

Abstract
Hematopoietic stem and progenitor cells (HPCs) are necessary for long-term survival. Genomic instability and persistent DNA damage may cause loss of adult stem cell function. The mismatch repair (MMR) pathway increases replication fidelity and defects have been implicated in malignant hematopoietic diseases. Little, however, is known about the role MMR pathway failure plays in the aging process of human HPCs. We hypothesized that loss of MMR occurs in HPCs as a process of human aging. We examined microsatellite instability and expression of the MMR genes MutL homologue 1 (MLH1) and MutS homologue 2 (MSH2) in HPCs and colony-forming cell-derived clones (CFCs) from human donors aged 0 to 86 years. CFCs from donors > 45 years had a greater frequency of microsatellite instability and CD34+ progenitors lacking MLH1 expression and protein than individuals ≤ 45 years. Loss of MSH2 did not correlate with age. Thus, a potentially early event in the normal human aging process is microsatellite instability accumulation in normal human HPCs associated with the loss of MLH1 protein expression.
Introduction
Hematopoietic stem and progenitor cells (HPCs) play a critical role in the regeneration and repopulation of the hematopoietic system.1 Over a lifetime of genomic replication, cells may accumulate genomic mutations, both from intrinsic replication errors and from extrinsic environmental factors, such as exposure to chemical mutagens or radiation. Absent DNA repair, these defects can lead to gene disruption, apoptosis, the accumulation of further genetic mutations, loss of differentiation plasticity, and eventually malignant transformation. Genomic maintenance in the multipotent progenitor is a crucial process to ensure long-term stability and population maintenance, especially during replication, proliferation, and differentiation into a functional hematopoietic system. Conversely, HPCs lose functionality and undergo malignant transformation at an increased rate in older individuals. These observations lead to the hypothesis that one component of normal aging involves loss of DNA damage repair that could, to some extent, give rise to genomic instability in the continually replicating HPC population.
Aberrant insertion or deletion loops and base:base mismatches are examples of replication-dependent genomic change that occur with considerable frequency. These mutational lesions result from the natural infidelity of DNA polymerase, counterbalanced by the DNA mismatch repair (MMR) pathway that is responsible for recognition and correction of these errors.2 This pathway consists of 3 major heterodimeric complexes, MutL homologue (MutL)α, MutS homologue (MutS)α, and MutSβ. MutLα is composed of MLH1 and PMS2, MutSα consists of MSH2 and MSH6, and MutSβ is formed by MSH2 and MSH3. The MutSα complex colocalizes to single mispaired bases, whereas MutSβ recognizes mispaired runs of 3 to 6 bases. MutLα is responsible for the recruitment of excision and repair machinery to the site of a noncomplimentary base marked by either MutSα or MutSβ.3,4 Lacking redundant partners in the formation of the MutL and MutS complexes, the MLH1 and MSH2 proteins are indispensable; thus, loss of either gene prevents mispaired base identification and processing by the MMR pathway.
Failure to identify or repair mispaired bases results in the replication-dependent accumulation of mutations and genomic instability, frequently at microsatellites. These short repeat sequences are scattered randomly throughout the genome and form stem loop structures during replication because of their repetitive nature. These secondary structures affect the precision with which DNA polymerases add bases and result in base:base mismatches that alter the microsatellite length. Microsatellite length changes are called microsatellite instability (MSI). Detection of MSI and loss of the MMR pathway are strongly associated with hereditary nonpolyposis colorectal cancer (HNPCC).5 Mutations in 1 allele of either the MLH1 or MSH2 genes account for roughly 90% of known HNPCC mutations, and MSI is frequently observed in tumors that arise in these patients after a subsequent mutation in the remaining allele.6 Individuals with homozygous mutations in MLH1 or MSH2 have a higher incidence of colorectal cancer, as well as increased risk of developing leukemia and lymphomas, implying that incompetent MMR also results in development of hematologic cancer.7,8 The presence of acquired MSI is observed in the progression of acute myelogenous leukemia (AML),9,10 myelodysplastic syndrome,11 as well as in therapy-related leukemias.12,13 Loss of MSH2 gene expression has further been observed in association with MSI in adult acute leukemia.14 Accumulation of genomic instability in these diseases is indicative of a potential role for MMR failure in the onset and progression of age-related hematopoietic disorders.
Although loss of MMR is associated with hematologic disorders, whether acquired MMR deficiency is an age-related phenomenon of the HPC population remains unclear. In this study, we hypothesized that subsets of HPCs from normal individuals acquire an age-dependent MMR pathway deficiency. We sought to determine whether an age-related MMR deficiency was detectable at the level of HPCs. To assess age-related loss of the MMR pathway in normal individuals, we analyzed MSI and MMR function from donors aged 0 to 86 years. We observed HPCs acquire an age-related MMR deficiency through analysis of MSI and MLH1 or MSH2 gene and protein expression. Methylation-specific sequencing of the MLH1 promoter region revealed CpG methylation correlated with the frequency of MSI-H colony-forming cell-derived clones (CFCs). These data provide direct evidence for an age-dependent loss of MMR function and of mutation accumulation in human HPCs.
Methods
Donor samples
Donated tissue and medical information were obtained from donors who accepted written informed consent in accordance with the Declaration of Helsinki regarding use of their donation under University Hospitals IRB protocol 3ZO3. Sixty-two 1-time samples were used in the MSI analysis of CFC-derived clones (supplemental Table 1 and supplemental Figure 1A-C; available on the Blood Web site; see the Supplemental Materials link at the top of the online article; Figure 1A-C), 9 donor samples were used in gene expression analysis of MLH1 and MSH2 by quantitative real-time PCR (QRT-PCR) in bulk isolations of CD34+ cells (Figure 2A-B), 38 samples were used in analysis of MLH1 and MSH2 protein in CD34+ cells by immunocytochemical staining (supplemental Figure 3 and Figure 4), 18 samples were used for analysis of MLH1 expression by QRT-PCR (Figure 5A-B), and 12 donor samples were used to perform methylation specific sequencing of the MLH1 promoter (Figure 6A-C). Samples were received as umbilical cord blood (UCB), iliac crest bone marrow aspirates (BMAs), or bone core samples obtained during orthopedic joint replacement procedures from otherwise healthy individuals. Sample size was limited, and often only single analyses were possible.
Figure 1.

MSI-positive CFUs accumulate with age. (A) Percentage of CFCs with low-grade instability with age (i; P = .30) and percentage of MSI-L CFCs from donors ≤ 45 years compared with those > 45 years by unpaired t test (ii; P = .17). (B) Percentage of MSI-H CFCs with donor age (i; P = .0032) and percentage of MSI-H CFCs from donors ≤ 45 years compared with those > 45 years by unpaired t test (ii; P = .0084). (C) Percentage of CFCs with any (MSI-H or MSI-L) detectable MSI with age (i; P = .017) and percentage of CFCs with any MSI from donors ≤ 45 years compared with those > 45 years by unpaired t test (ii; P = .015). Linear regression was performed with Welsh correction for variance. The solid line in the linear regression analysis represents a curve of best fit, and the dashed line indicates a 95% confidence interval.
Figure 2.

MSI fragment patterns show evidence of clonal evolution. Electropherograms depicting the consensus and MSI FPSs of D2S123 and D17S250 in the MSI-H CFCs of BMA 09 consensus (C9), C10, and C11 (A) or BMA 12 consensus (C06), C07, and C08 (B). Microsatellite loci not depicted are stable in these samples.
Figure 4.
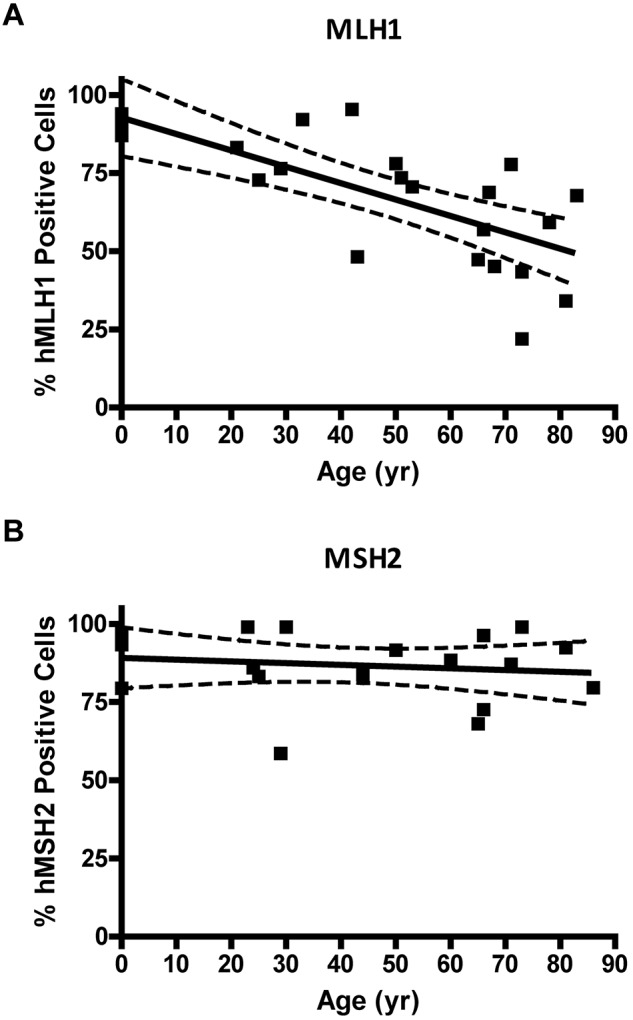
MLH1 but not MSH2 protein is lost in individual CD34+ cells with age. (A) Percent of MLH1-positive stained CD34+ cells versus donor age (P = .0001). (B) Percentage of MSH2-positive stained CD34+ cells versus donor age (P = .55). Solid lines represent the best fit linear regression, and dashed lines signify a 95% confidence interval.
Figure 5.
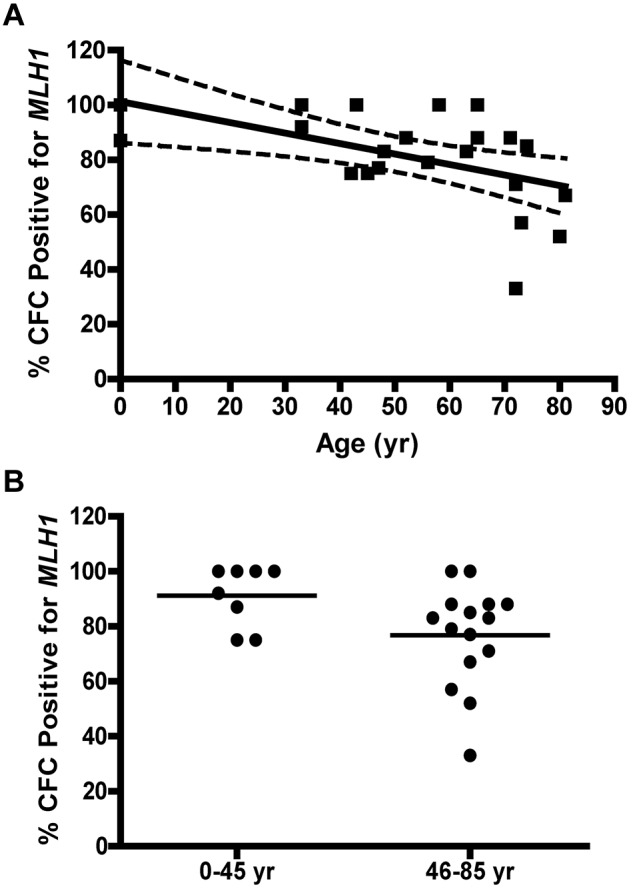
MLH1 gene expression is lost as a function of age. (A) Percentage of CFCs that lack detectable MLH1 gene expression by QRT-PCR with age. Solid lines represent the best fit linear regression, and dashed lines represent a 95% confidence interval (P = .0073). (B) Comparison of the percentage of CFUs expressing detectable MLH1 for donor samples ≤ 45 years with donors > 45 years old. Statistical analysis by 2-tailed unpaired t test (P = .029).
Figure 6.

Methylation of the MLH1 promoter. (A) Depicts the CpG sites with methylation observed in the CD34+ cells from 12 donors and the cell line RKO. Black diamonds represent all methylated cytosines observed in bacterial subclones of the 804 bp MLH1 promoter region fragment spanning from −938 bp to −134 bp of the minus strand generated by PCR amplification after bisulfite modification. (B) MLH1 gene promoter methylation observed in the bulk CD34+ cell population of individual donors shown as a percentage of CpG sites methylated for each donor sample. (C) Comparison of the percentage of CpG sites methylated in donor CD34+ cell samples ≤ 45 years to those of donors > 45 years by 2-tailed unpaired t test (P = .04). (D) Percentage of MSI-H CFCs seen in each individual donor compared with the percentage of CpG sites methylated in the MLH1 promoter region of total CD34+ cells from the same individual donor (P = .005).
CD34+ cell selection
Mononuclear cells were isolated from UCB, BMA, and bone core by the standard density gradient separation method modified from Schlenke et al.15 Mononuclear cells obtained from the interface and washed twice with PBS were then stained with anti-CD34+ antibody-conjugated magnetic beads as per the CD34+ isolation kit (Miltenyi Biotec) protocol. CD34+ cells were washed in PBS and resuspended in complete IMDM (HyClone Laboratories) supplemented with 10% fetal bovine serum (HyClone Laboratories).
Flow cytometric analysis of CD34+ cells
CD34+ cells obtained by immune-magnetic separation were stained with mouse monoclonal antibody AC136 to human CD34+ conjugated with PE (Miltenyi Biotec) or isotype control mouse serum as per the manufacturer's instructions. All samples were analyzed for CD34+ (supplemental Figure 2A-B) on an LSR I flow cytometer (BD Biosciences).
Colony-forming assay
Mononuclear or CD34+ cells were plated at clonal density (33 000 cells per mL of medium) in MethoCult H4434 Classic (StemCell Technologies). Colonies were grown, as per the manufacturer's instructions, for 12 to 16 days, after which time colonies were selected and DNA was extracted from each individually.
DNA isolation
DNA used in PCR amplifications for MSI analysis was obtained from single colonies washed twice with PBS and then incubated at 55°C for 2 hours in lysis buffer containing 10mM Tris-HCl (pH 8.0), 1mM EDTA, 1% Triton-X 100 buffer, and 10 mg/mL proteinase K. Proteinase was inactivated by a 10-minute 80°C incubation.
Microsatellite instability analysis
DNA from 12 to 24 CFCs from each of the individual normal donors was PCR amplified with primers for the microsatellite loci. Loci tested were BAT25, BAT26, D2S123, D17S250, and D5S346, originally described for assessment of HNPCC.6 Primer sequences and PCR conditions were as described in Umetani et al.16 For detection purposes, primers were labeled with 5′ fluorescent tags: 6-FAM, HEX, VIC, NED, or PET (Operon Biotechnologies). Detection of amplicon length was completed with a Typhoon 9200 scanner (GE Healthcare) or an ABI 3600 Genetic analyzer (Applied Biosystems). A consensus fragment pattern, for each donor, at each locus was determined by comparison of fragment length patterns of individual CFCs. Thus, a consensus pattern for each MSI locus was obtained for each donor and was based on the most common fragment pattern observed (supplemental Figure 1A-C). Each PCR reaction was then scored as either positive, negative, or no product. The relative frequency of MSI-L, -H or -Any CFCs for each patient sample was calculated by the formula [relative frequency = f/n], where f is the total number of all MSI-L, -H, or -Any CFCs for a single donor, and n is the total number of CFC analyzed for a single donor.
QRT-PCR analysis of MLH1 and MSH2 gene expression
RNA from CD34+ cells or from single CFCs was used as template for first strand synthesis using SuperScript III First-Strand Synthesis system (Invitrogen) by random hexameric priming. Amplification was performed in quadruplicate with a 7500 Fast Real-Time PCR system (Applied Biosystems) in standard mode, as per the manufacturer's protocol. MLH1 expression was assessed with TaqMan gene assays for human MLH1 (assay Hs00179866_m1; Applied Biosystems) or MSH2 (assay Hs0179887_m1; Applied Biosystems) and analyzed relative to human Actin-β (4352339E; Applied Biosystems) as an endogenous control. Relative quantitation (RQ) values were normalized to a single UCB sample (Figure 3A-B) or the K562 cell line. RQ values with nonoverlapping error bars are considered significantly different (P < .05). CFCs for which amplification of Actin-β was above the calculated threshold but lacked detectable MLH1 or MSH2 expression were scored as negative for either MLH1 or MSH2, respectively. The percentage of positive CFCs per sample was recorded.
Figure 3.
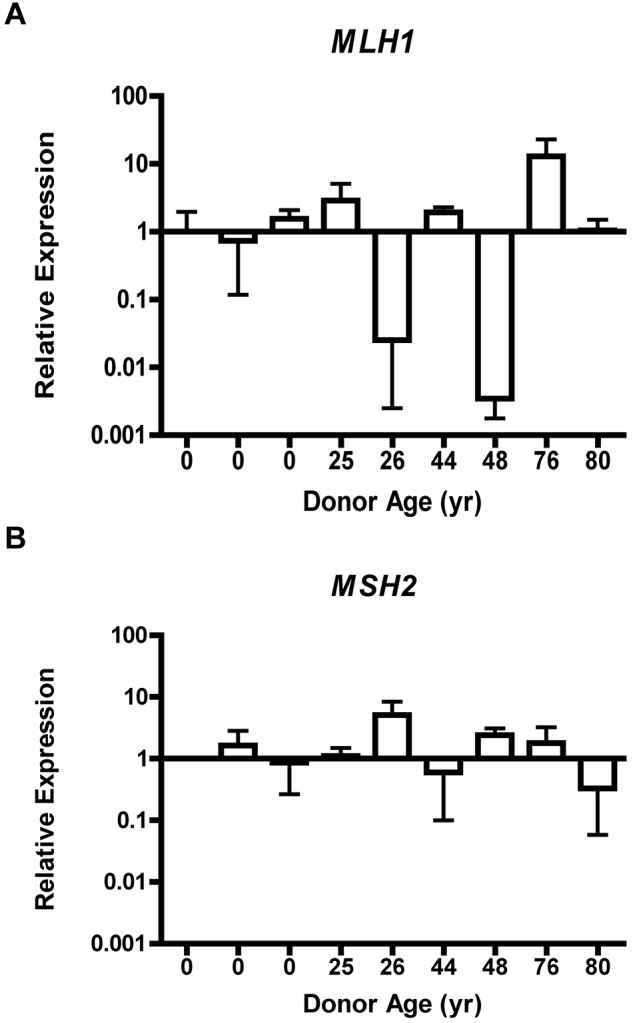
MLH1 gene expression is dysregulated, whereas MSH2 expression remains stable in CD34+ cells. Relative expression of MLH1 (A) or MSH2 (B) from CD34+ cells obtained from individuals of various ages by QRT-PCR. Error bars represent 95% confidence interval based on quadruplicate sampling.
Immunocytochemistry of MLH1 and MSH2
Fresh CD34+ cells or control cell lines were fixed (100 000 cells/slide) to gelatin-coated glass slides with 2% paraformaldehyde for 20 minutes, permeabilized with methanol, and used immediately or stored at −20°C. Before staining, slides were equilibrated to room temperature, dried, and rehydrated in PBS (pH 7.4). Slides were blocked in 10% normal goat serum (Sigma-Aldrich). Primary MLH1 (mouse anti–human MLH1; Invitrogen) and MSH2 (mouse anti–human MSH2; Invitrogen) monoclonal IgG1-κ antibodies were used at 1:100 and 1:250 dilution, respectively, in 1% BSA and PBS. Isotype negative controls for each slide were incubated with purified mouse IgG (Biocare Medical) as primary antibody. 4′,6-diamidino-2-phenylindole (DAPI) was used as a counterstain to illustrate nuclear DNA. Black-and-white images of stained cells were taken at ×20 magnification with an Eclipse Te200 microscope (Nikon), and images captured with a Spot RT Slider 2MP digital charge-coupled device camera (Diagnostic Instruments) for the blue and green channels and then merged with ImageJ 1.4 software (National Institutes of Health; supplemental Figure 3A-B).17
Receiver operating characteristic curve analysis
Green and blue fluorescence intensities were obtained from images of between 200 and 600 individual MLH1- and MSH2-stained control cells or CD34+ cells by ImageJ 1.4 software (supplemental Figure 3A-B).17 The fluorescence intensity ratio (FIR) of green (antibody) to blue (DAPI) was then calculated for individual control cells and used to create a receiver operator characteristic (ROC) curve based on assumed true positive and true negative FIR values derived from the control SW480, HCT116, or LOVO cell lines (supplemental Figure 3C). SW480 and HCT116 cells were selected for true positive and true negative values, respectively, for MLH1 staining, whereas staining SW480 and LOVO cells were considered true positive and true negatives for MSH2, respectively. The web-based ROC calculator developed by Johns Hopkins University was used to determine the true positive fraction and false positive fraction values from the ROC curve.18 The true positive fraction is defined as the fraction of positive control cells that have an FIR value above a theoretical FIR threshold value whereas the false positive fraction is the proportion of negative control cells that have a value, above that same theoretical threshold value. The true positive and false positive fraction values vary with the threshold value; thus, the optimal FIR threshold was calculated by determining the minimal value of ✓[(1 − true positive fraction)2 + (false positive fraction)2] over a range of potential threshold values spanning all true positive and false positive values observed, as described in Metz.19 Cells with an FIR value over this threshold were scored as positive for MLH1 or MSH2, whereas cells below this threshold were scored negative. Thus, for each microscopic field, the frequency of MLH1- or MSH2-positive cells could be determined.
Bisulfite modification and methylation-specific sequencing of the MLH1 promoter
Genomic DNA isolated from 100 000 CD34+ cells of individual donors (n = 12) or cells of the RKO colon tumor line were bisulfite modified using the EpiTect bisulfite kit (QIAGEN) as directed by the manufacturer's protocol. The primers MLH1-mF and MLH1-mR were used to amplify 804 bp of the reverse complimentary–converted promoter region of MLH1 from base pair −938 to −134. PCR amplification was carried out with Platinum Taq (Invitrogen), subcloned into the pCR4-TOPO TA vector. Ten colonies were then grown to confluence in 1 mL of LB media, and plasmid DNA was isolated with the PureLink Quick Plasmid Miniprep kit (Invitrogen) as per the product manual. Forward and reverse sequencing was performed with dye terminator PCR amplification by Biotic Solutions, using the M13-F and M13-R primers (supplemental Table 2). Methylation events were observed as A-> G conversions and recorded for each donor-specific bacterial clone. The percentage of CpG sites with methylation was calculated by dividing the number of CpG residues observed by the total number of CpG residues within the promoter region.
Statistical analysis
QRT-PCR error bars represent the SEM with a 95% confidence interval according to the manufacturer's instructions. Calculations for linear regressions, unpaired t tests, and Fisher exact test were performed with Prism Version 4.03 software (GraphPad).
Results
HPCs with high-grade MSI accumulate with age
MSI was measured in human CFCs across the human age span. Five microsatellite loci were chosen for classification of MSI in this study as proposed for the diagnosis of HNPCC.20 MSIs were identified by PCR product fragment patterns shifts (FPSs) for each of 5 microsatellite loci tested, all of which are standard sites used in the diagnosis of HNPCC. Examples of MSI electropherograms depicting products obtained at each microsatellite locus tested for each CFC obtained from 3 separate donors are shown in supplemental Figure 1A through C. A CFC is characterized as having low-grade MSI (MSI-L) when a single locus shows an aberrant FPS, high-grade MSI (MSI-H) when 2 or more loci are aberrant, and total MSI (MSI-Any) for the observation of FPSs at one or more locus within a single CFCs. Each CFC from an individual was analyzed for MSI at 2 mononucleotide repeat microsatellite loci (BAT25 and BAT26) and 3 dinucleotide repeat sequence loci (D2S123, D5S346, and D17S250)16 and then scored as having either stable microsatellites, MSI-L, or MSI-H.
The presence or absence of MSI was identified in individual CFCs grown from 62 normal donors: 15 samples from UCB, all 0 years; 29 samples from BMA taken from normal donors aged 23 to 84 years; and 18 samples from bone core samples taken at the time of an orthopedic joint replacement procedure from donors aged 48 to 85 years (supplemental Table 1). MSI was detected in individual CFC clones of most donors at all age groups; only 4 donors lacked any MSI, suggesting that MSI-L is common in HPCs. Donor samples displayed a diverse range of relative frequency of CFCs with evidence of MSI. The frequency of MSI at each locus across all samples ranged from 3.3% to 7.2%. MSI frequency at the BAT26 locus, 3.3%, was lower than that at the BAT25 and D2S123 and D17S250 loci (P < .05).
The relative frequency of CFCs with MSI at a single locus (MSI-L) did not increase with age (Figure 1Ai). Linear regression analysis of CFCs with MSI-H, however, indicated a significant increase in relative frequency as a function of age, with a positive rate of 0.16% per year of life from birth (P = .0032; Figure 1Bi). The relative frequency of MSI-Any CFCs also increased significantly at a rate of 0.22% per year of life (Figure 1Ci). The most striking differences were seen when the average relative frequency (μf) of MSI-H CFCs from all donors between 0 and 45 years were compared with the μf of MSI-H CFCs from donors > 45 years by unpaired t test (Figure 1Bii,Cii). The μf of MSI-H CFCs is significantly greater in the group of donors 46 to 84 years (μf = 12.9 ± 2.9% SEM) than from donors 0 to 45 years (μf = 4.2 ± 1.0% SEM) by unpaired t test (P = .0064; Table 1). The finding of MSI-H accumulation in CFCs is noteworthy, because instability at multiple microsatellite loci is associated with genetic defects of the MMR pathway.5,6,20–22
Table 1.
Unpaired t test of frequency of MSI-L or -H in CFC from donors in 2 age groups
| Age group, 0-45 y (n = 30)* |
Age group, 46-84 y (n = 32)* |
P† | |||
|---|---|---|---|---|---|
| μf,‡ % | ± SEM, % | μf,‡ % | ± SEM, % | ||
| MSI-L | 20.4 | ± 2.7 | 25.3 | ± 2.4 | .0894 |
| MSI-H | 4.2 | ± 1.0 | 12.9 | ± 2.9 | .0064* |
n is the number of donors in each group.
P value was calculated by comparison between the 2 age groups (0-45 and 46-84 y). Asterisk after a value denotes significance.
μf is the average relative frequency of MSI-H or MSI-L CFC from all donors within each age group.
An evolution of mutational events is indicative of clonal lineage expansion.23,24 FPSs are characteristics of mutational insertions and deletions at microsatellite loci. A survey of CFCs for evidence of identical FPSs at multiple loci identified 2 pairs of CFCs from 2 donors—BMA 09, a 29-year-old (C10 and C11) and BMA 12, a 33-year-old (C07 and C08)—that exhibit identical MSI FPSs at 2 of the same microsatellite loci, D2S123 and D17S250 loci (Figure 2A-B); however, all other loci are stable in these clones. The relative frequency of 2 CFCs from the same donor with the same MSI FPS at multiple loci (ie, MSI-H) was 1.1% of all CFC analyzed in this sample set. Thus, although it is possible these phenomenon represent clonal expansion, the frequency is low, and consequences of clonal evolution in the HPC population unknown.
Variable MLH1 gene expression with maintenance of MSH2 gene expression in CD34+ cells
Loss of MLH1 or MSH2 gene expression is an accepted cause of accumulation of MSI.25 Therefore, we tested expression of MLH1 and MSH2 by QRT-PCR in CD34+ progenitor cells isolated from 9 normal donors (3 UCB, 4 BMA, and 2 bone core; supplemental Figure 2A-B). A 10-fold decrease in the RQ of MLH1 expression relative to the normalized sample was identified in 2 of the 5 adult CD34+ samples (Figure 3C). Significant disparity in the RQ of MSH2 expression across age was not observed (Figure 3D). The standard deviation of MLH1 expression was 4.03 RQ over a range of 12.92 RQ. For MSH2 expression, the standard deviation was 1.45 RQ over a range of 4.77 RQ. Because only a proportion of CFCs had evidence of MSI-H, it was not expected that QT-PCR analysis of the entire CD34+ cell population from a single donor could detect complete loss of gene expression. Nonetheless, RQ of MLH1 and MSH2 gene expression within the CD34+ cell population did reveal a broad distribution of MLH1 expression levels between individuals, whereas MSH2 expression was relatively invariant.
MLH1, but not MSH2 protein, is lost in a subset of CD34+ cells
Immunocytochemical staining of CD34+ cells for MLH1 or MSH2 protein was performed to determine expressional status of the MLH1 (Figure 4A) or MSH2 (Figure 4B) genes in HPC. CD34+ cells were analyzed from 4 UCB samples (donor age, 0 years), 13 BMA samples from donors between 21 and 66 years, and 12 bone core samples from donors between 51 and 86 years (all from unique donors). The colon cancer SW480 cell line was used as the positive control for both MLH1 and MSH2 gene expression, the HCT116 cell line was used as the negative control for MLH1, and the LOVO cell line was used as the negative control for MSH2. Quantification of threshold values for expression of MLH1 or MSH2 proteins was based on an ROC curve derived from the FIR obtained from positive and negative cell lines (data not shown). The percentage of CD34+ cells with positive staining for MLH1 declined as a function of donor age (P = .0001; Figure 4C). No significant change was observed in the percentage of cells expressing MSH2 with age (P = .55; Figure 4D).
MLH1 gene expression is lost as a function of age in a subset of CFCs
Some HNPCC cases are associated with transcriptional down-regulation of MLH1.26,27 Loss of MLH1 transcription was determined by QRT-PCR analysis of MLH1 in CFC from 23 unique donor samples (3 UCB, age 0 years; 10 BMA or bone core, 33-60 years; and 10 bone core, 61-81 years). Amplification of both MLH1 and Actin-β could be detected in at least a portion of individual CFC samples from all donors; however, for specific CFC in which Actin-β was detected, no MLH1 transcripts could be detected. Therefore, these CFC were scored as having lost MLH1 expression. Across the donors analyzed, the percentage of CFC with MLH1 expression declined significantly as a function of age (P = .0073; Figure 5A). A significant proportion of CFCs with detectable MLH1 transcript was observed from donors between 0 and 45 years; however, donors > 45 years had fewer CFCs with detectable MLH1 transcript by unpaired t test (P = .029; Figure 5B). The mean relative expression of MLH1 in CFCs expressing MLH1 from individual donors was similar across all age groups (data not shown). A limited number of CFCs from donor samples also were analyzed by QRT-PCR for MSH2 gene expression and expression of the microRNA mir155. No correlation was observed between loss in MSH2 gene expression and donor age (data not shown). Similarly, expression of the microRNA mir155,28 known to reduce expression of MMR genes in other settings, was undetectable in CFCs (data not shown).
Methylation of MLH1 promoter is detectable in CD34+ genomic DNA
The 5′ promoter region −938 bp upstream of MLH1 transcriptional start site (+1 bp) is considered a CpG island with 63 potential CpG sites for 5-methyl-cytosine residues to occur. Presence of 5-methyl-cytosine residues in this region leads to transcriptional repression of the MLH1 gene.27 Individual donor total CD34+ cell samples with corresponding MSI-H CFC frequency data were bisulfite sequenced to determine whether methylation of CpG sites in the MLH1 promoter of the bulk CD34+ cell population correlated to the frequency of MSI-H CFCs in the same donor. DNA isolated from CD34+ cells obtained from 12 donors was tested for CpG methylation by methylation-specific sequencing. CpG methylation was observed in all samples examined (Figure 6A-B). Significantly more CpG methylation sites were observed in donors > 45 years than those ≤ 45 years (P < .05; Figure 6C). We observed a direct linear correlation between percentage of CpG sites methylated in the bulk CD34+ cell population and MSI-H CFC from the same donors (P < .05; Figure 6D).
Discussion
Our objective was to determine whether genomic instability in HPCs increased with donor age. HPCs from individuals between the ages of 0 and 84 years with normal blood counts, and normal hematopoiesis, were tested for MSI. The relative frequency of MSI-H CFCs increased with donor age. MSI-L in CFCs was common at all ages, likely spontaneous, and unrelated to an age associated acquired MMR deficiency. A population of CFC and CD34+ cells having lost MLH1 expression and protein was detected and observed to accumulate with age. This correlated with increased methylation of CpG in the MLH1 promoter, and with MSI-H. These data indicate that acquired MMR pathway deficiency involving loss of MLH1 expression occurs in the process of normal HPC aging.
The detection of MSI in HPCs is significant because mutations at microsatellite loci occur because of strand slippage during replication and are resolved through MMR.29,30 Analysis of MSI in tumor samples is complicated by tumor cell heterogeneity. Similar tissue heterogeneity in bone marrow is expected based on our observations of a subpopulation of HPC having lost MLH1. Our analysis of HPCs allowed analysis of MSI and MMR gene expression status in the genetic progeny of individual HPCs and provides an estimate of the rate with which HPCs acquire MMR pathway defects. Our discovery of an acquired MMR dysfunction in aging HPCs could not have been made in whole bone marrow extracts because of dilution of the MSI signal.
There have been no studies identifying loss of MMR in normal human HPCs with age. Cells from mice lacking MSH2 cannot recognize mispaired bases or initiate MMR, but they do retain an intact HPC population capable of self-renewal and regeneration, although they have significant competitive repopulation defects.31 MSI are not observed in the primary CFCs obtained from MSH2−/− mice; however, CFCs positive for MSI are detected in an HPC population forced to undergo regeneration during serial transplantation or selection by DNA-methylating agent treatment.31 Increased incidence of T-cell lymphoma in MSH2−/− mice is observed. Tumors from these mice are positive for MSI.32–35 In both repopulation and lymphomagenesis, the underlying MMR defect gives rise to MSI accumulation over time and is dependent on cell proliferation. In our study of MSI in human HPCs, MSI-H emerging in the HPCs of donors > 45 years is indicative of an acquired MMR deficiency and active mutation accumulation. This raises the possibility, as in the MSH2−/− mouse, that in HPCs of aging individuals the existence of MSI is a marker of accumulated mutation after proliferative or environmental stress.
The origin of this age-related MMR dysfunction seems to be loss of MLH1expression in the HPC of donors > 45 years of age. This was a surprising result because there are no prior data to suggest age-related loss of MLH1 in a subpopulation of HPCs in normal individuals. However, loss of MLH1 expression does occur. HNPCC is inherited in patients who inherit a mutation of MLH1 or MSH2. A second mutation then gives rise to MSI-H colorectal cancers, with loss of MMR observed in HNPCC.36
Another mechanism for the acquired loss of MLH1 or MSH2 gene expression in the normal allele leading to the tumors is promoter region CpG hypermethylation of the normal allele. The promoter sequence immediately upstream of MLH1 contains CpG islands for which methylation directly correlates with loss of MLH1 expression.27,37 We found an increasing percentage of CpG sites methylated in the promoter of the MLH1 gene in donor CD34+ cells correlated with an increasing percentage of MSI-H CFC obtained from the same donor. Based on the significant correlation between the degree of MLH1 promoter methylation and MSI-H in CFC, we propose that the cause of MSI and the loss of MLH1 expression could be CpG methylation of the MLH1 promoter in affected HPCs.
The increased frequency with which MSI-H CFCs are found in older individuals raises the question of the consequences of these events. We have not identified, within this population of individuals with normal blood counts, evidence to suggest that there is a progression of this MSI toward hematologic disease. In MSH2−/− mice, however, the underlying loss of MMR results in reduced hematopoietic stem cell engraftment potential and increased incidence of MSI-positive T-cell lymphomas.34 Loss of MMR and MSI therefore results in additional mutational events that culminate in hematopoietic failure and malignant transformation. This raises the possibility that genetic abnormalities accumulate and predispose aging HPCs to reduced function and increased malignant transformation. Human diseases such as AML, myelodysplasia, leukemia, lymphoma, anemias, and loss of broad based immunity all increase in incidence with age.38 In these diseases, 8% of primary MDS and 32% of AML patients show evidence of MSI, suggesting MMR failure is involved.9,11 Likewise, MSI also is frequently associated with cases of therapy-related secondary AML and myelodysplasia.12,39 As noted, there is already a clear association among loss of MMR, MSI, and hematologic malignancies. Only prospective analysis would uncover a prodromal loss of MMR and development of MSI before the onset of anemias, cytopenias, and leukemias.
This report is the first to observe that genomic instability increases in the normal human hematopoietic stem cell population with age. The observation of MSI in human CFCs, not associated with HNPCC syndrome, suggests a potential connection between genomic instability, marrow failure, and hematopoietic disease. The discovery of genetic mutations associated with aging in human HPCs is supported by the recent identification of clonal mosaicism in human somatic tissue over time and gives credence to the hypothesis that MMR failure and MSI accumulation may give rise to progressive hematopoietic disorders and be promoted by replicative, proliferative, or chemotherapeutic-induced stress.40,41 How loss of the MMR pathway in normal human hematopoietic stem cells gives rise to disease can now be addressed.
Supplementary Material
Acknowledgments
Bone core samples were obtained thanks to surgeons Drs Randall E. Marcus and Victor M. Goldberg.
These studies used the Cytometry Facility, Stem Cell Facility, and the Tissue Procurement and Histology Core Facility of the Case Comprehensive Cancer Center, Case Western Reserve University (P30 CA43703). This work was supported by grant AGR0124916 from the National Institutes of Aging.
Footnotes
There is an Inside Blood commentary on this article in this issue.
Presented in abstract form at the 51st Annual Meeting of the American Society of Hematology, New Orleans, LA, December 7, 2009.
The online version of the article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.K., K.L., E.T., A.S., G.S.G., and D.W. performed experiments; J.K. and P.F. analyzed results and prepared the figures; and J.K. and S.L.G. designed experiments and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Stanton L. Gerson, Wearn 151, University Hospitals Case Medical Center, Case Western Reserve University, 11100 Euclid Ave, Cleveland, OH 44106; e-mail: slg5@case.edu.
References
- 1.Kondo M, Wagers AJ, Manz MG, et al. Biology of hematopoietic stem cells and progenitors: implications for clinical application. Annu Rev Immunol. 2003;21(1):759–806. doi: 10.1146/annurev.immunol.21.120601.141007. [DOI] [PubMed] [Google Scholar]
- 2.Yang W. Structure and function of mismatch repair proteins. Mutat Res/DNA Repair. 2000;460(3-4):245–256. doi: 10.1016/s0921-8777(00)00030-6. [DOI] [PubMed] [Google Scholar]
- 3.Fukui K. DNA mismatch repair in eukaryotes and bacteria. J Nucleic Acids. 2010;2010 doi: 10.4061/2010/260512. Article ID 260512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kantelinen J, Kansikas M, Korhonen MK, et al. MutS[beta] exceeds MutS[alpha] in dinucleotide loop repair. Br J Cancer. 2010;102(6):1068–1073. doi: 10.1038/sj.bjc.6605531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lynch HT, Smyrk T, Lynch J. An update of HNPCC (Lynch syndrome). Cancer Genet Cytogenet. 1997;93(1):84–99. doi: 10.1016/s0165-4608(96)00290-7. [DOI] [PubMed] [Google Scholar]
- 6.Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(4):261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ricciardone M, Ozçelik T, Cevher B, et al. Human MLH1 deficiency predisposes to hematological malignancy and neurofibromatosis type 1. Cancer Res. 1999;59(2):290–293. [PubMed] [Google Scholar]
- 8.Whiteside D, McLeod R, Graham G, et al. A homozygous germ-line mutation in the human MSH2 gene predisposes to hematological malignancy and multiple café-au-lait spots. Cancer Res. 2002;62(2):359–362. [PubMed] [Google Scholar]
- 9.Herzog G, Lu-Hesselmann J, Zimmermann Y, Haferlach T, Hiddemann W, Dreyling M. Microsatellite instability and p53 mutations are characteristic of subgroups of acute myeloid leukemia but independent events. Haematologica. 2005;90(5):693–695. [PubMed] [Google Scholar]
- 10.Tasaka T, Lee S, Spira S, et al. Microsatellite instability during the progression of acute myelocytic leukaemia. Br J Haematol. 1997;98(1):219–221. doi: 10.1046/j.1365-2141.1997.1672985.x. [DOI] [PubMed] [Google Scholar]
- 11.Maeck L, Haase D, Schoch C, Hiddemann W, Alves F. Genetic instability in myelodysplastic syndrome: detection of microsatellite instability and loss of heterozygosity in bone marrow samples with karyotype alterations. Br J Haematol. 2000;109(4):842–846. doi: 10.1046/j.1365-2141.2000.02088.x. [DOI] [PubMed] [Google Scholar]
- 12.Olipitz W, Hopfinger G, Aguiar RCT, et al. Defective DNA-mismatch repair: a potential mediator of leukemogenic susceptibility in therapy-related myelodysplasia and leukemia. Genes Chromosomes Cancer. 2002;34(2):243–248. doi: 10.1002/gcc.10059. [DOI] [PubMed] [Google Scholar]
- 13.Ben Yehuda A, Globerson A, Krichevsky S, et al. Ageing and the mismatch repair system. Mechanisms of Ageing and Development. 2000;121(1-3):173–179. doi: 10.1016/s0047-6374(00)00208-6. [DOI] [PubMed] [Google Scholar]
- 14.Zhu YM, Das-Gupta EP, Russell NH. Microsatellite instability and p53 mutations are associated with abnormal expression of the MSH2 gene in adult acute leukemia. Blood. 1999;94(2):733–740. [PubMed] [Google Scholar]
- 15.Schlenke P, Kluter H, Muller-Steinhardt M, Hammers H-J, Borchert K, Bein G. Evaluation of a novel mononuclear cell isolation procedure for serological HLA typing. Clin Diagn Lab Immunol. 1998;5(6):808–813. doi: 10.1128/cdli.5.6.808-813.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Umetani N, Sasaki S, Watanabe T, Ishigami H, Ueda E, Nagawa H. Diagnostic primer sets for microsatellite instability optimized for a minimal amount of damaged DNA from colorectal tissue samples. Ann Surg Oncol. 2000;7(4):276–280. doi: 10.1007/s10434-000-0276-6. [DOI] [PubMed] [Google Scholar]
- 17.Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11(7):36–42. [Google Scholar]
- 18.Eng J. Baltimore, MD: Johns Hopkins University; 2010. ROC Analysis: Web-based Calculator for ROC Curves. [Google Scholar]
- 19.Metz CE. Basic principles of ROC analysis. Semin Nuclear Med. 1978;8(4):283–298. doi: 10.1016/s0001-2998(78)80014-2. [DOI] [PubMed] [Google Scholar]
- 20.Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248–5257. [PubMed] [Google Scholar]
- 21.Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138(6):2073–2087. doi: 10.1053/j.gastro.2009.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dietmaier W, Wallinger S, Bocker T, Kullmann F, Fishel R, Ruschoff J. Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res. 1997;57(21):4749–4756. [PubMed] [Google Scholar]
- 23.Cairns J. Mutation selection and the natural history of cancer. Nature. 1975;255(5505):197–200. doi: 10.1038/255197a0. [DOI] [PubMed] [Google Scholar]
- 24.Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68(4):820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baum CM, Weissman IL, Tsukamoto AS, Buckle AM, Peault B. Isolation of a candidate human hematopoietic stem-cell population. Proc Natl Acad Sci U S A. 1992;89(7):2804–2808. doi: 10.1073/pnas.89.7.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A. 1998;95(12):6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Veigl ML, Kasturi L, Olechnowicz J, et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci U S A. 1998;95(15):8698–8702. doi: 10.1073/pnas.95.15.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valeri N, Gasparini P, Fabbri M, et al. Modulation of mismatch repair and genomic stability by miR-155. Proc Natl Acad Sci U S A. 2010;107(15):6982–6987. doi: 10.1073/pnas.1002472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levinson G, Gutman GA. High frequencies of short frameshifts in poly-CA/TG tandem repeats borne by bacteriophage M13 in Escherichia coli K-12. Nucleic Acids Res. 1987;15(13):5323–5338. doi: 10.1093/nar/15.13.5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schlötterer C, Tautz D. Slippage synthesis of simple sequence DNA. Nucleic Acids Res. 1992;20(2):211–215. doi: 10.1093/nar/20.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reese JS, Liu L, Gerson SL. Repopulating defect of mismatch repair-deficient hematopoietic stem cells. Blood. 2003;102(5):1626–1633. doi: 10.1182/blood-2002-10-3035. [DOI] [PubMed] [Google Scholar]
- 32.de Wind N, Dekker M, Berns A, Radman M, te Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995;82(2):321–330. doi: 10.1016/0092-8674(95)90319-4. [DOI] [PubMed] [Google Scholar]
- 33.Reitmair AH, Redston M, Cai JC, et al. Spontaneous intestinal carcinomas and skin neoplasms in Msh2-deficient mice. Cancer Res. 1996;56(16):3842–3849. [PubMed] [Google Scholar]
- 34.Reitmair AH, Schmits R, Ewel A, et al. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat Genet. 1995;11(1):64–70. doi: 10.1038/ng0995-64. [DOI] [PubMed] [Google Scholar]
- 35.Zhang S, Lloyd R, Bowden G, Glickman BW, de Boer JG. Thymic lymphomas arising in Msh2 deficient mice display a large increase in mutation frequency and an altered mutational spectrum. Mutat Res/Fundam Mol Mech Mutagen. 2002;500(1-2):67–74. doi: 10.1016/s0027-5107(01)00297-4. [DOI] [PubMed] [Google Scholar]
- 36.Potocnik U, Glavac D, Golouh R, Ravnik-Glavac M. Causes of microsatellite instability in colorectal tumors: implications for hereditary non-polyposis colorectal cancer screening. Cancer Genet Cytogenet. 2001;126(2):85–96. doi: 10.1016/s0165-4608(00)00399-x. [DOI] [PubMed] [Google Scholar]
- 37.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A. 2006;103(5):1412–1417. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007;5(8):e201. doi: 10.1371/journal.pbio.0050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ben-Yehuda D, Krichevsky S, Caspi O, et al. Microsatellite instability and p53 mutations in therapy-related leukemia suggest mutator phenotype. Blood. 1996;88(11):4296–4303. [PubMed] [Google Scholar]
- 40.Laurie CC, Laurie CA, Rice K, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet. 2012;44(6):642–650. doi: 10.1038/ng.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jacobs KB, Yeager M, Zhou W, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet. 2012;44(6):651–658. doi: 10.1038/ng.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.