded from neurons in the control group. In the in vitro low Mg2+ hippocampal culture model similar results have been observed wherein [Ca2+]i remains elevated for several hours post-SE [33]. Longer durations of in vitro SE appear to cause more persistent elevations in [Ca2+]i. Further studies to characterize the Ca2+ plateau in this model are being conducted by our laboratory and will aid in our ability to better understand the mechanisms responsible for this novel observation.
Figure 3. The Ca2+ plateau following SE.
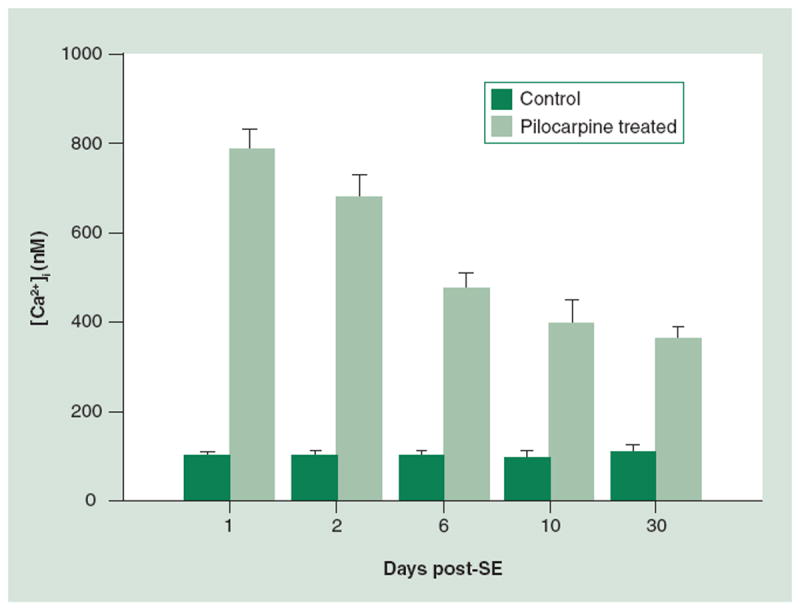
At one day post-SE, ([Ca2+]i) are significantly elevated when compared with controls, 790 ± 43 nM and 98 ± 10 nM, respectively (all data expressed as mean ± standard error of the mean). A total of 2 days post-SE the [Ca2+]i is 684 ± 47 nM, which is significantly higher than the 96 ± 14 nM observed in the control. At 6 days following SE, the [Ca2+]i is still markedly elevated when compared with controls, yet has fallen slightly from earlier timepoints, 475 ± 36 nM and 100 ± 10 nM, respectively. [Ca2+]i falls slightly at 10 days post-SE to 395 ± 56 nM but is still significantly elevated when compared with the controls with [Ca2+]i of 95 ± 14 nM. Finally, at 30 days post-SE the [Ca2+]i is 361 ± 28 nM versus 106 ± 16 nM in controls. The persistent elevation in [Ca2+]i represents the Ca2+ plateau.
[Ca2+]i: Intracellular Ca2+ concentration; SE: Status epilepticus.
Adapted with permission from [32].
SE causes long-term neuroplasticity changes including AE, elevated Ca2+ levels, & altered Ca2+ homeostatic mechanisms
In vitro studies
To evaluate Ca2+ dynamics following SE, cultured hippocampal neurons were patch-clamped to study electrophysiological activity and also imaged for evidence of alterations in fura-2 ratios 24 h post-low Mg2+ treatment [19,38]. When patched using whole-cell current clamp techniques, the low Mg2+-treated cells consistently displayed SREDs unlike control neurons, which failed to show SREDs (Figure 4). These SREDs resembled what is generally observed in clinical epilepsy, with the in vitro seizure episodes consisting of paroxysmal depolarization shifts and the cultures exhibiting similar anticonvulsant sensitivity. When cells were loaded with fura-2 and 340/380 ratios were recorded as a direct indicator of [Ca2+]i, ratio values appeared elevated 2.5-fold greater in low Mg2+ treated cells than in control neurons. This was indicated by an increase in fura-2 340/380 ratios from 0.20 ± 0.05 in control neurons to 0.5 ± 0.08 in low Mg2+ neurons (Figure 4).
Figure 4. Acquired epilepsy is associated with alterations in [Ca2+]i and Ca2+ homeostatic mechanisms 24 h post-in vitro SE.
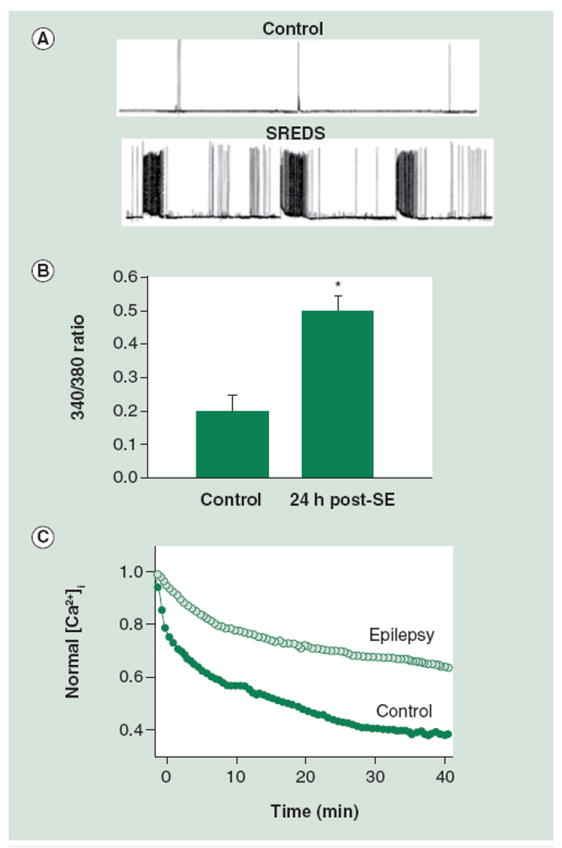
(A) Electrophysiological traces using current-clamp technique demonstrate baseline activity in control neurons and SREDs in neurons that had been treated in low Mg2+ for 3 h and then returned to normal maintenance media. Resting membrane potential = -61.5 mV; duration of shown trace = 30 min. (B) Bar graph comparing 340/380 ratio values in control versus SE groups 24 h after treatment with Mg2+-containing buffer solution or low Mg2+, respectively. Neurons in the SE group exhibit a 340/380 ratio of 0.5 ± 0.08 (mean ratio ± standard error of the mean). The ratio from control group neurons is significantly lower at 0.20 ± 0.05. (C) Glutamate stimulation causes increased [Ca2+]i. Peak ratios following glutamate are normalized to 1.0. Recovery from this challenge is hindered in epileptic neurons when compared with controls.
[Ca2+]i: Intracellular Ca2+ concentration; SE: Status epilepticus; SRED: Spontaneous, recurrent epileptiform discharge.
(A) reproduced with permission from [83] and (B) reproduced with permission from [37].
In addition, SE caused permanent changes in plasticity as demonstrated by alterations in Ca2+ homeostatic mechanisms. Neurons were briefly stimulated with 50 μM glutamate to test the ability of Ca2+ homeostatic mechanisms to compensate for brief excitatory challenges. These cells demonstrated a markedly diminished ability to recover from the insult (Figure 4), suggesting that the homeostatic mechanisms, which under normal conditions maintain tight control of Ca2+ influx, efflux and sequestration, are altered or diminished following SE. Stimulation with glutamate resulted in an increase in [Ca2+]i both in control and post-SE neurons; however, in neurons treated with low Mg2+, Ca2+ never returned to basal levels and showed a decreased rate of recovery when compared with controls.
In vitro analysis of in vivo model of SE-induced AE
To determine whether the changes in Ca2+ dynamics were transient or more long lasting, animals were treated with pilocarpine or vehicle to induce SE and were then maintained for 1 year post-SE [32]. These animals were evaluated for epilepsy using video-EEG and the spontaneous, recurrent seizures in the pilocarpine but not vehicle-treated group were observed, as shown in representative EEG recordings from control and epileptic animals (Figure 5). Hippocampal CA1 neurons isolated from epileptic animals 1 year post-pilocarpine demonstrated markedly elevated [Ca2+]i (325 ± 35 nM) – more than double the values observed in control neurons (Figure 5).
Figure 5. Acquired epilepsy is associated with alterations in [Ca2+]i and Ca2+ homeostatic mechanisms one year post-in vivo status epilepticus (SE).
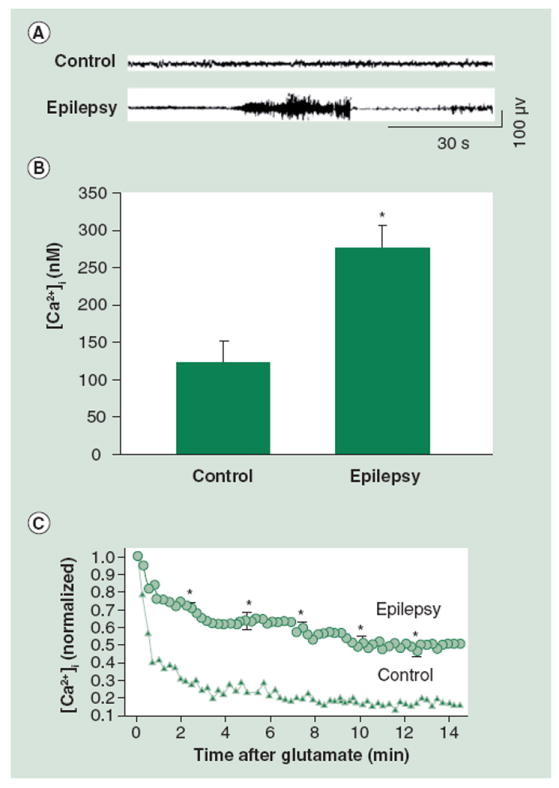
(A) Representative EEG recording from control (vehicle) rats and epileptic rats (following pilocarpine-induced SE). (B) Bar graph with neurons from epileptic animals at 1 year post-SE demonstrating [Ca2+]i of 325 ± 35 nM (data represented as mean ± standard error of the mean), which is significantly higher than neurons from control group. (C) Upon glutamate stimulation, the [Ca2+]i increases and then returns to baseline in neurons from the control group, unlike neurons from the epileptic group which also peak but then fail to return to baseline.
[Ca2+]i: Intracellular Ca2+ concentration.
(B) adapted with permission from [32] and (C) reproduced with permission from [35].
Finally, similarly to the in vitro culture model of post-SE AE, neurons from animals displaying the epileptic phenotype were acutely isolated at 1 year post-SE then challenged with glutamate to evaluate homeostatic mechanisms. These neurons were unable to restore Ca2+ as effectively as control neurons, therefore suggesting persistent alterations in homeostatic mechanisms at this time point (Figure 5). The observation that neurons in animals from the pilocarpine-treated group exhibited marked changes in both [Ca2+]i and Ca2+ homeostatic mechanisms 1 year after the initial injury suggests that Ca2+ dynamics post-SE affect plasticity changes and, ultimately, contribute to the maintenance of the chronic, epileptic phenotype.
These studies utilizing both in vivo and in vitro models demonstrated that well past the duration of neuronal injury, [Ca2+]i was elevated and Ca2+ homeostatic mechanisms were altered. These changes appear to play a significant role in the maintenance of the epileptic phenotype. The elucidation of the specific mechanisms behind such long-lasting alterations remains an important area of research; moreover, understanding how these changes lead to development or maintenance of chronic epilepsy could provide new information for the development of better treatments or new preventative measures to halt epileptogenesis.
Mechanisms underlying Ca2+ changes after SE
Glutamate & NMDA receptors
Status epilepticus is an excitotoxic injury, meaning that it triggers the release of large amounts of the amino acid neurotransmitter glutamate [39-41]. The degree and duration of SE directly affects the concentration of glutamate, which has been reported to exist at extracellular concentrations ranging from 1 to 5 μM under normal conditions. Excess glutamate following SE causes the over stimulation of glutamate receptors, including the NMDA receptor that is coupled with a Ca2+-permeable ion channel, thus leading to the movement of Ca2+ across the cell membrane into the cell. This was confirmed by the ability of NMDA antagonists to offer neuroprotection when administered prior to injury despite an undiminished SE [42-44]. Several studies have demonstrated that these elevations in glutamate cause neuronal death and that the inhibition of Ca2+ overload through the use of NMDA antagonists can prevent both the cognitive sequalae and epileptogenesis [43,45].
A majority of the work demonstrating that epileptogenesis is Ca2+-dependent was performed in the hippocampal neuronal culture model of epilepsy, since various pharmacological inhibitors could be utilized to tease out which systems were involved in the process. One study correlated electrophysiological activity with intracellular Ca2+ imaging in the presence of various Ca2+ channel inhibitors in order to determine if any of these channels were contributing to the development of SREDs (Table 1) [36]. During the 3-h low Mg2+ treatment, cells were in the presence of either low Ca2+ (0.2 mM), 1,2-bis(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetate (BAPTA) (100 μM), MK-801 (10 μM), [2R]-amino-5-phosphonovaleric acid (APV); 25 μM, 6-cyano-7-nitroquinoxaline-2,3-dione (10 μM), 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo [f]quinoxaline (10 μM) or nifedipine (5 μM). No significant changes in spike frequency, intensity or epileptic discharge duration were observed during in vitro SE under each condition [36]. However, both the Ca2+ chelating agent BAPTA and the low-Ca2+ conditions prevented the development of SREDs 48 h post-in vitro SE [36], demonstrating the overall importance of Ca2+ in causing SREDs. Furthermore, NMDA receptor antagonists, including MK-801 and APV, exhibited similar effects wherein SE was unaffected but SREDs failed to develop 48 h following low Mg2+ [36]. By contrast, the AMPA receptor antagonists 6-cyano-7-nitroquinoxaline-2,3-dione and 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo [f]quinoxaline, as well as the antagonist of L-type voltage gated Ca2+ channels, nifedipine, failed to inhibit SREDs at this time point [36]. Moreover, when Ca2+ imaging was performed on the neurons under each condition while the cells were in low Mg2+ it was demonstrated that only low extracellular Ca2+, NMDA antagonists and BAPTA were able to prevent or diminish Ca2+ entry during SE [36]. Thus, this study demonstrated in vitro that Ca2+ is a necessary component in the generation of SREDs post-SE and that the NMDA receptor in particular contributes to the Ca2+ entry involved in epileptogenesis.
Table 1.
Intracellular calcium levels and electrophysiological activity before and after low Mg2+ in the presence of various Ca2+ channel inhibitors to determine contribution of these channels to spontaneous, recurrent epileptiform discharges development.
| Condition | [Ca2+]i nM | Epilepsy | SE spike frequency |
|---|---|---|---|
| Control | 165 ± 35 | No | 0 |
| Low Mg2+ (SE) | 577 ± 35 | Yes | 12 ± 4.4 |
| + low Ca2+ | 177 ± 18 | No | 10 ± 3.2 |
| + BAPTA (100 μM) | 160 ± 8 | No | 14 ± 5.1 |
| + APV (25 μM) | 293 ± 24 | No | 16 ± 3.8 |
| + MK-801 (10 μM) | 287 ± 25 | No | 11 ± 2.9 |
| + CNQX (10 μM) | 433 ± 10 | Yes | 9 ± 3.0 |
| + NBQX (10 μM) | 422 ± 56 | Yes | 8 ± 2.4 |
| + Nifedipine (5 μM) | 441 ± 19 | Yes | 13 ± 4.1 |
| + TTX (1 μM) | 151 ± 17 | No | 0 |
The first column shows the various conditions including control and low Mg2+-treated neurons, as well as low Mg2+ neurons that were then treated with Ca2+ channel inhibitors on low Mg2+-treated cells. The second column shows [Ca2+]i levels observed in each condition. The third and fourth columns show whether or not the neurons developed SREDs (epilepsy) or whether the frequency of SE was changed under each condition, respectively. Data is represented as mean ± standard error for [Ca2+]i and SE spike frequency and as all or none effects for the development of epilepsy.
APV: (2R)-amino-5-phosphonovaleric acid; BAPTA: 1, 2-bis(2-aminophenoxy)ethane-N, N, N’, N’-tetraacetate ; [Ca2+]i: Intracellular calcium; CNQX: 6-cyano-7-nitroquinoxaline-2,3-dione;
NBQX: 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo[f]quinoxaline; SE: Status epilepticus; TTX: Tetrodotoxin.
Adapted with permission from [36].
Intracellular regulators of Ca2+
Neurons have a network of endoplasmic reticulum (ER) that contributes to the overall levels of [Ca2+]i by acting to either release or sequester Ca2+ [46]. Ca2+ that is stored in the ER can be released through activation of the inositol triphosphate receptors (IP3R) or ryanodine receptors [46,47]. These receptors gauge cytoplasmic Ca2+ and mediate Ca2+-induced Ca2+ release (CICR) from the ER; thus, the channels amplify the Ca2+ signal coming from outside the cell. IP3 and cyclic ADP ribose, levels of Ca2+ in the ER and other messengers can influence the sensitivity of these receptors. The ER also has a sequestration mechanism through the sarco/ER Ca2+ ATPase (SERCA), which pumps Ca2+ back into the ER from the cytosol [47]. The ER is able to sequester large amounts of Ca2+ under normal conditions.
Thus, SE causes glutamate release that stimulates Ca2+ entry into the cell, triggering the activation of CICR wherein ryanodine receptors and IP3R are activated to release more Ca2+ from ER stores [37,48]. During the Ca2+ plateau, epileptic neurons show upregulation of CICR mechanisms when compared with control neurons [37]. Interestingly, one study demonstrated that the intracellular injection of IP3 causes epileptiform discharges in hippocampal neurons [49]. Therefore, several groups have examined the ability of antiepileptic drugs to inhibit IP3 or IP3R as a means to prevent epileptiform activity [50,51]. Moreover, it has also been demonstrated that SERCA is unable to effectively sequester Ca2+ in ‘epileptic’ neurons [37], suggesting that this mechanism may contribute to the Ca2+ plateau. Future studies should further examine this relationship between SERCA uptake and CICR as they are interdependent processes.
Ca2+ binding proteins
Ca2+ binding proteins also play an important role in basic physiology further contributing to the maintenance of low [Ca2+]i. It has been hypothesized that the downregulation of Ca2+ binding proteins also contributes to the elevated [Ca2+]i maintained during the Ca2+ plateau following SE [52]. Several studies utilizing human epileptic hippocampal tissue showed that calbindin levels in the dentate granule cells were diminished [53,54]. The pilocarpine model has yielded similar results wherein it has been demonstrated that at 1 month after pilocarpine-induced SE epileptic animals demonstrated a 50% reduction in calbindin protein expression in the hippocampus [52]. These changes were observed up to 2 years after the initial SE [52]; moreover, calbindin mRNA expression was also decreased in these animals.
These observations have also been noted a few months post-initial injury in other models of epilepsy, including those utilizing kindling and kainic acid [55,56]. However, some studies have also shown that following kainic acid or electrically stimulated seizures, calbindin mRNA levels are unchanged or even increased [57-59]. Therefore, these data suggest that following a glutamate-induced Ca2+ influx, there is a transient compensatory upregulation in calbindin expression but the long-term elevations in [Ca2+]i observed in epileptic animals may be attributed to overall downregulation in protein expression. This suggests that the maintenance of the post-SE Ca2+ plateau may be mediated in part by a decrease in Ca2+ binding proteins, which normally buffer elevations in Ca2+.
Ca2+-regulated enzymes
Ca2+/calmodulin-dependent protein kinase II (CaM kinase II) is an enzyme that plays a major role in the modulation of neuronal excitability through its actions on Ca2+-dependent neuronal processes [60,61]. CaM kinase II has a widespread distribution throughout the nervous system and, interestingly, comprises up to 2% of total hippocampal protein [62].
Genetic studies have demonstrated that mice with a null mutation of the α-subunit of CaM kinase II show gene dose-dependent neuronal hyperexcitability and seizures [15]. In vitro molecular approaches have yielded supporting data by the demonstration that reduction of α-CaM kinase II levels through antisense oligonucleotides causes spontaneous epileptic discharges in culture [63]. While these studies have provided tremendous insight on CaM kinase II as a means of neuronal excitability, they have not examined whether CaM kinase II expression or activity was actually altered in neurons from epileptic animals or from cultures exhibiting in vitro epilepsy.
In whole animal and slice models that use repeated hippocampal stimulation to elicit SE, decreased CaM kinase II activity has been shown to trigger epileptiform activity [64]. Both pilocarpine and kainic acid models of SE-induced AE have shown decreased CaM kinase II activity and these results have also been observed in the low Mg2+ in vitro hippocampal culture model [65-67]. Interestingly, reduced CaM kinase II activity can be stopped by NMDA receptor antagonists administered prior to SE [61,67]. Thus, the change in CaM kinase II activity appears to be a consequence of NMDA receptor activation. Paradoxically, short-term NMDA receptor activation and the resulting elevations in intracellular Ca2+ lead to increased CaM kinase II activation and the translocation of the enzyme from the cytosol to excitatory synapses [68]. The precise role for CaM kinase II in the induction and development of epilepsy is being actively pursued and several studies have discussed in great detail the paradoxical nature of its interactions with NMDA receptors versus the hyperexcitability observed in CaM kinase II knockdown studies [68]. In pentylenetetrazol or electroconvulsive seizures (ECS) studies it has been demonstrated that seizures cause CaM kinase II to translocate from the synapse to the cytosol [65,69]. Therefore, in distancing the enzyme from its substrate, the translocation may serve to further reduce the effective enzymatic activity.
General effects of Ca2+ on cells
Cellular Ca2+ signaling is a complex process that is difficult to elucidate owing to the interdependence of Ca2+ handling/regulatory systems. However, clear links between Ca2+ levels and membrane excitability, protein phosphorylation and gene regulation have been demonstrated [70]. Moreover, elevated [Ca2+]i also has a profound effect on the neuronal circuit, cell death and glial function. Several studies have demonstrated that this increase in Ca2+ concentration triggers hypertrophy of glia and expression of proteins on glia that are not normally observed [71]. In addition, the elevations in [Ca2+]i have been shown to activate genes for growth factors including brain derived neurotrophic factor and FGF, induce changes in glutamate receptors and cause altered expression of cytoskeletal proteins [71]. All of these alterations significantly affect the development or maintenance of the epileptic circuit [72]. Developing efficacious therapeutic strategies may be aided by a more thorough understanding of Ca2+ dynamics in neurons and how these alterations in Ca2+ can affect glia and the neuronal circuit.
Generally, the degree of Ca2+ influx contributes greatly to the level of injury sustained by the neurons and impacts the overall fate of the cell. Severely injured cells exhibit an irreversible Ca2+ overload that leads to cell death through various means, some of which include: the disruption of cytoskeletal organization, formation of cell membrane blebs, activation of Ca2+-dependent endonuclease leading to chromatin cleavage and mitochondrial impairment resulting in loss of ATP synthesis [73]. Notably, cell death is a prominent manifestation immediately after SE where [Ca2+]i increases dramatically with a percentage cell death of approximately 10–15% in vivo [43] and 15% in vitro [20]. Interestingly, epilepsy itself did not cause more cell death in vitro, in vivo or in human studies [20,74-78]. Thus, although the elevated Ca2+ plateau is maintained, the levels are not high enough to induce significant cell death but can cause permanent alterations in neuronal homeostasis.
The neurons that survive SE undergo altered Ca2+ homeostasis but not necessarily an irreversible Ca2+ overload; therefore, since dead neurons do not seize, it is these injured but surviving neurons that serve as the substrate for epileptogenesis. These neurons sustain reversible elevations in [Ca2+]i that are maintained over days or weeks [32] – these increases mediate changes in synaptic plasticity, gene expression and neuronal function. Therefore, the overall increased [Ca2+]i directly affects nuclear Ca2+ levels and leads to the induction of gene transcription of several proapoptotic or epileptogenic genes and alters neuronal functions including excitability, transmitter release and neurotoxicity [79]. One effect is that increases in [Ca2+]i activate an enzyme, calcineurin, leading to a downstream cascade of events including an accelerated internalization of GABAA receptors resulting in decreased inhibitory neurotransmission [15,80]. Thus, it is the injured neurons and these molecular changes that become potential targets for future therapies to prevent AE. The ability to target these intracellular changes and reduce the Ca2+ load may prevent the development of the epileptic phenotype following SE or other neuronal injuries.
Expert commentary
Until now a majority of studies have focused on using new Ca2+ modulating drugs as prophylactic, neuroprotective agents to be administered prior to injury so as to prevent the development of AE or as anticonvulsive agents after the full development of AE. However, more work must be conducted on the clinically important period between the injury and the eventual development of AE as this time may represent a therapeutic window of opportunity wherein the long-term consequences of neuronal injury can be avoided through new pharmacological endeavors. The discovery of the Ca2+ plateau may represent a new therapeutic target to possibly treat and prevent some of the long-term effects of elevated Ca2+ on neuronal plasticity and the development of epilepsy.
It is important to develop new agents to prevent epileptogenisis. One study has successfully used pharmacological intervention following injury in the in vitro low Mg2+ model of SE to prevent the development of SREDs [81]. It was demonstrated that a new anticonvulsant drug [82,83], carisbamate, also exhibited anti-epileptogenic properties in vitro in hippocampal cultures [81]. This means that the drug was able to prevent the development of SREDs when given following in vitro SE and that it did not have to be present in the culture media for the SRED-free activity to persist, differentiating it from most anticonvulsants. It remains to be determined whether these properties exist in vivo, and currently the mechanism of action behind the antiepileptogenic properties is unknown. It will be interesting to examine how this drug affects Ca2+ dynamics. If alterations in Ca2+ are linked to epileptogenesis, modulation of Ca2+ immediately following SE may provide an important and interesting area for future research.
The screening of pharmacologic agents for various properties, including the ability to block epileptogenesis, is more reminiscent of approaches used in the discovery of antiepileptic (anticonvulsant) drugs (AEDs) than those more commonly utilized today in the search for antiepileptogenics. In the development of AEDs, high-throughput screening of various agents was performed in in vivo models in order to ascertain anticonvulsive properties. We have made a great deal of progress in the development of AEDs with both safety and efficacy profiles increasing tremendously in recent years. However, we are now starting to understand the subtleties with regards to the action of these drugs on particular channels, receptor expression and cellular processes; thus, we are just beginning to appreciate specific mechanisms of action.
By contrast, research groups that aim to develop antiepileptogenic agents to prevent the transformation to the epileptic condition have generally taken the approach of understanding mechanisms of plasticity and excitability and utilizing these observations to create new therapeutic targets [84]. While there are currently no antiepileptogenic drugs, much progress has been made in the elucidation of molecular mechanisms of epileptogenesis that will provide new approaches in the future [84]. The clinical implications of successfully treating patients following neuronal injuries, such as SE with pharmacologic agents that prevent the clinical sequelae, namely epilepsy, are great.
Thus, understanding the molecular changes that are responsible for altered Ca2+ homeostasis and increased [Ca2+]i during epileptogenesis may aid in the development of new drugs that can be administered immediately after brain injury in the hope of preventing the expression of epilepsy. Currently, studies have shown that various Ca2+ regulatory systems are modified or altered during SE and epileptogenesis. Increases in extracellular glutamate cause the massive flux of Ca2+ into the cell leading to increased CICR. These mechanisms, combined with later decreases in the ability of SERCA to pump Ca2+ back into the ER, as well as alterations in Ca2+ binding proteins and Ca2+-regulated enzymes, all contribute to changes in gene expression and the eventual development of the epileptic condition. Therefore, understanding the specific mechanisms behind the maintenance of the post-SE Ca2+ plateau and the designing of new drugs to target this plateau may provide much utility in the development of antiepileptogenic therapies.
Five-year view
In the pilocarpine model of SE-induced AEs it is well established that the elevations in intraneuronal Ca2+ are maintained post-injury in animals that develop epilepsy. However, other conditions, including stroke and TBI, can also lead to the development of epilepsy. Finding common pathophysiologic mechanisms behind how excitotoxic injuries can lead to the epileptic condition will bolster efforts to design new therapeutics to prevent epileptogenesis. Thus, the examination of Ca2+ dynamics in other models of injury-induced epilepsy may be of great clinical importance. As a ubiquitous second messenger, alterations in [Ca2+]i and Ca2+ homeostatic mechanisms have significant repercussions on gene expression, neurotransmitter release and, hence, neuronal plasticity. Therefore, the development and use of myriad models of AE to better understand the relationship between Ca2+ and epileptogenesis may offer an important future tool.
While many Ca2+ channel antagonists have been shown to exhibit anticonvulsive properties or potentiate the effects of AEDs both in experimental models and in clinical trials [85,86], there are also several reports that other Ca2+ channel inhibitors are completely ineffective [86]. Even less work has been done examining the effect of Ca2+-modifying agents in models of epileptogenesis, and the data demonstrating the anticonvulsive properties of these drugs suggests that numerous types of inhibitors will have to be screened. One study has shown that the NMDA antagonist, MK-801, was effective in decreasing the number of animals that developed chronic epilepsy following electrically-induced SE [45]. However, in the kainic acid-induced SE model, MK-801 did not prevent the development of epilepsy [87]. Conflicting results between different models of epileptogenesis have made the search for new therapeutics challenging [88] and have underscored the importance of utilizing several different models of epileptogenesis.
Studies aimed at discerning whether lowering Ca2+ to basal levels can diminish or halt the process of epileptogenesis may be of importance. The long-lasting Ca2+ plateau observed post-injury appears to be controlled by a multitude of channels, Ca2+ binding proteins and other regulators. Currently, Ca2+-modifying agents have been employed for other pathologies of the CNS. For example, the NMDA antagonist, memantine, has been utilized in patients with Alzheimer’s disease [89]. Interestingly, one Phase II clinical trial administered the Ca2+-chelating agent, DP-BAPTA, to patients following stroke and found that this drug improved the NIH stroke scores with no major cardiovascular or neurological side effects, even at the highest dose tested [101]. The ability to modulate Ca2+ without causing long-lasting, severe side effects is important. Since Ca2+ is found ubiquitously in the body and is necessary for many cellular processes, the potential side effects of altering Ca2+ pharmacologically could be tremendous and, thus, represent a major hurdle in the utilization of Ca2+-modifying drugs. The ability to synthesize these observations on altered Ca2+ dynamics with those from molecular–genetic studies identifying potential genes involved in epilepsy could provide novel insight in the generation of new antiepileptogenic targets.
Key issues.
Epilepsy affects approximately 2% of the world’s population, with acquired epilepsy constituting 40% of all cases of epilepsy.
Acquired epilepsy can be divided into three pathophysiologic stages: injury, latency/epileptogenesis and chronic epilepsy.
Intraneuronal calcium increases significantly during injury from status epilepticus.
In both in vivo and in vitro models of status epilepticus-induced acquired epilepsy, a significant calcium plateau is maintained during epileptogenesis.
As an important second messenger, sustained elevations in intraneuronal calcium can have tremendous implications on plasticity, gene expression, neurotransmitter release and the ultimate expression of epilepsy.
Understanding changes in expression/activity of various regulators of cellular calcium may provide new therapeutic targets through which we are able to lower intraneuronal calcium post-injury and, perhaps in doing so, prevent the development of epilepsy.
Acknowledgments
This research was funded by support from the National Institute of Neurological Disorders and Stroke grants RO1NS051505, RO1NS052529 and UO1NS058213.
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Contributor Information
Nisha Nagarkatti, Email: nagarkattin@vcu.edu, Departments of Neurology and Pharmacology and Toxicology, Virginia Commonwealth University, Richmond, VA, 23298, USA, Tel.: +1 804 828 3392, Fax: +1 804 828 6432.
Laxmikant S Deshpande, Email: deshpandels@vcu.edu, Department of Neurology, Virginia Commonwealth University, Richmond, VA, 23298, USA, Tel.: +1 804 828 3392, Fax: +1 804 828 6432.
Robert J DeLorenzo, Email: rdeloren@vcu.edu, Departments of Neurology, Pharmacology and Toxicology, and Molecular Biophysics and Biochemistry, Virginia Commonwealth University, School of Medicine, PO Box 980599, Richmond, VA 23298, USA, Tel.: +1 804 828 8969, Fax: +1 804 828 6432.
References
- 1.Delorenzo RJ, Sun DA, Deshpande LS. Cellular mechanisms underlying acquired epilepsy: the calcium hypothesis of the induction and maintainance of epilepsy. Pharmacol Ther. 2005;105(3):229–266. doi: 10.1016/j.pharmthera.2004.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Willmore LJ. Post-traumatic epilepsy: cellular mechanisms and implications for treatment. Epilepsia. 1990;31(Suppl. 3):S67–S73. doi: 10.1111/j.1528-1157.1990.tb05861.x. [DOI] [PubMed] [Google Scholar]
- 3.Cavalheiro EA, Leite JP, Bortolotto ZA, et al. Long-term effects of pilocarpine in rats: structural damage of the brain triggers kindling and spontaneous recurrent seizures. Epilepsia. 1991;32(6):778–782. doi: 10.1111/j.1528-1157.1991.tb05533.x. [DOI] [PubMed] [Google Scholar]
- 4.Lothman EW, Bertram EH., 3rd Epileptogenic effects of status epilepticus. Epilepsia. 1993;34(Suppl. 1):S59–S70. doi: 10.1111/j.1528-1157.1993.tb05907.x. [DOI] [PubMed] [Google Scholar]
- 5.Hesdorffer DC, Logroscino G, Cascino G, Annegers JF, Hauser WA. Risk of unprovoked seizure after acute symptomatic seizure: effect of status epilepticus. Ann Neurol. 1998;44(6):908–912. doi: 10.1002/ana.410440609. [DOI] [PubMed] [Google Scholar]
- 6.DeLorenzo RJ, Hauser WA, Towne AR, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology. 1996;46(4):1029–1035. doi: 10.1212/wnl.46.4.1029. [DOI] [PubMed] [Google Scholar]
- 7.Hauser WA. Status epilepticus: epidemiologic considerations. Neurology. 1990;40(5 Suppl. 2):9–13. [PubMed] [Google Scholar]
- 8.Wasterlain CG, Chen JW. Mechanistic and pharmacologic aspects of status epilepticus and its treatment with new antiepileptic drugs. Epilepsia. 2008;49(Suppl. 9):63–73. doi: 10.1111/j.1528-1167.2008.01928.x. [DOI] [PubMed] [Google Scholar]
- 9.DeLorenzo RJ, Pellock JM, Towne AR, Boggs JG. Epidemiology of status epilepticus. J Clin Neurophysiol. 1995;12(4):316–325. [PubMed] [Google Scholar]
- 10.Li JM, Chen L, Zhou B, Zhu Y, Zhou D. Convulsive status epilepticus in adults and adolescents of southwest China: mortality, etiology, and predictors of death. Epilepsy Behav. 2009;14(1):146–149. doi: 10.1016/j.yebeh.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 11.Drislane FW. Presentation, evaluation, and treatment of nonconvulsive status epilepticus. Epilepsy Behav. 2000;1(5):301–314. doi: 10.1006/ebeh.2000.0100. [DOI] [PubMed] [Google Scholar]
- 12.Fountain NB, Lothman EW. Pathophysiology of status epilepticus. J Clin Neurophysiol. 1995;12(4):326–342. [PubMed] [Google Scholar]
- 13.Deshpande LS, Lou JK, Mian A, et al. Time course and mechanism of hippocampal neuronal death in an in vitro model of status epilepticus: role of NMDA receptor activation and NMDA dependent calcium entry. Eur J Pharmacol. 2008;583(1):73–83. doi: 10.1016/j.ejphar.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fisher RS. Animal models of the epilepsies. Brain Res Brain Res Rev. 1989;14(3):245–278. doi: 10.1016/0165-0173(89)90003-9. [DOI] [PubMed] [Google Scholar]
- 15.McNamara JO, Huang YZ, Leonard AS. Molecular signaling mechanisms underlying epileptogenesis. Sci STKE. 2006;2006(356):re12. doi: 10.1126/stke.3562006re12. [DOI] [PubMed] [Google Scholar]
- 16.Klitgaard H, Matagne A, Vanneste-Goemaere J, Margineanu DG. Pilocarpine-induced epileptogenesis in the rat: impact of initial duration of status epilepticus on electrophysiological and neuropathological alterations. Epilepsy Res. 2002;51(1–2):93–107. doi: 10.1016/s0920-1211(02)00099-2. [DOI] [PubMed] [Google Scholar]
- 17.Pitkanen A, Kharatishvili I, Karhunen H, et al. Epileptogenesis in experimental models. Epilepsia. 2007;48(Suppl. 2):13–20. doi: 10.1111/j.1528-1167.2007.01063.x. [DOI] [PubMed] [Google Scholar]
- 18.Cavalheiro EA. The pilocarpine model of epilepsy. Ital J Neurol Sci. 1995;16(1–2):33–37. doi: 10.1007/BF02229072. [DOI] [PubMed] [Google Scholar]
- 19.Sombati S, Delorenzo RJ. Recurrent spontaneous seizure activity in hippocampal neuronal networks in culture. J Neurophysiol. 1995;73(4):1706–1711. doi: 10.1152/jn.1995.73.4.1706. [DOI] [PubMed] [Google Scholar]
- 20.Deshpande LS, Lou JK, Mian A, et al. In vitro status epilepticus but not spontaneous recurrent seizures cause cell death in cultured hippocampal neurons. Epilepsy Res. 2007;75(2–3):171–179. doi: 10.1016/j.eplepsyres.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeGiorgio CM, Tomiyasu U, Gott PS, Treiman DM. Hippocampal pyramidal cell loss in human status epilepticus. Epilepsia. 1992;33(1):23–27. doi: 10.1111/j.1528-1157.1992.tb02278.x. [DOI] [PubMed] [Google Scholar]
- 22.Sloviter RS. Status epilepticus-induced neuronal injury and network reorganization. Epilepsia. 1999;40(Suppl. 1):S34–S39. doi: 10.1111/j.1528-1157.1999.tb00876.x. discussion S40–S31. [DOI] [PubMed] [Google Scholar]
- 23.Tsuchida TN, Barkovich AJ, Bollen AW, Hart AP, Ferriero DM. Childhood status epilepticus and excitotoxic neuronal injury. Pediatr Neurol. 2007;36(4):253–257. doi: 10.1016/j.pediatrneurol.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 24.Pitkanen A. On the way to cure epilepsy. Expert Rev Neurother. 2004;4(6):917–920. doi: 10.1586/14737175.4.6.917. [DOI] [PubMed] [Google Scholar]
- 25.Deshpande LS, Sun DA, Sombati S, et al. Alterations in neuronal calcium levels are associated with cognitive deficits after traumatic brain injury. Neurosci Lett. 2008;441(1):115–119. doi: 10.1016/j.neulet.2008.05.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun DA, Deshpande LS, Sombati S, et al. Traumatic brain injury causes a long-lasting calcium (Ca2+)-plateau of elevated intracellular Ca levels and altered Ca2+ homeostatic mechanisms in hippocampal neurons surviving brain injury. Eur J Neurosci. 2008;27(7):1659–1672. doi: 10.1111/j.1460-9568.2008.06156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun DA, Sombati S, Blair RE, DeLorenzo RJ. Long-lasting alterations in neuronal calcium homeostasis in an in vitro model of stroke-induced epilepsy. Cell Calcium. 2004;35(2):155–163. doi: 10.1016/j.ceca.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 28.West AE, Chen WG, Dalva MB, et al. Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA. 2001;98(20):11024–11031. doi: 10.1073/pnas.191352298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mody I, MacDonald JF. NMDA receptor-dependent excitotoxicity: the role of intracellular Ca2+ release. Trends Pharmacol Sci. 1995;16(10):356–359. doi: 10.1016/s0165-6147(00)89070-7. [DOI] [PubMed] [Google Scholar]
- 30.Malenka RC, Nicoll RA. Long-term potentiation – a decade of progress? Science. 1999;285(5435):1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 31.Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7(2):357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raza M, Blair RE, Sombati S, et al. Evidence that injury-induced changes in hippocampal neuronal calcium dynamics during epileptogenesis cause acquired epilepsy. Proc Natl Acad Sci USA. 2004;101(50):17522–17527. doi: 10.1073/pnas.0408155101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pal S, Sombati S, Limbrick DD, Jr, DeLorenzo RJ. In vitro status epilepticus causes sustained elevation of intracellular calcium levels in hippocampal neurons. Brain Res. 1999;851(1–2):20–31. doi: 10.1016/s0006-8993(99)02035-1. [DOI] [PubMed] [Google Scholar]
- 34.Sun DA, Sombati S, Blair RE, DeLorenzo RJ. Calcium-dependent epileptogenesis in an in vitro model of stroke-induced “epilepsy”. Epilepsia. 2002;43(11):1296–1305. doi: 10.1046/j.1528-1157.2002.09702.x. [DOI] [PubMed] [Google Scholar]
- 35.Raza M, Pal S, Rafiq A, DeLorenzo RJ. Long-term alteration of calcium homeostatic mechanisms in the pilocarpine model of temporal lobe epilepsy. Brain Res. 2001;903(1–2):1–12. doi: 10.1016/s0006-8993(01)02127-8. [DOI] [PubMed] [Google Scholar]
- 36.DeLorenzo RJ, Pal S, Sombati S. Prolonged activation of the N-methyl-d-aspartate receptor-Ca2+ transduction pathway causes spontaneous recurrent epileptiform discharges in hippocampal neurons in culture. Proc Natl Acad Sci USA. 1998;95(24):14482–14487. doi: 10.1073/pnas.95.24.14482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pal S, Sun D, Limbrick D, Rafiq A, DeLorenzo RJ. Epileptogenesis induces long-term alterations in intracellular calcium release and sequestration mechanisms in the hippocampal neuronal culture model of epilepsy. Cell Calcium. 2001;30(4):285–296. doi: 10.1054/ceca.2001.0236. [DOI] [PubMed] [Google Scholar]
- 38.Pal S, Limbrick DD, Jr, Rafiq A, DeLorenzo RJ. Induction of spontaneous recurrent epileptiform discharges causes long-term changes in intracellular calcium homeostatic mechanisms. Cell Calcium. 2000;28(3):181–193. doi: 10.1054/ceca.2000.0146. [DOI] [PubMed] [Google Scholar]
- 39.Sherwin AL. Neuroactive amino acids in focally epileptic human brain: a review. Neurochem Res. 1999;24(11):1387–1395. doi: 10.1023/a:1022580506443. [DOI] [PubMed] [Google Scholar]
- 40.Liu Z, Stafstrom CE, Sarkisian MR, et al. Seizure-induced glutamate release in mature and immature animals: an in vivo microdialysis study. Neuroreport. 1997;8(8):2019–2023. doi: 10.1097/00001756-199705260-00043. [DOI] [PubMed] [Google Scholar]
- 41.Smolders I, Khan GM, Manil J, Ebinger G, Michotte Y. NMDA receptor-mediated pilocarpine-induced seizures: characterization in freely moving rats by microdialysis. Br J Pharmacol. 1997;121(6):1171–1179. doi: 10.1038/sj.bjp.0701231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meldrum BS. First Alfred Meyer Memorial Lecture. Epileptic brain damage: a consequence and a cause of seizures. Neuropathol Appl Neurobiol. 1997;23(3):185–201. discussion 201–202. [PubMed] [Google Scholar]
- 43.Rice AC, DeLorenzo RJ. NMDA receptor activation during status epilepticus is required for the development of epilepsy. Brain Res. 1998;782(1–2):240–247. doi: 10.1016/s0006-8993(97)01285-7. [DOI] [PubMed] [Google Scholar]
- 44.Hort J, Brozek G, Mares P, Langmeier M, Komarek V. Cognitive functions after pilocarpine-induced status epilepticus: changes during silent period precede appearance of spontaneous recurrent seizures. Epilepsia. 1999;40(9):1177–1183. doi: 10.1111/j.1528-1157.1999.tb00845.x. [DOI] [PubMed] [Google Scholar]
- 45.Prasad A, Williamson JM, Bertram EH. Phenobarbital and MK-801, but not phenytoin, improve the long-term outcome of status epilepticus. Ann Neurol. 2002;51(2):175–181. doi: 10.1002/ana.10085. [DOI] [PubMed] [Google Scholar]
- 46.Bardo S, Cavazzini MG, Emptage N. The role of the endoplasmic reticulum Ca2+ store in the plasticity of central neurons. Trends Pharmacol Sci. 2006;27(2):78–84. doi: 10.1016/j.tips.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 47.Friel D. Interplay between ER Ca2+ uptake and release fluxes in neurons and its impact on [Ca2+] dynamics. Biol Res. 2004;37(4):665–674. doi: 10.4067/s0716-97602004000400024. [DOI] [PubMed] [Google Scholar]
- 48.Lazarewicz JW, Rybkowski W, Sadowski M, et al. N-methyl-d-aspartate receptor-mediated, calcium-induced calcium release in rat dentate gyrus/CA4 in vivo. J Neurosci Res. 1998;51(1):76–84. doi: 10.1002/(SICI)1097-4547(19980101)51:1<76::AID-JNR8>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 49.Jin W, Sugaya A, Tsuda T, Ohguchi H, Sugaya E. Relationship between large conductance calcium-activated potassium channel and bursting activity. Brain Res. 2000;860(1–2):21–28. doi: 10.1016/s0006-8993(00)01943-0. [DOI] [PubMed] [Google Scholar]
- 50.Nagarkatti N, Deshpande LS, DeLorenzo RJ. Levetiracetam inhibits both ryanodine and IP3 receptor activated calcium induced calcium release in hippocampal neurons in culture. Neurosci Lett. 2008;436(3):289–293. doi: 10.1016/j.neulet.2008.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cataldi M, Lariccia V, Secondo A, di Renzo G, Annunziato L. The antiepileptic drug levetiracetam decreases the inositol 1,4,5-trisphosphate-dependent [Ca2+]i increase induced by ATP and bradykinin in PC12 cells. J Pharmacol Exp Ther. 2005;313(2):720–730. doi: 10.1124/jpet.104.079327. [DOI] [PubMed] [Google Scholar]
- 52.Carter DS, Harrison AJ, Falenski KW, Blair RE, DeLorenzo RJ. Long-term decrease in calbindin-D28K expression in the hippocampus of epileptic rats following pilocarpine-induced status epilepticus. Epilepsy Res. 2008;79(2–3):213–223. doi: 10.1016/j.eplepsyres.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Magloczky Z, Halasz P, Vajda J, Czirjak S, Freund TF. Loss of calbindin-D28K immunoreactivity from dentate granule cells in human temporal lobe epilepsy. Neuroscience. 1997;76(2):377–385. doi: 10.1016/s0306-4522(96)00440-x. [DOI] [PubMed] [Google Scholar]
- 54.Nagerl UV, Mody I, Jeub M, et al. Surviving granule cells of the sclerotic human hippocampus have reduced Ca2+ influx because of a loss of calbindin-D(28k) in temporal lobe epilepsy. J Neurosci. 2000;20(5):1831–1836. doi: 10.1523/JNEUROSCI.20-05-01831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baimbridge KG, Miller JJ. Hippocampal calcium-binding protein during commissural kindling-induced epileptogenesis: progressive decline and effects of anticonvulsants. Brain Res. 1984;324(1):85–90. doi: 10.1016/0006-8993(84)90624-3. [DOI] [PubMed] [Google Scholar]
- 56.Shetty AK, Turner DA. Intracerebroventricular kainic acid administration in adult rat alters hippocampal calbindin and non-phosphorylated neurofilament expression. J Comp Neurol. 1995;363(4):581–599. doi: 10.1002/cne.903630406. [DOI] [PubMed] [Google Scholar]
- 57.Lee S, Williamson J, Lothman EW, et al. Early induction of mRNA for calbindin-D28k and BDNF but not NT-3 in rat hippocampus after kainic acid treatment. Brain Res Mol Brain Res. 1997;47(1–2):183–194. doi: 10.1016/s0169-328x(97)00043-0. [DOI] [PubMed] [Google Scholar]
- 58.Sonnenberg JL, Frantz GD, Lee S, et al. Calcium binding protein (calbindin-D28k) and glutamate decarboxylase gene expression after kindling induced seizures. Brain Res Mol Brain Res. 1991;9(3):179–190. doi: 10.1016/0169-328x(91)90001-e. [DOI] [PubMed] [Google Scholar]
- 59.Lowenstein DH, Gwinn RP, Seren MS, Simon RP, McIntosh TK. Increased expression of mRNA encoding calbindin-D28K, the glucose-regulated proteins, or the 72 kDa heat-shock protein in three models of acute CNS injury. Brain Res Mol Brain Res. 1994;22(1–4):299–308. doi: 10.1016/0169-328x(94)90058-2. [DOI] [PubMed] [Google Scholar]
- 60.DeLorenzo RJ. The calmodulin hypothesis of neurotransmission. Cell Calcium. 1981;2(4):365–385. doi: 10.1016/0143-4160(81)90026-9. [DOI] [PubMed] [Google Scholar]
- 61.Blair RE, Sombati S, Churn SB, Delorenzo RJ. Epileptogenesis causes an N-methyl-d-aspartate receptor/Ca2+-dependent decrease in Ca2+/calmodulin-dependent protein kinase II activity in a hippocampal neuronal culture model of spontaneous recurrent epileptiform discharges. Eur J Pharmacol. 2008;588(1):64–71. doi: 10.1016/j.ejphar.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Erondu NE, Kennedy MB. Regional distribution of type II Ca2+/calmodulin-dependent protein kinase in rat brain. J Neurosci. 1985;5(12):3270–3277. doi: 10.1523/JNEUROSCI.05-12-03270.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carter DS, Haider SN, Blair RE, et al. Altered calcium/calmodulin kinase II activity changes calcium homeostasis that underlies epileptiform activity in hippocampal neurons in culture. J Pharmacol Exp Ther. 2006;319(3):1021–1031. doi: 10.1124/jpet.106.110403. [DOI] [PubMed] [Google Scholar]
- 64.Wasterlain CG, Farber DB. Kindling alters the calcium/calmodulin-dependent phosphorylation of synaptic plasma membrane proteins in rat hippocampus. Proc Natl Acad Sci USA. 1984;81(4):1253–1257. doi: 10.1073/pnas.81.4.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamagata Y, Imoto K, Obata K. A mechanism for the inactivation of Ca2+/calmodulin-dependent protein kinase II during prolonged seizure activity and its consequence after the recovery from seizure activity in rats in vivo. Neuroscience. 2006;140(3):981–992. doi: 10.1016/j.neuroscience.2006.02.054. [DOI] [PubMed] [Google Scholar]
- 66.Singleton MW, Holbert WH, 2nd, Ryan ML, et al. Age dependence of pilocarpine-induced status epilepticus and inhibition of CaM kinase II activity in the rat. Brain Res Dev Brain Res. 2005;156(1):67–77. doi: 10.1016/j.devbrainres.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 67.Churn SB, Sombati S, Jakoi ER, Severt L, DeLorenzo RJ. Inhibition of calcium/calmodulin kinase II α subunit expression results in epileptiform activity in cultured hippocampal neurons. Proc Natl Acad Sci USA. 2000;97(10):5604–5609. doi: 10.1073/pnas.080071697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Merrill MA, Chen Y, Strack S, Hell JW. Activity-driven postsynaptic translocation of CaMKII. Trends Pharmacol Sci. 2005;26(12):645–653. doi: 10.1016/j.tips.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 69.Dong Y, Rosenberg HC. Prolonged changes in Ca2+/calmodulin-dependent protein kinase II after a brief pentylenetetrazol seizure; potential role in kindling. Epilepsy Res. 2004;58(2–3):107–117. doi: 10.1016/j.eplepsyres.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 70.Friel DD, Chiel HJ. Calcium dynamics: analyzing the Ca2+ regulatory network in intact cells. Trends Neurosci. 2008;31(1):8–19. doi: 10.1016/j.tins.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 71.Ben-Ari Y. Cell death and synaptic reorganizations produced by seizures. Epilepsia. 2001;42(Suppl. 3):5–7. doi: 10.1046/j.1528-1157.2001.042suppl.3005.x. [DOI] [PubMed] [Google Scholar]
- 72.Morimoto K, Fahnestock M, Racine RJ. Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog Neurobiol. 2004;73(1):1–60. doi: 10.1016/j.pneurobio.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 73.Orrenius S, McCabe MJ, Jr, Nicotera P. Ca2+-dependent mechanisms of cytotoxicity and programmed cell death. Toxicol Lett. 1992;64–65(Spec. No):357–364. doi: 10.1016/0378-4274(92)90208-2. [DOI] [PubMed] [Google Scholar]
- 74.Fujikawa DG. Prolonged seizures and cellular injury: understanding the connection. Epilepsy Behav. 2005;7(Suppl. 3):S3–S11. doi: 10.1016/j.yebeh.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 75.Holmes GL. Seizure-induced neuronal injury: animal data. Neurology. 2002;59(9 Suppl. 5):S3–S6. doi: 10.1212/wnl.59.9_suppl_5.s3. [DOI] [PubMed] [Google Scholar]
- 76.Duncan JS. Seizure-induced neuronal injury: human data. Neurology. 2002;59(9 Suppl. 5):S15–S20. doi: 10.1212/wnl.59.9_suppl_5.s15. [DOI] [PubMed] [Google Scholar]
- 77.Liu RS, Lemieux L, Bell GS, et al. Cerebral damage in epilepsy: a population-based longitudinal quantitative MRI study. Epilepsia. 2005;46(9):1482–1494. doi: 10.1111/j.1528-1167.2005.51603.x. [DOI] [PubMed] [Google Scholar]
- 78.Pitkanen A, Nissinen J, Nairismagi J, et al. Progression of neuronal damage after status epilepticus and during spontaneous seizures in a rat model of temporal lobe epilepsy. Prog Brain Res. 2002;135:67–83. doi: 10.1016/S0079-6123(02)35008-8. [DOI] [PubMed] [Google Scholar]
- 79.Simpson PB, Challiss RA, Nahorski SR. Neuronal Ca2+ stores: activation and function. Trends Neurosci. 1995;18(7):299–306. doi: 10.1016/0166-2236(95)93919-o. [DOI] [PubMed] [Google Scholar]
- 80.Sanchez RM, Dai W, Levada RE, Lippman JJ, Jensen FE. AMPA/kainate receptor-mediated downregulation of GABAergic synaptic transmission by calcineurin after seizures in the developing rat brain. J Neurosci. 2005;25(13):3442–3451. doi: 10.1523/JNEUROSCI.0204-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Deshpande LS, Nagarkatti N, Ziobro JM, Sombati S, Delorenzo RJ. Carisbamate prevents the development and expression of spontaneous recurrent epileptiform discharges and is neuroprotective in cultured hippocampal neurons. Epilepsia. 2008;49(10):1795–1802. doi: 10.1111/j.1528-1167.2008.01667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grabenstatter HL, Dudek FE. A new potential AED, carisbamate, substantially reduces spontaneous motor seizures in rats with kainate-induced epilepsy. Epilepsia. 2008;49(10):1787–1794. doi: 10.1111/j.1528-1167.2008.01657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Deshpande LS, Nagarkatti N, Sombati S, DeLorenzo RJ. The novel antiepileptic drug carisbamate (RWJ 333369) is effective in inhibiting spontaneous recurrent seizure discharges and blocking sustained repetitive firing in cultured hippocampal neurons. Epilepsy Res. 2008;79(2–3):158–165. doi: 10.1016/j.eplepsyres.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pitkanen A, Lukasiuk K. Molecular and cellular basis of epileptogenesis in symptomatic epilepsy. Epilepsy Behav. 2009;14(Suppl. 1):16–25. doi: 10.1016/j.yebeh.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 85.Pisani A, Bonsi P, Martella G, et al. Intracellular calcium increase in epileptiform activity: modulation by levetiracetam and lamotrigine. Epilepsia. 2004;45(7):719–728. doi: 10.1111/j.0013-9580.2004.02204.x. [DOI] [PubMed] [Google Scholar]
- 86.Kulak W, Sobaniec W, Wojtal K, Czuczwar SJ. Calcium modulation in epilepsy. Pol J Pharmacol. 2004;56(1):29–41. [PubMed] [Google Scholar]
- 87.Brandt C, Potschka H, Loscher W, Ebert U. N-methyl-d-aspartate receptor blockade after status epilepticus protects against limbic brain damage but not against epilepsy in the kainate model of temporal lobe epilepsy. Neuroscience. 2003;118(3):727–740. doi: 10.1016/s0306-4522(03)00027-7. [DOI] [PubMed] [Google Scholar]
- 88.Loscher W. Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epilepsy Res. 2002;50(1–2):105–123. doi: 10.1016/s0920-1211(02)00073-6. [DOI] [PubMed] [Google Scholar]
- 89.Rogawski MA, Wenk GL. The neuropharmacological basis for the use of memantine in the treatment of Alzheimer’s disease. CNS Drug Rev. 2003;9(3):275–308. doi: 10.1111/j.1527-3458.2003.tb00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rice AC, DeLorenzo RJ. N-methyl-d-aspartate receptor activation regulates refractoriness of status epilepticus to diazepam. Neuroscience. 1999;93(1):117–123. doi: 10.1016/s0306-4522(99)00132-3. [DOI] [PubMed] [Google Scholar]
Website
- 101.Internet Stroke Center at Washington Univ. in St. Louis. The Stroke Trials Registry. www.strokecenter.org/trials/InterventionDetail.aspx?tid=95.