

Abstract
Repeated in vivo two-photon imaging of adult mammalian spinal cords, with subcellular resolution, would be crucial for understanding cellular mechanisms under normal and pathological conditions. Current methods are limited because they require surgery for each imaging session. Here we report a simple glass window methodology avoiding repeated surgical procedures and subsequent inflammation. We applied this strategy to follow axon integrity and the inflammatory response over months by multicolour imaging of adult transgenic mice. We found that glass windows have no significant effect on axon number or structure, cause a transient inflammatory response, and dramatically increase the throughput of in vivo spinal imaging. Moreover, we used this technique to track retraction/degeneration and regeneration of cut axons after a ‘pin-prick’ spinal cord injury with high temporal fidelity. We showed that regenerating axons can cross an injury site within 4 days and that their terminals undergo dramatic morphological changes for weeks after injury. Overall the technique can potentially be adapted to evaluate cellular functions and therapeutic strategies in the normal and diseased spinal cord.
Key points
Chronic in vivo imaging of cellular interactions within the adult spinal cord with subcellular resolution is important for understanding cellular physiology and disease progression.
Previous approaches for chronic in vivo spinal cord microscopy have required surgery for each imaging session.
Here we describe a novel method for implanting glass windows over the exposed spinal cords of adult mice for repeated in vivo microscopy.
We show that the windows remain clear for many months after implantation, do not damage axons or blood vessels, and are useful for studying cellular dynamics after spinal cord injury.
Our method represents an original technical breakthrough for scientists involved in spinal cord research and in vivo imaging, and is a useful tool for studying cellular physiology and disease progression.
Introduction
The spinal cord is the primary neurological link between the brain and other parts of the body, and damage to this system can have devastating sensory, motor and autonomic effects. Subcellular resolution imaging of the intact and pathological spinal cord over time in live mammals is a powerful way to understand its function and how it responds to injury or diseases. So far most studies using two-photon or epifluorescence microscopy imaging were performed by surgically exposing the spinal cords of fluorescent transgenic mice (Kerschensteiner et al. 2005; Dray et al. 2009; Ylera et al. 2009; Dibaj et al. 2010, 2011; Johannssen & Helmchen, 2010; Bareyre et al. 2011; Di Maio et al. 2011; Laffray et al. 2011; Nikićet al. 2011). Moreover, chronic observations required surgery for each imaging session (Kerschensteiner et al. 2005; Dray et al. 2009; Ylera et al. 2009; Bareyre et al. 2011; Di Maio et al. 2011; Nikićet al. 2011), which (i) increases the risk of damaging the site of interest, (ii) triggers an immune response, and (iii) reduces the throughput.
One approach to overcome these limitations is to implant glass windows over the exposed spinal cords. Methods for implanting glass windows over various regions of the adult mouse brain are well established (Misgeld & Kerschensteiner, 2006), and one recent study (Farrar et al. 2012) has proposed a method for implanting chambers over exposed spinal regions. These spinal chambers, however, require sophisticated implantation materials and surgical apparatus, and have a moderate success rate of ∼50%, thus limiting the accessibility and throughput of this approach. Our study aimed to develop an effective, low cost, and high throughput method for the implantation of glass windows for extended in vivo two-photon imaging studies. We show that implanted glass windows stabilize the spinal cord, remain clear for several weeks, do not damage axons, and result in only a transient immune response. Moreover, our data show that glass windows are useful for studying axon dynamics following an injury.
Methods
Surgery and glass window implantation
All surgical procedures were approved by the National Animal Studies Committee of France (authorization no. 13,300). A total of 43 adult mice (>8 weeks old and >19.6 g) were used to develop and validate the protocol. We used Thy1-cyan fluorescent protein (CFP)-23 (Feng et al. 2000), Thy1-green fluorescent protein (GFP)-M (Feng et al. 2000), LysM-GFP (Faust et al. 2000), CD11c-yellow fluorescent protein (YFP) (Lindquist et al. 2004) and Connexin43-CFP (Kunze et al. 2009) mice, which were backcrossed to C57/Bl6 mice and then crossbred to create mice with multiple fluorescent cell populations. Mice were deeply anaesthetized with ketamine–xylazine (120 mg kg−1; 12 mg kg−1), and supplemented hourly (40 mg kg−1; 4 mg kg−1). Ocry-gel (Laboratoire TVM, Lempdes, France) was applied to maintain eye moisture and surgical plane of anaesthesia was verified by a lack of toe pinch reflex. The fur around the surgical site was shaved, and the skin was cleaned with Vétédine iodine solution (Vétoquinol).
A dorsal midline incision of ∼1.5 cm was made over the vertebrae T12 to L2 and the muscles between the spinous and transverse processes were resected using a scalpel (see Supplementary Fig. 1). The animals were suspended from a spinal-fork stereotaxic apparatus (Harvard Apparatus) (Cunningham et al. 2005), between T12 and L2 (± one vertebra). Using a surgical microscope, the muscles covering the lateral aspects of the transverse processes of T12 and L2 were identified and gently resected. Prior to surgery standard staples were shaped as shown in Fig. 1A, placed for 20 min in 95% ethanol, then rinsed in phosphate buffered saline (PBS) with dexamethasone (0.1%), penicillin (1000 U ml−1), and streptomycin (1 mg ml−1). The distances between the tips of the staples were adjusted to fit closely along the sides of the pedicles of the vertebral arches of T12 and L2, respectively. The tips were inserted along the edges of their respective vertebrae and glued into place with cyanoacrylate. These staples served as the anchoring points for the remainder of the glass window set-up. The animals were released from the forks and cyanoacrylate was applied to all tissue from the edges of the exposed vertebrae to the edges of the cut skin. Special care was taken to avoid applying glue directly on the vertebrae. Dental cement was applied over the cyanoacrylate layer, to form a rigid ring around the vertebrae. A paperclip, bent to the shape shown in Fig. 1B, was implanted to serve as a holding point for surgery and imaging. A drop of PBS was placed on the exposed vertebrae to keep them moist until the dental cement hardened.
Figure 1. Spinal glass window implantation procedure.
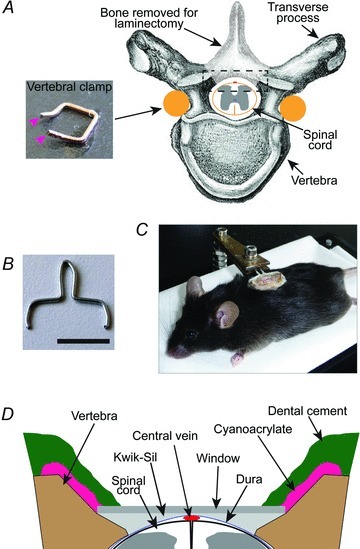
A, schematic diagram showing the vertebral clamp and laminectomy. The tips of the vertebral clamp (inset; magenta arrowheads) were inserted under the vertebral transverse processes and glued into place with cyanoacrylate. B, image showing a modified paperclip. The lower parts, including the tips, were implanted into the dental cement surrounding the window. The upper part protruded from dental cement and was used to clamp the animal for surgery and imaging. Scale bar, 1 cm. C, image of an anaesthetized mouse with an implanted window. The window structure is being supported by a clamp attached to a plastic base. The animal is freely breathing while in the support, but the window remains stationary relative to the clamp. D, detailed view of spinal glass window (from boxed region in A). The edges of the vertebrae were shaped so that the window rests on both sides of the vertebral opening without compressing the spinal cord. Kwik-Sil was used as a physical barrier preventing the infiltration of opaque tissue between the window and spinal cord.
The mice were attached to a holder (Fig. 1C), and a laminectomy was carried out on 2 vertebrae using a high-speed drill (Dremel) with a carbide bur (size = round 1/4; World Precision Instruments, Hitchin, England) (Fig. 1A). The vertebrae were kept cool and free of debris by repeated rinsing with PBS. Prior to surgery glass windows of various sizes (1.8–2.1 mm wide; 3.8–4.5 mm long) were cut from cover glass (no. 1.5 thickness) using a surgical microscope and a carbide burred drill tip glued to a handle. Once the spinal cord was exposed, the glass window that best fitted the laminectomy opening was chosen for implantation. The lateral edges of the vertebral openings were shaped so that the glass rested on the edges of the bone and was flat relative to the spinal cord, but did not touch the spinal cord, thus avoiding axon damage when pressure was applied (Fig. 1D). The glass window was cleaned and dried, and the dura mater was rinsed with PBS, cleared of any debris, and allowed to dry until tacky (Dombeck et al. 2007, 2009, 2010). A line of liquid Kwik-Sil (World Precision Instruments) was applied to the dura mater surface along the midline of the spinal cord, and the glass window was immediately placed over the spinal cord. Gentle pressure was applied to maintain the glass in place while the Kwik-Sil cured to a gel (∼60 s). Cyanoacrylate was then applied to the edges of the glass and the surrounding bone and muscle. Finally, a generous layer of dental cement was applied over the cyanoacrylate to reinforce the structure. Post-operative analgesia was obtained by administration of dexamethazone (0.2 mg kg−1) and rimadyl (5 mg kg−1) s.c. Animals were then imaged, returned to their cage with tissue for nesting, and kept warm until they recovered from anaesthesia.
Spinal cord injury
Seven animals received a unilateral pin-prick spinal cord injury, as described previously (Dray et al. 2009), before glass window implantation. Briefly, a 26G needle was mounted to a microdrive (Wold Precision Instruments) and positioned over the animal to pierce the spinal cord along the dorsoventral axis at the border between the gracile and cuneate fasciculi. Injuries were made roughly in the middle of the exposed spinal cord rostrocaudally. The needle was slowly inserted until half of the bevel was below the surface of the spinal cord (∼500 μm) and immediately withdrawn. The injury sometimes caused a small amount of bleeding, which usually stopped within a few seconds. Blood was rinsed away with PBS before glass window implantation.
Imaging
For each imaging session mice were lightly anaesthetized with ∼1.75% isoflurane (Baxter) (v/v) in air for ∼2 min, followed by ketamine–xylazine (100 mg kg−1; 10 mg kg−1). Light gaseous anaesthetic was used to reduce animal anxiety and the physical stress on the window associated with restraining the animal. For long sessions (>1 h), animals were supplemented with ∼0.4–1.0% isoflurane (v/v) in air from ∼45 min after the start of the session until completion. In all animals either 120 μl of Rhodamine B isothiocyanate-dextran 70 kDa (Sigma) (20 mg ml−1 in PBS) or 40 μl of QDot-655 (Qtracker 655 non-targeted quantum dots; Invitrogen) (50 μl ml−1 in PBS) was injected i.v. immediately before each imaging session. Most animals were administered dexamethazone (0.2 mg kg−1) and rimadyl (5 mg kg−1) s.c. prior to imaging. Similar to other studies in the brain (Drew et al. 2010), we observed no differences between treated and untreated mice. Animals were connected to a clamp by the embedded paperclip. This clamp was mounted to a small table that fitted under the objective lens of the microscope. An immersion pool was made around the glass window by applying warm liquid agar (3%; Sigma) to the outer edges of the dental cement. Throughout imaging the animals were freely breathing and the microscope chamber was warmed to ∼32°C. Following each imaging session the animals were returned to their cage with a piece of tissue for nesting and kept warm until they recovered from anaesthesia. Of the 43 animals included in this study, only two animals were found dead in their cage despite no previous indications of distress or sickness (22 and 50 days post-implantation, and 8 and 14 imaging sessions respectively). Otherwise, animals were killed by cervical dislocation under deep anaesthesia once window clarity was impeded mainly due to tissue growth between the spinal cord and Kwik-Sil or the formation of bubbles between the Kwik-Sil and the window. Similar to other studies we observed no detrimental effects of frequent repeated anaesthesia (Butterfield et al. 2004) or fluorophore injection (Dray et al. 2009; Di Maio et al. 2011).
A tunable femtosecond pulsed laser (Mai-Tai, Spectra Physics, Évry, France) was coupled to a Zeiss two-photon microscope (LSM 7 MP) equipped with a 20× water immersion objective lens (NA = 1.0) and five non-descanned detectors. The laser was tuned to 840 nm, 945 nm, or 985 nm to optimize excitation according to the fluorophore combination to be examined. At 840 nm CFP, Rhodamine, and QDot-655 were bright, whereas GFP and YFP were faint. At 945 nm all of the fluorophores except Rhodamine were bright. At 985 nm all of the fluorophores except CFP were bright. Filter sets were designed to optimize the separation of the emission spectra of multiple fluorophores including CFP (SP 490), GFP (BP 500/24), YFP (BP 534/30), Rhodamine-B (BP 587/35), and QDot-655 (BP 641/75). For each image stack laser intensity, and sometimes master gain, were adjusted according to imaging depth in order to maximize image intensity while minimizing saturation throughout the image stack.
We re-imaged the same regions in successive sessions using two strategies. First, the window was epi-illuminated with LED lights on the side of the objective. A gross positioning of the spinal cord was achieved with the motorized stage using visual cues such as large blood vessels or cemented borders of the window. Fine positioning was achieved in the two-photon mode until the pattern of the real-time image matched the pattern of the image acquired on the previous session. Small diameter blood vessels with specific branching patterns provided the most easily recognizable and reliable anatomical markers.
Images were acquired at a resolution of 0.83 μm or 0.41 μm per pixel to improve our ability to detect fine cellular processes. Optical sectioning was either 5 or 2.4 μm. Field of view was 425 μm × 425 μm, but stacks consisting of 2 × 4 fields of view were typically acquired.
Analysis
Images were analysed using ZEN light edition (Zeiss) and ImageJ software. Analysis was performed on raw data, but all presented images are pseudo-coloured and contrast enhanced for clarity. Occasionally, there was a drift in the field of view for an image stack, which was realigned using the StackReg plugin on ImageJ (Thévenaz et al. 1998). Close appositions between cells and axon trajectories were confirmed by scrolling through 3D image stacks. Quantitative data from adjacent days were binned for calculating averages.
For LysM(+) and CD11c(+) cell counts, we used image stacks acquired at 425 μm × 425 μm and optical sectioning every 2.4 μm. To start, one box of 291 μm × 291 μm was placed over image stacks acquired on day 0 (d0). The box was positioned to include part of the central vein and at least one side-branch blood vessel. Cell bodies were counted by scanning through the depth of the image stack from the surface of the spinal cord to roughly 30 μm deep. Cells located within blood vessels were not counted, and care was taken to count only cell bodies and not brightly labelled side branches. For each subsequent imaging session, the box was realigned to the same x–y position using blood vessels as anatomical markers.
For axon counts in animals without injury, we used image stacks acquired at 425 μm × 425 μm and optical sectioning every 2.4 μm. To start, two boxes with and area of 166 μm × 21 μm were positioned over regions containing axons and not obscured by blood vessels, of the d0 image stacks. The numbers of axons within the boxes were counted from the dorsal surface to roughly 20 μm deep. For each subsequent imaging session, the boxes were realigned to the same x–y position.
For axon counts in animals with spinal cord injury, we used tiled image stacks with an acquisition area of 850 μm × 1700 μm and optical sectioning every 5.0 μm. To start, five boxes with an area of 200 μm × 21 μm, aligned mediolaterally and separated by 200 μm rostrocaudally, were positioned so that the middle box was located at the injury site on the d0 image stacks. A sixth box of the same size was positioned contralateral of the spinal cord injury site. The number of axons within each box was counted from the dorsal surface to roughly 20 μm deep. For each subsequent imaging session, the boxes were realigned to the same x–y position.
Results
Spinal glass window as a tool for chronic in vivo imaging
Throughout the design process, we aimed for the windows to be very well tolerated by the animals, high throughput, long-lasting, and have a high success rate. We found that mice with implanted windows were highly mobile several hours after implantation (Supplementary video 1), showed no signs of pain, and continued to gain weight for the duration of the experiment (Fig. 2A). After implantation, the prep-time for each imaging session was ∼10–15 min, compared to 1–2.5 h using the earlier surgical approaches (Dray et al. 2009). Moreover, implanted glass windows reduced the rostrocaudal curvature of the spinal cord and reduced the amplitude of respiratory and cardiac movement artifacts, thus improving overall image quality (Fig. 2B and C). Similar to windows implanted over brain regions, a layer of vascular tissue developed between the window and spinal cord in all animals. However, the tissue developed slowly, was thin and translucent, and did not prevent high quality imaging with subcellular resolution (Fig. 2D–F). Glass windows remained clear and allowed subcellular resolution imaging and tracking of individual axons for up to 350 days after implantation (average = 83 ± 20, mean ± SEM, n = 22 mice), and 22 imaging sessions (average = 12 ± 1, mean ± SEM, n = 22 mice) (Fig. 2G–I, see Supplementary Fig. 2 for all time points). Over 86% of windows remained clear at 2 weeks post-implantation, and more than 77% of windows remained clear at 5 weeks post-implantation.
Figure 2. Spinal glass window viability and basic properties.
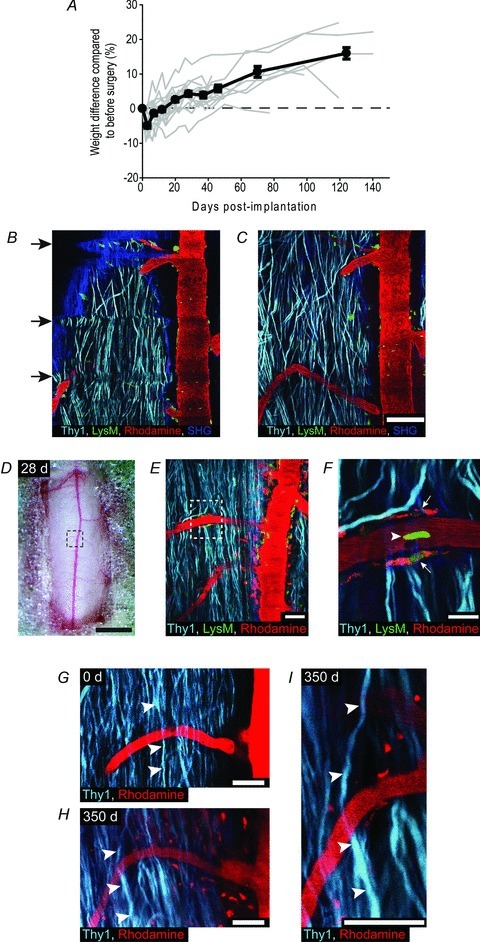
A, plots showing animal weight over time as a percentage change from before surgery. Average (black lines) and data from individual animals (grey lines) (n = 17). Error bars, SEM. B and C, representative images showing the same spinal cord region immediately before (B), and after (C) window implantation. The blue arch in the top of B (SHG, second harmonic generation from collagen in dura mater) highlights the curvature of the spinal cord before window implantation. Arrows in B show the horizontal distortions in the image caused by respiratory movements before window implantation. Scale bar, 100 μm. Excitation wavelength 945 nm. D–F, images of a spinal cord through a spinal glass window 28 days after implantation. D, image showing exposed spinal cord through window. Dashed box outlines the area shown in E. Scale bar, 1 mm. E, representative images of axons (Thy1), LysM(+) cells, and blood vessels (Rhodamine). Dashed box outlines the area shown in F. Scale bar, 50 μm. F, higher magnification image showing detailed structure of individual axons, blood vessels, a LysM(+) cell within the blood vessel (arrow head), and perivascular LysM(+) cells (arrows). Notice that the perivascular cells also contain Rhodamine, likely to have been engulfed following previous imaging sessions. Scale bar, 20 μm. G and H, images showing axons and blood vessels from the same spinal region at 0 days and 350 days after window implantation (n = 22 imaging sessions). The same axon (arrowheads) was identified at each subsequent post-implantation interval. Scale bar, 50 μm. I, high power view of identified axon at 350 days post-implantation. Scale bar, 50 μm. Excitation wavelength 945 nm. In all panels Caudal is up, Rostral is down.
Minimal effect on the cellular environment of the spinal cord
A transient immune response in the spinal cord is expected after surgery whereas a prolonged and severe response would be consistent with spinal cord damage and glass window rejection. To quantify the immune response triggered by glass window implantation, we generated two mouse lines. We crossed (i) transgenic Thy1-CFP mice expressing CFP in most DRG axons (Feng et al. 2000) with LysM-GFP mice expressing enhanced GFP (EGFP) in myelo-monocytic cells, including granulocytes and most macrophages (Faust et al. 2000; Geissmann et al. 2010), to yield Thy1-CFP//LysM-GFP mice, and (ii) Thy1-CFP mice with CD11c-YFP mice expressing enhanced YFP (EYFP) in subpopulations of dendritic cells (Lindquist et al. 2004), microglia (Lindquist et al. 2004), and macrophages (Wilson & O’Neill, 2003; Geissmann et al. 2010; Hume, 2011) to yield Thy1-CFP//CD11c-YFP mice.
Glass windows were implanted over the exposed spinal cords of both Thy1-CFP//LysM-GFP and Thy1-CFP//CD11c-YFP mice and we observed the recruitment dynamics of LysM and CD11c expressing immune cells, respectively. Given the non-invasive nature of the imaging protocol, and to observe cellular dynamics with high temporal fidelity in the same animals, the spinal cords were imaged every 2–6 days for the first 6 weeks and every 1–2 weeks thereafter (Fig. 3A and B). LysM(+) and CD11c(+) cells were initially found in perivascular zones and within the white mater away from blood vessels (Fig. 3C and D). There was a rapid yet modest increase in the number of LysM(+) cells within the first 3 days post-implantation, with the numbers dropping towards baseline by 12 days (Fig. 3E). Compared to LysM(+) cells, the number of CD11c(+) cells increased with a slightly higher magnitude and had a more protracted time course before dropping towards baseline (Fig. 3F). The immune response within the spinal cord was transient, which supports the conclusion that it is likely due to the initial surgery rather than window implantation or rejection.
Figure 3. Long-term spinal cord integrity after glass window implantation.
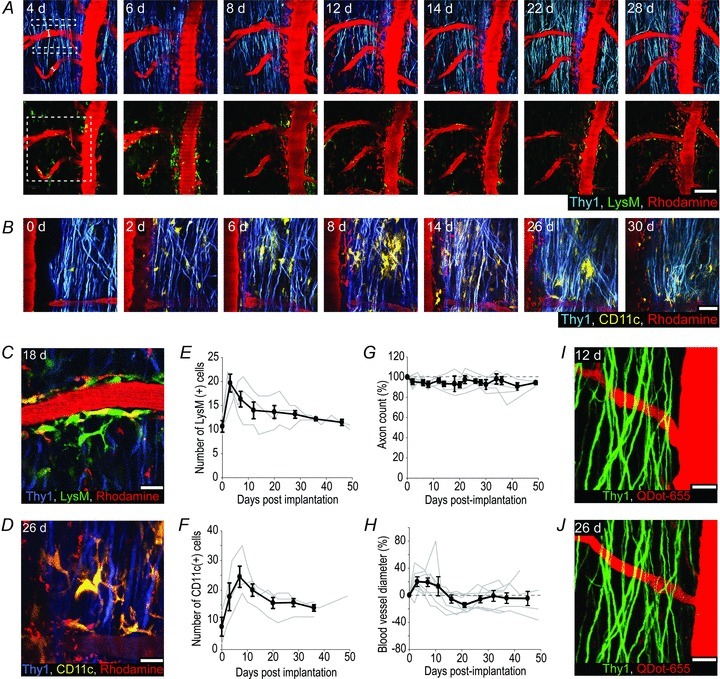
A, representative images of the same region of a Thy1-CFP//LysM-GFP mouse spinal cord (4–28 days) after window implantation. Upper left dashed boxes indicate the regions used for axon counts for all subsequent imaging sessions (see G). The white lines across the blood vessels outline the site of blood vessel diameter measurements (see H). The same blood vessels were measured at 5 locations for every session. Bottom left dashed box shows the region used to count LysM(+) cells (see E and F). Excitation wavelength, 840 nm (top row) and 985 nm (bottom row). Scale bar, 100 μm. B, representative images showing the same regions of a Thy1-CFP//CD11c-YFP mouse spinal cord (0–30 days) after window implantation. Excitation wavelength, 945 nm. Scale bar, 50 μm. C and D, representative images showing the morphological characteristics of LysM(+) (C) and CD11c(+) (D) cells at 18d and 26d respectively. Excitation wavelength, 985 nm (C) 945 nm (D). Scale bars, 20 μm. E and F, average number (black line) of LysM(+) (E) and CD11c(+) (F) cells within a predefined zone of the spinal cord (dashed box in lower left image of A) over time. Grey lines, data from individual animals. Error bars, SEM (n = 3 LysM-GFP; n = 3 CD11c-YFP). G, mean number of axons (black line) within predefined zones of the spinal cord (dashed boxes in upper left image of A) over time. Grey lines, data from individual animals (n = 6). Error bars, SEM. H, plots showing the average diameter of central vein side branches (black line) over time. Grey lines, data from individual animals (n = 6). Error bars, SEM. I and J, representative images showing the same regions of a Thy1-GFP mouse after multiple imaging sessions using QDot-655 to visualize the vascular compartment. Notice the lack of QDot-655 labelled perivascular cells after 5 (I) and 8 (J) imaging sessions (cf. A–D). Excitation wavelength, 840 nm. Scale bars, 50 μm. In all panels Caudal is up, Rostral is down.
A key assessment of whether implanted glass windows cause damage to the spinal cord is to examine axon integrity and astrogliosis. With the rare exception of a few swellings, axons had normal cylindrical morphologies and followed typical DRG axon trajectories. As a quantitative measure, we counted the number of axons for each imaging session and found that axon numbers remained constant over time (d0 = 145 ± 13 axons, mean ± SEM, n = 6; 34d to 36d = 135 ± 11 axons, mean ± SEM, n = 6) (Fig. 3G). In addition, using Connexin43-CFP mice expressing CFP in astrocytes (n = 4), we found that astrocyte distributions were similar in regions directly below the window compared to regions in the ventral spinal cord or regions rostral and caudal of the window (Supplementary Fig. 3).
Previous work has shown that cranial glass windows can have effects on vasculature (Drew et al. 2010). To test whether this is the case for our glass windows, Rhodamine-dextran (70 kDa) was injected intravenously prior to every imaging session to visualize the dorsal spinal vasculature. Visual inspection of blood vessels over time revealed very few changes in their structure or trajectory (Fig. 3A). Quantitative analysis of secondary vessels that branch from the central vein over time showed a transient increase in vascular diameter concomitant with the timeline of immune cell recruitment (6–8d = 19 ± 6%, mean ± SEM, n = 9; 42–49d = −5 ± 10%, mean ± SEM, n = 6) (Fig. 3H). However, after several imaging sessions, many perivascular cells were also labelled with Rhodamine. Therefore we tested whether QDots are a viable alternative to dextran-coupled fluorophores for repeated vascular labelling. Following several imaging sessions using QDot-655 to label the vasculature (n = 5), we found that there were virtually no perivascular cells with QDot-655 (Fig. 3I and J), and that QDots were non-toxic and well tolerated by the animals.
Collectively, our data show that implanted spinal glass windows have little effect on spinal cord cytoarchitecture, and are a useful tool for studying cell behaviour in the adult spinal cord for extended periods of time with high temporal fidelity.
Imaging axons after spinal cord injury
We then tested if spinal glass windows are an appropriate tool for studying cellular dynamics in diseased spinal cord. As a model we chose to study axon degeneration/retraction and growth dynamics following ‘pin-prick’ spinal cord injuries (Dray et al. 2009). Although there were almost no axons at the injury site at day 0, by 46 days their numbers recovered to 58% of the density observed on the uninjured side of the spinal cord (Fig. 4A and B). Rostral and caudal of the injury site there was a rapid loss of axons by 3 days, followed by a gradual increase in their numbers. By 46 days, depending on the distance to the injury site, their numbers increased to 71% (400 μm caudal; Fig. 4C), 62% (200 μm caudal; Fig. 4D), 34% (200 μm rostral; Fig. 4E) and 46% (400 μm rostral; Fig. 4F) compared to control side.
Figure 4. Axon dynamics after spinal cord injury.
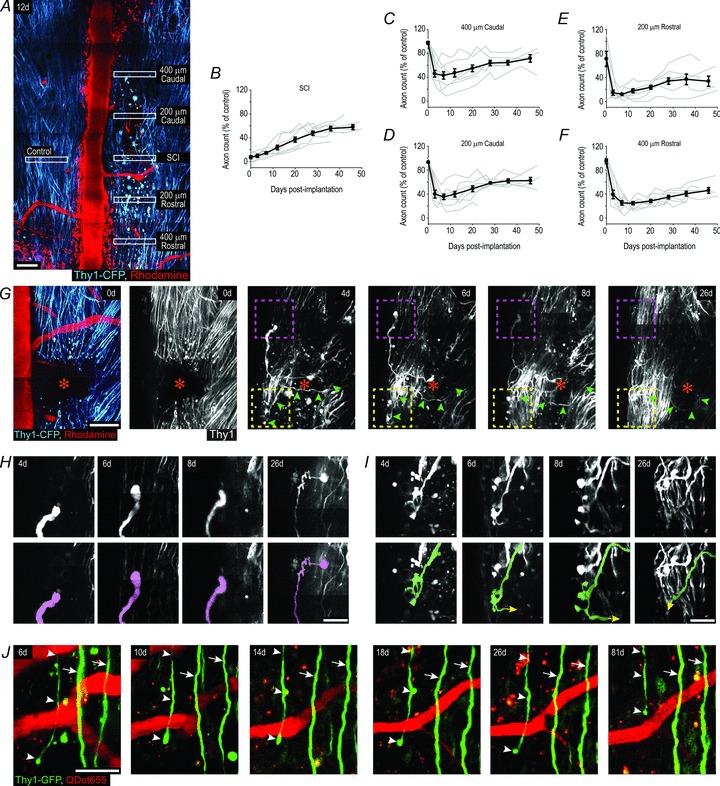
A, representative image showing axons and blood vessels 12 days after injury in a Thy1-CFP mouse. Scale bar, 100 μm. B–F, quantification of axon loss and recruitment at injury sites (B), two zones caudal (C and D), and two zones rostral (E and F) of the injury sites. Averages across animals (black lines); data from individual animals (grey lines). Error bars, SEM (n = 7). G, image stacks showing an injury site (orange star) in a Thy1-CFP mouse over time. Green arrowheads at 4d, 6d, 8d, and 26d mark the trajectory of an axon that crossed the injury site between 0d and 4d (green and red shaded axons in lower panels of I respectively). Scale bar, 100 μm. H and I, image stacks showing a cut axon terminal elongating caudally over time (H; outlined by the magenta box in G), and a cut axon whose primary shaft crossed the injury site between 0d and 4d (green arrowheads in G; green shaded axon in I). Lower panels show coloured tracings of the axons shown in dashed boxes of the upper panels. Yellow arrows (I) indicate that the axon continued beyond the end of the tracing, but was too deep within the tissue or entangled within other CFP axons to be resolved. Scale bars, 40 μm. J, image stacks showing two uninjured axons (arrows) and a cut axon terminal (arrowheads) caudal of an injury site in a Thy1-GFP mouse over time. This terminal remains swollen, has no branches, does not regenerate, and progressively retracts over time. Scale bars, 50 μm. In all panels Caudal is up, Rostral is down.
Closer examination of the injury sites over time revealed that they were repopulated by two distinct types of axons. The first set had morphological features typical of uninjured axons. These were located on the edges of the injury sites, and projected rostrocaudally (Fig. 4G). The second set of axons occupied all parts of the injury sites, often followed tortuous trajectories (Fig. 4G), and could sometimes be traced to abrupt terminations both within and beyond the injury sites (Fig. 4H and I). Moreover, we could reliably identify and track the growth and morphological features of individual cut axon terminals over the period of days and weeks (Fig. 4G–I). However, the density of Thy1-CFP stained axons sometimes impeded the identification of specific axon terminals for analysis. To test whether a mouse line with fewer stained axons is more suitable to individual axon analysis, we used Thy1-GFP mice (Fig. 4J). In these mice individual axons and cut axon terminals were easily and reliably identified from one imaging session to another, but the probability of observing specific events within any given experiment (e.g. an axon that regenerated through the injury site) were reduced. Collectively, these results demonstrate that implanted glass windows are a useful tool for examining large populations of axons after spinal cord injury as well as the precise growth dynamics of individual axon terminals in the same animals for extended periods of time.
Discussion
The use of a high-throughput, low cost dorsal glass window method that is suitable for extended subcellular two-photon microscopy investigations of normal and diseased spinal cord represents a technical breakthrough for scientists exploring a diverse range of spinal cord physio-pathologies and in vivo imaging. We showed that it is now possible to achieve imaging with micrometric repositioning and resolution, as well as excellent temporal fidelity over days and multiple weeks with a high probability of success. The method overcomes the hurdle of multiple surgeries which dramatically limits the number of sessions and influences the inflammatory status of the animal and significantly improves the experimental rate. By comparison with the recently developed glass chamber implantation technique, which requires a range of specialized custom made equipment for implementation, the glass window technique has a higher probability of success (cf. ∼50% success at 5 weeks for implanted chambers versus >77% success at 5 weeks for implanted glass windows), and is viable for longer periods of time (cf. maximum 140d for implanted chambers versus 350d for implanted glass windows) (Farrar et al. 2012). Moreover, the glass window approach overcomes other key problems such as motion artifacts due to respiratory or heart beat movements, which will complement existing methods that require a stable spinal cord, such as calcium imaging of spinal cord network activity (Johannssen & Helmchen, 2010; Laffray et al. 2011).
Importantly, the longevity of the glass windows opens new opportunities for chronic imaging of disease progression at subcellular resolution in the same animal, which is un-attainable with any other approaches. For example, we tracked spinal cord injury progression for up to 22 imaging sessions for 50 weeks. These data also confirm the already reported repopulation of the lesion site 1 month after pin-prick lesion (Dray et al. 2009). Yet, in support of neuronal tracing and histochemical data pointing to neuronal plasticity after injury (Bareyre, 2008), our high temporal fidelity experiments revealed that some of the axons located within spinal cord injury sites at later post-injury times are likely to be spared axons repopulating the site, whereas others are likely to be due to regenerative sprouting. Our observations of individual cut axon terminals are consistent with other histological neuronal tracing studies showing that cut axon terminals with more complex branch structures have a higher growth capacity compared to cut axon terminals with a simple terminal swelling (cf. Fig. 4I and J) (Fenrich & Rose, 2009, 2011). Taken together, our approach allowed us to differentiate between spared axons and bona fide regenerating axons over time, which is important for examining the cellular mechanisms of potential therapies (Steward et al. 2003).
Given that glass windows are well suited for repeated imaging sessions and that dextran-coupled fluorophores accumulate in phagocytic perivascular cells after repeated imaging sessions (Dray et al. 2009; Farrar et al. 2012), we identified QDot-655 as a viable alternative to dextran-coupled fluorophores for staining blood vessels. This approach improves contrast of the vascular compartment and would ultimately facilitate automated identification and segmentation of the vascular compartment for neurovascular and tumour vascularization studies.
We also used spinal glass windows to characterize inflammation following spinal surgery. Using transgenic mice with fluorescent axons (CFP), myelo-monocytes (GFP), and microglia/macrophage cells (YFP), we tracked the differential and transient recruitment dynamics of immune cells of different lineages as part of the healing process following spinal cord surgery. These mice will be instrumental for future studies aimed to investigate inflammatory responses in nervous system pathologies.
The mouse spinal cord is an important site for autoimmune and injury models. Our methodology is suitable for answering questions dealing with various aspects of spinal cord physiology and pathologies, provided that appropriate multicolour transgenic mice are used. Moreover, in a preclinical research context, cell graft therapies are often considered as a valuable therapeutic approach without having a clear understanding of the fate of these cells for extended periods of time (Li et al. 2010). Our dynamic and cell-resolved imaging approach will certainly be a useful tool for tracking the fates of grafted cells and clarifying the mechanisms responsible for their beneficial effects. Finally, with this method the efficacy and target selectivity of new therapeutic molecules can be assessed in real time at a microscopic scale in vivo, and their therapeutic time-lines optimized.
Acknowledgments
We thank Dr C. Ricard and S. Meijia-Gervacio for helpful discussions; the staff of the animal and PicSIL imaging facilities of the IBDML for technical support; Drs G. Chemini and H. Luche at the Centre d’Immunologie de Marseille-Luminy (CIML) for the LysM-GFP and CD11c-YFP mouse lines; and Dr M. Theis at the Institute of Cellular Neuroscience, University of Bonn for the Connexin43-CFP mice. This work was supported by an institutional grant from Centre National de la Recherche Scientifique, and by grants from the Association de Recherche sur la Sclérose en Plaque (ARSEP) (to G.R. and K.K.F), Agence Nationale de la Recherche (ANR JCJC), Fédération de Recherche sur le Cerveau (FRC) Institut de Recherche sur la Moelle Epinière (IRME) (to F.D.). K.K.F. was supported by an ARSEP fellowship. Imaging was performed on the PIcSIL imaging facility of the IBDML.
Glossary
- CFP
cyan fluorescent protein
- DRG
dorsal root ganglion
- GFP
green fluorescent protein
- LysM
lysozyme M
- QDot
quantum dot
- YFP
yellow fluorescent protein
Author contributions
K.K.F. and F.D. conceived the study and analysed the data; K.K.F, P.W. and F.D designed the window implantation protocol; K.K.F.and P.W. carried out the experiments; M.H. did the genotyping and helped generate the transgenic animals; M.Z. helped collect and analyse the data; K.K.F, G.R., and F.D. interpreted the data and wrote the paper. All authors approved the final version of the publication.
Supplementary material
Supplemental Table 1
Supplemental Table 2
Supplemental Figure 1
Supplemental Figure 2
References
- Bareyre FM. Neuronal repair and replacement in spinal cord injury. J Neurol Sci. 2008;265:63–72. doi: 10.1016/j.jns.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Garzorz N, Lang C, Misgeld T, Büning H, Kerschensteiner M. In vivo imaging reveals a phase-specific role of STAT3 during central and peripheral nervous system axon regeneration. Proc Natl Acad Sci U S A. 2011;108:6282–6287. doi: 10.1073/pnas.1015239108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield NN, Graf P, Ries CR, MacLeod BA. The effect of repeated isoflurane anesthsia on spatial and psychomotor performance in young and aged mice. Anesth Analg. 2004;98:1305–1311. doi: 10.1213/01.ane.0000108484.91089.13. [DOI] [PubMed] [Google Scholar]
- Cunningham MG, Donalds RA, Scouten CW, Tresch MC. A versatile, low-cost adaptor for stereotaxic and electrophysiologic spinal preparations in juvenile and adult rodents. Brain Res Bull. 2005;68:157–162. doi: 10.1016/j.brainresbull.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Dibaj P, Nadrigny F, Steffens H, Scheller A, Hirrlinger J, Schomburg ED, Neusch C, Kirchhoff F. NO mediates microglial response to acute spinal cord injury under ATP control in vivo. Glia. 2010;58:1133–1144. doi: 10.1002/glia.20993. [DOI] [PubMed] [Google Scholar]
- Dibaj P, Steffens H, Zschüntzsch J, Nadrigny F, Schomburg ED, Kirchhoff F, Neusch C. In vivo imaging reveals distinct inflammatory activity of CNS microglia versus PNS macrophages in a mouse model for ALS. PLoS One. 2011;6:e17910. doi: 10.1371/journal.pone.0017910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maio A, Skuba A, Himes BT, Bhagat SL, Hyun JK, Tessler A, Bishop D, Son Y-J. In vivo imaging of dorsal root regeneration: rapid immobilization and presynaptic differentiation at the CNS/PNS border. J Neurosci. 2011;31:4569–4582. doi: 10.1523/JNEUROSCI.4638-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombeck DA, Graziano MS, Tank DW. Functional clustering of neurons in motor cortex determined by cellular resolution imaging in awake behaving mice. J Neurosci. 2009;29:13751–13760. doi: 10.1523/JNEUROSCI.2985-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombeck DA, Harvey CD, Tian L, Looger LL, Tank DW. Functional imaging of hippocampal place cells at cellular resolution during virtual navigation. Nat Neurosci. 2010;13:1433–1440. doi: 10.1038/nn.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombeck DA, Khabbaz AN, Collman F, Adelman TL, Tank DW. Imaging large-scale neural activity with cellular resolution in awake, mobile mice. Neuron. 2007;56:43–57. doi: 10.1016/j.neuron.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray C, Rougon G, Debarbieux F. Quantitative analysis by in vivo imaging of the dynamics of vascular and axonal networks in injured mouse spinal cord. Proc Natl Acad Sci U S A. 2009;106:9459–9464. doi: 10.1073/pnas.0900222106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew PJ, Shih AY, Driscoll JD, Knutsen PM, Blinder P, Davalos D, Akassoglou K, Tsai PS, Kleinfeld D. Chronic optical access through a polished and reinforced thinned skull. Nat Methods. 2010;7:981–984. doi: 10.1038/nmeth.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar MJ, Bernstein IM, Schlafer DH, Cleland TA, Fetcho JR, Schaffer CB. Chronic in vivo imaging in the mouse spinal cord using an implanted chamber. Nat Methods. 2012;9:297–302. doi: 10.1038/nmeth.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust N, Varas F, Kelly LM, Heck S, Graf T. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood. 2000;96:719–726. [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fenrich KK, Rose PK. Spinal interneuron axons spontaneously regenerate after spinal cord injury in the adult feline. J Neurosci. 2009;29:12145–12158. doi: 10.1523/JNEUROSCI.0897-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenrich KK, Rose PK. Axons with highly branched terminal regions successfully regenerate across spinal midline transections in the adult cat. J Comp Neurol. 2011;519:3240–3258. doi: 10.1002/cne.22686. [DOI] [PubMed] [Google Scholar]
- Geissmann F, Gordon S, Hume DA, Mowat AM, Randolph GJ. Unravelling mononuclear phagocyte heterogeneity. Nat Rev Immunol. 2010;10:453–460. doi: 10.1038/nri2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume DA. Applications of myeloid-specific promoters in transgenic mice support in vivo imaging and functional genomics but do not support the concept of distinct macrophage and dendritic cell lineages or roles in immunity. J Leukoc Biol. 2011;89:525–538. doi: 10.1189/jlb.0810472. [DOI] [PubMed] [Google Scholar]
- Johannssen HC, Helmchen F. In vivo Ca2+ imaging of dorsal horn neuronal populations in mouse spinal cord. J Physiol. 2010;588:3397–3402. doi: 10.1113/jphysiol.2010.191833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat Med. 2005;11:572–577. doi: 10.1038/nm1229. [DOI] [PubMed] [Google Scholar]
- Kunze A, Congreso MR, Hartmann C, Wallraff-Beck A, Hüttmann K, Bedner P, Requardt R, Seifert G, Redecker C, Willecke K, Hofmann A, Pfeifer A, Theis M, Steinhäuser C. Connexin expression by radial glia-like cells is required for neurogenesis in the adult dentate gyrus. Proc Natl Acad Sci U S A. 2009;106:11336–11341. doi: 10.1073/pnas.0813160106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laffray S, Pagès S, Dufour H, De Koninck P, De Koninck Y, Côté D. Adaptive movement compensation for in vivo imaging of fast cellular dynamics within a moving tissue. PLoS One. 2011;6:e19928. doi: 10.1371/journal.pone.0019928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SC, Tachiki LML, Luo J, Dethlefs BA, Chen Z, Loudon WG. A biological global positioning system: considerations for tracking stem cell behaviors in the whole body. Stem Cell Rev. 2010;6:317–333. doi: 10.1007/s12015-010-9130-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist RL, Shakhar G, Dudziak D, Wardemann H, Eisenreich T, Dustin ML, Nussenzweig MC. Visualizing dendritic cell networks in vivo. Nat Immunol. 2004;5:1243–1250. doi: 10.1038/ni1139. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M. In vivo imaging of the diseased nervous system. Nat Rev Neurosci. 2006;7:449–463. doi: 10.1038/nrn1905. [DOI] [PubMed] [Google Scholar]
- Nikić I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, Bareyre FM, Brück W, Bishop D, Misgeld T, Kerschensteiner M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med. 2011;17:495–499. doi: 10.1038/nm.2324. [DOI] [PubMed] [Google Scholar]
- Steward O, Zheng B, Tessier-Lavigne M. False resurrections: distinguishing regenerated from spared axons in the injured central nervous system. J Comp Neurol. 2003;459:1–8. doi: 10.1002/cne.10593. [DOI] [PubMed] [Google Scholar]
- Thévenaz P, Ruttimann UE, Unser M. A pyramid approach to subpixel registration based on intensity. IEEE Trans Image Process. 1998;7:27–41. doi: 10.1109/83.650848. [DOI] [PubMed] [Google Scholar]
- Wilson HL, O’Neill HC. Identification of differentially expressed genes representing dendritic cell precursors and their progeny. Blood. 2003;102:1661–1669. doi: 10.1182/blood-2002-08-2426. [DOI] [PubMed] [Google Scholar]
- Ylera B, Ertürk A, Hellal F, Nadrigny F, Hurtado A, Tahirovic S, Oudega M, Kirchhoff F, Bradke F. Chronically CNS-injured adult sensory neurons gain regenerative competence upon a lesion of their peripheral axon. Curr Biol. 2009;19:930–936. doi: 10.1016/j.cub.2009.04.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.