

Abstract
The corticospinal tract (CST) is a major descending pathway contributing to the control of voluntary movement in mammals. During the last decades anatomical and electrophysiological studies have demonstrated significant reorganization in the CST after spinal cord injury (SCI) in animals and humans. In animal models of SCI, anatomical evidence showed corticospinal sprouts rostral and caudal to the lesion and their integration into intraspinal axonal circuits. Electrophysiological data suggested that indirect connections from the primary motor cortex to forelimb motoneurons, via brainstem nuclei and spinal cord interneurons, or direct connections from slow uninjured corticospinal axons, might contribute to the control of movement after a CST injury. In humans with SCI, post mortem spinal cord tissue revealed anatomical changes in the CST some of which were similar but others markedly different from those found in animal models of SCI. Human electrophysiological studies have provided ample evidence for corticospinal reorganization after SCI that may contribute to functional recovery. Together these studies have revealed a large plastic capacity of the CST after SCI. There is also a limited understanding of the relationship between anatomical and electrophysiological changes in the CST and control of movement after SCI. Increasing our knowledge of the role of CST plasticity in functional restoration after SCI may support the development of more effective repair strategies.

Martin Oudega received his PhD from the University of Leiden in The Netherlands. Currently, he is faculty at the Departments of Physical Medicine and Rehabilitation, Neurobiology and Bioengineering at the University of Pittsburgh, USA. He directs the Spinal Cord Injury and Repair Laboratory and studies the efficacy of cellular and non-cellular transplants, alone or in combination with axon growth-supporting interventions, to elicit anatomical and/or functional restoration after spinal cord injury. He also investigates which genes are crucial for successful axon regeneration and functional restoration in zebrafish with spinal cord injury. The overall goal of his studies is to develop spinal cord repair strategies for clinical translation. Monica A. Perez received her PhD from the University of Miami, FL, USA. She is a faculty member at the Departments of Physical Medicine and Rehabilitation and Physical Therapy and a member of the Systems Neuroscience Institute at the University of Pittsburgh, USA. Her research examines how the brain and spinal cord contribute to the control of movements in healthy humans and in individuals with spinal cord injury. This theme is mainly investigated from a neurophysiological point of view, using a combination of transcranial magnetic and electrical stimulation, and peripheral nerve stimulation techniques. The overall goal of her studies is to develop strategies that can be used to maximize function of partially paralysed muscles after spinal cord injury.
Introduction
There are over 400,000 persons with spinal cord injury (SCI) in the United States and several millions worldwide. SCI impairs motor and sensory function resulting in disabilities that seriously diminish the quality of life (Hill et al. 2009; Herrmann et al. 2011). Numerous repair strategies have been tested in a laboratory environment but treatments that improved function have not yet been translated successfully into the clinic. At present, rehabilitation-based approaches are most widely used to promote recovery after SCI and these probably depend on the recruitment of descending motor pathways including the corticospinal tract (CST). The CST contributes significantly to the control of motor skilled movements in mammals (Lemon, 2008). During the last decades the CST has been a prominent target for investigating injury-induced plasticity and motor recovery after SCI.
This review discusses anatomical and electrophysiological reorganization in the CST after SCI in animals and humans. In different species, the CST originates from a variety of cortical areas and terminates in different regions of the spinal grey matter (Lemon & Griffiths, 2005). In the macaque monkey, the CST originates from nine different cortical regions, and the primary motor cortex contributes to about 30% of the axons (Galea & Darian-Smith, 1994). Each cortical subdivision has its own unique pattern of termination within the spinal grey matter, indicating different functional roles for each of the different subdivisions. Indeed, CST projections originating from primary motor cortex are of major importance for voluntary movement, while those from dorsal and ventral premotor areas are involved in the sensory guidance of movement, axons from the supplementary motor area in the planning and coordination of movement sequences, and axons from cingulate motor areas in emotional aspects of movement (Lemon & Griffiths, 2005; Lemon, 2008). This review focuses on corticospinal axons originating in the primary motor cortex but these issues will be considered in the summary of our conclusions. In animal models of SCI we will focus on rodents, cats and macaque monkeys. Rodents offer the possibility for detailed quantitative studies of SCI-induced changes in CST anatomy using tracing and histological techniques. Cats contribute to our understanding of the reorganization and development of corticospinal neurons (Martin, 2005). Monkeys have anatomical structures, motor apparatus and motor behaviour comparable to humans (Lemon & Griffiths, 2005; Darian-Smith, 2007). In humans, anatomical and electrophysiological changes in CST organization have been demonstrated in numerous studies revealing a large plastic ability that may contribute to functional recovery after SCI.
Anatomical reorganization of the CST after SCI
Rodents
The CST in rodents consists of a major crossed dorsal and minor uncrossed ventral component (Fig. 1A; Joosten et al. 1987; Terashimi, 1995). About 95% of all corticospinal axons are located in the ventral aspect of the dorsal columns and about 3–5% in the medial aspect of the ventral columns (Fig. 1A; Joosten et al. 1992; Brösamle & Schwab, 1997). Two minor CST components are located laterally and dorsolaterally in the lateral columns (Joosten et al. 1992). The defined location of the dorsal CST allows focal lesions without damaging other descending tracts. However, focal dorsal CST lesions will leave the ventral and lateral CST intact, which is important to consider in the interpretation of anatomical and electrophysiological data.
Figure 1. Corticospinal tract anatomy in rodents and cats/monkeys/humans.
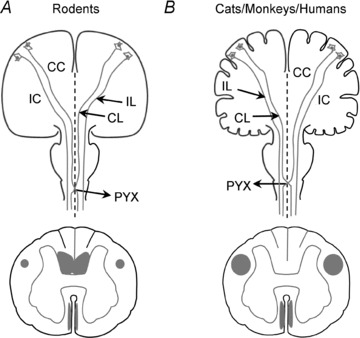
Schematic drawings of the brain and spinal cord illustrating the course of the corticospinal tract (CST) in adult rodents and cats/monkeys/humans. A, in adult rodents, corticospinal axons deriving from neurons in the motor cortex converge in the corpus callosum (CC), course through the internal capsule (IC), and cross the midline (dashed line) in the pyramidal decussation (PYX) and descend in the spinal cord. Most corticospinal axons cross the midline (>90%; contralateral (CL) crossed axons). The remaining corticospinal axons follow the same trajectory but do not cross the midline (<10%; ipsilateral (IL) uncrossed axons). The CST has three bilateral components that course in the ventral part of the dorsal columns, the dorsal aspect of the lateral columns, and the medial aspect of the ventral columns. B, in adult cats, monkeys and humans, corticospinal axons from motor cortex neurons course through the corpus callosum (CC) and internal capsule (IC) to cross the midline (dashed line) in the pyramidal decussation (PYX) and descend in the spinal cord. Most corticospinal axons cross the midline (75–95%; contralateral (CL) crossed axons). The remaining corticospinal axons have a similar route but do not cross the midline (5–25%, ipsilateral (IL) uncrossed axons). In these species, the CST has two bilateral components coursing in the dorsal aspect of the lateral columns (the lateral cerebrospinal fascicles) and the medial aspect of the ventral columns (the ventral cerebrospinal fascicles). For clarity, the illustrations of brain and spinal cord are not to scale.
The rodent's motor cortex experiences dynamic structural changes after SCI. Studies by Kalil and collaborators demonstrated severe shrinkage (atrophy) of layer 5 corticospinal neurons as early as 2 weeks after a lesion at the pyramidal decussation (Kalil & Schneider, 1975). In agreement, retrograde tracing (McBride et al. 1989; Tang et al. 2004) and imaging (Carter et al. 2008) studies showed atrophy of corticospinal neurons after SCI. After axotomy at the spinal cord level, some corticospinal axons retrogradely degenerate while others die back over a few millimeters (Oudega et al. 1999). The distal parts of the axotomized corticospinal axons undergo progressive fragmentation and Wallerian degeneration starting within days after injury (Hill et al. 2001). Death of corticospinal neurons resulting from axotomy after SCI remains controversial. Some studies have confirmed SCI-induced loss of corticospinal neurons (Hains et al. 2003; Sasaki et al. 2006) but others are in variance (Kalil & Schneider, 1975; Barron et al. 1988; McBride et al. 1989; Nielson et al. 2010). The disagreement may be due to differences in methodology, the ability of surviving axons to reorganize (Li et al. 1994; Hill et al. 2001), time of injury (Leenen et al. 1989; Merline & Kalil, 1990; Oudega et al. 1994), or injury location. For example, injuries close to the pyramids result in pronounced death of corticospinal neurons (Bonatz et al. 2000) whereas the loss is much less with injuries at the spinal cord level (Kalil & Schneider, 1975), although this still remains controversial (Hains et al. 2003). The morphology of dendritic spines of corticospinal neurons also changes after SCI with a marked decrease in postsynaptic spine density and an increase in spine length and head diameter (Kim et al. 2006). The remarkable increase in spine length suggests that corticospinal neurons acquire an immature and modifiable pattern of synaptic connectivity after SCI (Kim et al. 2006).
Many studies have demonstrated that the CST has the capacity to spontaneously sprout rostrally and caudally to a spinal cord lesion. Injury-induced sprouting has been shown from injured and uninjured corticospinal axons, days and weeks after injury, and near and away from the injury site (Fouad et al. 2001, 2011; Hill et al. 2001; Bareyre et al. 2004; Ghosh et al. 2009; Onifer et al. 2011). A lesion of the dorsal CST only in the thoracic spinal cord resulted in corticospinal sprouts which established synaptic connections with cervical propriospinal neurons projecting to adjacent or distant spinal cord segments (Fig. 2A; Fouad et al. 2001; Bareyre et al. 2004; Ghosh et al. 2009). Interestingly, with longer post-lesion survival times, synaptic connections of corticospinal sprouts on propriospinal neurons with short projections were lost whereas those on propriospinal neurons with long projections remained (Bareyre et al. 2004). This refinement of newly established indirect corticospinal connections ensured an influence onto the spinal cord below the level of the injury, which may have important functional implications. These data suggest that remodeling of corticospinal neurons after SCI occurs in two phases: initially injured corticospinal axons contact neurons in a non-specific fashion which is followed later by refinement of the established connections (Bareyre et al. 2004). A lesion of the dorsal CST also leads to substantial spontaneous sprouting from the ventral (undamaged) CST (Fig. 2A) and a lesion of both the dorsal and ventral CST eliminated all sprouting (Fig. 2B; Weidner et al. 2001). If the ventral CST lesion was completed after the dorsal CST lesion animals did not show functional improvements, suggesting that sprouting from ventral CST axons was involved in functional recovery (Weidner et al. 2001).
Figure 2. Corticospinal sprouting after SCI.
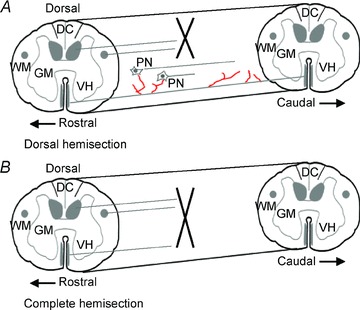
Schematic drawings of the CST response to SCI in adult rats/mice. A, a dorsal hemisection transecting the dorsal and lateral CST results in extensive sprouting (in red) of the unlesioned ventral component of the CST rostral and caudal to the lesion. Rostral CST sprouts connect with propriospinal neurons (PN) with local or distant projections. The former connections are transient while the latter remain, thereby establishing CST influences over the caudal spinal cord segments. Note that rostral is to the left as indicated in both figures. B, CST sprouting is absent after a complete spinal cord transection in adult rats/mice. Abbreviations: DC, dorsal columns; GM, grey matter; VH, ventral horn; WM, white matter.
Corticospinal sprouting can also be elicited by experimental interventions including the application of brain-derived neurotrophic factor (BDNF) into the cortex (Namiki et al. 2000; Hiebert et al. 2002; Zhou & Shine, 2003), transplantation of bone marrow-derived mesenchymal stem cells genetically modified to express higher levels of BDNF (Sasaki et al. 2009), neutralization of the neurite outgrowth inhibitory protein Nogo-A (Schwab, 2004; Gonzenbach & Schwab, 2008), electrical stimulation of the CST (Brus-Ramer et al. 2007; Carmel et al. 2010) and rehabilitative training paradigms (Fouad et al. 2000; Girgis et al. 2007; Krajacic et al. 2010).
It is important to consider that the sprouting capacity of the CST, whether spontaneous or induced, may be influenced by factors such as Wallerian degeneration, changes in other injured or intact axonal tracts (Ballermann & Fouad, 2006), lesion severity (Hill et al. 2001), development (Kalil & Reh, 1982) and external factors such as aging (Jaerve et al. 2011). Methodological aspects may also affect CST sprouting. For example, a single neurotrophin injection may increase (Schnell et al. 1994) but prolonged neurotrophin infusion may decrease (Hagg et al. 2005) collateral sprouting.
Cats
The CST in cats contains a larger dorsolateral and a smaller ventral component. At the cervical level, the dorsolateral CST contains about 92% and the ventral CST about 63% crossed axons (Fig. 1B; Chambers & Liu, 1957; Flindt-Egebak, 1979; Martin, 2005).
Diffusion tensor imaging analysis showed Wallerian degeneration of descending tracts several centimeters caudal to a unilateral hemisection 3–21 days post-lesion (Cohen-Adad et al. 2011). Although, specific descending tracts were not identified in this study, the regions of interest covered the location of the CST. As in rodents, the CST in cats has the capacity to sprout spontaneously. Evidence has shown that the density of corticospinal axon terminals increases after a unilateral CST lesion and that the sprouting capacity of the CST varied with development (Gómez-Pinilla et al. 1986; Murray & Goldberger, 1986; Martin et al. 1999). In neonatal cats, the density of corticospinal axon terminals increased ipsilateral to the lesion whereas in adult cats most terminals were found contralateral to the lesion (Gómez-Pinilla et al. 1986). During the postnatal corticospinal critical period, competitive or cooperative interactions between corticospinal neurons have been reported on the lesioned and intact side, suggesting that activity-dependent refinement of axon terminals is a mechanism of anatomical and functional CST reorganization (Martin et al. 1999).
The sprouting capacity of the CST in cats can be enhanced under experimental conditions. Electrical stimulation of CST axons rescues existing and promotes growth of transient corticospinal terminals, suggesting that both orthodromic and antidromic effects of stimulation could lead to further sprouting in the developing and mature CST (Salimi & Martin, 2004). Also, blockage of CST activity on one side resulted in novel spinal grey matter axon terminals from the unaffected CST (Chakrabarty & Martin, 2010), which might be used to enhance the potential of the damaged corticospinal system after SCI.
Non-human primates
The majority of CST axons in non-human primates descend in the dorsal aspect of the lateral columns (Fig. 1B; Lawrence & Kuypers, 1968) and consist of around 90% crossed axons at the cervical enlargement (Galea & Darian-Smith, 1994; Lacroix et al. 2004). A minor portion of corticospinal axons descend in the medial aspect of the ventral columns (Fig. 1B; Lawrence & Kuypers, 1968). In macaques, CST neurons with monosynaptic connections with spinal motoneurons are located in the caudal but not rostral region of the motor cortex (Rathelot & Strick, 2009), which may provide an important part of the neural substrate for the enhanced manual dexterity in macaques and humans.
Past and more recent anatomical studies showed that unilateral CST lesions in the cervical spinal cord of macaque monkeys results in atrophy of corticospinal neurons (Wannier et al. 2005). Wallerian degeneration of the distal part of axotomized corticospinal axons starts hours after SCI (Bresnahan, 1978). As in rodents, in macaque monkeys there is some controversy regarding the effect of the distance between a lesion and the motor cortex on the death of corticospinal neurons. Substantial loss of corticospinal neurons was reported after a unilateral pyramidotomy (Pernet & Hepp-Reymond, 1975) while the loss after a cervical spinal cord lesion was minimal (Wannier et al. 2005). However, other studies showed large survival of corticospinal neurons irrespective of the location of the damage (Lassek, 1946; Bronson et al. 1978). It should be kept in mind that due to body size, lesions at a specific spinal cord level reflect larger distances in monkeys than in rodents, which might affect the probability of survival and thus the comparison between the two species.
Earlier studies with macaques suggested pronounced spontaneous sprouting of corticospinal axons after SCI, but the non-specificity of the histological methods limited the interpretation (McCough et al. 1958). In later studies, tracing techniques were used to show corticospinal projections to the hemicord caudal to a cervical (Galea & Darian-Smith, 1997) and lumbar (Aoki et al. 1986) spinal cord lesion. Galea and Darian-Smith (1997) used retrograde and anterograde tracing to demonstrate a lack of substantial anatomical reorganization of corticospinal projections over a recovery period of over 2 years after a C3 hemisection. In contrast, Aoki and colleagues (1997) used retrograde tracing unilaterally to a hemisection before, directly after and up to 38 months after the lesion to show a significant increase in retrogradely labelled neurons in the precentral motor cortex ipsilateral to the lesion, suggesting that corticospinal projections to the spinal cord caudal to the lesion were newly formed on the side of the spinal cord hemisection, possibly by collateral sprouting. Similarly, in adult rhesus monkeys, extensive spontaneous sprouting of spared corticospinal axons was reported after a low cervical hemisection (Fig. 3; Rosenzweig et al. 2010). In this study, 24 weeks post-lesion, corticospinal axon density unilateral to the lesion was restored up to 60% of the pre-lesion density most probably due to sprouting of unlesioned contralateral corticospinal axons (Rosenzweig et al. 2009, 2010).
Figure 3. Extensive compensatory plasticity of the lesioned corticospinal tract in monkeys.
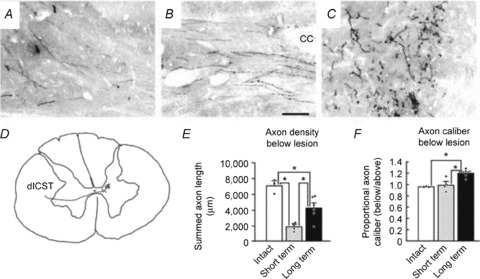
Corticospinal tract axons were labelled with D-A488. Axon density in intact (A), short-term lesioned (B), and long-term lesioned (C) monkey. In D, a single axon is reconstructed showing its origin from the left dorsolateral CST. E, axon density quantification revealed that CST density was reduced ∼75% 2 weeks after injury and recovered to more than half of pre-lesion axon density by 24 weeks post-lesion. F, quantification of axon thickness revealed that long-term lesioned animals exhibited a 20% increase in axon calibre below the lesion. In E and F, dots denote individual animals’ data points, *P < 0.05. Scale bar in A–C, 100 μm. Error bars indicate SEM. Figure modified from Fig. 4 in Rosenzweig et al. 2010.
Some experimental interventions were shown to facilitate corticospinal sprouting in the spinal cord. Neutralization of the neurite outgrowth inhibitory protein Nogo-A leads to extensive axonal sprouting caudal (Fig. 4; Freund et al. 2006) and rostral (Freund et al. 2007) to the lesion. Importantly, these observations for the first time provided evidence for a proof-of-principle in different animal models of SCI which might facilitate the development of new therapies for SCI in humans.
Figure 4. Nogo-A-specific antibody enhanced corticospinal axon sprouting.
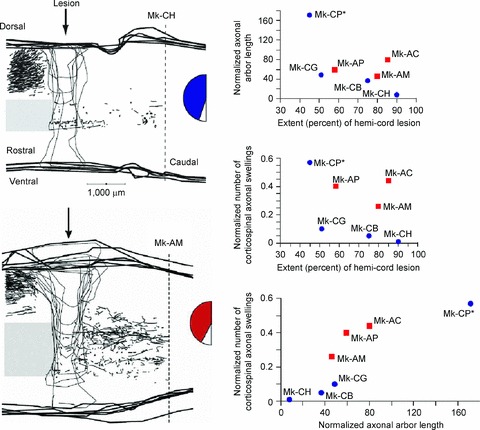
Corticospinal axons were labelled using biotinylated dextran amine (BDA) injections in the contralateral motor cortex. BDA-labelled axonal arbors caudal to the lesion were more numerous in monkeys treated with Nogo-A-specific antibody (Mk-AM; left, bottom) than with control antibody (Mk-CH; left, top). Seven monkeys were used to determine the normalized cumulative corticospinal axonal arbor length (in mm; right, top) and the normalized number of axonal swellings by corticospinal axons (i.e. putative re-established contacts with interneurons or motoneurons; right, middle), which were plotted as a function of lesion extent. Monkey Mk-CP had profuse sprouting but was incompletely lesioned. The number of corticospinal axonal swellings was also plotted as a function of the cumulative corticospinal axonal arbor length (in mm) (right, bottom). In all graphs, blue circles represent control antibody-treated monkeys and red squares represent Nogo-A-specific antibody-treated monkeys. The extent of the blue and red zones in the semicircular figures represents the extent of the hemicord lesion. Figure modified from Fig. 2 in Freund et al. 2006.
Humans
The location of the CST in humans and its division in a dorsal and ventral component is similar to that found in non-human primates. The CST consists of about 75% crossed axons in the dorsal aspect of the lateral columns and 25% uncrossed axons in the medial aspect of the ventral columns (Fig. 1B; Nyberg-Hansen & Rinvik, 1963; Lemon & Griffiths, 2005).
Early after SCI, necrosis and apoptosis are responsible for the death of neurons and glia near and far from the lesion. At later stages, the lesion commonly consists of a multilocular cavity traversed by vascular–glial bundles accompanied by regenerated nerve roots (Kakulas, 2004). Post mortem human spinal cord tissue revealed Wallerian degeneration in the CST as early as 12 days (Becerra et al. 1995; Buss et al. 2004) and by magnetic resonance imaging (MRI) analysis as early as 4–10 weeks post-lesion (Terae et al. 1993; Becerra et al. 1995; Quencer & Bunge, 1996). The areas of Wallerian degeneration were found to include progressive astrogliosis (Bunge et al. 1993; Puckett et al. 1997). Interestingly, in the chronically injured human spinal cord, the number of reactive astrocytes around the lesion cavities was small (Bunge et al. 1993; Puckett et al. 1997) in comparison to that found in rodent models of SCI (Murray et al. 1990). This finding may have significant implications for the regenerative ability of axons in the injured human spinal cord as they may not be exposed to the growth-inhibitory molecules expressed by reactive astrocytes to the same degree as in rodents. Other histological data showed that myelin loss during Wallerian degeneration of the CST after SCI is gradual with complete removal from the degenerated white matter over 8 years after injury (Buss et al. 2004).
In agreement, diffusion tensor imaging analysis indicated loss of CST axons and/or myelin in humans with chronic complete thoracic SCI (Wrigley et al. 2009). The water diffusion changes were also observed in the corticopontine tract which was not damaged by the spinal injury, suggesting that in humans, as in animal models of SCI, uninjured tracts undergo reorganization after the lesion. Evidence suggests that corticospinal neurons undergo atrophy after SCI (Yamamoto et al. 1989). Despite ample evidence for the presence of CST sprouting in animal models of SCI, evidence in humans remains sparse and indirect. A few studies showed a reduced number of myelinated corticospinal axons and retrograde degeneration in post mortem material after chronic SCI (Hunt, 1904; Bronson et al. 1978; Fishman, 1987). A marked depletion of CST axons was reported at the injury site and close to normal numbers of CST axons at a distance from the injury regardless of the injury duration, proposing that degenerated axons were replaced by collateral sprouts of surviving axons (Fishman, 1987).
Electrophysiological changes in the CST after SCI
Rodents
After SCI, transcranial electrical (TES; Fouad et al. 2001; Iyer et al. 2010; Nordblom et al. 2012) and magnetic (TMS; Magnusson et al. 1999; Chiba et al. 2003; Fujiki et al. 2004; Nielsen et al. 2007; Zhang et al. 2008) stimulation techniques have been used to examine the electrophysiology of descending pathways including the CST. TES applied over the rat motor cortex elicited different patterns of electromyographic (EMG) responses in forelimb muscles 4 weeks after a mid-thoracic CST lesion, suggesting the presence of corticospinal axon sprouting (Fouad et al. 2001). More recently, TES applied over the rat motor cortex 20 weeks after a complete thoracic spinal cord lesion with subsequent peripheral nerve transplantation revealed motor-evoked potentials (MEPs) in hindlimb muscles (Nordblom et al. 2012). In this study, the reappearance of MEPs post-lesion was paralleled by traced CST axons caudally to the lesion site. It is possible that some CST axons conducted signals that contributed to the evoked responses. However, other pathways from the brainstem and propriospinal axons in the spinal cord also may have been involved. Indeed, some controversy exists regarding which pathways are responsible for the evoked responses elicited by motor cortical stimulation in rats (Kamida et al. 1998; Luft et al. 2001, 2002; Chiba et al. 2003; Kaga et al. 2003). It has been shown that electrical stimulation of the CST at the pyramids elicits excitatory post-synaptic potentials (EPSPs) in forelimb motoneurons mediated by di- and trisynaptic excitatory corticofugal pathways and not exclusively by CST axons (Alstermark et al. 2004). Furthermore, a localized lesion of the rat CST did not affect the size of short-latency MEPs in forelimb and hindlimb muscles elicited by TMS over the motor cortex and mixed descending inputs contribute to the longer latency MEPs (Fig. 5; Nielsen et al. 2007). It also remains unclear how sprouting of CST axons (Hill et al. 2001) and of other descending pathways (Ballermann & Fouad, 2006) might contribute to the evoked responses.
Figure 5. The effect of dorsal CST lesion on biceps brachii MEPs evoked by TMS in the rat.
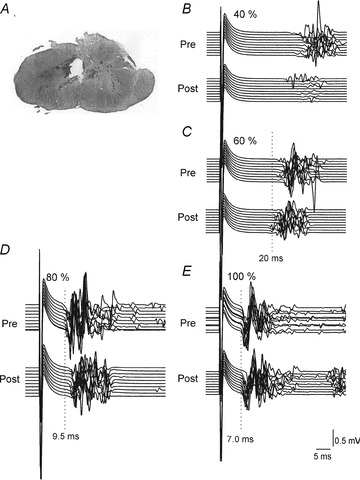
A, location and extent of the CST lesion in the dorsal funiculus. B–E, MEPs before (Pre; upper traces) and after (Post; lower traces) the lesion. The TMS stimulation intensity was 40, 60, 80 and 100% of maximum stimulator output in B, C, D and E, respectively. Ten traces are shown for each condition separated slightly in order to better visualize the different latencies of the MEPs. At low TMS intensities MEPs with long (15–20 ms) latencies (B and C, Pre) were found and these were suppressed after the lesion (B and C, Post). MEPs with 10 ms latencies were evoked at 80% of the maximum TMS intensity (D, Pre) and these were significantly depressed (D, Post) but not to the same extent as the MEPs with longer latencies. Maximum TMS intensities evoked MEPs at latencies around 7 ms (E, Pre) and these were not depressed following the CST lesion (E, Post). These observations show that it is unlikely that activation of motor cortex CST neurons contributes significantly to at least the earliest occurring MEPs evoked by TMS. Figure from Nielsen et al. 2007.
Cats
Compared to the other species, fewer data about electrophysiological changes in the CST after SCI are known from cats. Electrophysiological recordings of CST volleys after SCI have been employed mostly for assessing the location and completeness of the injury (Alstermark et al. 1986; Perfiliev et al. 1998). These studies revealed that after a complete CST transection at different levels of the cervical spinal cord, C3–C4 propriospinal neurons are probably involved in mediating signals to motoneurons required for reaching movements (Alstermark et al. 1986). Also, it was found that after a complete CST lesion the angular movement in the proximal interphalangeal joint of the paw was disrupted, indicating that the integrity of the CST is important for this particular behaviour (Perfiliev et al. 1998).
Non-human primates
In monkeys, changes in direct CST volleys in the injured spinal cord and monosynaptic field potentials in forelimb motor nuclei on the lesion side have been used to examine the severity of the lesion (Sasaki et al. 2004). In this study, stimulation of the contralateral pyramid after a complete CST transection resulted in disynaptic EPSPs in forelimb motoneurons (Sasaki et al. 2004). Importantly, the EPSPs were not observed in intact animals unless the feedforward inhibition of the C3–C4 system was decreased (Alstermark et al. 1999). These data suggested that a lesion of the CST can affect the output from the propriospinal system which might have important implications in humans. However, there is also evidence of a lack of a significant contribution of the C3–C4 system after CST injury to EPSPs in forelimb motoneurons, suggesting that the observed EPSPs could also result from monosynaptic corticomotoneuronal projections from slow uninjured corticospinal neurons (Maier et al. 1998; Lemon et al. 2004), which might be relevant because fast axons appear to be more susceptible to injury (Quencer et al. 1992).
After a thoracic spinal cord contusion, wide-spread TES of the brain, probably including the motor cortex, resulted in D-wave responses with long latencies and small amplitudes recorded by epidural electrodes near the injury (Arunkumar et al. 2001). The changes in both measurements correlated with motor function but were not predictive of the magnitude of recovery. The fact that in most cases the latency and amplitude returned back to baseline values within hours after the contusive injury suggests that the initial results were due to spinal shock rather than CST damage. A more recent study showed that MEPs could be elicited in leg muscles by applying TMS over the motor cortex 22 weeks after a complete thoracic spinal cord transection (Hernandez-Lain et al. 2011). Intriguingly, the intensity required for eliciting MEPs was similar pre-lesion and immediately post-lesion which is in disagreement with most studies in humans with SCI. Even though the authors argued that these results could represent a sign of CST regeneration, no evidence for this possibility was provided. Here, as in rodents, there are methodological considerations that need to be taken into account in the interpretation of the results. Without simultaneous recordings of MEPs, epidural descending volleys and recordings from spinal motoneurons, it remains uncertain that the changes in muscle MEPs reflect changes in the CST. Furthermore, the responses elicited by TES and TMS over the motor cortex in monkeys activate corticospinal axons projecting to spinal motoneurons in a complex fashion that is influenced by the size of the cell body, conduction velocity, location of the neurons and their level of excitability (Edgley et al. 1997). In monkeys, as in other animal models, it is also unclear how sprouting of CST axons, and other descending axons (Rosenzweig et al. 2010), might affect the specificity of the responses evoked by cortex stimulation.
Humans
Since the late twentieth century electrophysiological changes in the human CST after SCI have been studied primarily with TMS (Levy et al. 1990; Topka et al. 1991; Brouwer et al. 1992). In the acute and chronic SCI, MEPs elicited by TMS over the motor cortex have provided a measure of functional integrity of the CST.
Regardless of the time after injury, the majority of studies have shown a delay in the MEP latency in individuals with incomplete SCI compared to controls (Alexeeva et al. 1997, 1998; Curt et al. 1998; Davey et al. 1999; Smith et al. 2000; Ellaway et al. 2007). MEP latencies were found to be delayed by 2–15 ms in arm and leg muscles after cervical SCI for 1 day and for up to 6.5 years (Alexeeva et al. 1998; Davey et al. 1998; Smith et al. 2000). A delay in MEP latency of 2–3 ms was also found in scalenes and parasternal intercostals muscles during inspiration and expiration (Lissens & Vanderstraten, 1996). Individuals with thoracic SCI show MEP latency delays of 7–8 ms in paravertebral muscles at post-lesion times between 3 months and 19.9 years (Cariga et al. 2002). In some studies, it is difficult to precisely estimate changes in MEPs latencies over time because of methodological aspects such as combining data from individuals with acute and chronic SCI in the same analysis, and the use of different TMS stimulus intensities or contraction strengths. Nevertheless, there is clear consensus that MEP latencies are more prolonged after SCI and that the time after injury does not correlate to the MEP latency in the muscles tested (Smith et al. 2000). More recent studies continue to report similarly delayed latencies of MEPs in leg (Barthélemy et al. 2010) and arm (Roy et al. 2011) muscles in individuals with cervical, thoracic and lumbar SCI with post-lesion times of over 1 year.
One possible explanation for the delays in MEP latencies is the demyelination of corticospinal axons. This is typically observed in the injured human spinal cord (Bunge et al. 1993) and especially affects large-diameter axons (Quencer et al. 1992), which to some extent are activated by TMS (Petersen et al. 2003, 2010). Demyelination in the injured spinal cord occurs gradually over an extended period of time (Buss et al. 2004) which, unless the immediate injury-induced loss of myelin is at play, would not fully explain the acute presence (as early as 1 day post-lesion) of the delay in MEP latencies. It is then most likely that other factors also contribute to this particular deficit. The reduced number of myelinated CST axons and retrograde degeneration of injured CST axons after SCI (Hunt, 1904; Bronson et al. 1978; Fishman, 1987; Yamamoto et al. 1989) may be possible candidates. The lack of changes in MEP latency over time after injury despite the fact that patients improved their EMG recruitment patterns (Alexeeva et al. 1997; Smith et al. 2000) argues against the possibility that remyelination is an important contributor to the recovery process.
Most studies also showed that the threshold for evoking an MEP is increased after SCI and that this parameter is less affected by time after injury (Davey et al. 1998, 1999; Smith et al. 2000; Cariga et al. 2002; Freund et al. 2011). A longitudinal study in individuals with incomplete SCI demonstrated that motor thresholds examined at rest or during small voluntary contractions were significantly increased from 1 to 300 days after injury compared to controls (Smith et al. 2000). In agreement, individuals with post-lesion times between 90 to 852 days showed increased resting and active motor thresholds in finger muscles compared to controls (Davey et al. 1998, 1999). The increase in MEP threshold after SCI may be related to a decrease in corticospinal axons reaching the spinal motoneurons involved in the specific motor task. This is also consistent with the loss of innervations from segments above the lesion, which can contribute to MEP activation. It is important to consider that synaptic relays at the cortical and spinal cord level contribute to the formation of an MEP (Siebner & Rothwell, 2003). Thus, after incomplete SCI, threshold magnitude may be determined by the excitability of synaptic relays and the synaptic activity in the cortex (Roy et al. 2011).
The functional integrity of the CST has also been monitored intraoperatively during spine surgeries by measuring changes in MEPs elicited by TES (Deletis & Sala, 2008). Calancie et al. (1998) suggested that intraoperative changes in the threshold of MEPs elicited by TES accurately predict postoperative motor outcome. However, Sala et al. (2006) reported complete abolishment of MEPs during surgery in patients that recovered completely in hours or weeks after surgery. Together, these data suggest that monitoring of MEPs alone may not be sufficient for predicting motor outcome after an injury to the CST and that simultaneous recording of other measurements are necessary to determine that changes in muscle MEPs reflect changes in the CST.
More limited studies have investigated transmission in the CST during motor tasks after SCI. Evidence has shown that MEP latencies during static and dynamic tasks correlate with maximal movement velocity (Wirth et al. 2008), opening the likelihood that the sensitivity of TMS responses might be different during voluntary movement after SCI. A recent study showed that MEPs elicited in a resting limb were not modulated by contralateral strong voluntary contractions in individuals with SCI even in those that were able to perform the same level of voluntary force as controls (Fig. 6; Bunday & Perez, 2012). These last findings suggest that recovery of motor function and aspects of corticospinal function, such as crossed facilitatory effects, may be differentially affected by the injury. Deficits in CST transmission during voluntary activity after SCI have also been shown using coherence analysis. Coherence is a standard technique used to measure the strength of correlations between two signals in the frequency domain (Baker, 2007). Muscle-to-muscle coherence is thought to arise from last-order branches from corticospinal projections and probably reflects common corticospinal synaptic drive (Farmer et al. 1993; Conway et al. 1995; Baker et al. 1997). Studies have shown that coherence in the 10–20 Hz frequency band between populations of tibialis anterior motor units was absent during the swing phase of locomotion in individuals with SCI (Hansen et al. 2005; Barthélemy et al. 2010). Also, intermuscular coherence measured between hamstrings and vastus lateralis EMGs during locomotion was found to be decreased in individuals with SCI compared to controls (Norton & Gorassini, 2006). The results from coherence analysis indicate that after SCI changes in corticospinal transmission are present during the natural execution of a motor task. Although the combination of MEP and coherence measurements during voluntary activity have provided evidence about different aspects of corticospinal transmission after SCI, due to differences in the source of both signals, a direct comparison between these measurements remain difficult.
Figure 6. MEPs in individuals with and without cervical SCI.
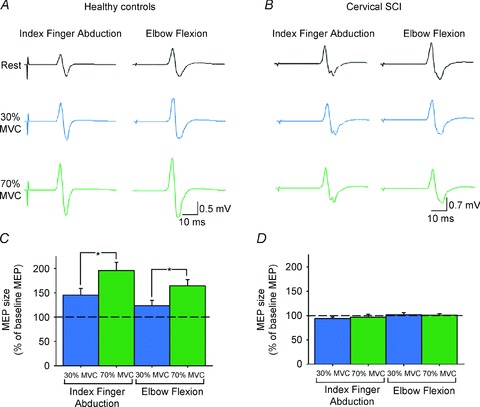
MEPs recorded from the resting first dorsal interosseous muscle (FDI) of a representative healthy control (A) and an individual with cervical SCI (B) while the other side remained at rest or performed 30% (blue traces) or 70% (green traces) of maximal voluntary contraction (MVC) into index finger abduction or elbow flexion. Group data (C, healthy controls, n = 10; D, cervical SCI, n = 14). The abscissa shows the MVC levels tested (30% of MVC, blue bars; 70% of MVC, green bars). The ordinate shows the size of the FDI MEP as a percentage of the baseline FDI MEP. Note the increase in FDI MEP size during contralateral index finger abduction and elbow flexion in healthy controls but not in individuals with cervical SCI. Error bars indicate SEMs. *P < 0.05. Figure modified from Fig. 1 in Bunday & Perez, 2012.
Training interventions after SCI
Rodents
Rehabilitative training alone or in combination with other therapies was shown to promote reorganization in the CST after SCI (Fouad et al. 2000; Girgis et al. 2007; Krajacic et al. 2010; Starkey et al. 2011). Training effects appear to depend, at least in part, on the location of the SCI and in the specificity and onset time of training. For example, 6 weeks of training a single-pellet-grasping task after a lesion of the dorsal CST or the dorsal and lateral CST resulted in functional improvements and CST sprouting (Krajacic et al. 2010). However, when the injury involved the lateral CST and the rubrospinal tract, which is located in the dorsal aspect of the lateral columns, no changes were observed in both motor behaviour and collateral sprouting. This surprising result might indicate that differences in the role of the injured descending tracts may have an impact on the behavioural and anatomical effects of training. In addition, training adult rats to perform a single-pellet-grasping task or horizontal ladder walking after a pyramidotomy resulted in specific improvements in the practiced task and some recovery in the untrained task (Starkey et al. 2011). Interestingly, in this study, only practice of the grasping task resulted in an increase in CST sprouting, suggesting that the complexity and demands of the training protocol are important factors that may impact the anatomical effects of training. Training efficacy appears to also be affected by the onset time of training. While acute training onset, as early as 4 days after injury, elicited task-specific improvements in motor recovery in a single-pellet-grasping task it also resulted in impairments in an untrained motor task (Girgis et al. 2007). The impairment in an untrained task was not present when training onset was shortly delayed, starting 12 days after injury (Krajacic et al. 2009).
When rehabilitative training strategies are combined with other interventions such as chondroitinase ABC (ChABC, a bacterial enzyme that digests extracellular axonal growth-inhibitory chondroitin sulfate proteoglycans) treatment functional recovery improved in acute (Garcia-Alias et al. 2009) and chronic (Wang et al. 2011) SCI. It is thought that CST sprouting above the lesion contributed to the observed functional improvements (Garcia-Alias et al. 2009; Wang et al. 2011). However, training-induced CST sprouting does not always come with behavioural improvements. After a thoracic lesion of the CST in rats, Nogo-A antibody treatment was found to improve the step cycle and CST sprouting. On the other hand, rats that trained on a treadmill showed improvements in locomotor function that were different to those seen after Nogo-A antibody treatment and also did not show signs of CST sprouting (Maier et al. 2009). The combination of Nogo-A antibody treatment and treadmill training resulted in increased CST sprouting but also in interference and poorer locomotor performance (Maier et al. 2009). These results emphasized the need to understand single treatments well before implementing them into combinatorial strategies because the ultimate strength of the combination depends on the characteristics (i.e. mechanism of action, timing, etc.) of the individual components. Also, the data underscore the possibility that a combinatorial approach might not always be the best option to maximize functional outcomes.
Cats
Experiments in cats have demonstrated that the CST is not essential for a motor task such as locomotion but important in situations in which adjustments of gait control are required such as walking on a horizontal ladder or during modifications of walking patterns (Drew, 1993; Drew et al. 2002). In cats, a lesion of the CST leads to drop foot, suggesting that corticospinal neurons are actively involved in muscle activity during undemanding walking (Drew et al. 2002). Despite the fact that CST injuries have been associated with specific locomotor deficits, intensive treadmill training of cats resulted in bilateral hindlimb locomotion within hours of a complete spinal cord lesion (Barrière et al. 2008). Even untrained cats recovered quadrupedal locomotion, albeit more delayed than trained cats. Training mostly affects spinal locomotor networks, but there are specific features such as corrective responses to the loss of ground support that depend to a large extent in the integrity of supraspinal input (Hiebert et al. 1994) which may involve the CST. These results demonstrate the necessity of considering the variations in corticospinal function and organization between species (Lemon & Griffiths, 2005) before extrapolation of experimental data.
Non-human primates
To our knowledge no evidence for the effect of rehabilitative training has been reported in non-human primates with SCI.
Humans
Evidence of the involvement of the CST in functional recovery after SCI has been demonstrated by studies using TMS and coherence analysis. In individuals with incomplete SCI, the size of MEPs in leg muscles elicited by TMS over the leg motor cortex was increased after 3–5 months of daily locomotor training (Thomas & Gorassini, 2005). The changes in MEP size were significantly correlated to the degree of locomotor recovery, suggesting that the CST was involved, at least in part, in the functional recovery of walking after training. Additionally, coherence measured between hamstrings and vastus lateralis in the 24–40 Hz frequency band increased after locomotor training in individuals with incomplete SCI, suggesting an increased corticospinal drive to leg muscles during walking (Norton & Gorassini, 2006). More direct evidence for a correlation between functional outcome and electrophysiological measures of corticospinal function was recently reported by Barthélemy and collaborators (2010). This study showed in individuals with SCI that 10–20 Hz coherence between paired tibialis anterior EMG recordings obtained during the swing phase of walking was positively correlated with the magnitude of foot drop, measured by toe elevation and ankle angle excursion during the swing phase of locomotion.
The involvement of the CST in functional recovery after injury has been also suggested for upper limb muscles. Individuals with cervical SCI demonstrate an increase in the size of the maximum MEPs elicited in the biceps brachii and extensor carpis radialis brevis and longus after intensive exercise therapy combined with functional electrical stimulation (Ellaway et al. 2011). A case report, in an individual with chronic cervical SCI, showed that after bimanual massed practice training combined with somatosensory stimulation, the MEP motor map measured in the biceps brachii muscle shifted anteriorly and increased in area and volume (Hoffman & Field-Fote, 2007). Furthermore, the threshold to elicit MEPs during a small voluntary contraction in an intrinsic finger muscle increased after the use of 5 Hz repetitive TMS (rTMS) over the hand representation in the motor cortex for five consecutive sessions in individuals with cervical SCI (Kuppuswamy et al. 2011). This unexpected result, suggesting lowering of corticospinal excitability after high-frequency rTMS, was attributed to some extent to a different balance of corticospinal excitability to different muscles after rTMS in individuals with SCI. Thus, if an isolated muscle contraction was needed individuals might have generated less corticospinal drive targeting other muscles. However, this interesting hypothesis remains to be tested. Together these data not only support the view of the involvement of the CST in recovery of motor function after SCI but also confirm that transmission in the CST is of relevance for human motor behaviour. Electrophysiological techniques can also be used to explore the impact of SCI on descending tracts other than the CST (Marchand-Pauvert et al. 2001; Pötter et al. 2008), which remain poorly understood and can represent a challenge for future studies.
Summary
During the last decades a large number of studies involving animal models of SCI have provided detailed evidence for anatomical changes in the CST after injury. In rodents, cats and monkeys SCI results in atrophy or death of corticospinal neurons, degeneration or dieback of CST axons and spontaneous CST sprouting including the formation of synaptic connections in the spinal cord. An important conclusion that can be drawn from these investigations is that ultimately CST reorganization represents the balance between lost and new synaptic connections and may differ per type of injury. Fewer studies have examined electrophysiological changes in the CST after SCI and much of the data are based on indirect methods to identify corticospinal neurons such as comparisons of MEP sizes or motor responses evoked by motor cortical stimulation. Thus, a clear gap exists in our knowledge of physiological effects of CST stimulation on spinal motoneurons after SCI. Some of the pressing questions that need to be understood are: How do residual corticospinal–motoneuronal synapses behave after SCI? How do residual descending inputs drive motoneurons? What are the effects of CST sprouting on spinal inter- and motoneurons? What is the functional role of new synaptic connections? Why do some CST axons sprout while others remain dormant or degenerate? Answering these questions represents challenges for future new investigations and they might be tackled by the use of a multidisciplinary approach, by further refinement of the methodology used to characterize the CST after SCI, and by considering these issues and their potential problems in the early stages of experimental designs.
In humans, studies using post-mortem spinal cord tissue revealed that some anatomical changes in the CST after SCI are similar and others markedly different from those reported in animals. For example, the number of astrocytes around injury-induced cystic cavities is small in humans but high in rodents. Also, while in animals the presence of collateral CST sprouting has been well demonstrated, their empirical demonstration in humans with SCI still remains circumstantial. In contrast to animal models of SCI, most of our information about CST reorganization after SCI in humans derives from electrophysiological studies. Although there is a consensus that MEP latencies and motor thresholds are increased regardless of the time after SCI, emerging studies are focusing on better understanding the involvement of the reorganized corticospinal pathway during functionally relevant motor tasks, which might be especially important for elucidating mechanism of recovery after SCI.
After decades of research, an intriguing unanswered question is why a large gap still exists in the extrapolation of knowledge from animal models into human SCI and repair. The answer to this question might be difficult to obtain mostly because of the complexity of the problem and to the differences in CST organization and transmission to spinal motoneurons between species. For example, the CST originates from many different functional areas making it unlikely that its contribution will involve a single role. In addition, the CST terminates extensively within the spinal grey matter, which might be reflected in its functions in the control of nociceptive inputs (Wall & Lidierth, 1997), somatosensory and reflex inputs (Pierrot-Deseilligny & Burke, 2005), autonomic (Bacon & Smith, 1993) and trophic (Martin et al. 1999) functions. In this review, we discussed in depth anatomical and electrophysiological consequences of SCI affecting CST projections from primary motor cortex. However, the consequences of damage to CST projections from other cortical regions might be reflected in behavioural aspects of movement such as the ability to plan a movement, ability to use sensory signals for movement corrections, ability to coordinate sequential actions and many others. These regions can play compensatory roles which, if properly understood, might be used as targets for clinical-related therapies. Therefore, one of the biggest challenges for future studies is to establish the relationship between anatomical and electrophysiological changes in the CST after SCI considering the diversity and complexity of CST projections to further understand their impact in reorganization. Future outcomes might benefit from expanding our knowledge in all these specific areas, which might support the development of the greatly needed strategies to restore function after human SCI.
Glossary
- BDNF
brain-derived neurotrophic factor
- CST
corticospinal tract
- SCI
spinal cord injury
- MEP
motor-evoked potentials
- TES
transcranial electrical stimulation
- TMS
transcranial magnetic stimulation
References
- Alexeeva N, Broton JG, Calancie B. Latency of changes in spinal motoneuron excitability evoked by transcranial magnetic brain stimulation in spinal cord injured individuals. Electroencephalogr Clin Neurophysiol. 1998;109:297–303. doi: 10.1016/s0924-980x(98)00021-6. [DOI] [PubMed] [Google Scholar]
- Alexeeva N, Broton JG, Suys S, Calancie B. Central cord syndrome of cervical spinal cord injury: widespread changes in muscle recruitment studied by voluntary contractions and transcranial magnetic stimulation. Exp Neurol. 1997;148:399–406. doi: 10.1006/exnr.1997.6689. [DOI] [PubMed] [Google Scholar]
- Alstermark B, Górska T, Johannisson T, Lundberg A. Effects of dorsal column transection in the upper cervical segments on visually guided forelimb movements. Neurosci Res. 1986;3:462–466. doi: 10.1016/0168-0102(86)90039-8. [DOI] [PubMed] [Google Scholar]
- Alstermark B, Isa T, Ohki Y, Saito Y. Disynaptic pyramidal excitation in forelimb motoneurons mediated via C3-C4 propriospinal neurons in the Macaca fuscata. J Neurophysiol. 1999;82:3580–3585. doi: 10.1152/jn.1999.82.6.3580. [DOI] [PubMed] [Google Scholar]
- Alstermark B, Ogawa J, Isa T. Lack of monosynaptic corticomotoneuronal EPSPs in rats: disynaptic EPSPs mediated via reticulospinal neurons and polysynaptic EPSPs via segmental interneurons. J Neurophysiol. 2004;91:1832–1839. doi: 10.1152/jn.00820.2003. [DOI] [PubMed] [Google Scholar]
- Aoki T, Matsunaga T, Misaki K, Watanabe Y, Terashima T. Abnormal distributions of callosal commissural and corticothalamic neurons in the cerebral neocortex of Shaking Rat Kawasaki. Neuroscience. 2002;114(2):427–438. doi: 10.1016/s0306-4522(02)00303-2. [DOI] [PubMed] [Google Scholar]
- Aoki M, Fujito Y, Satomi H, Kurosawa Y, Kasaba T. The possible role of collateral sprouting in the functional restitution of corticospinal connections after spinal hemisection. Neurosci Res. 1986;3:617–627. doi: 10.1016/0168-0102(86)90058-1. [DOI] [PubMed] [Google Scholar]
- Arunkumar MJ, Srinivasa Babu K, Chandy MJ. Motor and somatosensory evoked potentials in a primate model of experimental spinal cord injury. Neurol India. 2001;49:219–224. [PubMed] [Google Scholar]
- Bacon SJ, Smith AD. A monosynaptic pathway from an identified vasomotor centre in the medial prefrontal cortex to an autonomic area in the thoracic spinal cord. Neuroscience. 1993;54(3):719–728. doi: 10.1016/0306-4522(93)90242-8. [DOI] [PubMed] [Google Scholar]
- Baker SN. Oscillatory interactions between sensorimotor cortex and the periphery. Curr Opin Neurobiol. 2007;6:649–655. doi: 10.1016/j.conb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SN, Olivier E, Lemon RN. Coherent oscillations in monkey motor cortex and hand muscle EMG show task-dependent modulation. J Physiol. 1997;501:225–241. doi: 10.1111/j.1469-7793.1997.225bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballermann M, Fouad K. Spontaneous locomotor recovery in spinal cord injured rats is accompanied by anatomical plasticity of reticulospinal fibers. Eur J Neurosci. 2006;23:1988–1996. doi: 10.1111/j.1460-9568.2006.04726.x. [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Kerschensteiner M, Raineteau O, Mettenleiter TC, Weinmann O, Schwab ME. The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat Neurosci. 2004;7:269–277. doi: 10.1038/nn1195. [DOI] [PubMed] [Google Scholar]
- Barrière G, Leblond H, Provencher J, Rossignol S. Prominent role of the spinal central pattern generator in the recovery of locomotion after partial spinal cord injuries. J Neurosci. 2008;28:3976–3987. doi: 10.1523/JNEUROSCI.5692-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron KD, Dentinger MP, Popp AJ, Mankes R. Neurons of layer Vb of rat sensorimotor cortex atrophy but do not die after thoracic cord transection. J Neuropathol Exp Neurol. 1988;47:62–74. doi: 10.1097/00005072-198801000-00008. [DOI] [PubMed] [Google Scholar]
- Barthélemy D, Willerslev-Olsen M, Lundell H, Conway BA, Knudsen H, Biering-Sørensen F, Nielsen JB. Impaired transmission in the corticospinal tract and gait disability in spinal cord injured persons. J Neurophysiol. 2010;104:1167–1176. doi: 10.1152/jn.00382.2010. [DOI] [PubMed] [Google Scholar]
- Becerra JL, Puckett WR, Hiester ED, Quencer RM, Marcillo AE, Post MJ, Bunge RP. MR-pathologic comparisons of wallerian degeneration in spinal cord injury. AJNR Am J Neuroradiol. 1995;16:125–133. [PMC free article] [PubMed] [Google Scholar]
- Bonatz H, Rohrig S, Mestres P, Meyer M, Giehl KM. An axotomy model for the induction of death of rat and mouse corticospinal neurons in vivo. J Neurosci Methods. 2000;100:105–115. doi: 10.1016/s0165-0270(00)00238-7. [DOI] [PubMed] [Google Scholar]
- Bresnahan JC. An electron-microscopic analysis of axonal alterations following blunt contusion of the spinal cord of the rhesus monkey (Macaca mulatta. J Neurol Sci. 1978;37:59–82. doi: 10.1016/0022-510x(78)90228-9. [DOI] [PubMed] [Google Scholar]
- Bronson R, Gilles FH, Hall J, Hedley-Whyte ET. Long term post-traumatic retrograde corticospinal degeneration in man. Human Path. 1978;9:602–607. doi: 10.1016/s0046-8177(78)80143-9. [DOI] [PubMed] [Google Scholar]
- Brösamle C, Schwab ME. Cells of origin, course, and termination patterns of the ventral, uncrossed component of the mature rat corticospinal tract. J Comp Neurol. 1997;386:293–303. doi: 10.1002/(sici)1096-9861(19970922)386:2<293::aid-cne9>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Brouwer B, Bugaresti J, Ashby P. Changes in corticospinal facilitation of lower limb spinal motor neurons after spinal cord lesions. J Neurol Neurosurg Psychiatry. 1992;55:20–24. doi: 10.1136/jnnp.55.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brus-Ramer M, Carmel JB, Chakrabarty S, Martin JH. Electrical stimulation of spared corticospinal axons augments connections with ipsilateral spinal motor circuits after injury. J Neurosci. 2007;27:13793–13801. doi: 10.1523/JNEUROSCI.3489-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunday KL, Perez MA. Impaired crossed facilitation of the corticospinal pathway after cervical spinal cord injury. J Neurophysiol. 2012;107:2901–2911. doi: 10.1152/jn.00850.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunge RP, Puckett WR, Becerra JL, Marcello A, Quencer RM. Observation on the pathology of human spinal cord injury. A review and classification of 22 new cases with details from a case of chronic cord compression with extensive focal demyelinization. In: Seil FJ, editor. Advances in Neurology. New York: Raven Press; 1993. pp. 75–89. 1993. [PubMed] [Google Scholar]
- Buss A, Brook GA, Kakulas B, Martin D, Franzen R, Schoenen J, Noth J, Schmitt AB. Gradual loss of myelin and formation of an astrocytic scar during Wallerian degeneration in the human spinal cord. Brain. 2004;127:34–44. doi: 10.1093/brain/awh001. [DOI] [PubMed] [Google Scholar]
- Calancie B, Harris W, Broton JG, Alexeeva N, Green BA. “Threshold-level” multipulse transcranial electrical stimulation of motor cortex for intraoperative monitoring of spinal motor tracts: description of method and comparison to somatosensory evoked potential monitoring. J Neurosurg. 1998;88:457–470. doi: 10.3171/jns.1998.88.3.0457. [DOI] [PubMed] [Google Scholar]
- Cariga P, Catley M, Nowicky AV, Savic G, Ellaway PH, Davey NJ. Segmental recording of cortical motor evoked potentials from thoracic paravertebral myotomes in complete spinal cord injury. Spine (Phila Pa 1976) 2002;27:1438–1443. doi: 10.1097/00007632-200207010-00013. [DOI] [PubMed] [Google Scholar]
- Carmel JB, Berrol LJ, Brus-Ramer M, Martin JH. Chronic electrical stimulation of the intact corticospinal system after unilateral injury restores skilled locomotor control and promotes spinal axon outgrowth. J Neurosci. 2010;30:10918–10926. doi: 10.1523/JNEUROSCI.1435-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter LM, Starkey ML, Akrimi SF, Davies M, McMahon SB, Bradbury EJ. The yellow fluorescent protein (YFP-H) mouse reveals neuroprotection as a novel mechanism underlying chondroitinase ABC-mediated repair after spinal cord injury. J Neurosci. 2008;28:14107–14120. doi: 10.1523/JNEUROSCI.2217-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty S, Martin JH. Postnatal development of a segmental switch enables corticospinal tract transmission to spinal forelimb motor circuits. J Neurosci. 2010;30:2277–2288. doi: 10.1523/JNEUROSCI.5286-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers WW, Liu CN. Corticospinal tract of the cat: an attempt to correlate the pattern of degeneration with deficits in reflex activity following neocortical lesions. J Comp Neurol. 1957;108:23–55. doi: 10.1002/cne.901080103. [DOI] [PubMed] [Google Scholar]
- Chiba A, Oshio K, Inase M. Magnetically evoked EMGs in rats. Neurol Res. 2003;25:87–89. doi: 10.1179/016164103101200987. [DOI] [PubMed] [Google Scholar]
- Cohen-Adad J, Leblond H, Delivet-Mongrain H, Martinez M, Benali H, Rossignol S. Wallerian degeneration after spinal cord lesions in cats detected with diffusion tensor imaging. Neuroimage. 2011;57:1068–1076. doi: 10.1016/j.neuroimage.2011.04.068. [DOI] [PubMed] [Google Scholar]
- Conway BA, Halliday DM, Farmer SF, Shahani U, Maas P, Weir AI, Rosenberg JR. Synchronization between motor cortex and spinal motoneuronal pool during the performance of a maintained motor task in man. J Physiol. 1995;489:917–924. doi: 10.1113/jphysiol.1995.sp021104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curt A, Keck ME, Dietz V. Functional outcome following spinal cord injury: significance of motor-evoked potentials and ASIA scores. Arch Phys Med Rehabil. 1998;79:81–86. doi: 10.1016/s0003-9993(98)90213-1. [DOI] [PubMed] [Google Scholar]
- Darian-Smith C. Monkey models of recovery of voluntary hand movement after spinal cord and dorsal root injury. ILAR J. 2007;48:396–410. doi: 10.1093/ilar.48.4.396. [DOI] [PubMed] [Google Scholar]
- Davey NJ, Smith HC, Savic G, Maskill DW, Ellaway PH, Frankel HL. Comparison of input-output patterns in the corticospinal system of normal subjects and incomplete spinal cord injured patients. Exp Brain Res. 1999;127:382–390. doi: 10.1007/s002210050806. [DOI] [PubMed] [Google Scholar]
- Davey NJ, Smith HC, Wells E, Maskill DW, Savic G, Ellaway PH, Frankel HL. Responses of thenar muscles to transcranial magnetic stimulation of the motor cortex in patients with incomplete spinal cord injury. J Neurol Neurosurg Psychiatry. 1998;65:80–87. doi: 10.1136/jnnp.65.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deletis V, Sala F. Intraoperative neurophysiological monitoring of the spinal cord during spinal cord and spine surgery: a review focus on the corticospinal tracts. Clin Neurophysiol. 2008;119:248–264. doi: 10.1016/j.clinph.2007.09.135. [DOI] [PubMed] [Google Scholar]
- Drew T. Motor cortical activity during voluntary gait modifications in the cat. I. Cells related to the forelimbs. J Neurophysiol. 1993;70:179–199. doi: 10.1152/jn.1993.70.1.179. [DOI] [PubMed] [Google Scholar]
- Drew T, Jiang W, Widajewicz W. Contributions of the motor cortex to the control of the hindlimbs during locomotion in the cat. Brain Res Brain Res Rev. 2002;40:178–191. doi: 10.1016/s0165-0173(02)00200-x. [DOI] [PubMed] [Google Scholar]
- Edgley SA, Eyre JA, Lemon RN, Miller S. Comparison of activation of corticospinal neurons and spinal motor neurons by magnetic and electrical transcranial stimulation in the lumbosacral cord of the anaesthetized monkey. Brain. 1997;120:839–853. doi: 10.1093/brain/120.5.839. [DOI] [PubMed] [Google Scholar]
- Ellaway PH, Catley M, Davey NJ, Kuppuswamy A, Strutton P, Frankel HL, Jamous A, Savic G. Review of physiological motor outcome measures in spinal cord injury using transcranial magnetic stimulation and spinal reflexes. J Rehabil Res Dev. 2007;44:69–76. doi: 10.1682/jrrd.2005.08.0140. [DOI] [PubMed] [Google Scholar]
- Ellaway PH, Kuppuswamy A, Balasubramaniam AV, Maksimovic R, Gall A, Craggs MD, Mathias CJ, Bacon M, Prochazka A, Kowalczewski J, Conway BA, Galen S, Catton CJ, Allan DB, Curt A, Wirth B, van Hedel HJ. Development of quantitative and sensitive assessments of physiological and functional outcome during recovery from spinal cord injury: a clinical initiative. Brain Res Bull. 2011;84:343–357. doi: 10.1016/j.brainresbull.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Farmer SF, Bremner FD, Halliday DM, Rosenberg JR, Stephens JA. The frequency content of common synaptic inputs to motoneurones studied during voluntary isometric contraction in man. J Physiol. 1993;470:127–155. doi: 10.1113/jphysiol.1993.sp019851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman PS. Retrograde changes in the corticospinal tract of posttraumatic paraplegics. Arch Neurol. 1987;44:1082–1084. doi: 10.1001/archneur.1987.00520220078021. [DOI] [PubMed] [Google Scholar]
- Flindt-Egebak P. The corticofugal projections from the sensorimotor cortex to the spinal cord. A neuroanatomical and autoradiographical study in the cat with some methodological comments. J Hirnforsch. 1979;20:363–373. [PubMed] [Google Scholar]
- Fouad K, Krajacic A, Tetzlaff W. Spinal cord injury and plasticity: opportunities and challenges. Brain Res Bull. 2011;84:337–342. doi: 10.1016/j.brainresbull.2010.04.017. [DOI] [PubMed] [Google Scholar]
- Fouad K, Metz GA, Merkler D, Dietz V, Schwab ME. Treadmill training in incomplete spinal cord injured rats. Behav Brain Res. 2000;115:107–113. doi: 10.1016/s0166-4328(00)00244-8. [DOI] [PubMed] [Google Scholar]
- Fouad K, Pedersen V, Schwab ME, Brosamle C. Cervical sprouting of corticospinal fibers after thoracic spinal cord injury accompanies shifts in evoked motor responses. Curr Bio. 2001;11:1766–1770. doi: 10.1016/s0960-9822(01)00535-8. [DOI] [PubMed] [Google Scholar]
- Freund P, Rothwell J, Craggs M, Thompson AJ, Bestmann S. Corticomotor representation to a human forearm muscle changes following cervical spinal cord injury. Eur J Neurosci. 2011;34:1839–1846. doi: 10.1111/j.1460-9568.2011.07895.x. [DOI] [PubMed] [Google Scholar]
- Freund P, Schmidlin E, Wannier T, Bloch J, Mir A, Schwab ME, Rouiller EM. Nogo-A-specific antibody treatment enhances sprouting and functional recovery after cervical lesion in adult primates. Nat Med. 2006;12:790–792. doi: 10.1038/nm1436. [DOI] [PubMed] [Google Scholar]
- Freund P, Wannier T, Schmidlin E, Bloch J, Mir A, Schwab ME, Rouiller EM. Anti-Nogo-A antibody treatment enhances sprouting of corticospinal axons rostral to a unilateral cervical spinal cord lesion in adult macaque monkey. J Comp Neurol. 2007;502:644–659. doi: 10.1002/cne.21321. [DOI] [PubMed] [Google Scholar]
- Fujiki M, Kobayashi H, Inoue R, Ishii K. Immediate plasticity in the motor pathways after spinal cord hemisection: implications for transcranial magnetic motor-evoked potentials. Exp Neurol. 2004;187:468–477. doi: 10.1016/j.expneurol.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Galea MP, Darian-Smith I. Multiple corticospinal neuron populations in the macaque monkey are specified by their unique cortical origins, spinal terminations, and connections. Cereb Cortex. 1994;4:166–194. doi: 10.1093/cercor/4.2.166. [DOI] [PubMed] [Google Scholar]
- Galea MP, Darian-Smith I. Corticospinal projection patterns following unilateral section of the cervical spinal cord in the newborn and juvenile macaque monkey. J Comp Neurol. 1997;381:282–306. [PubMed] [Google Scholar]
- García-Alías G, Barkhuysen S, Buckle M, Fawcett JW. Chondroitinase ABC treatment opens a window of opportunity for task-specific rehabilitation. Nat Neurosci. 2009;12:1145–1151. doi: 10.1038/nn.2377. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Sydekum E, Haiss F, Peduzzi S, Zörner B, Schneider R, Baltes C, Rudin M, Weber B, Schwab ME. Functional and anatomical reorganization of the sensory-motor cortex after incomplete spinal cord injury in adult rats. J Neurosci. 2009;29:12210–12219. doi: 10.1523/JNEUROSCI.1828-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girgis J, Merrett D, Kirkland S, Metz GA, Verge V, Fouad K. Reaching training in rats with spinal cord injury promotes plasticity and task specific recovery. Brain. 2007;130:2993–3003. doi: 10.1093/brain/awm245. [DOI] [PubMed] [Google Scholar]
- Gómez-Pinilla F, Villablanca JR, Sonnier BJ, Levine MS. Reorganization of pericruciate cortical projections to the spinal cord and dorsal column nuclei after neonatal or adult cerebral hemispherectomy in cats. Brain Res. 1986;385:343–355. doi: 10.1016/0006-8993(86)91081-4. [DOI] [PubMed] [Google Scholar]
- Gonzenbach RR, Schwab ME. Disinhibition of neurite growth to repair the injured adult CNS: focusing on Nogo. Cell Mol Life Sci. 2008;65:161–176. doi: 10.1007/s00018-007-7170-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagg T, Baker KA, Emsley JG, Tetzlaff W. Prolonged local neurotrophin-3 infusion reduces ipsilateral collateral sprouting of spared corticospinal axons in adult rats. Neuroscience. 2005;130:875–887. doi: 10.1016/j.neuroscience.2004.10.018. [DOI] [PubMed] [Google Scholar]
- Hains BC, Black JA, Waxman SG. Primary cortical motor neurons undergo apoptosis after axotomizing spinal cord injury. J Comp Neurol. 2003;462:328–341. doi: 10.1002/cne.10733. [DOI] [PubMed] [Google Scholar]
- Hansen NL, Conway BA, Halliday DM, Hansen S, Pyndt HS, Biering-Sørensen F, Nielsen JB. Reduction of common synaptic drive to ankle dorsiflexor motoneurons during walking in patients with spinal cord lesion. J Neurophysiol. 2005;94:934–942. doi: 10.1152/jn.00082.2005. [DOI] [PubMed] [Google Scholar]
- Hernández-Laín A, Piedras MJ, Cavada C. Functional evaluation of paraplegic monkeys (Macaca mulatta) over fourteen months post-lesion. Neurosci Res. 2011;69:144–153. doi: 10.1016/j.neures.2010.11.003. [DOI] [PubMed] [Google Scholar]
- Herrmann KH, Kirchberger I, Biering-Sørensen F, Cieza A. Differences in functioning of individuals with tetraplegia and paraplegia according to the International Classification of Functioning, Disability and Health (ICF) Spinal Cord. 2011;49:534–543. doi: 10.1038/sc.2010.156. [DOI] [PubMed] [Google Scholar]
- Hiebert GW, Gorassini MA, Jiang W, Prochazka A, Pearson KG. Corrective responses to loss of ground support during walking. II. Comparison of intact and chronic spinal cats. J Neurophysiol. 1994;71:611–622. doi: 10.1152/jn.1994.71.2.611. [DOI] [PubMed] [Google Scholar]
- Hiebert GW, Khodarahmi K, McGraw J, Steeves JD, Tetzlaff W. Brain-derived neurotrophic factor applied to the motor cortex promotes sprouting of corticospinal tract fibers but not regeneration into a peripheral nerve transplant. J Neurosci Res. 2002;69:160–168. doi: 10.1002/jnr.10275. [DOI] [PubMed] [Google Scholar]
- Hill CE, Beattie MS, Bresnahan JC. Degeneration and sprouting of identified descending supraspinal axons after contusive spinal cord injury in the rat. Exp Neurol. 2001;171:153–169. doi: 10.1006/exnr.2001.7734. [DOI] [PubMed] [Google Scholar]
- Hill MR, Noonan VK, Sakakibara BM, Miller WC. Quality of life instruments and definitions in individuals with spinal cord injury: a systematic review. Spinal Cord. 2009;48:438–450. doi: 10.1038/sc.2009.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LR, Field-Fote EC. Cortical reorganization following bimanual training and somatosensory stimulation in cervical spinal cord injury: a case report. Phys Ther. 2007;87:208–223. doi: 10.2522/ptj.20050365. [DOI] [PubMed] [Google Scholar]
- Hunt RH. The retrograde atrophy of the pyramidal tracts. J Nerv Mental Dis. 1904;31:504–512. [Google Scholar]
- Iyer S, Maybhate A, Presacco A, All AH. Multi-limb acquisition of motor evoked potentials and its application in spinal cord injury. J Neurosci Methods. 2010;193:210–216. doi: 10.1016/j.jneumeth.2010.08.017. [DOI] [PubMed] [Google Scholar]
- Jaerve A, Schiwy N, Schmitz C, Mueller HW. Differential effect of aging on axon sprouting and regenerative growth in spinal cord injury. Exp Neurol. 2011;231:284–294. doi: 10.1016/j.expneurol.2011.07.002. [DOI] [PubMed] [Google Scholar]
- Joosten EA, Gribnau AA, Dederen PJ. An anterograde tracer study of the developing corticospinal tract in the rat: three components. Brain Res. 1987;433:121–130. doi: 10.1016/0165-3806(87)90070-8. [DOI] [PubMed] [Google Scholar]
- Joosten EA, Schuitman RL, Vermelis ME, Dederen PJ. Postnatal development of the ipsilateral corticospinal component in rat spinal cord: a light and electron microscopic anterograde HRP study. J Comp Neurol. 1992;326:133–146. doi: 10.1002/cne.903260112. [DOI] [PubMed] [Google Scholar]
- Kaga A, Fujiki M, Hori S, Nakano T, Isono M. Motor evoked potentials following transcranial magnetic stimulation after middle cerebral artery and/or basilar artery occlusions in rats. J Clin Neurosci. 2003;10:470–475. doi: 10.1016/s0967-5868(03)00082-1. [DOI] [PubMed] [Google Scholar]
- Kakulas BA. Neuropathology: the foundation for new treatments in spinal cord injury. Spinal Cord. 2004;42:549–563. doi: 10.1038/sj.sc.3101670. [DOI] [PubMed] [Google Scholar]
- Kalil K, Reh T. A light and electron microscopic study of regrowing pyramidal tract fibers. J Comp Neurol. 1982;211:265–275. doi: 10.1002/cne.902110305. [DOI] [PubMed] [Google Scholar]
- Kalil K, Schneider GE. Retrograde cortical aand axonal changes following lesions of the pyramidal tract. Brain Res. 1975;89:15–27. doi: 10.1016/0006-8993(75)90130-4. [DOI] [PubMed] [Google Scholar]
- Kamida T, Fujiki M, Hori S, Isono M. Conduction pathways of motor evoked potentials following transcranial magnetic stimulation: a rodent study using a ‘figure-8’ coil. Muscle Nerve. 1998;21:722–731. doi: 10.1002/(sici)1097-4598(199806)21:6<722::aid-mus3>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Kim BG, Dai HN, McAtee M, Vicini S, Bregman BS. Remodeling of synaptic structures in the motor cortex following spinal cord injury. Exp Neurol. 2006;198:401–415. doi: 10.1016/j.expneurol.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Krajacic A, Weishaupt N, Girgis J, Tetzlaff W, Fouad K. Training-induced plasticity in rats with cervical spinal cord injury: effects and side effects. Behav Brain Res. 2010;214:323–331. doi: 10.1016/j.bbr.2010.05.053. [DOI] [PubMed] [Google Scholar]
- Kuppuswamy A, Balasubramaniam AV, Maksimovic R, Mathias CJ, Gall A, Craggs MD, Ellaway PH. Action of 5 Hz repetitive transcranial magnetic stimulation on sensory, motor and autonomic function in human spinal cord injury. Clin Neurophysiol. 2011;122:2452–2461. doi: 10.1016/j.clinph.2011.04.022. [DOI] [PubMed] [Google Scholar]
- Lacroix S, Havton LA, McKay H, Yang H, Brant A, Roberts J, Tuszynski MH. Bilateral corticospinal projections arise from each motor cortex in the macaque monkey: a quantitative study. J Comp Neurol. 2004;473:147–161. doi: 10.1002/cne.20051. [DOI] [PubMed] [Google Scholar]
- Lassek AM. The pyramidal tract; speed of degeneration in axons following ablation of cells of origin in the monkey. J Comp Neurol. 1946;85:45–51. doi: 10.1002/cne.900850105. [DOI] [PubMed] [Google Scholar]
- Lawrence DG, Kuypers HG. The functional organization of the motor system in the monkey. II. The effects of lesions of the descending brain-stem pathways. Brain. 1968;91:15–36. doi: 10.1093/brain/91.1.15. [DOI] [PubMed] [Google Scholar]
- Leenen LP, Meek J, Posthuma PR, Nieuwenuys R. Differences in the fiber composition of the pyramidal tract in two- and 14-month-old rats. Neuroscience. 1989;28:635–643. doi: 10.1016/0306-4522(89)90010-9. [DOI] [PubMed] [Google Scholar]
- Lemon RN. Descending pathways in motor control. Annu Rev Neurosci. 2008;31:195–218. doi: 10.1146/annurev.neuro.31.060407.125547. [DOI] [PubMed] [Google Scholar]
- Lemon RN, Griffiths J. Comparing the function of the corticospinal system in different species: organizational differences for motor specialization? Muscle Nerve. 2005;32:261–279. doi: 10.1002/mus.20333. [DOI] [PubMed] [Google Scholar]
- Lemon RN, Kirkwood PA, Maier MA, Nakajima K, Nathan P. Direct and indirect pathways for corticospinal control of upper limb motoneurones in the primate. Prog Brain Res. 2004;143:263–279. doi: 10.1016/S0079-6123(03)43026-4. [DOI] [PubMed] [Google Scholar]
- Levy WJ, Jr, Amassian VE, Traad M, Cadwell J. Focal magnetic coil stimulation reveals motor cortical system reorganized in humans after traumatic quadriplegia. Brain Res. 1990;510:130–134. doi: 10.1016/0006-8993(90)90738-w. [DOI] [PubMed] [Google Scholar]
- Li WY, Yew DW, Chuah MI, Leung PC, Tsang DC. Axonal sprouting in the hemisected adult rat spinal cord. Neuroscience. 1994;61:133–139. doi: 10.1016/0306-4522(94)90066-3. [DOI] [PubMed] [Google Scholar]
- Lissens MA, Vanderstraeten GG. Motor evoked potentials of the respiratory muscles in tetraplegic patients. Spinal Cord. 1996;34:673–678. doi: 10.1038/sc.1996.122. [DOI] [PubMed] [Google Scholar]
- Luft AR, Kaelin-Lang A, Hauser TK, Buitrago MM, Thakor NV, Hanley DF, Cohen LG. Modulation of rodent cortical motor excitability by somatosensory input. Exp Brain Res. 2002;142:562–569. doi: 10.1007/s00221-001-0952-1. [DOI] [PubMed] [Google Scholar]
- Luft AR, Kaelin-Lang A, Hauser TK, Cohen LG, Thakor NV, Hanley DF. Transcranial magnetic stimulation in the rat. Exp Brain Res. 2001;140:112–121. doi: 10.1007/s002210100805. [DOI] [PubMed] [Google Scholar]
- McBride RL, Feringa ER, Garver MK, Williams JK., Jr Prelabeled red nucleus and sensorimotor cortex neurons of the rat survive 10 and 20 weeks after spinal cord transection. J Neuropathol Exp Neurol. 1989;48:568–576. doi: 10.1097/00005072-198909000-00007. [DOI] [PubMed] [Google Scholar]
- McCouch GP, Austin GM, Liu CN, Liu CY. Sprouting as a cause of spasticity. J Neurophysiol. 1958;21:205–216. doi: 10.1152/jn.1958.21.3.205. [DOI] [PubMed] [Google Scholar]
- Magnuson DS, Trinder TC, Zhang YP, Burke D, Morassutti DJ, Shields CB. Comparing deficits following excitotoxic and contusion injuries in the thoracic and lumbar spinal cord of the adult rat. Exp Neurol. 1999;156:191–204. doi: 10.1006/exnr.1999.7016. [DOI] [PubMed] [Google Scholar]
- Maier IC, Ichiyama RM, Courtine G, Schnell L, Lavrov I, Edgerton VR, Schwab ME. Differential effects of anti-Nogo-A antibody treatment and treadmill training in rats with incomplete spinal cord injury. Brain. 2009;132:1426–1440. doi: 10.1093/brain/awp085. [DOI] [PubMed] [Google Scholar]
- Maier MA, Illert M, Kirkwood PA, Nielsen J, Lemon RN. Does a C3–C4 propriospinal system transmit corticospinal excitation in the primate? An investigation in the macaque monkey. J Physiol. 1998;511:191–212. doi: 10.1111/j.1469-7793.1998.191bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand-Pauvert V, Mazevet D, Pradat-Diehl P, Alstermark B, Pierrot-Deseilligny E. Interruption of a relay of corticospinal excitation by a spinal lesion at C6-C7. Muscle Nerve. 2001;24:1554–1561. doi: 10.1002/mus.1183. [DOI] [PubMed] [Google Scholar]
- Martin JH. The corticospinal system: from development to motor control. Neuroscientist. 2005;11:161–173. doi: 10.1177/1073858404270843. [DOI] [PubMed] [Google Scholar]
- Martin JH, Kably B, Hacking A. Activity-dependent development of cortical axon terminations in the spinal cord and brain stem. Exp Brain Res. 1999;125:184–199. doi: 10.1007/s002210050673. [DOI] [PubMed] [Google Scholar]
- Merline M, Kalil K. Cell death of corticospinal neurons is induced by axotomy before but not after innervation of spinal targets. J Comp Neurol. 1990;296:506–516. doi: 10.1002/cne.902960313. [DOI] [PubMed] [Google Scholar]
- Murray M, Goldberger ME. Replacement of synaptic terminals in lamina II and Clarke's nucleus after unilateral lumbosacral dorsal rhizotomy in adult cats. J Neurosci. 1986;6:3205–3217. doi: 10.1523/JNEUROSCI.06-11-03205.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray M, Wang SD, Goldberger ME, Levitt P. Modification of astrocytes in the spinal cord following dorsal root or peripheral nerve lesions. Exp Neurol. 1990;110:248–257. doi: 10.1016/0014-4886(90)90036-r. [DOI] [PubMed] [Google Scholar]
- Namiki J, Kojima A, Tator CH. Effect of brain-derived neurotrophic factor, nerve growth factor, and neurotrophin-3 on functional recovery and regeneration after spinal cord injury in adult rats. J Neurotrauma. 2000;17:1219–1231. doi: 10.1089/neu.2000.17.1219. [DOI] [PubMed] [Google Scholar]
- Nielsen JB, Perez MA, Oudega M, Enriquez-Denton M, Aimonetti JM. Evaluation of transmission in descending motor tracts by transcranial magnetic stimulation in the rat. Eur J Neurosci. 2007;25:805–814. doi: 10.1111/j.1460-9568.2007.05326.x. [DOI] [PubMed] [Google Scholar]
- Nielson JL, Sears-Kraxberger I, Strong MK, Wong JK, Willenberg R, Steward O. Unexpected survival of neurons of origin of the pyramidal tract after spinal cord injury. J Neurosci. 2010;30:11516–11528. doi: 10.1523/JNEUROSCI.1433-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordblom J, Persson JK, Aberg J, Blom H, Engqvist H, Brismar H, Sjödahl J, Josephson A, Frostell A, Thams S, Brundin L, Svensson M, Mattsson P. FGF1 containing biodegradable device with peripheral nerve grafts induces corticospinal tract regeneration and motor evoked potentials after spinal cord resection. Restor Neurol Neurosci. 2012;30:91–102. doi: 10.3233/RNN-2011-0623. [DOI] [PubMed] [Google Scholar]
- Norton JA, Gorassini MA. Changes in cortically related intermuscular coherence accompanying improvements in locomotor skills in incomplete spinal cord injury. J Neurophysiol. 2006;95:2580–2589. doi: 10.1152/jn.01289.2005. [DOI] [PubMed] [Google Scholar]
- Nyberg-Hansen R, Rinvik E. Some comments on the pyramidal tract, with special reference to its individual variations in man. Acta Neurol Scand. 1963;39:1–30. [Google Scholar]
- Onifer SM, Smith GM, Fouad K. Plasticity after spinal cord injury: relevance to recovery and approaches to facilitate it. Neurotherapeutics. 2011;8:283–293. doi: 10.1007/s13311-011-0034-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudega M, Varon S, Hagg T. Distribution of corticospinal motor neurons in the postnatal rat: quantitative evidence for massive collateral elimination and modest cell death. J Comp Neurol. 1994;347:115–126. doi: 10.1002/cne.903470109. [DOI] [PubMed] [Google Scholar]
- Oudega M, Vargas CG, Weber AB, Kleitman N, Bunge MB. Long-term effects of methylprednisolone following transection of adult rat spinal cord. Eur J Neurosci. 1999;11:2453–2464. doi: 10.1046/j.1460-9568.1999.00666.x. [DOI] [PubMed] [Google Scholar]
- Perfiliev S, Pettersson LG, Lundberg A. Food-taking in the cat investigated with transection of the rubro- and corticospinal tracts. Neurosci Res. 1998;32:181–184. doi: 10.1016/s0168-0102(98)00073-x. [DOI] [PubMed] [Google Scholar]
- Pernet U, Hepp-Reymond MC. Retrograde degeneration of the pyramidal cells in the motor cortex of apes (Macaca fascicularis) (in German) Acta Anat (Basel) 1975;91:552–561. [PubMed] [Google Scholar]
- Petersen NC, Butler JE, Taylor JL, Gandevia SC. Probing the corticospinal link between the motor cortex and motoneurones: some neglected aspects of human motor cortical function. Acta Physiol (Oxf) 2010;198:403–416. doi: 10.1111/j.1748-1716.2009.02066.x. [DOI] [PubMed] [Google Scholar]
- Petersen NT, Pyndt HS, Nielsen JB. Investigating human motor control by transcranial magnetic stimulation. Exp Brain Res. 2003;152:1–16. doi: 10.1007/s00221-003-1537-y. [DOI] [PubMed] [Google Scholar]
- Pierrot-Deseilligny E, Burke D. The Circuitry of the Human Spinal Cord: Its Role in Motor Control and Movement Disorders. Cambridge, UK: Cambridge University Press; 2005. [Google Scholar]
- Pötter M, Herzog J, Siebner HR, Kopper F, Steigerwald F, Deuschl G, Volkmann J. Subthalamic nucleus stimulation modulates audiospinal reactions in Parkinson disease. Neurology. 2008;70:1445–1451. doi: 10.1212/01.wnl.0000310422.49977.ea. [DOI] [PubMed] [Google Scholar]
- Puckett WR, Hiester ED, Norenberg MD, Marcillo AE, Bunge RP. The astroglial response to Wallerian degeneration after spinal cord injury in humans. Exp Neurol. 1997;148:424–432. doi: 10.1006/exnr.1997.6692. [DOI] [PubMed] [Google Scholar]
- Quencer RM, Bunge RP. The injured spinal cord: imaging, histopathologic clinical correlates, and basic science approaches to enhancing neural function after spinal cord injury. Spine (Phila Pa 1976) 1996;21:2064–2066. doi: 10.1097/00007632-199609150-00002. [DOI] [PubMed] [Google Scholar]
- Quencer RM, Bunge RP, Egnor M, Green BA, Puckett W, Naidich TP, Post MJ, Norenberg M. Acute traumatic central cord syndrome: MRI-pathological correlations. Neuroradiology. 1992;34:85–94. doi: 10.1007/BF00588148. [DOI] [PubMed] [Google Scholar]
- Rathelot JA, Strick PL. Subdivisions of primary motor cortex based on cortico-motoneuronal cells. Proc Natl Acad Sci U S A. 2009;106:918–923. doi: 10.1073/pnas.0808362106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig ES, Brock JH, Culbertson MD, Lu P, Moseanko R, Edgerton VR, Havton LA, Tuszynski MH. Extensive spinal decussation and bilateral termination of cervical corticospinal projections in rhesus monkeys. J Comp Neurol. 2009;513:151–163. doi: 10.1002/cne.21940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig ES, Courtine G, Jindrich DL, Brock JH, Ferguson AR, Strand SC, Nout YS, Roy RR, Miller DM, Beattie MS, Havton LA, Bresnahan JC, Edgerton VR, Tuszynski MH. Extensive spontaneous plasticity of corticospinal projections after primate spinal cord injury. Nat Neurosci. 2010;13:1505–1510. doi: 10.1038/nn.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy FD, Zewdie ET, Gorassini MA. Short-interval intracortical inhibition with incomplete spinal cord injury. Clin Neurophysiol. 2011;122:1387–1395. doi: 10.1016/j.clinph.2010.11.020. [DOI] [PubMed] [Google Scholar]
- Sala F, Palandri G, Basso E, Lanteri P, Deletis V, Faccioli F, Bricolo A. Intraoperative motor evoked potential monitoring improves outcome after surgery of intramedullary spinal cord tumor: a historical control study in 50 patients. Neurosurgery. 2006;58:1129–1143. doi: 10.1227/01.NEU.0000215948.97195.58. [DOI] [PubMed] [Google Scholar]
- Salimi I, Martin JH. Rescuing transient corticospinal terminations and promoting growth with corticospinal stimulation in kittens. J Neurosci. 2004;24:4952–4961. doi: 10.1523/JNEUROSCI.0004-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Hains BC, Lankford KL, Waxman SG, Kocsis JD. Protection of corticospinal tract neurons after dorsal spinal cord transection and engraftment of olfactory ensheathing cells. Glia. 2006;53:352–359. doi: 10.1002/glia.20285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Radtke C, Tan AM, Zhao P, Hamada H, Houkin K, Honmou O, Kocsis JD. BDNF-hypersecreting human mesenchymal stem cells promote functional recovery, axonal sprouting, and protection of corticospinal neurons after spinal cord injury. J Neurosci. 2009;29:14932–14941. doi: 10.1523/JNEUROSCI.2769-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Isa T, Pettersson LG, Alstermark B, Naito K, Yoshimura K, Seki K, Ohki Y. Dexterous finger movements in primate without monosynaptic corticomotoneuronal excitation. J Neurophysiol. 2004;92:3142–3147. doi: 10.1152/jn.00342.2004. [DOI] [PubMed] [Google Scholar]
- Siebner HR, Rothwell J. Transcranial magnetic stimulation: new insights into representational cortical plasticity. Exp Brain Res. 2003;148:1–16. doi: 10.1007/s00221-002-1234-2. [DOI] [PubMed] [Google Scholar]
- Starkey ML, Bleul C, Maier IC, Schwab ME. Rehabilitative training following unilateral pyramidotomy in adult rats improves forelimb function in a non-task-specific way. Exp Neurol. 2011;232:81–89. doi: 10.1016/j.expneurol.2011.08.006. [DOI] [PubMed] [Google Scholar]
- Schnell L, Schneider R, Kolbeck R, Barde YA, Schwab ME. Neurotrophin-3 enhances sprouting of corticospinal tract during development and after adult spinal cord lesion. Nature. 1994;367:170–173. doi: 10.1038/367170a0. [DOI] [PubMed] [Google Scholar]
- Schwab ME. Nogo and axon regeneration. Curr Opin Neurobiol. 2004;14:118–124. doi: 10.1016/j.conb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Smith HC, Savic G, Frankel HL, Ellaway PH, Maskill DW, Jamous MA, Davey NJ. Corticospinal function studied over time following incomplete spinal cord injury. Spinal Cord. 2000;38:292–300. doi: 10.1038/sj.sc.3100994. [DOI] [PubMed] [Google Scholar]
- Tang XQ, Wang Y, Huang ZH, Han JS, Wan Y. Adenovirus-mediated delivery of GDNF ameliorates corticospinal neuronal atrophy and motor function deficits in rats with spinal cord injury. Neuroreport. 2004;15:425–429. doi: 10.1097/00001756-200403010-00009. [DOI] [PubMed] [Google Scholar]
- Terae S, Taneichi H, Abumi K. MRI of wallerian degeneration of the injured spinal cord. J Comput Assist Tomogr. 1993;17:700–703. doi: 10.1097/00004728-199309000-00006. [DOI] [PubMed] [Google Scholar]
- Terashima T. Anatomy, development and lesion-induced plasticity of rodent corticospinal tract. Neurosci Res. 1995;22:139–161. doi: 10.1016/0168-0102(95)00895-9. [DOI] [PubMed] [Google Scholar]
- Thomas SL, Gorassini MA. Increases in corticospinal tract function by treadmill training after incomplete spinal cord injury. J Neurophysiol. 2005;94:2844–2855. doi: 10.1152/jn.00532.2005. [DOI] [PubMed] [Google Scholar]
- Topka H, Cohen LG, Cole RA, Hallett M. Reorganization of corticospinal pathways following spinal cord injury. Neurology. 1991;41:1276–1283. doi: 10.1212/wnl.41.8.1276. [DOI] [PubMed] [Google Scholar]
- Wall PD, Lidierth M. Five sources of a dorsal root potential: their interactions and origins in the superficial dorsal horn. J Neurophysiol. 1997;78:860–871. doi: 10.1152/jn.1997.78.2.860. [DOI] [PubMed] [Google Scholar]
- Wang D, Ichiyama RM, Zhao R, Andrews MR, Fawcett JW. Chondroitinase combined with rehabilitation promotes recovery of forelimb function in rats with chronic spinal cord injury. J Neurosci. 2011;31:9332–9344. doi: 10.1523/JNEUROSCI.0983-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wannier T, Schmidlin E, Bloch J, Rouiller EM. A unilateral section of the corticospinal tract at cervical level in primate does not lead to measurable cell loss in motor cortex. J Neurotrauma. 2005;22:703–717. doi: 10.1089/neu.2005.22.703. [DOI] [PubMed] [Google Scholar]
- Weidner N, Ner A, Salimi N, Tuszynski MH. Spontaneous corticospinal axonal plasticity and functional recovery after adult central nervous system injury. Proc Natl Acad Sci U S A. 2001;98:3513–3518. doi: 10.1073/pnas.051626798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth B, van Hedel HJ, Curt A. Ankle paresis in incomplete spinal cord injury: relation to corticospinal conductivity and ambulatory capacity. J Clin Neurophysiol. 2008;25:210–217. doi: 10.1097/WNP.0b013e318183f4e3. [DOI] [PubMed] [Google Scholar]
- Wrigley PJ, Gustin SM, Macey PM, Nash PG, Gandevia SC, Macefield VG, Siddall PJ, Henderson LA. Anatomical changes in human motor cortex and motor pathways following complete thoracic spinal cord injury. Cereb Cortex. 2009;19:224–232. doi: 10.1093/cercor/bhn072. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Yamasaki M, Imai T. Retrograde pyramidal tract degeneration in a patient with cervical haematomyelia. J Neurol Neurosurg Psychiatry. 1989;52:382–486. doi: 10.1136/jnnp.52.3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YP, Burke DA, Shields LB, Chekmenev SY, Dincman T, Zhang Y, Zheng Y, Smith RR, Benton RL, DeVries WH, Hu X, Magnuson DS, Whittemore SR, Shields CB. Spinal cord contusion based on precise vertebral stabilization and tissue displacement measured by combined assessment to discriminate small functional differences. J Neurotrauma. 2008;25:1227–1240. doi: 10.1089/neu.2007.0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Shine HD. Neurotrophic factors expressed in both cortex and spinal cord induce axonal plasticity after spinal cord injury. J Neurosci Res. 2003;74:221–226. doi: 10.1002/jnr.10718. [DOI] [PubMed] [Google Scholar]