

Abstract
Diketopiperazines (DKPs) are a common motif in various biologically active natural products, and hence they may be useful scaffolds for the rational design of receptor probes and therapeutic agents. We constructed a new bicyclic scaffold that combines a DKP bridged with a 10-membered ring. In this way we obtained a three-dimensional molecular skeleton, with several amendable sites that provide a starting point to design a new combinatorial library having diverse substituent groups. Structural variation is based upon the flexibility of alkylation of the nitrogen atoms of the DKP and on the side-chain olefin. We obtained a 10-membered secondary ring through a ring-closure metathesis reaction using the second generation Grubbs catalyst. Rings containing both O-ethers and S-ethers were compared. N-Alkyl or arylalkyl groups were introduced optionally at the two Nα-atoms. This is a general scheme that will allow us to test rings of varying sizes, linkages, and stereochemical parameters. The DKP derivatives were tested for activity in astrocytoma cells expressing receptors coupled to phospholipase C. Inhibitory effects were observed for signaling elicited by activation of human nucleotide P2Y receptors but not m3 muscarinic receptors. Compound 20 selectively inhibited calcium mobilization (IC50 value of 486 ± 16 nM) and phosphoinositide turnover elicited by a selective P2Y1 receptor agonist, but this compound did not compete for binding of a radiolabeled nucleotide-competitive receptor antagonist. Therefore, the new class of DKP derivatives shows utility as pharmacological tools for P2Y receptors.
Introduction
The diketopiperazine (DKP) ring has been noted as a motif in various biologically active natural products. Examples include the commercial antibiotic bicyclomycin,1 the fungal metabolites TAN-14 A, C and E,2 and the thaxtomin A and B.3 DKPs may be useful scaffolds for the rational design of receptor probes and drugs. Owing to the presence of multiple H-bond acceptors and donors and the side chains of the component amino acids, DKPs have multiple sites for structural elaboration of defined stereochemistry. Others have explored the synthesis of DKPs of varied structure,4–8 and combinatorial/polymer-supported syntheses of DKPs have been reported.9–12
One of the limitations of conventional medicinal chemical agents, as applied to the new domain of chemical genetics,13,14 is the typical planarity found in most known pharmaceutical substances discovered through organic synthesis. We desired to explore a three-dimensional molecular skeleton, which could be regarded as a starting point to design a new combinatorial library having diverse substituent groups. In the present study we have built a new bicyclic scaffold that combines a DKP additionally bridged with a 10-membered ring. A similar approach to synthesize a series of bridged DKPs, which were desired as mimics of a peptide β-turn found in peptide hormones, was introduced by Reitz and co-workers.15
Construction of the DKP skeleton was achieved with amino acid derivatives N-Boc-L-serine and L-allylglycine, thus providing great flexibility for structural variation, including substitution at the nitrogen atoms of the DKP and on the side-chain olefin. The 10-membered ring was obtained through a ring-closure metathesis (RCM) reaction using the second generation Grubbs catalyst.16,17 This methodology provides a bicyclic DKP template with ether and olefinic groups present. This is a general scheme that will allow us to test larger and smaller rings, vary the ether linkage and manipulate stereochemical parameters. The ether and olefinic functionality is desired for both chemical advantages (i.e. the ability to derivatize at these positions) and biological advantages (i.e. to provide additional recognition elements for interaction with receptors and other biopolymers).
Selected bicyclic DKP analogues were subjected to biological assays in a variety of receptor-based systems. Several of the derivatives were found to inhibit the activity of extracellular nucleotides through G protein-coupled P2Y receptors and subsequent biochemical steps. Eight mammalian subtypes of 18–21 such receptors (P2Y1,2,4,6,11–14) are currently sequence-defined. The P2Y receptors are present in the cardiovascular, immune, and nervous systems, as well as in other systems. They are currently the focus of intense drug discovery efforts to design selective agonists and antagonists, which are lacking for most of the subtypes. There are numerous therapeutic possibilities under exploration for P2Y receptor modulators. For example, antagonists of the P2Y1 and P2Y12 receptors are of interest as antithrombotic agents.22–32 Agonists of the P2Y2 receptor are of interest in the treatment of pulmonary diseases, including cystic fibrosis. The P2Y6 receptor has recently been implicated in protection against apoptosis induced by TNFα.33
Results
Chemical synthesis
A new bicyclic scaffold that combines a DKP bridged with a 10-membered ring was prepared. This 10-membered secondary ring was obtained through a RCM reaction using the second generation Grubbs catalyst. The starting materials in this synthesis (Schemes 1–3) were two amino acid derivatives of defined stereochemistry, N-Boc-L-serine and L-allylglycine, which provided the bicyclic DKP template with both ether and olefinic groups present. In order to substitute the O-ether moiety with an S-ether, L-cysteine 6 was used as the starting material instead of N-Boc-L-serine 1.
Scheme 1.

Preparation of protected Ser and Cys derivatives. Reagents and conditions: (i) NaH, allylbromide, DMF, rt, 94%; (ii) TMSCHN2, Et2 O–MeOH 1: 1, rt, 78%; (iii) TFA, CH2 Cl2, rt, 93% (5); (iv) EtONa, allylbromide, EtOH, rt, 76%; (v) TMSCHN2, Et2 O–CH2 Cl2 –MeOH 1: 5: 5, rt, 33% (13), 32% (14), 55% (15), 22% (16); (vi) 2,4-dimethoxybenzaldehyde or 2,4,6-trimethoxybenzaldehyde, NaBH3 CN, MeOH, rt, 53% (9), 53% (10), 41% (11), 41% (12); (vii) 2,4-dimethoxybenzaldehyde or 2,4,6-trimethoxybenzaldehyde, NaBH(OAc)3, CH2 Cl2, rt, 85% (13), 82% (14), 74% (15), 58% (16), *partial racemization was obtained in this reaction.
Scheme 3.

Synthesis of bicyclic DKP derivatives. The closure of the DKP ring preceded the RCM reaction. DMB = 2,4-(MeO)2 PhCH2. Reagents and conditions: (i) piperidine–CH2 Cl2 1: 4, rt, 42%; (ii) 37 (0.2 mM), second generation Grubbs catalyst, CH2 Cl2, rt, 21% (20), 43% (38); 37 (40 mM), 79% (38).
The first step of the synthesis was the introduction of the allyl group in the N-Boc-L-serine and L-cysteine using the methods reported in the literature.34,35 Esterification of compound 2 with TMSCHN2 followed by removal of the Boc group with TFA afforded the amino ester 5. The intermediate 8 was obtained after esterification of the amino acid 7 (Scheme 1).
The amino methyl ester 5 was subjected to reductive amination in the presence of 2,4-dimethoxybenzaldehyde or 2,4,6-trimethoxybenzaldehyde to give compounds 13 and 14, respectively. Following the same procedure, the thio derivatives 15 and 16 were obtained from 8 (Scheme 1). Our initial procedure, in order to minimize racemization, involved mixing the amine, NaBH(OAc)3 and the aldehyde simultaneously in CH2Cl2. 15,36,37 The reactions carried at room temperature for 2 h afforded the desired compounds but with racemization that was partial (70: 30) for the dimethoxybenzyl derivatives 13 and 15 and total (50: 50) for the trimethoxybenzyl derivatives 14 and 16. An alternative route consisted of the reductive amination followed by esterification. The amino acid derivative 4 was subjected to reductive amination using NaBH3CN in methanol at room temperature for 24 h to obtain 9, which after esterification with TMSCHN2 provided 13 with less than 5% of racemization. Following the same procedure, 14–16 were obtained from 4 or 7 with a similar degree of racemization.
Fmoc-L-allylglycine (17) was coupled with the secondary amine 13 using O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl-uronium hexafluorophosphate (HATU), 1-hydroxy-7-azabenzo-triazole (HOAt) and N-ethylmorpholine (NEM) in a mixture of CH2Cl2–DMF (9: 1) to obtain the dipeptide 18 (Scheme 2). RCM of 18 (1 mM) using the second generation Grubbs catalyst in dichloromethane at room temperature provided 19. When the first generation Grubbs catalyst was used with 18 no reaction was observed. Finally, removal of the Fmoc group of 19 using a mixture of piperidine–CH2Cl2 (1: 4) was accompanied by in situ formation of the DKP, and compound 20 was obtained in high yield (93%).
Scheme 2.

Synthesis of bicyclic DKP derivatives. The RCM reaction preceded the closure of the DKP ring. Reagents and conditions: (i) HATU, HOAt, NEM, CH2 Cl2 –DMF 9: 1, rt, 53% (18), 40% (23), 37% (31), 41% (34); (ii) second generation Grubbs catalyst, CH2 Cl2, rt, 60% (19), 57% (24), 57% (32), 71% (35); (iii) piperidine–CH2 Cl2 1: 4, rt, 93% (20), 73% (25), 73% (33), 60% (36); (iv) CH2 Cl2 –TFA 2: 1, rt, 96%; (v) MeI or BnBr, NaH, THF, rt, 63% (21), 60% (22), 96% (27), 85% (28); (vi) CH2 Cl2 –TFA 2: 1, rt, 63% (29), 89% (30).
Similar reaction sequences starting from compounds 14, 15 and 16, instead of 13, provided the bicyclic DKP templates 25, 33 and 36.
In order to introduce more diversity in the scaffold, the amide in the DKP was alkylated with methyl and benzyl groups.38 Compounds 20 and 25 were treated with NaH and the resulting anions reacted with methyl iodide and benzyl bromide to give 21, 22, 27 and 28.
The trimethoxybenzyl group in 25, 27 and 28 was removed easily under acidic conditions; however, the normally acid-labile dimethoxybenzyl group in 20 was stable under these conditions.36
By this route the new bicyclic scaffold was obtained through RCM followed by the formation of the DKP ring (Scheme 2). The possibility was explored to obtain these derivatives through a new route, by which the DKP ring was built first and then the 10-membered ring was formed (Scheme 3). In this case, compound 18 reacted with the mixture of piperidine–CH2Cl2 (1: 4) to obtain the DKP 37. The RCM reaction of 37 using first generation Grubbs catalyst was not satisfactory. When the second generation catalyst was used and the reaction was performed in conditions of high dilution (0.2 mM), 20 (21%) was obtained with the dimer 38 (43%). However, with higher concentrations (40 mM) only the dimer 38 was obtained (79%).
Pharmacological activity
Compounds 20–22, 25–30, 33, and 36 were screened at a 10 lM concentration in an assay of phospholipase C in 1321N1 human astrocytoma cells stably expressing the human P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11 receptors. The biological characterization of the dimeric 38 will be reported elsewhere. In P2Y1 receptor-expressing cells, inhibition of agonist (30 nM 2-MeSADP)-induced phospholipase C (PLC) was observed only for 20 (45 ± 10%), 33 (33 ± 4%), and 36 (18 ± 4%). At the other P2Y receptor subtypes, substantial inhibition of PLC activity (ca. 50% at 10 μM) was observed only for compounds 21 and 36 at the P2Y4 receptor and compound 20 at the P2Y6 receptors. Compound 20 and other DKPs (Fig. 1) inhibited the mobilization of intracellular [Ca2+] in astrocytoma cells stably expressing the human P2Y1 receptor, with concentration-dependent reduction of the maximal effect. Compound 20 was the most potent with complete inhibition of [Ca2+] mobilization at 10 μM. Increasing concentrations of compound 20 progressively attenuated the 2MeSADP effect (Fig. 2). Compound 20 was found to inhibit [Ca2+]i transients elicited by 2MeSADP with an IC50 value of 486 ± 16 nM. Control experiments in which PLC was stimulated via an endogenous m3 muscarinic receptor (using carbachol) in the control astrocytoma cells or via heterologously expressed P2Y2,4,6 receptors (using the appropriate nucleotide) demonstrated insignificant or weak inhibition by compound 20. Thus, the inhibitory effects of 20, albeit insurmountable, were selective for signaling induced by P2Y1 receptor activation. However, in a radioreceptor binding assay compound 20 at 10 μM failed to compete for the binding of a specific P2Y1 antagonist radioligand, [3H]MRS2279.39 Therefore, the binding of the DKP was not occurring at the principal nucleotide binding site of this receptor.
Fig. 1.
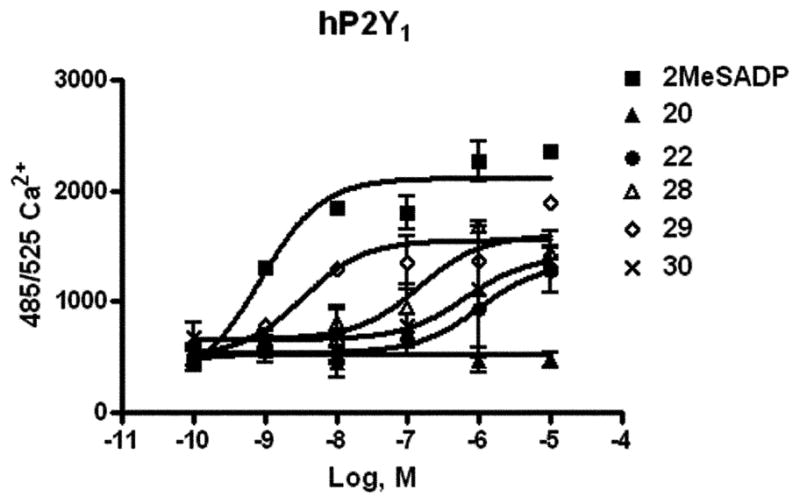
Effects of DKP derivatives (10 μM, structures in Scheme 2) on concentration–response curves for intracellular Ca2+ changes induced by 2-MeSADP acting at P2Y1 receptors expressed in 1321N1 astrocytoma cells. The cells were pre-treated for 20 min at room temperature with antagonist before application of the agonist 2-MeSADP.
Fig. 2.
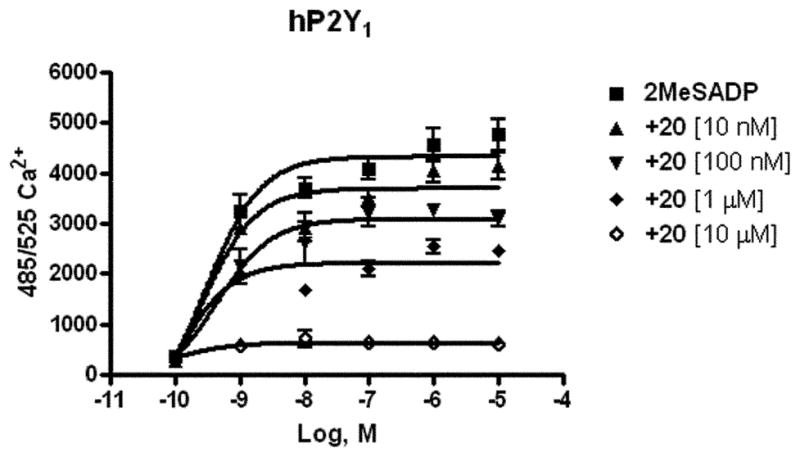
Effect of compound 20 on the concentration–response curve of the [Ca2+]i transient induced by 2MeSADP in P2Y1 receptors expressed in 1321N1 astrocytoma cells. The cells were pre-treated for 20 min at room temperature with different concentrations of compound 20 before application of the agonist 2-MeSADP.
As an indication of the selectivity of these derivatives for the P2Y receptor pathway, interactions with a limited number of other receptors were examined (data not shown). Binding experiments at the human A1 AR expressed in CHO cell membranes40 indicated that compound 20 at 10 μM displayed no significant inhibition of radioligand binding. Similarly, this compound failed to displace binding at the mouse TRH receptors, or to either activate or antagonize functional effects of the m3 muscarinic receptor or the human calcium sensing receptor.
By means of a molecular dynamics simulation we carried out a conformational analysis of compound 20. The DKP ring-puckering coordinates (Θ≅ −80°; P2 ≅ −30°; Q ≅ 40°) indicated a boat–screw/boat conformation.
Two different stable conformations were found for the 10-membered ring: in the first conformation the ring bends toward the dimethoxybenzyl group, while in the second one it bends in the opposite direction. Two diametrically opposite stable conformations were also found for the dimethoxybenzyl group. The coordinates of two representative conformations, in PDB format, are supplied as ESI.†
Consistent with the non-competitive nature of these ligands, the stable conformations of compound 20 do not resemble the bound conformation of the competitive P2Y1 antagonist MRS2279, neither by steric nor electronic criteria.41
Discussion
These DKP scaffold molecules were intended for broad screening with no a priori bias for any particular protein target. Among the targets thus far examined, inhibition was associated exclusively with the P2Y receptors. These receptors are diverse in the nature of their endogenous nucleotide ligands. Adenine nucleotides are required for activation of the P2Y1, P2Y11, P2Y12, and P2Y13 subtypes, and uracil nucleotides activate P2Y2, P2Y4, and P2Y6 subtypes. The P2Y2 receptor is equipotently activated by UTP and ATP. The most recently identified subtypes are the P2Y13 receptor, which is present in human monocytes and lymphocytes,42 and the P2Y14 receptor, which is activated by UDP-glucose.43 P2Y receptor ligands are being investigated for therapeutic applications in the cardiovascular, endocrine, and other systems. Exploration of selective agonists and antagonists modulators is most advanced at the P2Y1 and P2Y12 receptors, but at most of the P2Y receptors selective pharmacological probes are lacking.22–32 Compound 20 was shown to antagonize signaling of the P2Y1 receptor selectively and with moderate potency. At the P2Y1 receptor, the previously described high affinity antagonists are nucleotide derivatives and therefore highly charged, which is highly limiting in pharmacological studies due to low bioavailability and stability.
Therefore, owing to the ability to modify the chemical functionality of this new class of DKP derivatives and the already demonstrated biological activity, it shows promise as an approach to designing new pharmacological tools. Further study will be required to explore the mechanism of inhibition of P2Y receptor-induced effects and to determine the overall pharmacological selectivity of this class of compounds. It must still be determined if manipulation of functional groups on the DKP derivatives is able to vary the spectrum of interaction of these scaffold molecules with signaling of specific P2Y receptor subtypes, and among varied biochemical target proteins in general. If the inhibition by the DKP is found to occur in direct association with P2Y receptors, e.g. at an allosteric site, several of the compounds already prepared, such as compound 20, will provide insurmountable antagonists with receptor selectivity.
Experimental
Chemical synthesis
Reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO). Compounds 2 and 7 were synthesized as reported.34,35
1H NMR spectra were obtained with a Varian Gemini 300 spectrometer using CDCl3, CD3OD or D2O as solvents. The chemical shifts are expressed as ppm downfield from TMS. All melting points were determined with a Thomas-Hoover apparatus (A.H. Thomas Co.) and are uncorrected.
Purity of the compounds was checked using a Hewlett-Packard 1100 HPLC equipped with a Luna 5 μm RP-C18(2) analytical column (250 × 4.6 mm; Phenomenex, Torrance, CA). System A: linear gradient solvent system: H2O–CH3CN from 80: 20 to 0: 100 in 20 min, then isocratic for 5 min; the flow rate was 1 mL min−1. System B: linear gradient solvent system: 5 mM TBAP–CH3CN from 80: 20 to 20: 80 in 20 min, then isocratic for 2 min; the flow rate was 1 mL min−1. Peaks were detected by UV absorption with a diode array detector. All derivatives tested for biological activity showed >98% purity in the HPLC systems.
TLC analysis was carried out on aluminium sheets precoated with silica gel F254 (0.2 mm) from Aldrich. Low-resolution CI–NH3 (chemical ionization) mass spectra were measured with a Finnigan 4600 mass spectrometer, and low-resolution FAB (fast atom bombardment) mass spectrometry was performed with a JEOL SX102 spectrometer with 6 kV Xe atoms following desorption from a glycerol matrix or on LC/MS 1100 Agilent, 1100 MSD, with a Waters Atlantis C18 column. High-resolution mass measurements were performed on a Micromass/Waters LCT Premier Electrospray Time of Flight (TOF) mass spectometer coupled with a Waters HPLC system.
(2S)-3-Allyloxy-2-tert-butoxycarbonylamino-propionic acid methyl ester (3)
To a solution of compound 2 (4.48 g, 18.3 mmol) in 130 mL of Et2O–MeOH (1: 1) was added TMSCHN2 (2 M in Et2O) dropwise until a yellow tint persisted. The reaction mixture was stirred at room temperature for 30 min. The solvent was removed in vacuo, and the residue was purified by silica gel column chromatography (petroleum ether–ethyl acetate, 15: 1) to afford 3 (3.71 g, 78%). 1H NMR (CDCl3) δ 5.83 (m, 1H, CH =CH2), 5.38 (br d, J = 8.8 Hz, 1H, NH), 5.21 (m, 2H, CH=CH 2), 4.43 (m, 1H, H-2), 3.98 (m, 2H, CH 2CH=CH2), 3.85 (m, 1H, 1H-3), 3.77 (s, 3H, OCH3), 3.65 (m, 1H, 1H-3), 1.46 [s, 9H, C(CH3)3]; MS (FAB) m/z 260 (M + H)+.
(2S)-3-Allyloxy-2-amino-propionic acid (4)
To a solution of compound 2 (242 mg, 0.99 mmol) in CH2Cl2 (15 mL) was added TFA (1 mL). The reaction mixture was stirred at room temperature for 2 h. The solvent was removed in vacuo to obtain compound 4 as a crude solid (520 mg), which then was used directly in the next step without further purification. 1H NMR (D2O) δ 5.96 (m, 1H, CH =CH2), 5.33 (m, 2H, CH=CH 2), 4.11 (m, 2H, CH 2CH=CH2), 3.93 (m, 3H, H-2, H-3).
(2S)-3-Allyloxy-2-amino-propionic acid methyl ester (5)
To a solution of compound 3 (1.02 g, 3.93 mmol) in CH2Cl2 (60 mL) was added TFA (10 mL). The reaction mixture was stirred at room temperature for 30 min, and then was neutralized with the addition of saturated NaHCO3 (225 mL). The product was extracted with CH2Cl2, dried over Na2SO4, and evaporated to obtain compound 5 (585 mg, 93%). 1H NMR (CDCl3) δ 5.87 (m, 1H, CH=CH2), 5.22 (m, 2H, CH=CH 2), 4.01 (m, 2H, CH 2CH=CH2), 3.75 (s, 3H, OCH3), 3.68 (m, 3H, H-2, H-3).
(2R)-3-Allylsulfanyl-2-amino-propionic acid methyl ester (8)
To a suspension of compound 7 (1.21 g, 7.49 mmol) in 55 mL of Et2O–MeOH–CH2Cl2 (1:5:5), was added TMSCHN2 (2 M in Et2O) dropwise until a yellow tint persisted. The reaction mixture was stirred at room temperature for 2 h. The solvent was removed in vacuo to obtain compound 8 as a crude solid (1.46 g), which then was used directly in the next step without further purification. 1H NMR (CDCl3) δ 5.78 (m, 1H, CH =CH2), 5.12 (m, 2H, CH=CH 2), 3.75 (s, 3H, OCH3), 3.64 (m, 1H, H-2), 3.15 (m, 2H, CH 2CH=CH2), 2.88 (m, 1H, 1H-3), 2.71 (m, 1H, 1H-3); MS (CI) m/z 176 (M + H)+.
(2S)-3-Allyloxy-2-(2,4-dimethoxybenzylamino)-propionic acid (9)
(Procedure A) To a solution containing the crude solid 4 (260 mg) in MeOH (20 mL) was added NaBH3CN (38 mg, 0.58 mmol) and 2,4-dimethoxybenzaldehyde (75 mg, 0.45 mmol). The reaction mixture was stirred at room temperature for 24 h. Water (3 mL) was added, and the product was extracted with CH2Cl2 and dried over Na2SO4. After removal of the solvent, the residue was purified by column chromatography on silica gel (CH2Cl2–MeOH, 92: 8) to give 9 (77 mg, 53% from 2) as a white solid. 1H NMR (CDCl3) δ 7.25 (d, J = 8.7 Hz, 1H, Ar–H), 6.47 (m, 2H, Ar–H), 5.83 (m, 1H, CH=CH2), 5.19 (m, 2H, CH=CH 2), 4.76 (br s, 1H, NH), 4.39 (d, J = 13.0 Hz, 1H, HCHAr), 4.24 (d, J = 13.0 Hz, 1H, HCHAr), 4.01–3.69 (m, 4H, CH 2CH=CH2, H-3), 3.85 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 3.56 (m, 1H, H-2). HRMS m/z found 296.1501 (M + H)+. C15H22NO5 requires 296.1498. Mp 135–136 °C.
(2S)-3-Allyloxy-2-(2,4,6-trimethoxybenzylamino)-propionic acid (10)
(Procedure A) Compound 10 (84 mg, 53% from 2) was obtained from a solution of crude solid 4 (260 mg), NaBH3CN (38 mg, 0.58 mmol) and 2,4,6-trimethoxybenz-aldehyde (89 mg, 0.45 mmol) in MeOH (20 mL). 1H NMR (CDCl3) δ 6.11 (s, 2H, Ar–H), 5.79 (m, 1H, CH =CH2), 5.54 (br s, 1H, NH), 5.17 (m, 2H, CH=CH 2), 4.49 (d, J = 13.2 Hz, 1H, HCHAr), 4.40 (d, J = 13.2 Hz, 1H, HCHAr), 3.98–3.74 (m, 3H, CH 2CH=CH2, 1H-3), 3.82 (s, 9H, OCH3), 3.68 (m, 1H, 1H-3), 3.54 (m, 1H, H-2). HRMS m/z found 326.1588 (M +H)+. C16H24NO6 requires 326.1604.
(2R)-3-Allylsulfanyl-2-(2,4-dimethoxybenzylamino)-propionic acid (11)
(Procedure A) Compound 11 (150 mg, 41%) as a white solid was obtained from 7 (206 mg, 1.28 mmol), NaBH3CN (100 mg, 1.51 mmol) and 2,4-dimethoxybenzaldehyde (197 mg, 1.16 mmol) in MeOH (40 mL). 1H NMR (CDCl3) δ 7.30 (d, J = 8.7 Hz, 1H, Ar–H), 6.81 (br s, 1H, NH), 6.48 (m, 2H, Ar–H), 5.64 (m, 1H, CH=CH2), 5.05 (m, 2H, CH=CH 2), 4.45 (d, J = 13.2 Hz, 1H, HCHAr), 4.18 (d, J = 13.2 Hz, 1H, HCHAr), 3.87 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 3.41 (m, 1H, H-2), 3.16 (m, 1H, HCHCH=CH2), 2.92 (m, 3H, HCH CH=CH2, H-3). HRMS m/z found 312.1265 (M + H)+. C15H22NO4S requires 312.1270. Mp 150–151 °C.
(2R)-3-Allylsulfanyl-2-(2,4,6-trimethoxybenzylamino)-propionic acid (12)
(Procedure A) Compound 12 (168 mg, 41%) was obtained from 7 (214 mg, 1.33 mmol), NaBH3CN (104 mg, 1.57 mmol) and 2,4,6-trimethoxybenzaldehyde (242 mg, 1.21 mmol) in MeOH (40 mL). 1H NMR (CDCl3) δ 6.13 (s, 2H, Ar–H), 6.06 (br s, 1H, NH), 5.61 (m, 1H, CH=CH2), 5.06 (m, 2H, CH=CH 2), 4.49 (d, J = 13.3 Hz, 1H, H CHAr), 4.41 (d, J = 13.3 Hz, 1H, HCHAr), 3.85 (s, 6H, OCH3), 3.82 (s, 3H, OCH3), 3.34 (m, 1H, H-2), 3.17 (m, 1H, HCHCH=CH2), 2.84 (m, 3H, HCHCH=CH2, H-3). MS (API-ES) m/z 342 (M + H)+.
(2S)-3-Allyloxy-2-(2,4-dimethoxybenzylamino)-propionic acid methyl ester (13)
(Procedure B) To a solution of compound 5 (169 mg, 1.06 mmol) in CH2Cl2 (30 mL) was added NaBH(OAc)3 (280 mg, 1.26 mmol) and 2,4-dimethoxybenzaldehyde (164 mg, 0.97 mmol). The reaction mixture was stirred at room temperature for 2 h. Water (30 mL) was added, the product was extracted with CH2Cl2 and dried over Na2SO4. After removal of the solvent, the residue was purified by column chromatography on silica gel (petroleum ether–ethyl acetate, 2: 1) to give 13 (253 mg, 85%). (Procedure C) To a suspension of compound 9 (23 mg, 0.08 mmol) in 5.5 mL of Et2O–MeOH–CH2Cl2 (1: 5: 5) was added TMSCHN2 (2 M in Et2O) until a yellow tint persisted. The reaction mixture was stirred at room temperature for 30 min. After removal of the solvent, the product was purified by preparative thin-layer chromatography (petroleum ether–ethyl acetate, 2: 1) to give 13 (8 mg, 33%). 1H NMR (CDCl3) δ 7.14 (d, J = 8.7 Hz, 1H, Ar–H), 6.42 (m, 2H, Ar–H), 5.84 (m, 1H, CH=CH2), 5.19 (m, 2H, CH=CH 2), 3.95 (m, 2H, CH 2CH=CH2), 3.80 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 3.75 (d, J = 8.4 Hz, 2H, CH 2Ar), 3.69 (s, 3H, OCH3), 3.63 (m, 2H, H-3), 3.49 (m, 1H, H-2), 2.12 (br s, 1H, NH); MS (FAB) m/z 310 (M + H)+.
(2S)-3-Allyloxy-2-(2,4,6-trimethoxybenzylamino)-propionic acid methyl ester (14)
(Procedure B) Compound 14 (934 mg, 82%) was obtained from 5 (585 mg, 3.68 mmol), NaBH(OAc)3 (970 mg, 4.35 mmol) and 2,4,6-trimethoxybenzaldehyde (670 mg, 3.34 mmol) in CH2Cl2 (70 mL). (Procedure C) Compound 14 (12 mg, 32%) was obtained from 10 (36 mg, 0.11 mmol) and TMSCHN2 (2 M in Et2O) in Et2O–MeOH–CH2Cl2 (1: 5: 5) (5.5 mL). 1H NMR (CDCl3) δ 6.09 (s, 2H, Ar–H), 5.83 (m, 1H, CH=CH2), 5.17 (m, 2H, CH=CH 2), 3.92 (m, 2H, CH 2CH=CH2), 3.83 (d, J = 2.1 Hz, 2H, CH 2Ar), 3.80 (s, 3H, OCH3), 3.79 (s, 6H, OCH3), 3.63 (s, 3H, OCH3), 3.59 (m, 2H, H-3), 3.50 (m, 1H, H-2), 2.71 (br s, 1H, NH); MS (FAB) m/z 340 (M + H)+.
(2R)-3-Allylsulfanyl-2-(2,4-dimethoxybenzylamino)-propionic acid methyl ester (15)
(Procedure B) Compound 15 (797 mg, 74% from 7) was obtained from a solution containing crude solid 8 (731 mg), NaBH(OAc)3 (988 mg, 4.43 mmol) and 2,4-dimethoxybenzaldehyde (578 mg, 3.41 mmol) in CH2Cl2 (65 mL). The product was purified by column chromatography on silica gel (petroleum ether–ethyl acetate, 3: 1). (Procedure C) Compound 15 (76 mg, 55%) was obtained from 11 (132 mg, 0.42 mmol) and TMSCHN2 (2 M in Et2O) in Et2O–MeOH (1: 1) (10 mL). The product was purified by column chromatography on silica gel (petroleum ether–ethyl acetate, 4: 1). 1H NMR (CDCl3) δ 7.12 (d, J = 8.7 Hz, 1H, Ar–H), 6.42 (m, 2H, Ar–H), 5.73 (m, 1H, CH=CH2), 5.06 (m, 2H, CH=CH 2), 3.81 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 3.76 (m, 2H, CH 2Ar), 3.69 (s, 3H, OCH3), 3.41 (m, 1H, H-2), 3.05 (m, 2H, CH 2CH=CH2), 2.74 (m, 2H, H-3); MS (CI) m/z 326 (M + H)+.
(2R)-3-Allylsulfanyl-2-(2,4,6-trimethoxybenzylamino)-propionic acid methyl ester (16)
(Procedure B) Compound 16 (697 mg, 58% from 7) was obtained from a solution containing crude solid 8 (731 mg), NaBH(OAc)3 (988 mg, 4.43 mmol) and 2,4,6-trimethoxybenzaldehyde (682 mg, 3.41 mmol) in CH2Cl2 (65 mL). The product was purified by column chromatography on silica gel (petroleum ether–ethyl acetate, 2: 1). (Procedure C) Compound 16 (35 mg, 22%) was obtained from 12 (154 mg, 0.45 mmol) and TMSCHN2 (2M in Et2O) in Et2O–MeOH (1: 1) (12 mL). The product was purified by column chromatography on silica gel (petroleum ether–ethyl acetate, 4: 1). 1H NMR (CDCl3) δ 6.10 (s, 2H, Ar–H), 5.72 (m, 1H, CH =CH2), 5.05 (m, 2H, CH=CH 2), 3.81 (m, 2H, CH 2Ar), 3.80 (s, 3H, OCH3), 3.79 (s, 6H, OCH3), 3.65 (s, 3H, OCH3), 3.37 (m, 1H, H-2), 3.03 (m, 2H, CH 2CH=CH2), 2.72 (m, 2H, H-3), 2.42 (br s, 1H, NH); MS (CI) m/z 356 (M + H)+.
(2S)-(9H -Fluoren-9-ylmethoxycarbonylamino)-pent-4-enoic acid (17)
A solution of Fmoc-Cl (900 mg, 3.38 mmol) in dioxane (5 mL) was added, at 0 °C, to a suspension of L-allylglycine (299 mg, 2.60 mmol) and NaHCO3 (655 mg, 7.79 mmol) in 16.8 mL H2O–dioxane (1.4: 1). The mixture was stirred for 15 min at 0 °C and then at room temperature for 4.5 h. The pH was adjusted to approximately 9 with the addition of solid NaHCO3 and the mixture was diluted with H2O (120 mL) and washed with Et2O. The aqueous layer was acidified to pH = 3 with an aqueous HCl solution (6 M). The product was extracted with EtOAc, dried over Na2SO4 and the solvent was removed under reduced pressure. The resulting residue was purified by column chromatography on silica gel (CH2Cl2–MeOH, 97: 3) to afford 17 (615 mg, 70%). 1H NMR (CDCl3) δ 7.75 (d, J = 7.5 Hz, 2H, Ar–H), 7.57 (d, J = 6.9 Hz, 2H, Ar–H), 7.39 (t, J = 7.4 Hz, 2H, Ar–H), 7.29 (t, J = 7.4 Hz, 2H, Ar–H), 5.71 (m, 1H, H-4), 5.34 (br d, J = 7.8 Hz, 1H, NH), 5.17 (m, 2H, H-5), 4.47 (m, 1H, H-2), 4.39 (d, J = 6.9 Hz, 2H, CHCH 2), 4.21 (t, J = 6.9 Hz, 1H, CH CH2), 2.58 (m, 2H, H-3); MS (FAB) m/z 338 (M + H)+.
(2S,2′S)-3-Allyloxy-2-{(2,4-dimethoxybenzyl)-[2′-(9H-fluoren-9-ylmethoxycarbonylamino)-pent-4′-enoyl]-amino}-propionic acid methyl ester (18)
(Procedure D) A solution containing compound 17 (638 mg, 1.89 mmol), HATU (742 mg, 1.89 mmol), HOAt (3.79 mL, 1.89 mmol, solution 0.5–0.7 M in DMF) and NEM (0.49 mL, 3.79 mmol) in CH2Cl2 (34.2 mL) was added to a solution of compound 13 (117 mg, 0.38 mmol) in 15.5 mL CH2Cl2–DMF (9: 1). The reaction mixture was stirred at room temperature overnight. The solvent was removed in vacuo, and the residue was purified by silica gel column chromatography (petroleum ether–ethyl acetate, 2: 1) to afford 18 (126 mg, 53%). 1H NMR (CDCl3) δ 7.77 (d, J = 7.5 Hz, 2H, Ar–H), 7.61 (d, J = 7.2 Hz, 2H, Ar–H), 7.40 (t, J = 7.4 Hz, 2H, Ar–H), 7.31 (t, J = 7.4 Hz, 2H, Ar–H), 7.17 (d, J = 7.8 Hz, 1H, Ar–H), 6.43 (m, 2H, Ar–H), 5.74 (m, 2H, CH=CH2), 5.14 (m, 4H, CH=CH 2), 4.80 (m, 1H, H CHAr), 4.53–4.02 (m, 6H, HCHAr, CHCH 2OCO, CH CH2OCO, CH CH2CH=CH2, H-2), 3.88 (m, 2H, OCH 2CH=CH2), 3.84–3.55 (m, 2H, H-3), 3.79 (s, 3H, OCH3), 3.77 (s, 3H, OCH3), 3.59 (s, 3H, OCH3), 2.48 (m, 2H, CH 2CH=CH2); MS (FAB) m/z 629 (M + H)+.
(3S,6S)-4-(2,4-Dimethoxybenzyl)-6-(9H -fluoren-9-ylmethoxy-carbonylamino)-5-oxo-3,4,5,6,7,10-hexahydro-2H-[1,4]oxazecine-3-carboxylic acid methyl ester (19)
(Procedure E) To a solution of compound 18 (70 mg, 0.11 mmol) in CH2Cl2 (500 mL) was added second generation Grubbs catalyst (35 mg, 0.04 mmol). The reaction mixture was stirred at room temperature for 48 h. The solvent was removed in vacuo, and the residue was purified by column chromatography on silica gel (petroleum ether–ethyl acetate, 2: 1) to afford 19 (40 mg, 60%). 1H NMR (CDCl3) δ 7.78 (d, J = 7.2 Hz, 2H, Ar–H), 7.63 (d, J = 7.2 Hz, 2H, Ar–H), 7.41 (t, J = 7.2 Hz, 2H, Ar–H), 7.32 (t, J = 7.1 Hz, 2H, Ar–H), 7.08 (d, J = 8.1 Hz, 1H, Ar–H), 6.40 (m, 2H, Ar–H), 6.07 (d, J = 7.5 Hz, 1H, NH), 5.83 (m, 2H, H-8, H-9), 5.25 (m, 1H, H-6), 4.94 (d, J = 15.0 Hz, 1H, HCHAr), 4.51–4.34 (m, 3H, CHCH 2OCO, 1H-10), 4.29–4.08 (m, 3H, CH CH2OCO, H-2), 3.90 (m, 2H, HCHAr, 1H-10), 3.79 (s, 3H, OCH3), 3.75 (m, 1H, H-3), 3.69 (s, 3H, OCH3), 3.43 (s, 3H, OCH3), 2.68 (m, 1H, 1H-7), 2.53 (m, 1H, 1H-7); MS (FAB) m/z 601 (M + H)+.
(1S,8S)-11-(2,4-Dimethoxybenzyl)-3-oxa-9,11-diazabicyclo-[6.2.2]dodec-5-ene-10,12-dione (20)
(Procedure F) Compound 19 (24 mg, 0.04 mmol) was stirred at room temperature in 5 mL of CH2Cl2–piperidine (4: 1) for 2 h. After the solvent was removed, the resulting residue was purified by silica gel column chromatography (CH2Cl2–MeOH, 96: 4) to obtain 20 (13 mg, 93%) as a white solid. 1H NMR (CDCl3) δ 7.24 (m, 1H, Ar–H), 6.45 (m, 2H, Ar–H), 5.93 (br s, 1H, NH), 5.62 (m, 2H, H-5, H-6), 4.98 (d, J = 14.3 Hz, 1H, HCHAr), 4.25 (m, 3H, H-4, H-8), 4.14 (d, J = 14.3 Hz, 1H, HCHAr), 3.89 (m, 3H, H-1, H-2), 3.79 (s, 6H, OCH3), 3.11 (m, 1H, 1H-7), 2.83 (m, 1H, 1H-7); 13C NMR (CDCl3) δ 169.5, 168.1, 161.0, 158.9, 132.0, 131.4, 125.9, 116.4, 104.9, 98.6, 73.2, 71.5, 61.5, 55.6, 53.8, 41.6, 34.0; HRMS m/z found 347.1591 (M + H)+. C18H23N2O5 requires 347.1607; HPLC (System A) 8.1 min (99%), (System B) 9.1 min (99%). Mp 155–156 °C.
(1S,8S)-11-(2,4-Dimethoxybenzyl)-9-methyl-3-oxa-9,11-diaza-bicyclo[6.2.2]dodec-5-ene-10,12-dione (21)
(Procedure G) A solution of compound 20 (10 mg, 0.03 mmol) in THF (1 mL) was added, at 0 °C, to a suspension of NaH (3.5 mg, 0.09 mmol, 60% dispersion in mineral oil) in THF (1 mL). After stirring at room temperature for 30 min, the reaction mixture was cooled to 0 °C and CH3I (18 μL, 0.29 mmol) was added. The reaction mixture was stirred at room temperature for 5 h, followed by quenching with H2O. The product was extracted with CH2Cl2, dried over Na2SO4, and the solvent was removed under reduced pressure. The resulting residue was purified by preparative thin-layer chromatography (CH2Cl2–MeOH, 96: 4) to obtain 21 (6.6 mg, 63%). 1H NMR (CDCl3) δ 7.25 (m, 1H, Ar–H), 6.44 (m, 2H, Ar–H), 5.67 (m, 1H, H-6), 5.55 (m, 1H, H-5), 4.95 (d, J = 14.4 Hz, 1H, HCHAr), 4.21 (m, 2H, H-4), 4.16 (d, J = 14.4 Hz, 1H, HCHAr), 4.15 (m, 1H, H-8), 3.90 (m, 3H, H-1, H-2), 3.79 (s, 6H, OCH3), 3.07 (m, 1H, 1H-7), 2.91 (s, 3H, NCH3), 2.88 (m, 1H, 1H-7); 13C NMR (CDCl3) δ 168.1, 167.3, 161.0, 158.9, 132.1, 130.8, 125.7, 116.5, 104.9, 98.6, 72.8, 71.4, 61.7, 61.1, 55.6, 41.3, 33.9, 32.9; HRMS m/z found 377.1777 (M+ H)+. C19H25N2O5 requires 361.1763; HPLC (System A) 9.1 min (98%), (System B) 10.4 min (98%).
(1S,8S)-9-Benzyl-11-(2,4-dimethoxybenzyl)-3-oxa-9,11-diaza-bicyclo[6.2.2]dodec-5-ene-10,12-dione (22)
(Procedure G) Compound 22 (7.5 mg, 60%) was obtained as a white solid from 20 (10 mg, 0.03 mmol), NaH (4.2 mg, 0.10 mmol, 60% dispersion in mineral oil) and BnBr (54 μL, 0.45 mmol) in THF (2 mL) after for 8 h stirring at room temperature. The product was purified by preparative thin-layer chromatography (CH2Cl2–MeOH, 97: 3). 1H NMR (CDCl3) δ 7.29 (m, 3H, Ar–H), 7.19 (m, 3H, Ar–H), 6.44 (m, 2H, Ar–H), 5.68 (m, 1H, H-6), 5.55 (m, 1H, H-5), 5.29 (d, J = 14.7 Hz, 1H, HCHPh), 4.96 (d, J = 14.7 Hz, 1H, HCHAr), 4.20 (m, 2H, H-4), 4.16 (m, 2H, H-8, HCHAr), 3.99 (m, 2H, H-1, 1H-2), 3.80 (s, 3H, OCH3), 3.79 (m, 2H, 1H-2, HCH Ph), 3.77 (s, 3H, OCH3), 2.89 (m, 2H, H-7); 13C NMR (CDCl3) δ 167.9, 167.4, 161.1, 158.9, 135.7, 131.8, 130.5, 129.1, 128.4, 128.1, 126.3, 116.4, 104.9, 98.7, 72.8, 71.5, 61.7, 57.8, 55.6 (2C), 48.7, 41.7, 32.9; HRMS m/z found 437.2095 (M + H)+. C25H29N2O5 requires 437.2076; HPLC (System A) 13.4 min (98%), (System B) 16.2 min (98%). Mp 129–130 °C.
(2S,2′S)-3-Allyloxy-2-[[2′-(9H -fluoren-9-ylmethoxycarbonyl-amino)-pent-4′-enoyl]-(2,4,6-trimethoxybenzyl)-amino]-propionic acid methyl ester (23)
(Procedure D) Compound 23 (278 mg, 40%) was obtained from 14 (357 mg, 1.05 mmol) in 45 mL CH2Cl2–DMF (9: 1), and 17 (1.77 g, 5.26 mmol), HATU (2.06 g, 5.26 mmol), HOAt (10.5 mL, 5.26 mmol, solution 0.5–0.7 M in DMF) and NEM (1.35 mL, 10.5 mmol), in CH2Cl2 (99 mL) and DMF (0.47 mL). 1H NMR (CDCl3) δ 7.76 (d, J = 7.5 Hz, 2H, Ar–H), 7.61 (d, J = 7.5 Hz, 2H, Ar–H), 7.40 (t, J = 7.2 Hz, 2H, Ar–H), 7.30 (t, J = 7.2 Hz, 2H, Ar–H), 6.10 (s, 2H, Ar–H), 5.84 (m, 2H, CH=CH2), 5.15 (m, 4H, CH=CH 2), 4.61 (m, 2H, CH 2Ar), 4.49–4.07 (m, 5H, CHCH 2OCO, CH CH2OCO, CH CH2CH=CH2, H-2), 3.98 (m, 2H, OCH 2CH=CH2), 3.81 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 3.70 (m, 2H, H-3), 3.50 (s, 3H, OCH3), 2.58 (m, 1H, HCHCH=CH2), 2.44 (m, 1H, HCH CH=CH2); MS (API-ES) m/z 681 (M + Na)+.
(3S,6S)-6-(9H -Fluoren-9-ylmethoxycarbonylamino)-5-oxo-4-(2,4,6-trimethoxybenzyl)-3,4,5,6,7,10-hexahydro-2H -[1,4]oxa-zecine-3-carboxylic acid methyl ester (24)
(Procedure E) Compound 24 (5.5 mg, 57%) was obtained from 23 (10 mg, 0.015 mmol) and second generation Grubbs catalyst (4 mg, 0.005 mmol) in CH2Cl2 (60 mL). The product was purified by preparative thin-layer chromatography (petroleum ether–ethyl acetate, 2: 1). 1H NMR (CDCl3) δ 7.78 (d, J = 7.5 Hz, 2H, Ar–H), 7.63 (d, J = 7.5 Hz, 2H, Ar–H), 7.41 (t, J = 7.4 Hz, 2H, Ar–H), 7.32 (t, J = 7.4 Hz, 2H, Ar–H), 6.08 (m, 3H, Ar–H, NH), 5.83 (m, 2H, H-8, H-9), 5.33 (m, 1H, H-6), 4.78–4.37 (m, 5H, CH 2Ar, CHCH 2OCO, 1H-10), 4.29–4.08 (m, 3H, CHCH2OCO, H-2), 3.92 (m, 2H, H-3, 1H-10), 3.80 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 3.74 (s, 3H, OCH3), 3.40 (s, 3H, OCH3), 2.70 (m, 1H, 1H-7), 2.49 (m, 1H, 1H-7); MS (API-ES) m/z 653 (M + Na)+.
(1S,8S)-11-(2,4,6-Trimethoxybenzyl)-3-oxa-9,11-diazabicyclo-[6.2.2]dodec-5-ene-10,12-dione (25)
(Procedure F) Compound 25 (35 mg, 73%) was obtained as a white solid from 24 (80 mg, 0.13 mmol) and 15 mL of CH2Cl2–piperidine (4: 1). 1H NMR (CDCl3) δ 6.17 (br s, 1H, NH), 6.09 (s, 2H, Ar–H), 5.63 (m, 2H, H-5, H-6), 5.26 (d, J = 14.1 Hz, 1H, HCHAr), 4.29 (m, 2H, H-4), 4.24 (m, 1H, H-8), 4.10 (d, J = 14.1 Hz, 1H, HCHAr), 4.04 (m, 1H, 1H-2), 3.81 (s, 3H, OCH3), 3.77 (s, 6H, OCH3), 3.74 (m, 1H, 1H-2), 3.64 (m, 1H, H-1), 3.11 (m, 1H, 1H-7), 2.81 (m, 1H, 1H-7); 13C NMR (CDCl3) δ 169.9, 167.7, 161.8, 160.3, 131.5, 126.0, 103.2, 90.7, 73.0, 71.4, 59.7, 55.9, 55.6, 53.8, 35.5, 34.1; HRMS m/z found 377.1705 (M + H)+. C19H25N2O6 requires 377.1713; HPLC (System A) 8.3 min (99%), (System B) 9.3 min (99%). Mp 123–124 °C.
(1S,8S)-3-Oxa-9,11-diazabicyclo[6.2.2]dodec-5-ene-10,12-dione (26)
(Procedure H) To a solution of compound 25 (10 mg, 0.03 mmol) in CH2Cl2 (1.5 mL) was added TFA (0.75 mL). After stirring at room temperature for 7 h, the solvent was removed under reduced pressure. The residue was purified by preparative thin-layer chromatography (CH2Cl2–MeOH, 94: 6) to afford 26 (5 mg, 96%) as a white solid. 1H NMR (CD3OD) δ 5.59 (m, 2H, H-5, H-6), 4.33 (m, 2H, H-4), 4.06 (m, 1H, H-8), 3.95 (m, 1H, 1H-2), 3.77 (m, 2H, H-1, 1H-2), 3.04 (m, 1H, 1H-7), 2.77 (m, 1H, 1H-7); 13C NMR (CD3OD) δ 173.0, 171.1, 132.5, 126.2, 76.0, 74.9, 59.1, 54.4, 35.0; HRMS m/z found 197.0932 (M + H)+. C9H13N2O3 requires 197.0926.
(1S,8S)-9-Methyl-11-(2,4,6-trimethoxybenzyl)-3-oxa-9,11-diazabicyclo[6.2.2]dodec-5-ene-10,12-dione (27)
(Procedure G) Compound 27 (10 mg, 96%) was obtained from 25 (10 mg, 0.03 mmol), NaH (3.5 mg, 0.09 mmol, 60% dispersion in mineral oil) and CH3I (18 μL, 0.29 mmol) in THF (2 mL) after 7 h of stirring at room temperature. The product was purified by preparative thin-layer chromatography (CH2Cl2–MeOH, 96: 4). 1H NMR (CDCl3) δ 6.08 (s, 2H, Ar–H), 5.68 (m, 1H, H-6), 5.57 (m, 1H, H-5), 5.23 (d, J = 13.8 Hz, 1H, HCHAr), 4.24 (m, 2H, H-4), 4.15 (m, 1H, H-8), 4.10 (d, J = 13.8 Hz, 1H, HCHAr), 3.96 (m, 1H, 1H-2), 3.80 (s, 3H, OCH3), 3.78 (m, 1H, 1H-2), 3.77 (s, 6H, OCH3), 3.69 (m, 1H, H-1), 3.08 (m, 1H, 1H-7), 2.89 (s, 3H, NCH3), 2.86 (m, 1H, 1H-7); 13C NMR (CDCl3) δ 167.7, 167.6, 161.8, 160.3, 130.7, 126.0, 103.2, 90.7, 72.5, 71.2, 61.1, 60.0, 55.9, 55.6, 35.2, 33.8, 32.9; HRMS m/z found 391.1867 (M + H)+. C20H27N2O6 requires 391.1869; HPLC (System A) 9.1 min (98%), (System B) 10.6 min (98%). Mp 158–159 °C.
(1S,8S)-9-Benzyl-11-(2,4,6-trimethoxybenzyl)-3-oxa-9,11-diazabicyclo[6.2.2]dodec-5-ene-10,12-dione (28)
(Procedure G) Compound 28 (10.6 mg, 85%) was obtained from 25 (10 mg, 0.03 mmol), NaH (4 mg, 0.1 mmol, 60% dispersion in mineral oil) and BnBr (64 μL, 0.53 mmol) in THF (2 mL) after 24 h of stirring at room temperature. The product was purified by preparative thin-layer chromatography (CH2Cl2–MeOH, 97: 3). 1H NMR (CDCl3) δ 7.28 (m, 3H, Ar–H), 7.16 (m, 2H, Ar–H), 6.10 (s, 2H, Ar–H), 5.70 (m, 1H, H-6), 5.60 (m, 1H, H-5), 5.33 (d, J = 15.3 Hz, 1H, HCHPh), 5.20 (d, J = 14.0 Hz, 1H, HCHAr), 4.27 (m, 2H, H-4), 4.15 (m, 1H, H-8), 4.13 (d, J =14.0 Hz, 1H, HCHAr), 3.89 (m, 2H, H-2), 3.82 (s, 3H, OCH3), 3.75 (s, 6H, OCH3), 3.73 (m, 1H, H-1), 3.70 (d, J = 15.3 Hz, 1H, HCHPh), 2.98 (m, 1H, 1H-7), 2.86 (m, 1H, 1H-7); 13C NMR (CDCl3) δ 167.9, 167.7, 161.8, 160.3, 135.8, 130.6, 128.9, 128.1, 128.0, 126.5, 103.1, 90.7, 72.4, 71.1, 59.9, 57.7, 55.9, 55.6, 48.4, 35.3, 32.8; HRMS m/z found 467.2208 (M + H)+. C26H31N2O6 requires 467.2182; HPLC (System A) 13.3 min (98%), (System B) 16.3 min (98%).
(1S,8S)-9-Methyl-3-oxa-9,11-diazabicyclo[6.2.2]dodec-5-ene-10,12-dione (29)
(Procedure H) Compound 29 (1.7 mg, 63%) was obtained from 27 (5 mg, 0.01 mmol) and TFA (0.35 mL) in CH2Cl2 (0.7 mL). 1H NMR (CDCl3) δ 6.12 (br s, 1H, NH), 5.62 (m, 2H, H-5, H-6), 4.32 (m, 2H, H-4), 4.07 (m, 2H, 1H-2, H-8), 3.96 (m, 1H, H-1), 3.73 (m, 1H, 1H-2), 3.14 (m, 1H, 1H-7), 2.95 (s, 3H, NCH3), 2.88 (m, 1H, 1H-7); HRMS m/z found 211.1086 (M+ H)+. C10H15N2O3 requires 211.1083.
(1S,8S)-9-Benzyl-3-oxa-9,11-diazabicyclo[6.2.2]dodec-5-ene-10,12-dione (30)
(Procedure H) Compound 30 (2.7 mg, 89%) was obtained from 28 (5 mg, 0.01 mmol) and TFA (0.35 mL) in CH2Cl2 (0.7 mL). The product was purified by preparative thin-layer chromatography (CH2Cl2–MeOH, 96: 4). 1H NMR (CDCl3) δ 7.31 (m, 3H, Ar–H), 7.25 (m, 2H, Ar–H), 6.22 (br s, 1H, NH), 5.64 (m, 2H, H-5, H-6), 5.38 (d, J = 15.0 Hz, 1H, HCHPh), 4.35 (m, 2H, H-4), 4.11 (m, 2H, 1H-2, H-8), 4.04 (m, 1H, H-1), 3.73 (d, J = 15.0 Hz, 1H, HCHPh), 3.70 (m, 1H, 1H-2), 3.01 (m, 1H, 1H-7), 2.86 (m, 1H, 1H-7); HRMS m/z found 287.1391 (M + H)+. C16H19N2O3 requires 287.1396.
(2R,2′S)-3-Allylsulfanyl-2-{(2,4-dimethoxybenzyl)-[2′-(9H -fluoren-9 - ylmethoxycarbonylamino) - pent - 4′ - enoyl] - amino } -propionic acid methyl ester (31)
(Procedure D) Compound 31 (107 mg, 37%) was obtained from 15 (146 mg, 0.45 mmol) in 20 mL CH2Cl2–DMF (9: 1), and 17 (755 mg, 2.24 mmol), HATU (878 mg, 2.24 mmol), HOAt (4.48 mL, 2.24 mmol, solution 0.5–0.7 M in DMF) and NEM (0.58 mL, 4.48 mmol) in CH2Cl2 (40.3 mL). The product was purified by column chromatography on silica gel (petroleum ether–ethyl acetate, 3: 1). 1H NMR (CDCl3) δ 7.77 (d, J = 7.2 Hz, 2H, Ar–H), 7.61 (d, J = 7.2 Hz, 2H, Ar–H), 7.40 (t, J = 7.4 Hz, 2H, Ar–H), 7.31 (t, J = 7.4 Hz, 2H, Ar–H), 7.16 (d, J = 8.7 Hz, 1H, Ar–H), 6.43 (m, 2H, Ar–H), 5.75 (m, 2H, CH=CH2), 5.11 (m, 4H, CH=CH 2), 4.78 (d, J = 15.6 Hz, 1H, H CHAr), 4.49–4.18 (m, 5H, HCHAr, CHCH 2OCO, CH CH2OCO, CH CH2CH=CH2), 4.07 (m, 1H, H-2), 3.78 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 3.55 (s, 3H, OCH3), 3.10 (m, 3H, SCH 2CH=CH2, 1H-3), 2.94 (m, 1H, 1H-3), 2.61 (m, 1H, HCHCH=CH2), 2.46 (m, 1H, HCHCH=CH2); MS (API-ES) m/z 667 (M + Na)+.
(3R,6S)-4-(2,4-Dimethoxybenzyl)-6-(9H -fluoren-9-ylmethoxy-carbonylamino)-5-oxo-3,4,5,6,7,10-hexahydro-2H-[1,4]thiazecine-3-carboxylic acid methyl ester (32)
(Procedure E) Compound 32 (52 mg, 57%) was obtained from 31 (95 mg, 0.15 mmol) and second generation Grubbs catalyst (46 mg, 0.05 mmol) in CH2Cl2 (148 mL). The product was purified by column chromatography on silica gel (petroleum ether–ethyl acetate, 2.5: 1). 1H NMR (CDCl3) δ 7.77 (d, J = 7.5 Hz, 2H, Ar–H), 7.63 (d, J = 7.5 Hz, 2H, Ar–H), 7.41 (t, J = 7.4 Hz, 2H, Ar–H), 7.32 (t, J = 7.5 Hz, 2H, Ar–H), 7.12 (d, J = 7.5 Hz, 1H, Ar–H), 6.41 (m, 2H, Ar–H), 6.15 (br d, J = 6.3 Hz, 1H, NH), 5.67 (m, 2H, H-8, H-9), 5.30 (m, 1H, H-6), 5.05 (d, J = 14.4 Hz, 1H, HCHAr), 4.43 (m, 2H, CHCH 2OCO), 4.29–4.07 (m, 2H, CHCH2OCO, HCH Ar), 3.79 (s, 3H, OCH3), 3.76–3.63 (m, 1H, H-3), 3.71 (s, 3H, OCH3), 3.52–3.28 (m, 2H, H-10), 3.37 (s, 3H, OCH3), 3.02–2.72 (m, 3H, H-2, 1H-7), 2.45 (m, 1H, 1H-7); MS (API-ES) m/z 639 (M + Na)+.
(1R,8S)-11-(2,4-Dimethoxybenzyl)-3-thia-9,11-diazabicyclo-[6.2.2]dodec-5-ene-10,12-dione (33)
(Procedure F) Compound 33 (19 mg, 73%) was obtained from 32 (45 mg, 0.07 mmol) and 5 mL of CH2Cl2–piperidine (4: 1). The product was purified by preparative thin-layer chromatography (CH2Cl2–MeOH, 96: 4). 1H NMR (CDCl3) δ 7.26 (d, J = 8.4 Hz, 1H, Ar–H), 6.45 (m, 3H, Ar–H, NH), 5.71 (m, 2H, H-5, H-6), 5.21 (d, J = 14.6 Hz, 1H, HCHAr), 4.33 (m, 1H, H-1), 4.28 (d, J = 14.6 Hz, 1H, HCHAr), 4.24 (m, 1H, H-2), 3.79 (s, 6H, OCH3), 3.45 (m, 1H, 1H-4), 3.25–3.02 (m, 4H, H-2, 1H-4, 1H-7), 2.55 (m, 1H, 1H-7); 13C NMR (CDCl3) δ 169.6, 167.0, 161.1, 159.1, 132.5, 130.7, 127.4, 116.2, 104.7, 98.7, 60.1, 56.9, 55.6, 55.2, 45.4, 32.4, 30.4, 29.3; HRMS m/z found 363.1371 (M + H)+. C18H23N2O4S requires 363.1379; HPLC (System A) 9.8 min (99%), (System B) 11.4 min (99%).
(2R,2′S)-3-Allylsulfanyl-2-[[2′ -(9H -fluoren-9-ylmethoxycar-bonylamino)-pent-4′-enoyl]-(2,4,6-trimethoxybenzyl)-amino]-propionic acid methyl ester (34)
(Procedure D) Compound 34 (124 mg, 41%) was obtained from 16 (159 mg, 0.45 mmol) in 20 mL CH2Cl2–DMF (9: 1), and 17 (755 mg, 2.24 mmol), HATU (878 mg, 2.24 mmol), HOAt (4.48 mL, 2.24 mmol, solution 0.5–0.7 M in DMF) and NEM (0.58 mL, 4.48 mmol) in CH2Cl2 (40.3 mL). The product was purified by column chromatography on silica gel (petroleum ether–ethyl acetate, 2.5: 1). 1H NMR (CDCl3) δ 7.76 (d, J = 7.5 Hz, 2H, Ar–H), 7.62 (m, 2H, Ar–H), 7.40 (t, J = 7.2 Hz, 2H, Ar–H), 7.30 (m, 2H, Ar–H), 6.10 (s, 2H, Ar–H), 5.82 (m, 2H, CH=CH2), 5.13 (m, 4H, CH=CH 2), 4.72–4.18 (m, 5H, CH 2Ar, CHCH 2OCO, CHCH2OCO), 3.89–3.66 (m, 2H, CH CH2CH=CH2, H-2), 3.81 (s, 3H, OCH3), 3.78 (s, 6H, OCH3), 3.47 (s, 3H, OCH3), 3.15 (m, 3H, SCH 2CH=CH2, 1H-3), 2.98 (m, 1H, 1H-3), 2.65 (m, 1H, HCHCH=CH2), 2.48 (m, 1H, HCH CH=CH2); MS (API-ES) m/z 697 (M + Na)+.
(3R,6S)-6-(9H -Fluoren-9-ylmethoxycarbonylamino)-5-oxo-4-(2,4,6-trimethoxybenzyl)-3,4,5,6,7,10-hexahydro-2H -[1,4]thiaze-cine-3-carboxylic acid methyl ester (35)
(Procedure E) Compound 35 (30 mg, 71%) was obtained from 34 (44 mg, 0.07 mmol) and second generation Grubbs catalyst (17 mg, 0.02 mmol) in CH2Cl2 (65 mL). The product was purified by preparative thin-layer chromatography (petroleum ether–ethyl acetate, 2: 1). 1H NMR (CDCl3) δ 7.77 (d, J = 7.2 Hz, 2H, Ar–H), 7.63 (m, 2H, Ar–H), 7.40 (t, J = 7.4 Hz, 2H, Ar–H), 7.31 (t, J = 7.2 Hz, 2H, Ar–H), 6.16 (d, J = 6.9 Hz, 1H, NH), 6.09 (m, 2H, Ar–H), 5.65 (m, 2H, H-8, H-9), 5.36 (m, 1H, H-6), 4.70 (m, 2H, CH 2Ar), 4.43 (m, 2H, CHCH 2OCO), 4.24 (t, J = 7.2 Hz, 1H, CHCH2OCO), 3.84–3.62 (m, 1H, H-3), 3.80 (s, 3H, OCH3), 3.77 (s, 6H, OCH3), 3.52–3.27 (m, 2H, H-10), 3.36 (s, 3H, OCH3), 3.05–2.65 (m, 3H, H-2, 1H-7), 2.43 (m, 1H, 1H-7); MS (API-ES) m/z 669 (M + Na)+.
(1R,8S)-11-(2,4,6-Trimethoxybenzyl)-3-thia-9,11-diazabicyclo-[6.2.2]dodec-5-ene-10,12-dione (36)
(Procedure F) Compound 36 (9 mg, 60%) was obtained from 35 (25 mg, 0.04 mmol) and 3.75 mL of CH2Cl2–piperidine (4: 1). The product was purified by preparative thin-layer chromatography (CH2Cl2–MeOH, 96: 4). 1H NMR (CDCl3) δ 6.09 (s, 3H, Ar–H, NH), 5.71 (m, 2H, H-5, H-6), 5.39 (d, J = 13.8 Hz, 1H, HCHAr), 4.41 (d, J = 13.8 Hz, 1H, HCHAr), 4.25 (m, 1H, H-8), 4.07 (m, 1H, H-1), 3.80 (s, 3H, OCH3), 3.78 (s, 6H, OCH3), 3.45 (m, 1H, 1H-4), 3.26–3.01 (m, 4H, H-2, 1H-4, 1H-7), 2.54 (m, 1H, 1H-7); 13C NMR (CDCl3) δ 169.6, 166.4, 161.8, 160.6, 130.7, 127.5, 103.2, 90.7, 60.1, 56.0, 55.6, 55.3, 55.2, 38.2, 32.9, 30.6, 29.5; HRMS m/z found 393.1495 (M + H)+. C19H25N2O5S requires 393.1484; HPLC (System A) 9.7 min (99%), (System B) 11.3 min (99%).
(3S,6S)-3-Allyl-6-allyloxymethyl-1-(2,4-dimethoxybenzyl)-piperazine-2,5-dione (37)
(Procedure F) Compound 37 (52 mg, 42%) was obtained from 18 (206 mg, 0.33 mmol) and 10 mL of CH2Cl2–piperidine (4: 1). The product was purified by column chromatography on silica gel (petroleum ether–ethyl acetate, 1: 2). 1H NMR (CDCl3) δ 7.24 (d, J = 8.2 Hz, 1H, Ar–H), 6.45 (m, 2H, Ar–H), 6.19 (br s, 1H, NH), 5.82 (m, 2H, CH=CH2), 5.22 (m, 4H, CH=CH 2), 5.10 (d, J = 14.7 Hz, 1H, H CHAr), 4.12 (d, J = 14.7 Hz, 1H, HCHAr), 4.01 (m, 1H, H-6), 3.98 (m, 2H, OCH 2CH=CH2), 3.93 (m, 1H, H-3), 3.82 (m, 2H, CHCH 2O), 3.79 (2s, 6H, OCH3), 2.90 (m, 1H, H CHCH=CH2), 2.60 (m, 1H, HCHCH=CH2); 13C NMR (CDCl3) δ 166.9, 166.4, 161.0, 158.8, 134.1, 133.7, 131.9, 120.1, 118.2, 116.3, 104.8, 98.6, 72.6, 67.8, 60.1, 55.6, 55.4, 41.7, 40.6; MS (FAB) m/z 375 (M + H)+.
(1S,8S,11S,18S)-21,23-Bis-(2,4-dimethoxybenzyl)-3,13-dioxa-9,19,21,23-tetraazatricyclo[16.2.2.28,11]tetracosa-5,15-diene-10, 20,22,24-tetraone (38)
(Procedure E) To a solution of compound 37 (15 mg, 0.04 mmol) in CH2Cl2 (1 mL) was added second generation Grubbs catalyst (5 mg, 0.006 mmol). The reaction mixture was stirred at room temperature for 16 h. The solvent was removed in vacuo, and the residue was purified by preparative thin-layer chromatography (CHCl3–MeOH, 97: 3) to afford 38 (11 mg, 79%). 1H NMR (CDCl3) δ 9.18 (m, 2H, NH), 7.24 (d, J = 8.3 Hz, 2H, Ar–H), 6.45 (m, 4H, Ar–H), 5.65 (m, 4H, H-5, H-6, H-15, H-16), 5.18 (d, J = 14.5 Hz, 2H, HCHAr), 4.33 (m, 2H, 1H-4, 1H-14), 4.25 (m, 2H, 1H-2, 1H-12), 4.11 (d, J = 14.5 Hz, 2H, HCHAr), 3.99 (m, 2H, H-8, H-18), 3.93 (m, 2H, H-1, H-11), 3.79 (s, 6H, OCH3), 3.78 (s, 6H, OCH3), 3.76 (m, 2H, 1H-2, 1H-12), 3.67 (m, 2H, 1H-4, 1H-14), 2.90 (m, 2H, 1H-7, 1H-17), 2.68 (m, 2H, 1H-7, 1H-17); 13C NMR (CDCl3) δ 169.5, 166.3, 161.0, 158.8, 131.8, 130.6, 127.9, 116.2, 104.9, 98.6, 72.5, 69.8, 60.5, 55.6, 55.3, 40.8, 40.0; HRMS m/z found 693.3149 (M + H)+. C36H45N4O10 requires 693.3136.
Pharmacological analyses
Cell culture and membrane preparation
Human 1321N1 astrocytoma cells transfected individually with the hP2Y1,2,4,6,11 receptors26,33,40 were grown at 37 °C in a humidified incubator with 5% CO2–95% air in Dulbecco’s modified Eagle’s medium (JRH Biosciences, Inc.) supplemented with 10% fetal bovine serum (FBS), 100 Units mL−1 penicillin, 100 μg mL−1 streptomycin and 2 mM L-glutamine. The cells were grown to ca. 60% confluence for the experiments.
For membrane preparation, human astrocytoma cells expressing human P2Y1 receptors were grown to approximately 80% confluence and then harvested. The cells were homogenized and suspended and then centrifuged at 100 g for 5 min at room temperature. The pellet was resuspended in 50 mM tris(hydroxymethyl)aminomethane (Tris) hydrochloride buffer (pH 7.4). The suspension was homogenized with a Polytron homogenizer (Brinkmann) for 10 s and was then recentrifuged at 20 000 g for 20 min at 4 °C. The resultant pellets were resuspended in Tris buffer (pH 7.4), and the suspension was stored at −80 °C until the binding experiments. The protein concentration was measured with the Bradford assay.44
Storage of test substances
Agonists were dissolved as stock solutions in Tris buffer (pH 7.4), and the DKP derivatives were stored as frozen stock solutions in DMSO (5 mM) at −20 °C. Prior to use the DMSO solutions were warmed briefly to 50–60 °C.
Determination of inositol phosphates
The quantity of inositol phosphates was measured essentially as reported.25,26,33,41 The P2Y1,2,4,6,11-1321N1 cells were grown to confluence in 6-well plates in the presence of myo-[3H]inositol (2 μCi mL−1) for 24 h. Cells were then treated for 30 min at 37 °C with antagonists or buffer in the presence of 20 mM LiCl, followed by another 30 min of incubation at 37 °C with the appropriate agonist. Agonists used were: P2Y1, 2-MeSADP; hP2Y2, UTP; hP2Y4, UTP; hP2Y6, UDP; hP2Y11, ATP. The reaction was terminated upon aspiration of the medium and addition of cold formic acid (20 mM). After 30 min, supernatants were neutralized with NH4OH, and applied to Bio-Rad Dowex AG 1-X8 anion exchange columns. All of the columns were then washed with water followed by a 60 mM sodium formate solution containing 5 mM sodium tetraborate. Total inositol phosphates were eluted with 1 M ammonium formate containing 0.1 M formic acid, and radioactivity values were measured using a liquid scintillation counter.
Radioligand binding assay
P2Y1 receptor binding experiments were performed as previously described.39 Briefly, membranes (40 μg protein) from astrocytoma cells stably expressing human P2Y1 receptors were incubated with [3H]MRS2279 (8 nM) for 30 min at 4 °C in a total assay volume of 200 μL. For adenosine A1 receptor binding, an agonist radioligand [3H]R-PIA (2.0 nM) was incubated with membranes (40 lg protein per tube) from CHO cells stably expressing human adenosine A1 receptors for 60 min at 25 °C.40 Radiolabeled ligand concentrations used in all assays approximated the Kd values of the receptor. Binding reactions were terminated by filtration through Whatman GF/B glass-fiber filters under reduced pressure with an MT-24 cell harvester (Brandel), and radioactivity was determined with a 1414 liquid scintillation counter (Wallac, Win Spectral, Perkin Elmer Life Sciences).
Calcium mobilization assay
Human astrocytoma cells stably expressing human P2Y1 receptors were cultured in Dulbecco’s modified Eagle’s medium (DMEM, JRH Biosciences, Inc., Lenexa, KS, USA) and F12 (1: 1) supplemented with 10% fetal bovine serum, 100 units penicillin mL−1, 100 μg streptomycin mL−1, 2 μmol glutamine mL−1, and 500 μg geneticin mL−1. For the assay, cells were grown overnight in 100 μL of media in 96-well flat-bottom plates at 37°C at 5% CO2 or until approximately 60–80% confluency. The calcium assay kit (Molecular Devices, Sunnyvale, CA) was used as directed without washing of the cells, and with probenecid added to the loading dye at a final concentration of 2.5 mM to increase dye retention. Cells were loaded with 50 μL of dye with probenecid to each well and incubated for 45 min at room temperature. The compound plate was prepared using dilutions of various compounds in Hank’s buffer. For antagonist studies, both agonist and antagonist were added to the sample plate. Samples were run in duplicate using a Molecular Devices Flexstation I at room temperature. Cell fluorescence (excitation = 485 nm, emission = 525 nm) was monitored following exposure to the compound. Increases in intracellular calcium are reported as the maximum fluorescence value after exposure minus the basal fluorescence value before exposure.
Data analysis
IC50 values obtained in radioligand binding assays and in assays of inhibition of agonist-stimulated inositol phosphate accumulation were calculated by a four-parameter logistic equation and the GraphPad software package (GraphPad, San Diego, CA). All concentration–effect curves were repeated in at least three separate experiments, carried out in duplicate or triplicate with different membrane preparations or different 1321N1 cell cultures.
Molecular modeling
Molecular modeling was carried out with the module Discover3 of InsightII (Accelrys, Inc.) on Silicon Graphics Origin 200 computer, using the CFF91 forcefield.45
An NVT (constant-volume/constant-temperature) molecular dynamics simulation was carried out for 500 ns at 298 K, with a time step of 1 fs. Conformations were sampled at regular intervals of 100 ns and energy minimized employing the BFGS Newton method, until an RMS gradient lower than 0.00001 kcal mol−1 Å −1 was reached.
For a description of the puckering of the DKP ring we used the coordinates Θ (phase angle of symmetrical interconversion), P2 (phase angle of pseudorotation), and Q (total puckering amplitude) as defined by Haasnoot,46 utilizing the final formulae that we recently reported.40
Supplemental Material
Acknowledgments
Dr Pedro Besada thanks the Cystic Fibrosis Foundation for financial support. Mass spectral measurements were carried out by Dr Victor Livengood and NMR by Wesley White (NIDDK). We thank Heng T. Duong and Brian Harris (NIDDK) for proofreading the manuscript, and Dr Zhan-Guo Gao, Lanh Bloodworth, Dr Jianxin Hu, and Dr Xinping Lu (NIDDK) for biological assays.
Footnotes
Electronic supplementary information (ESI) available: coordinates of two representative conformations, and molecular model of compound 20 (in PDB format). See http://www.rsc.org/suppdata/ob/b4/b416349d/
References
- 1.Williams RM, Durham CA. Bicyclomycin: synthetic, mechanistic, and biological studies. Chem Rev. 1988;88:511–540. [Google Scholar]
- 2.Funabashi Y, Horiguchi T, Iinuma S, Tanida S, Harada S. TAN-1496 A, C and E, diketopiperazine antibiotics with inhibit activity against mammalian DNA topoisomerase I. J Antibiot. 1994;47:1202–18. doi: 10.7164/antibiotics.47.1202. [DOI] [PubMed] [Google Scholar]
- 3.Moyroud J, Gelin J, Chene A, Mortier J. Synthese d’analogues structuraux de Thaxtomines, phytotoxines responsables de la gale de la pomme de terre. Tetrahedron. 1996;52:8525–8534. [Google Scholar]
- 4.Szardenings AK, Burkoth TS. A simple procedure for the solid phase synthesis of diketopiperazine and diketomorpholine derivatives. Tetrahedron. 1997;53:6573–6593. [Google Scholar]
- 5.Alsina J, Jensen KJ, Albericio F, Barany G. Solid-phase synthesis with tris(alkoxy)benzyl backbone amide linkage (BAL) Chem Eur J. 1999;5:2787–2795. [Google Scholar]
- 6.Perrotta E, Altamura M, Barani T, Bindi S, Giannotti D, Harmat NJS, Nannicini R, Maggi CA. 2,6-Diketopiperazines from amino acids, from solution-phase to solid-phase organic synthesis. J Comb Chem. 2001;3:453–460. doi: 10.1021/cc0000904. [DOI] [PubMed] [Google Scholar]
- 7.Dinsmore CJ, Beshore DC. Recent advances in the synthesis of diketopiperazines. Tetrahedron. 2002;58:3297–3312. [Google Scholar]
- 8.Guo T, Dong G, Fitzpatrick D, Geng P, Ho KK, Jibilian CH, Kultgen SG, Liu R, McDonald E, Saionz KW, Valenzano KJ, Xie D, Adang AEP, van Straten NCR, Webb ML. Discovery of potent biaryl diketopiperazine FSH receptor agonists: rapid lead optimization through parallel synthesis. Poster of the 228th ACS National Meeting; Philadelphia, PA. 2004. [Google Scholar]
- 9.Szardenings AK, Harris D, Lam S, Shi L, Tien D, Wang Y, Patel DV, Navre M, Campbell DA. Rational design and combinatorial evaluation of enzyme inhibitor scaffolds: identification of novel inhibitors of matrix metalloproteinases. J Med Chem. 1998;41:2194–2200. doi: 10.1021/jm980133j. [DOI] [PubMed] [Google Scholar]
- 10.Szardenings AK, Antonenko V, Campbell DA, DeFrancisco N, Ida S, Shi L, Sharkov N, Tien D, Wang Y, Navre M. Identification of highly selective inhibitors of collagenase-1 from combinatorial libraries of diketopiperazines. J Med Chem. 1999;42:1348–1357. doi: 10.1021/jm980475p. [DOI] [PubMed] [Google Scholar]
- 11.Bianco A, Sonksen CP, Roepstorff P, Briand J-P. Solid-phase synthesis and structural characterization of highly substituted hydroxyproline-based 2,5-diketopiperazines. J Org Chem. 2000;65:2179–2187. doi: 10.1021/jo991818+. [DOI] [PubMed] [Google Scholar]
- 12.Bianco A, Furrer J, Limal D, Guichard G, Elbayed K, Raya J, Piotto M, Briand J-P. Multistep synthesis of 2,5-diketopiperazines on different solid supports monitored by high resolution magic angle spinning NMR spectroscopy. J Comb Chem. 2000;2:681–690. doi: 10.1021/cc0000489. [DOI] [PubMed] [Google Scholar]
- 13.Koehler AN, Shamji AF, Schreiber SL. Discovery of an inhibitor of a transcription factor using small molecule microarrays and diversity-oriented synthesis. J Am Chem Soc. 2003;125:8420–8421. doi: 10.1021/ja0352698. [DOI] [PubMed] [Google Scholar]
- 14.Shokat K, Velleca M. Novel chemical genetic approaches to the discovery of signal transduction inhibitors. Drug Discov Today. 2002;7:872–879. doi: 10.1016/s1359-6446(02)02391-7. [DOI] [PubMed] [Google Scholar]
- 15.Creighton CJ, Leo GC, Du Y, Reitz AB. Design, synthesis, and conformational analysis of eight-membered cyclic peptidomimetics prepared using ring closing metathesis. Bioorg Med Chem. 2004;12:4375–4385. doi: 10.1016/j.bmc.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 16.Scholl M, Ding S, Lee CW, Grubbs RH. Synthesis and activity of a new generation of ruthenium-basedolefin metathesis catalysts coordinated with 1,3-dimesityl-4,5-dihydroimidazol-2-ylidene ligands. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 17.Connon SJ, Blechert S. Recents developments in olefin cross-metathesis. Angew Chem, Int Ed. 2003;42:1900–1923. doi: 10.1002/anie.200200556. [DOI] [PubMed] [Google Scholar]
- 18.von Kügelgen I, Wetter A. Molecular pharmacology of P2Y- receptors. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:310–323. doi: 10.1007/s002100000310. [DOI] [PubMed] [Google Scholar]
- 19.King BF, Townsend-Nicholson A. Recombinant P2Y receptors: the UCL experience. J Auton Nerv Syst. 2000;81:164–170. doi: 10.1016/s0165-1838(00)00134-x. [DOI] [PubMed] [Google Scholar]
- 20.Hollopeter G, Jantzen HM, Vincent D, Li G, England L, Ramakrishnan V, Yang RB, Nurden P, Nurden A, Julius D, Conley PB. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature. 2001;409:202–207. doi: 10.1038/35051599. [DOI] [PubMed] [Google Scholar]
- 21.Communi D, Suarez Gonzalez N, Detheux M, Brézillon S, Lannoy V, Parmentier M, Boeynaems J-M. Identification of a novel human ADP receptor goupled to Gi. J Biol Chem. 2001;276:41 479–41 485. doi: 10.1074/jbc.M105912200. [DOI] [PubMed] [Google Scholar]
- 22.Jacobson KA. Purine and pyrimidine nucleotide (P2) receptors. Ann Rep Med Chem. 2002;37:75–84. doi: 10.2174/1568026043450961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nandanan E, Camaioni E, Jang SY, Kim Y-C, Cristalli G, Herdewijn P, Secrist JA, Tiwari KN, Mohanram A, Harden TK, Boyer JL, Jacobson KA. Structure–activity relationships of bisphosphate nucleotide derivatives as P2Y1 receptor antagonists and partial agonists. J Med Chem. 1999;42:1625–1638. doi: 10.1021/jm980657j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nandanan E, Jang SY, Moro S, Kim H, Siddiqi MA, Russ P, Marquez VE, Busson R, Herdewijn P, Harden TK, Boyer JL, Jacobson KA. Synthesis, biological activity, and molecular modeling of ribose-modified adenosine bisphosphate analogues as P2Y1 receptor ligands. J Med Chem. 2000;43:829–842. doi: 10.1021/jm990249v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim HS, Barak D, Harden TK, Boyer JL, Jacobson KA. Acyclic and cyclopropyl analogues of adenosine bisphosphate antagonists of the P2Y1 receptor: structure activity relationships and receptor docking. J Med Chem. 2001;44:3092–3108. doi: 10.1021/jm010082h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boyer J, Adams M, Ravi RG, Jacobson KA, Harden TK. 2-Chloro-N6-methyl-(N)-methanocarba-2′-deoxyadenosine-3′,5′-bisphosphate is a selective high affinity P2Y1 receptor antagonist. Br J Pharmacol. 2002;135:2004–2010. doi: 10.1038/sj.bjp.0704673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fischer B, Boyer JL, Hoyle CHV, Ziganshin AU, Brizzolara AL, Knight GE, Zimmet J, Burnstock G, Harden TK, Jacobson KA. Identification of potent, selective P2Y-purinoceptor agonists: structure–activity relationships for 2-thioether derivatives of adenosine 5′-triphosphate. J Med Chem. 1993;36:3937–3946. doi: 10.1021/jm00076a023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boyer JL, Siddiqi S, Fischer B, Romera-Avila T, Jacobson KA, Harden TK. Identification of potent P2Y purinoceptor agonists that are derivatives of adenosine 5′-monophosphate. Br J Pharmacol. 1996;118:1959–1964. doi: 10.1111/j.1476-5381.1996.tb15630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fischer B, Chulkin A, Boyer JL, Harden KT, Gendron FP, Beaudoin AR, Chapal J, Hillaire-Buys D, Petit P. 2-Thioether 5′-O-(1-thiotriphosphate)adenosine derivatives as new insulin secretagogues acting through P2Y-Receptors. J Med Chem. 1999;42:3636–3646. doi: 10.1021/jm990158y. [DOI] [PubMed] [Google Scholar]
- 30.Raboisson P, Baurand A, Cazenave JP, Gachet C, Retat M, Spiess B, Bourguignon JJ. Novel antagonists acting at the P2Y1 purinergic receptor: synthesis and conformational analysis using potentiometric and nuclear magnetic resonance titration techniques. J Med Chem. 2002;45:962–972. doi: 10.1021/jm0104062. [DOI] [PubMed] [Google Scholar]
- 31.Raboisson P, Baurand A, Cazenave JP, Gachet C, Schultz D, Spiess B, Bourguignon JJ. A general approach toward the synthesis of C-nucleoside pyrazolo[1,5-a]-1,3,5-triazines and their 3′,5′-bisphosphate C-nucleotide analogues as the first reported in vivo stable P2Y1 -receptor antagonists. J Org Chem. 2002;67:8063–8071. doi: 10.1021/jo026268l. [DOI] [PubMed] [Google Scholar]
- 32.Ingall AH, Dixon J, Bailey A, Coombs ME, Cox D, McInally JI, Hunt SF, Kindon ND, Theobald BJ, Willis PA, Humphries RG, Leff P, Clegg JA, Smith JA, Tomlinson W. Antagonists of the platelet P2T receptor: a novel approach to antithrombotic therapy. J Med Chem. 1999;42:213–220. doi: 10.1021/jm981072s. [DOI] [PubMed] [Google Scholar]
- 33.Kim SG, Soltysiak KA, Gao ZG, Chang TS, Chung E, Jacobson KA. Tumor necrosis factor α-induced apoptosis in astrocytes is prevented by the activation of P2Y6, but not P2Y4 nucleotide receptors. Biochem Pharmacol. 2003;65:923–931. doi: 10.1016/s0006-2952(02)01614-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glenn MP, Pattenden LK, Reid RC, Tyssen DP, Tyndall JDA, Birch CJ, Fairlie DP. β-Strand mimicking macrocyclic amino acids: templates for protease inhibitors with antiviral activity. J Med Chem. 2002;45:371–381. doi: 10.1021/jm010414i. [DOI] [PubMed] [Google Scholar]
- 35.Koch I, Keusgen M. Diastereoselective synthesis of alliin by an asymmetric sulfur oxidation. Pharmazie. 1998;53:668–671. [Google Scholar]
- 36.Boojamra CG, Burow KM, Thompson LA, Ellman JA. Solid-phase synthesis of 1,4-benzodiazepine-2,5-diones. Library preparation and demonstration of synthesis generality. J Org Chem. 1997;62:1240–1256. [Google Scholar]
- 37.Jensen KJ, Alsina J, Songster MF, Vagner J, Albericio F, Barany G. Backbone amide linker (BAL) strategy for solid-phase synthesis of C-terminal-modified and cyclic peptides. J Am Chem Soc. 1998;120:5441–5452. [Google Scholar]
- 38.Fresno M, Alsina J, Royo M, Barany G, Albericio F. Solid-phase synthesis of diketopiperazine, useful scaffolds for combinatorial chemistry. Tetrahedron Lett. 1998;39:2639–2642. [Google Scholar]
- 39.Waldo GL, Corbitt J, Boyer JL, Ravi G, Kim HS, Ji XD, Lacy J, Jacobson KA, Harden TK. Quantitation of the P2Y1 receptor with a high affinity radiolabeled antagonist. Mol Pharmacol. 2002;62:1249–1257. doi: 10.1124/mol.62.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohno M, Costanzi S, Kim HS, Kempeneers V, Vastmans K, Herdewijn P, Maddileti S, Gao ZG, Harden TK, Jacobson KA. Nucleotide analogues containing 2-oxa-bicyclo[2.2.1]heptane and L-α-threofuranosyl ring rystems: interactions with P2Y receptors. Bioorg Med Chem. 2004;12:5619–5630. doi: 10.1016/j.bmc.2004.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Costanzi S, Mamedova L, Gao Z-G, Jacobson KA. Architecture of P2Y nucleotide receptors: structural comparison based on sequence analysis, mutagenesis, and homology modeling. J Med Chem. 2004;47:5393–5404. doi: 10.1021/jm049914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L, Jacobsen SEW, Bengtsson A, Erlinge D. P2 receptor mRNA expression profiles in human lymphocytes, monocytes and CD34+ stem and progenitor cells. BMC Immunol. 2004;5:16. doi: 10.1186/1471-2172-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chambers JK, Macdonald LE, Sarau HM, Ames RS, Freeman K, Foley JJ, Zhu Y, McLaughlin MM, Murdock P, McMillan L, Trill J, Swift A, Aiyar N, Taylor P, Vawter L, Naheed S, Szekeres P, Hervieu G, Scott C, Watson JM, Murphy AJ, Duzic E, Klein C, Bergsma DJ, Wilson S, Livi GP. A G protein-coupled receptor for UDP-glucose. J Biol Chem. 2000;275:10 767–10 771. doi: 10.1074/jbc.275.15.10767. [DOI] [PubMed] [Google Scholar]
- 44.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem. 1976;76:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 45.Maple JR, Hwang MJ, Stockfisch TP, Dinur U, Waldman M, Ewig CS, Hagler AT. Derivation of Class II force fields. 1. Methodology and quantum force field for the alkyl functional group and alkane molecules. J Comput Chem. 1994;15:162–182. [Google Scholar]
- 46.Haasnoot CAG. Conformational analysis of six-membered rings in solution: ring-puckering coordinates derived from vicinal NMR proton–proton coupling constants. J Am Chem Soc. 1993;115:1460–1468. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.