

Abstract
The hypothalamus is a critical controller of homeostatic responses and plays a fundamental role in reward-seeking behaviour. Recently, hypothalamic neurones in the perifornical/lateral hypothalamic area (PF/LHA) have also been implicated in drug-seeking behaviour through projections to extra-hypothalamic sites such as the ventral tegmental area. For example, a population of neurones that expresses the peptide orexin has been strongly implicated in addiction-relevant behaviours. To date, the effect of addictive drugs on synaptic properties in the hypothalamus remains largely unexplored. Previous studies focusing on the PF/LHA neurones, however, have shown that the orexin system exhibits significant plasticity in response to food or sleep restriction. This neuroadaptive ability suggests that PF/LHA neurones could be highly susceptible to modifications by drug exposure. Here, we sought to determine whether cocaine produces synaptic plasticity in PF/LHA neurones. Whole-cell patch-clamp techniques were used to examine the effects of experimenter-administered (passive) or self-administered (SA) cocaine on glutamatergic synaptic transmission in PF/LHA neurones. These experiments demonstrate that both passive and SA cocaine exposure increases miniature excitatory postsynaptic current (mEPSC) frequency in PF/LHA neurones. In addition, SA cocaine reduced the paired-pulse ratio but the AMPA/NMDA ratio of evoked excitatory inputs was unchanged, indicative of a presynaptic locus for synaptic plasticity. Dual-labelling for orexin and excitatory inputs using the vesicular glutamate transporter (VGLUT2), showed that passive cocaine exposure increased VGLUT2-positive appositions onto orexin neurones. Further, a population of recorded neurones that were filled with neurobiotin and immunolabelled for orexin confirmed that increased excitatory drive occurs in this PF/LHA population. Given the importance of the PF/LHA and the orexin system in modulating drug addiction, we suggest that these cocaine-induced excitatory synapse-remodelling events within the hypothalamus may contribute to persistence in drug-seeking behaviour and relapse.
Key points
Drugs of addiction are well-established in their capacity to alter brain reward pathways.
The perifornical/lateral hypothalamus has previously been shown to be drug responsive, participate in relapse to drug taking, and project to key reward pathway structures.
This study demonstrates that cocaine enhances excitatory drive to perifornical/lateral hypothalamic neurones, and these changes involve altered presynaptic function. Orexin-positive neurones were among the populations that underwent these presynaptic changes.
The results indicate that a greater understanding of the drug-induced synaptic changes in perifornical/lateral hypothalamus may instruct future pharmacotherapies aimed at preventing drug relapse.
Introduction
The perifornical/lateral hypothalamic area (PF/LHA) regulates reward-seeking behaviour. Seminal studies first alluded to this role by demonstrating that lesions of the lateral hypothalamus affected feeding behaviour, and that rats would press a lever to obtain electrical stimulation of this region (Anand & Brobeck, 1952; Olds & Milner, 1954). More recent anatomical studies have established that the PF/LHA strongly projects to important brain regions responsible for controlling motivation and reward-seeking behaviour, including the prefrontal cortex (PFC), nucleus accumbens (NAC) and ventral tegmental area (VTA) (Petrovich et al. 2005; Borgland et al. 2006; Gonzalez et al. 2012). Despite these earlier studies describing a key role for the lateral hypothalamus in natural reward-seeking behaviour (e.g. for food), it has only been in the last decade that significant progress has been made in understanding the specific neurotransmitter systems responsible for these behaviours (DiLeone et al. 2003). Research has now begun to address how lateral hypothalamic ‘feeding peptides’ interface with brain regions known to drive reward-seeking behaviours and how these hypothalamic systems might be ‘hijacked’ by highly potent non-conventional chemical rewards such as drugs of abuse.
It is now well established that in addition to triggering food-seeking behaviour, PF/LHA neurones are also recruited by stimuli associated with addictive drugs (Dayas et al. 2008; Hamlin et al. 2008). Furthermore, drug-seeking is abolished by inactivation of the PF/LHA (Marchant et al. 2010) indicating that the neurones in this region contribute to the control of ‘motivational action’ and produce both conventional and ‘pathological’ reward-seeking behaviour. A number of neuropeptide transmitters have been implicated in both natural and drug-seeking behaviours. Of these, neurones expressing the neuropeptide orexin (hypocretin) found within the PF/LHA have received significant attention. Specifically, Harris & Aston-Jones (2006) showed that an orexin-1 receptor (OrxR-1) antagonist SB-334867 reduced preference for a morphine-paired environment (Harris & Aston-Jones, 2006). Further, in rodent models of relapse, systemic SB-334867 administration reduces drug-seeking behaviour (Boutrel et al. 2005; Harris & Aston-Jones, 2006; Lawrence et al. 2006). Recent data suggest that orexins are likely to influence drug-seeking through their projections to the VTA (Wang et al. 2009; James et al. 2011, 2012; Mahler et al. 2012). Importantly, orexins may also influence drug-seeking through projections to cortical regions (Hollander et al. 2008; Jupp & Lawrence, 2009).
Despite this evidence of PF/LHA involvement in drug-seeking, very few studies have addressed how the functional properties of hypothalamic neurones are altered by drug exposure. Thus, while drug-induced synaptic plasticity is well characterized in both the VTA and NAC (Borgland et al. 2006; Mameli et al. 2009), it is not known whether similar modifications occur at synaptic inputs onto PF/LHA neurones. There is, however, some evidence to suggest that hypothalamic neurones are likely to be susceptible to drug-induced plasticity. For example, Ahmed et al. (2005) showed that the lateral hypothalamus exhibits substantial gene expression changes for markers of both pre- and postsynaptic signalling molecules in animals with escalated cocaine intake. Evidence from other fields also supports a hypothesis that the PF/LHA orexin neurones are easily ‘rewired’ and likely to be susceptible to drug-induced plasticity (Horvath & Gao, 2005). For example, overnight food restriction increased the frequency of miniature excitatory postsynaptic currents (mEPSCs) in orexin neurones and promoted the formation of new excitatory, vesicular glutamate transporter 2 (VGLUT2)-positive inputs onto these cells (Horvath & Gao, 2005). Similarly, sleep deprivation also promotes plasticity at hypothalamic glutamatergic synapses, increasing both the frequency and amplitude of mEPSCs in orexin neurones (Rao et al. 2007). These data indicate that hypothalamic circuits may be ‘soft-wired’, a characteristic that could endow a vulnerability to repeated drug exposure and confer increased addiction and relapse vulnerability. Whether similar changes occur within PF/LHA circuitry that could affect orexin neurone recruitment (or other hypothalamic cell populations that regulate drug-seeking) in response to drug exposure is yet to be determined (Chung et al. 2009).
The purpose of the present study was therefore to provide evidence that the PF/LHA itself is subject to synaptic modifications in response to drug exposure. Accordingly, we used experimenter-administered (i.e. passive) and self-administration (SA) drug exposure procedures in rats and tested whether cocaine altered excitatory drive to PF/LHA using patch-clamp recording techniques. To determine if cocaine-induced changes in excitatory drive in the PF/LHA were likely to affect the recruitment of orexin neurones, we used immunohistochemistry for vesicular glutamate transporter 2 (VGLUT2) and vesicular GABA transporter (VGAT), markers of excitatory versus inhibitory inputs, respectively. Further, a population of recorded neurones was filled with neurobiotin and immunolabelled to identify orexin neurones post hoc.
Methods
Animals
All experimental procedures were approved by the University of Newcastle Animal Care and Ethics Committee and performed in accordance with the New South Wales Animal Research Act. Male Sprague–Dawley rats (n = 58, Animal Resource Centre, Perth, Australia) aged 3–5 weeks were housed two per cage in a temperature- and humidity-controlled room on a reversed 12 h–12 h light–dark cycle (lights off at 10:00 am) with ad libitum access to food and water. All the experimental procedures (i.e electrophysiological recordings and immunohistochemistry) were carried out 24 h after the last passive cocaine or cocaine self-administration session.
Cocaine exposure procedures
Animals were subjected to one of two methods of drug exposure: passive or SA cocaine. In passive cocaine experiments, animals were weighed and separated into two groups; one group received cocaine injections (n = 14), and the other group received saline injections (vehicle, n = 15). Prior to treatment with cocaine, 6-week-old animals were conditioned to daily handling (1 h per day for 3 days) and were subjected to single-daily sham injections and placed in an enclosed arena (50 cm × 50 cm) for 1 h. Over the next 7 days, animals received an injection of either saline or cocaine hydrochloride (15 mg kg−1; i.p.) and placed in the enclosed arena for an hour before they were returned to their home cage.
For cocaine self-administration experiments, animals (n = 15) underwent catheter surgery as previously described (James et al. 2011). Briefly, a Silastic catheter was surgically implanted into the right jugular vein under isofluorane anaesthesia (1–3%). Cocaine self-administration was conducted in standard operant conditioning chambers equipped with two retractable levers controlled by a Windows-based PC, using MED-PC IV software (Med Associates, St Albans, VT, USA). Cocaine was delivered via a syringe pump (5 rpm motor, Med Associates) located on the outside of the cubicle (dose of 0.25 mg per infusion i.v.). At 6 weeks of age animals were trained (2 h sessions, 7 days) to press on the right lever for a cocaine reward on a fixed ratio 1 (FR1) schedule. Infusions were followed by a white cue light above the active lever signalling a 20 s time-out period and rewards were limited to 20 infusions per session. Following training, animals self-administered cocaine for an additional 7 days with the cocaine infusion limit of 20 rewards removed.
In control experiments, a food dispenser mounted to the exterior of the operant chambers delivered food rewards directly into a food hopper inside the operant chamber. During the first 7 days of self-administration training, animals (n = 14) were food restricted in their home cage to 20 g of Purina rat chow per day. Animals were trained to press the right lever (FR1) to receive a 45 mg Noyes food pellet (TestDiet, Richmond, IN, USA). This was followed by a 20 s time-out period as above, but food rewards were limited to 200 pellets per sessions. Following the 7 day training protocol, animals were returned to unrestricted feeding conditions in their home cage and allowed to self-administer food-pellets for an additional 7 days. Importantly, this avoided any possible role for food restriction-related alterations in the PF/LHA as previous work has demonstrated that neuronal properties, which can be altered by food restriction, normalize within 24 h of re-feeding (Horvath & Gao, 2005).
Electrophysiology
Rats were deeply anaethesized with ketamine (1 ml kg−1) and decapitated. Brains were rapidly removed and immersed in ice-cold oxygenated (95% O2, 5% CO2) sucrose substituted artificial cerebrospinal fluid (S-ACSF, containing in mm: 236.2 sucrose, 25 NaHCO3, 13.6 glucose, 2.5 KCl, 2.5 CaCl2, 1 NaH2PO4 and 1 MgCl2). Transverse slices (300 μm) containing PF/LHA were obtained using a vibrating-blade microtome (Leica, VT-1000S, Heidelberg, Germany), then transferred to an oxygenated chamber containing artificial cerebrospinal fluid (ACSF; 119.4 mm NaCl substituted for sucrose) and incubated for 1 h at room temperature. Slices were transferred to a recording chamber and continually superfused with oxygenated ACSF (∼32°C). Neurones were visualized using infrared differential interference contrast microscopy. Whole-cell recordings were made using a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA), digitized at 10 kHz, via an ITC-18 computer interface (Instrutech, Long Island, NY, USA) and recorded onto a Macintosh computer running AxoGraph X software (Axograph, Berkeley, CA, USA). All recordings were restricted to the PF/LHA region. After obtaining the whole-cell recording configuration, series resistance and input resistance were calculated based on the response to a –5 mV voltage step from a holding potential of –70 mV. These values were monitored at the beginning and end of each recording and data were rejected if values changed by more than 20%. Miniature excitatory postsynaptic currents (mEPSCs) were recorded in voltage clamp at a holding potential of –70 mV with series resistance of <25 MΩ, in the presence of tetrodotoxin (1 μm) and picrotoxin (50 μm). Recording pipettes (2–4 MΩ) were filled with an internal solution containing (in mm): 135 KCH3SO4, 6 NaCl, 2 MgCl2, 10 Hepes, 0.1 EGTA, 2 MgATP, 0.3 NaGTP (pH 7.3 with KOH) and in some cases 0.1% Neurobiotin. In some experiments a bipolar stimulating electrode was positioned immediately medial and dorsal to the PF/LHA to stimulate excitatory inputs (0.1 ms pulse duration, 1.2 × threshold stimulus intensity, range = 120–140 μA). Evoked EPSCs (eEPSCs) were recorded with an internal solution containing (in mm): 120 caesium methanesulfonate, 20 CsCl, 10 Hepes, 4 Mg-ATP, 0.3 Na3-GTP, 0.2 EGTA, 10 sodium phosphocreatine, 5 QX-314 (pH 7.3 with CsOH). These recordings assessed both the paired-pulse, and AMPA/NMDA ratio of eEPSCs. Neurones were voltage clamped at –70 mV, in the presence of picrotoxin (50 μm), to record the paired pulse ratio (50 ms interstimulus interval) of AMPAR-mediated EPSCs. NMDAR-mediated EPSCs were recorded at a holding potential of +40 mV with the addition of CNQX (10 μm). A minimum of 5–10 eEPSCs was collected for analysis, with a 30 s interval between protocols. In a subset of experiments we assessed the ability of picrotoxin (50 μm) to abolish inhibitory postsynaptic currents (IPSCs) evoked by a bipolar stimulating electrode. This confirmed that bath application of picrotoxin provided a full block of evoked IPSCs (n = 6, data not shown). Following recording, some slices were fixed in 4% PFA for an hour and stored in 0.1 m phosphate-buffered saline (PBS). Slices were processed for immunohistochemical detection of orexin (as described below) with neurobiotin secondary (3 h, 1:500, Streptavidin-Cy3; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA), and visualized for immunohistochemistry to assess neurobiotin/orexin co-labelling.
Data analysis
mEPSCs were detected and captured using a sliding template method, along with a minimum amplitude threshold criterion of 10 pA (Axograph software). Under these conditions, extremely small mEPSCs could escape detection because they could not be resolved from background recording noise. This could potentially introduce errors when assessing mEPSC amplitude and frequency across conditions. In order to assess the effect of these detection parameters, we constructed histograms from 5 ms of baseline data preceding each mEPSC in all recordings. In all cases the noise distributions were Gaussian with means clustered around 0 pA. Importantly, the mean value for peak to peak noise was not different across recording conditions (passive cocaine vs. saline, 10.13 ± 0.79 pA vs. 11.48 ± 0.55 pA, P = 0.27; self administration cocaine vs. food, 10.09 ± 0.47 pA vs. 9.75 ± 0.43 pA; P = 0.6), suggesting that the effect of noise was the same across our dataset. We cannot, however, exclude the possibility that very small mEPSCs may have gone undetected and were more frequent in one particular condition.
Captured mEPSCs were individually inspected and excluded from the analysis if they included overlapping events or had an unstable baseline before the rise or after the decay phase of the mEPSC. Data were rejected if a significant trend was evident in either mEPSC amplitude or instantaneous frequency over the course of the experiment. Analyses were performed on averaged mEPSCs, generated by aligning the rising phase of all accepted events. Peak amplitude, rise-time (calculated over 10–90% of peak amplitude) and decay time constant (calculated over 20–80% of the decay phase) were obtained using automated procedures (Axograph software). Average mEPSC frequency was obtained by dividing the number of captured events by the analysis duration in seconds. Peak amplitude, rise-time and decay time constant were also evaluated for eEPSCs. Paired-pulse ratio (PPR) was calculated by dividing the mean peak amplitude of the second eEPSC by the mean peak amplitude of the first. AMPA/NMDA ratios were calculated by dividing the mean peak amplitude of evoked AMPA EPSCs (recorded at −70 mV) by the mean peak amplitude of evoked NMDA EPSCs (recorded in CNQX at +40 mV). For statistical analyses, the Shapiro–Wilk test of normality was initially used to determine if data were normally distributed. Normally distributed data were compared using Student's t tests. In cases where the assumption of normality was violated, data were analysed using non-parametric Mann–Whitney U t tests (indicated in text). All data are expressed as means ± SEM.
Immunohistochemistry
Animals were deeply anaesthesized and transcardially perfused with 0.1 m phosphate buffered saline, followed by 4% paraformaldehyde (PFA). Brains were removed, postfixed (PFA, 1 h, at 4°C), and cryoprotected (15% sucrose in 0.1 m phosphate buffer, pH7.4 at 4°C). Serial 25 μm coronal sections of the forebrain were cut on a freezing microtome (Leica), and sections containing the PF/LHA were obtained and processed for immunohistochemistry. Free floating brain sections were incubated in the following series of antibodies: primary orexin antibody (24 h, 1:1000, goat polyclonal, Santa Cruz Biotechnology, Santa Cruz, CA, USA) with either vesicular glutamate transporter (VGLUT2) antibody (1:5000, rabbit polyclonal, Synaptic Systems Goettingen, Germany) or vesicular GABA transporter (VGAT) antibody (1:250, rabbit polyclonal; Millipore, Temecula, CA, USA). Sections were then incubated in a cocktail of secondary antibodies (3 h, 1:500, AMCA anti-goat, Jackson Immunoresearch, and 1:500, Alexa Fluor 488 donkey anti-rabbit, Molecular Probes). Sections were mounted onto gelatin-coated slides and coverslipped with gelvatol.
In a separate experiment, dual-immunolabelling for orexin and neuronal specific nuclear protein (NeuN) was carried out to assess the proportion of orexin-positive neurones in the PF/LHA region where the majority of recordings were made. Brain sections were incubated with primary orexin antibody (as above) and NeuN antibody (1:200, mouse polyclonal, Millipore, Temecula, CA, USA). Sections were then incubated in AMCA anti-goat and Alexa Fluor 488 anti-mouse (1:500 Molecular Probes, Invitrogen). Controls for antibody specificity and cross reactivity were performed for all experiments by primary antibody omission.
Labelled PF/LHA sections were examined using confocal microscopy (Nikon Eclipse 80i Microscope attached to Eclipse C1 confocal system). Z-series were taken (60× objective) and images were imported into ImageJ version 1.37 (National Institute of Health, USA) for analysis. Images were background subtracted and the z-series reconstructed. Counts of VGLUT2 or VGAT puncta in close apposition to orexin neurones (<1 μm) were made within the PF/LHA. Data were first analysed using the Shapiro–Wilk test to determine if the data were normally distributed, followed by Student's t test. All data are expressed as means ± SEM.
Drugs
Cocaine hydrochloride (GlaxoSmithKline, Victoria, Australia) was dissolved in sterile physiological saline: 15 mg ml−1 for intraperitoneal injection, or 2.5 mg ml−1 for self-administration. Drugs for electrophysiology were made at 1000 times stock concentrations and then diluted to the final concentration in bath superfusate. Picrotoxin and CNQX were purchased from Sigma-Aldrich (St Louis, MO, USA), and TTX was obtained from Alomone Laboratories (Jerusalem, Israel).
Results
This study includes two sets of experiments designed to assess the effect of cocaine on excitatory drive to neurones in the PF/LHA. In experiment 1 we exposed animals to passive cocaine injections (Fig. 1A). In experiment 2 we used a self-administration procedure to expose animals to cocaine (Fig. 1B) and as a control, a group of animals were trained to lever press for food rewards. Food-trained rats exhibited higher levels of rewarded lever pressing than cocaine rats (69.3 ± 9.3 vs. 30.3 ± 2.0). Similarly, inactive (unrewarded) lever presses were higher in the food versus cocaine groups (7.2 ± 2.8 vs. 2.5 ± 1.8). Differences between the pharmacological effects of cocaine versus the shorter satiating actions of a food reward are likely to account for these behavioural differences and are consistent with previous studies (Borgland et al. 2009).
Figure 1. Cocaine-exposure procedures.
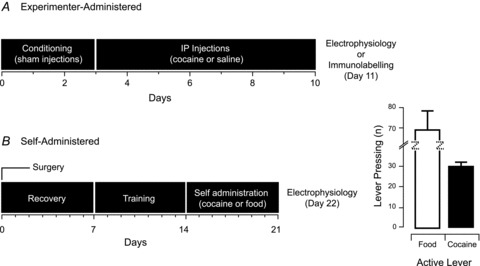
A, timeline summarizing the cocaine and saline experimenter-administered injection regimes. B, timeline summarizing cocaine and food (control) self-administration regime. Bar plots show mean active (rewarded) lever pressing over the final 3 days of food or cocaine self-administration.
Experimenter-administered cocaine potentiates excitatory synaptic input to PF/LHA neurones
To determine whether experimenter-administered cocaine potentiates excitatory synaptic input to PF/LHA neurones, we recorded AMPAR-mediated mEPSCs from PF/LHA neurones in saline- and cocaine-exposed rats (Fig. 2). Cocaine treatment had no effect on the input resistance (cocaine vs. saline, 182.19 ± 19.22 MΩvs. 227.94 ± 34.30 MΩ; P > 0.05) and the series resistance of recorded neurones (cocaine vs. saline, 9.70 ± 0.96 MΩvs. 11.87 ± 1.77 MΩ; P > 0.05). mEPSC frequency, however, was significantly increased in cocaine- compared to saline-exposed rats (21.90 ± 5.13 Hz versus 8.31 ± 1.95 Hz, P < 0.05, Mann–Whitney U test; Fig. 2A and B). In contrast, mEPSC amplitude (22.48 ± 2.17 pA vs. 28.74 ± 4.82 pA), rise time (0.53 ± 0.04 ms vs. 0.53 ± 0.07 ms), and decay time constant (2.39 ± 0.32 ms vs. 2.57 ± 0.43 ms) were similar in cocaine and saline groups. Together, these results suggest that experimenter-administered cocaine may preferentially alter presynaptic function onto PF/LHA neurones without affecting postsynaptic receptor density or kinetic properties.
Figure 2. Presynaptic plasticity in PF/LHA after passive cocaine injections.
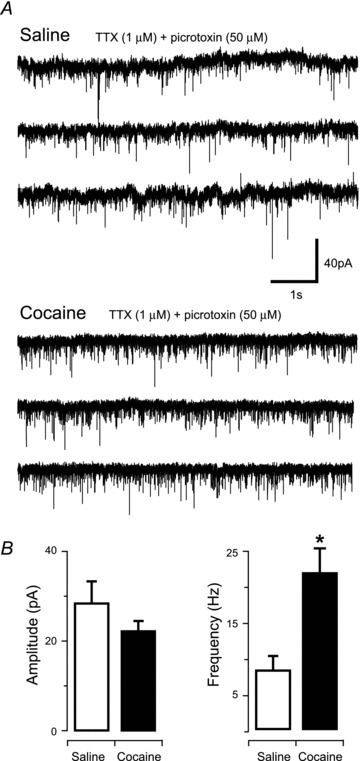
A, traces of representative mEPSC recordings from PF/LHA neurones in saline- (upper) and cocaine-treated rats (lower). B, group data plots show mEPSC amplitude was similar in recordings from cocaine and saline exposed animals; n = 7 and n = 6, respectively. In contrast, cocaine exposure significantly increased mEPSC frequency (*P < 0.05).
Cocaine self-administration substantially potentiates excitatory synaptic input to PF/LHA neurones
Previous work has shown that passive cocaine exposure can have differential effects on synaptic plasticity within some brain regions compared to self-administered cocaine (Chen et al. 2008). Therefore, in a second series of experiments we recorded mEPSCs from PF/LHA neurones in animals following 2 weeks of cocaine self-administration (Fig. 3). Similar to the experimenter-administered cocaine effects (above), cocaine self-administration had no effect on the input resistance (cocaine vs. food, 160.58 ± 17.01 MΩvs. 147.75 ± 11.39 MΩ; P > 0.05) and the series resistance of recorded neurons (cocaine vs. saline, 7.94 ± 0.56 MΩvs. 7.12 ± 0.39 MΩ; P > 0.05), but mEPSC frequency was substantially elevated in cocaine-trained rats compared to food-trained controls (22.46 ± 2.30 Hz vs. 10.46 ± 1.77 Hz, P < 0.01, Mann–Whitney U test, Fig. 3A and B). In contrast, mEPSC amplitude (24.32 ± 0.99 pA vs. 23.27 ± 2.06 pA), rise time (0.51 ± 0.02 ms vs. 0.56 ± 0.04 ms), and decay time constant (1.51 ± 0.09 ms vs. 1.72 ± 0.10 ms) remained similar between cocaine- and food-trained rats. Thus, in our hands the cocaine-induced effects on excitatory drive to PF/LHA neurones are similar under passive and self-administered drug exposure regimens. Interestingly, two-way analysis of variance (ANOVA) revealed a significant main effect of treatment (i.e. passive vs. self-administration) on input resistance (205.61 ± 14.35 MΩvs. 154.42 ± 10.34 MΩ, F1,113 = 8.79, P < 0.01). These data suggest that the act of training animals to self-administer drug and food may have altered the input resistance of PF/LHA neurones.
Figure 3. Cocaine self-administration increases excitatory synaptic input to PF/LHA neurones.
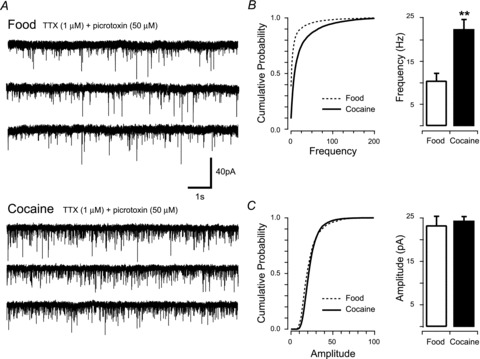
A, traces show representative recordings in PF/LHA neurones from rats that self-administered food (upper traces) or cocaine (lower traces). B, cumulative probability (from representative recordings, left) and bar plots (group data, right) of mEPSC frequency highlight a significant increase in events recorded from cocaine- compared to food-trained rats; n = 18 and n = 16, respectively (**P < 0.01). C, in contrast, cumulative probability (from representative recordings, left) and bar plots (group data, right) of mEPSC amplitude indicate no difference in the distribution or mean values between food- and cocaine-trained rats.
Cocaine self-administration alters paired-pulse but not AMPA/NMDA ratio
In order to further explore the effects of cocaine on excitatory synapses in PF/LHA neurones, a bipolar stimulating electrode was used to electrically activate synaptic inputs. These experiments compared synaptic function in cocaine- and food-trained rats. First, short-term synaptic plasticity and presynaptic release probability were assessed using a paired-pulse protocol (50 ms inter-stimulus interval) to determine paired- pulse ratios (Fig. 4). The outcome of these experiments showed that neurones from rats that self-administered cocaine had significantly lower paired-pulse ratios, i.e. paired-pulse depression, compared to food-rewarded controls (cocaine vs. food, 0.82 ± 0.07 vs. 1.05 ± 0.1, P < 0.05, Fig. 4A). We also recorded evoked EPSCs (eEPSCs) to assess whether cocaine self-administration causes postsynaptic changes to excitatory synapses in the PF/LHA. Responses were recorded at −70 mV or +40 mV holding potentials to isolate the AMPA- and NMDA-mediated components of eEPSCs, respectively (Fig. 4B). A comparison of the properties of AMPA and NMDA eEPSCs showed that amplitude (AMPA, 271.38 ± 53.95 pA vs. 260.08 ± 53.65 pA; NMDA, 102.97 ± 18.17 pA vs. 139.47 ± 40.57 pA), rise time (AMPA, 1.57 ± 0.22 ms vs. 1.64 ± 0.29 ms; NMDA, 4.45 ± 0.37 ms vs. 6.92 ± 2.17 ms), and decay time constant (AMPA, 5.61 ± 0.73 ms vs. 7.74 ± 1.05 ms; NMDA, 95.65 ± 10.46 ms vs. 140.62 ± 37.74 ms) were all similar in cocaine- versus food-trained animals. The peak amplitude of AMPA- and NMDA-mediated eEPCSs was also used to determine the AMPA/NMDA ratio, which also remained similar between cocaine- and food-rewarded animals (2.64 ± 0.55 vs. 2.50 ± 0.57, Fig. 4B). These results suggest that cocaine does not alter the postsynaptic receptor composition of excitatory synapses.
Figure 4. Cocaine self-administration causes paired-pulse depression but does not alter the AMPA/NMDA ratio.
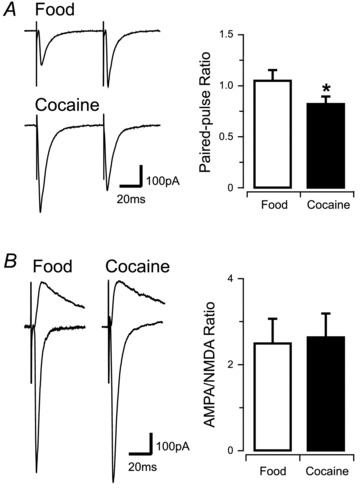
A, traces show representative evoked excitatory postsynaptic currents (eEPSCs) recorded in response to paired stimuli in PF/LHA neurones from food- (upper traces) or cocaine- (lower traces) trained rats. Bar plot (right) compares group data indicating that food-trained rats exhibited paired-pulse facilitation, whereas cocaine-trained rats exhibited paired-pulse depression (*P < 0.05). B, traces show an averaged inward eEPSC recorded at −70 mV (AMPA receptor-mediated) and overlayed averaged outward eEPSCs recorded at +40 mV in the presence of 1 μm CNQX (NMDA receptor-mediated). Left traces are from food-trained and right traces are from cocaine-trained rats. Bar plot (right) of AMPA/NMDA ratio compares group data, indicating no differences between food- and cocaine-trained rats.
Anatomical evidence that experimenter-administered cocaine alters excitatory input to orexin neurones
To determine whether any of the cocaine-induced changes in excitatory drive to PF/LHA neurones may affect orexin neurones, we assessed the density of putative glutamatergic (VGLUT2-apposition) and GABAergic (VGAT-apposition) synapses onto orexin-positive neurones (Fig. 5A). No differences were detected in VGAT-positive puncta closely apposed to orexin neurones in cocaine- versus saline-exposed rats (21.15 ± 1.52 vs. 22.64 ± 0.87, Fig. 5B). Rats exposed to cocaine, however, showed a significant increase in VGLUT2-positive puncta closely apposed to orexin neurones as compared to saline-treated rats (23.74 ± 0.68 vs. 19.77 ± 1.52, P < 0.01, Fig. 5B).
Figure 5. Cocaine treatment increases putative apposition of excitatory VGLUT2 puncta.
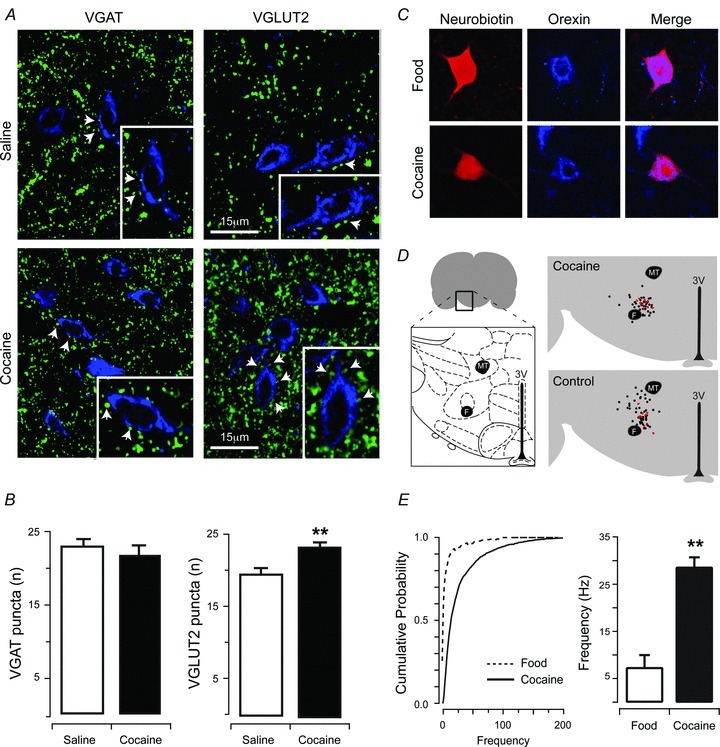
A, dual-labelling for VGAT and orexin (left), or VGLUT2 and orexin (right). Upper panels show saline-treated animals and lower panels show cocaine-treated animals. B, cocaine exposure did not increase putative apposition of inhibitory VGAT puncta onto orexin neurones. In contrast, cocaine exposure significantly increased putative apposition of excitatory VGLUT2 puncta onto orexin neurones (**P < 0.01). C, image panels show immunolabelling for neurobiotin (left), or orexin (middle), and merged pictures (right) from a recorded neurone confirming that population of recordings were from orexin positive neurones. Upper panels are from food-trained controls and lower panels are from cocaine-trained rats. D, schematic diagram showing the collated locations of all recordings in cocaine-exposed and control animals, with red dots indicating confirmed orexin positive recordings (Bregma 2.76–Bregma 3.24). Note that recovery of neurobiotin and orexin labelling was only performed for cocaine self-administration experiments. E, cumulative probability of orexin neurones (from representative recordings, left) and bar plots (group data, right) of mEPSC frequency highlight a significant increase in events recorded from cocaine- compared to food-trained rats (**P < 0.01).
Functional evidence that self-administered cocaine alters excitatory input to orexin neurones
In order to directly assess whether orexin neurones in the lateral hypothalamus undergo enhanced excitatory drive, neurobiotin-filled recorded neurones were dual-immunolabelled for orexin/neurobiotin (Fig. 5C), and data from orexin/neurobiotin-positive neurones were grouped. A small proportion of recorded slices produced reliable orexin/neurobiotin immunostaining (control n = 9; cocaine n = 10), with only a subgroup of these confirmed as containing orexin/neurobiotin-positive (control n = 4, cocaine, n = 3) neurones that yielded data included in the final analysis (thus approximately 36% of PF/LHA neurones were orexin-positive). Despite these limited recoveries, mEPSC frequency in orexin/neurobiotin-positive neurones was increased by cocaine self-administration (28.63 ± 2.05 Hz vs. 7.34 ± 2.63 Hz, P < 0.01, Fig. 5E) while amplitude (−27.05 ± 2.13 pA vs.−18.80 ± 2.42 pA), rise time (0.49 ± 0.04 ms vs. 0.60 ± 0.05 ms) and decay time constant (1.72 ± 0.36 ms vs. 1.84 ± 0.16 ms) remained similar between cocaine- and food-trained rats (P > 0.05). To assess whether the proportion of neurones recovered in our recordings simply reflects the overall proportion of PH/LHA neurones that express orexin within the area we sampled, we combined immunolabelling for orexin and NeuN, a marker of post-mitotic neurones. This analysis revealed that orexin neurones accounted for approximately 43% (48 of 112 cells counted in 6 sections) of the total population of PF/LHA neurones in the region we used for our electrophysiological analyses.
Discussion
In the present study we provide the first anatomical and functional evidence that cocaine exposure induces plasticity in the PF/LHA. Our data are consistent with evidence that plasticity occurs in PF/LHA neurones after acute non-drug environmental challenges and the emerging role of a number of hypothalamic neuropeptide systems in drug-motivated behaviours. Taken together we believe these data indicate that hypothalamic circuitry is highly ‘plastic’, and thus susceptible to cocaine-induced maladaptations. These changes in hypothalamic circuitry may contribute to increased addiction and relapse vulnerability. Interestingly, although we only recovered a small population of recorded neurones, it appears clear from our data that one of the PF/LHA populations that are vulnerable to drug-induced plasticity expresses the orexin neuropeptides.
The present findings indicate that cocaine specifically alters presynaptic inputs onto PF/LHA neurones. This conclusion was supported by our findings that both methods of cocaine exposure produced an identical signature of changes to mEPSCs, i.e. passive or self-administered cocaine increased the frequency of mEPSCs, but not amplitude. One caveat to this finding is that we cannot exclude the possibility that some very small mEPSCs (<10 pA) may have gone undetected in control or cocaine treatment groups. This has the potential to influence our measurements of mEPSC amplitude and frequency, but our capacity to resolve all events above 10 pA and the comparison of baseline noise amongst groups suggests this effect should be minimal. It is interesting to note that our data indicate that self-administration training may have reduced the input resistance of PF/LHA neurones. This is probably unsurprising given that acquiring the operant task is a learning process that could evoke changes in PF/LHA cells that are different from passive drug injections. Further studies will be required to understand the significance of this finding with respect to addiction and to identify the putative mechanisms responsible, e.g. potential training-induced changes in channel expression in PF/LHA neurones.
Consistent with cocaine having effects at hypothalamic synaptic terminals, we observed no change in the AMPA/NMDA ratio in animals trained to self-administer cocaine. The AMPA/NMDA ratio is a common measure of changes in postsynaptic receptor composition and increases in this ratio have been shown to occur in other addiction relevant brain regions such as the NAC and VTA (Kauer & Malenka, 2007; Mameli et al. 2009; Moussawi et al. 2011). Importantly, other studies have shown that PF/LHA neurones do demonstrate postsynaptic changes in the AMPA/NMDA ratio in response to pharmacological (modafinil) and environmental (sleep deprivation) challenges (Horvath & Gao, 2005; Rao et al. 2007). These data indicate that our failure to detect a change in this parameter is not due to an inability of this cell population to undergo measurable changes in postsynaptic strength. Presynaptic plasticity was also supported by our paired-pulse experiments, which demonstrated a cocaine-induced paired-pulse depression. This finding is widely interpreted as indicative of increased release probability (Debanne et al. 1996), and consistent with a cocaine-induced increase in mEPSC frequency.
With respect to the likely identity of the PF/LHA neurones that undergo cocaine-induced presynaptic plasticity, increased numbers of VGLUT2-positive puncta were observed closely apposed to orexin neurones. Further supporting this finding was the observation that mEPSC frequency increased in a small group of neurobiotin-positive neurones confirmed as immunopositive for orexin. While we did not functionally test cocaine-induced changes to inhibitory GABAergic inputs onto PF/LHA neurones, we did not observe a change in the number of VGAT-positive inputs onto orexin neurones after cocaine exposure, making it unlikely that the effects we report are due to decreased inhibitory inputs from local GABAergic neurones. It is noteworthy that the recovery of orexin neurones in the PF/LHA using neurobiotin filling and orexin immunohistochemistry was somewhat lower than we expected (approximately 36%). Therefore, to determine if we are likely to have underestimated the proportion of orexin neurones in our recordings, we assessed the proportion of orexin neurones relative to the total number of neurones by labelling PF/LHA tissue for orexin and NeuN. We found that orexin neurones account for approximately 43% of the total neuronal population in the PF/LHA region. These data suggest that our recordings did in fact include a representative proportion of orexin neurones and reinforces our conclusion that cocaine-induced synaptic plasticity in PF/LHA does include orexin neurones.
The identity of non-orexin PF/LHA neurones that presumably also undergo cocaine-induced plasticity was not pursued in this study. Melanin concentrating hormone (MCH)-positive neurones are a separate, non-overlapping population of cells that are anatomically adjacent to orexin neurones in the PF/LHA (Elias et al. 2001). Importantly, like orexin neurones, this population has recently been implicated in drug-motivated behaviours and thus may also participate in cocaine-induced synaptic remodelling in the PF/LHA. Chung et al. (2009) recently showed that MCH-1 receptor knockout mice display reduced place preference and sensitization for cocaine and MCH1-R antagonism reduced cue-, and cocaine- (but interestingly not stress) elicited drug-seeking behaviour (Chung et al. 2009). These effects appear to be mediated by a direct projection to the NAC shell (Georgescu et al. 2005; Bittencourt, 2011). It will be interesting for future studies to determine whether cocaine-induced presynaptic plasticity occurs in MCH PF/LHA neurones.
It is also worth noting the evidence of an important contribution for local, intra-hypothalamic glutamate neurones in providing positive feedback that exists in the PF/LHA (Acuna-Goycolea et al. 2004). In light of this circuitry, it is possible that the increased presynaptic plasticity we observed was caused by an increase in both the number of release sites and the probability of release from local glutamate interneurones. As the PF/LHA also receives a significant glutamatergic projection from other addiction-relevant brain regions, e.g. prefrontal cortex and basolateral amygdala (BLA), the possibility that cocaine-induced plasticity occurred at excitatory terminals originating from these sites cannot be ruled out (Henny & Jones, 2006; Morshedi & Meredith, 2008). For example, Morshedi & Meredith (2008) have recently shown that repeated injections of amphetamine up-regulate Fos immunoreactivity in medial PFC (mPFC) neurones that project to the LHA. (Morshedi & Meredith, 2008). Further, Petrovich et al. (2002, 2005) demonstrated that a BLA to PF/LHA circuit controls cue-potentiated feeding behaviour (Petrovich et al. 2002, 2005). The NAC shell a part of the brain ‘reward’ circuitry may also influence the recruitment of hypothalamic neurones in response to drug-relevant stimuli (Marchant et al. 2009; Millan et al. 2010). Because the NAC projection neurones are presumably GABAergic, any influence over hypothalamic circuits with relevance to the present findings would presumably involve a polysynaptic pathway leading to the disinhibition of glutamatergic interneurones. It will be important for future studies to identify the source of these glutamatergic inputs that are altered by cocaine exposure.
With regard to potential mechanisms for the cocaine-induced increase to excitatory input, previous studies have demonstrated that Group III metabotropic glutamate receptors (mGluRs) maintain tonic inhibition of excitatory synaptic input onto hypothalamic orexin neurones (Acuna-Goycolea et al. 2004). Group III mGluRs normally reduce neurotransmitter release either by inhibiting presynaptic voltage-dependent calcium channels or through direct effects on release machinery (Kuzmiski & Bains, 2010). Cocaine-induced loss of mGluR function has been observed in other brain regions such as the NAC (Moussawi et al. 2011) and it is therefore tempting to speculate that mGluR dysfunction may contribute to the elevated synaptic input we observed onto hypothalamic neurones. Future experiments that employ selective mGluR antagonists will be required to further test this possibility.
In summary, we show for the first time that cocaine induces presynaptic plasticity in hypothalamic neurones, and that orexin neurones are one population of PF/LHA neurones that are susceptible to these changes. These findings are particularly relevant to the accumulating evidence that lateral hypothalamic orexin neurones play an important role in the expression of addiction-like behaviours including drug-seeking (Harris & Aston-Jones, 2006; Lawrence et al 2006; James et al. 2011). Orexins appear to mediate these effects, at least in part, through projections to the VTA. For example, systemic SB-334867, an orexin-1 receptor antagonist, prevented cocaine-induced increases in the AMPA/NMDA ratio in VTA dopamine neurones and blocked cocaine sensitization (Borgland et al. 2006). Further, we recently showed that intra-VTA SB-334867 attenuates cue-induced cocaine-seeking (James et al. 2011). Enhanced excitatory drive to the VTA from the over-excited PF/LHA orexin neurones could initiate and contribute to the persistent plasticity within dopamine neurones following long-term cocaine self-administration and increase relapse vulnerability (Chen et al. 2008).
The present findings are interesting in light of the recent studies demonstrating that food restriction, sleep deprivation and now cocaine increases evidence of presynaptic plasticity in PF/LHA and orexin neurones. These data indicate an intrinsic capacity of PF/LHA circuits to rapidly rewire in response to acute challenges to homeostasis. In the case of natural stimuli, e.g. food, this presynaptic plasticity is reversed within 24 h after re-feeding (Horvath & Gao, 2005). Thus, increases in excitatory inputs may be an important mechanism via which PF/LHA circuits respond to changes in energy status and/or levels of arousal to appropriately regulate behavioural output. Presumably cocaine ‘hijacks’ these same homeostatic mechanisms. Future studies will need to determine how these effects are conferred and whether cocaine persistently corrupts these pre-synaptic plasticity mechanisms, or might also produce postsynaptic effects. In addition to the obvious implications for addiction, these studies will advance our understanding of ‘normal’ homeostatic plasticity within the PF/LHA. Understanding these processes may provide new insight into the development of pharmacological treatment options that reduce pathological reward-seeking behaviour.
Acknowledgments
We thank E. M. Levi and L. Hickey for their excellent technical assistance. J.W.Y. is supported by a Priority Reseach Centre in Translational Neuroscience and Mental Health scholarship. This research was supported by NHMRC grants DC011080 (C.V.D), and DC011080 (B.A.G); and HMRI grant DC011080 (C.V.D. and B.A.G.).
Glossary
- NAC
nucleus accumbens
- PF/LHA
perifornical/lateral hypothalamic area
- PFC
prefrontal cortex
- VTA
ventral tegmental area
Author contributions
C.V.D., B.A.G and J.W.Y. conceived and designed the experiments. J.W.Y collected the data. J.W.Y., C.V.D., B.A.G., M.H.J. and P.J. analysed the data. J.W.Y., C.V.D., B.A.G., M.H.J., P.J. and J.S.B. drafted the article. Experiments were performed in the laboratories of C.V.D. and B.A.G. at the University of Newcastle. All authors approved the final version of this manuscript.
References
- Acuna-Goycolea C, Li Y, Van Den Pol AN. Group III metabotropic glutamate receptors maintain tonic inhibition of excitatory synaptic input to hypocretin/orexin neurons. J Neurosci. 2004;24:3013–3022. doi: 10.1523/JNEUROSCI.5416-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed SH, Lutjens R, van der Stap LD, Lekic D, Romano-Spica V, Morales M, Koob GF, Repunte-Canonigo V, Sanna PP. Gene expression evidence for remodeling of lateral hypothalamic circuitry in cocaine addiction. Proc Natl Acad Sci U S A. 2005;102:11533–11538. doi: 10.1073/pnas.0504438102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand BK, Brobeck JR. Food intake and spontaneous activity of rats with lesions in the amygdaloid nuclei. J Neurophysiol. 1952;15:521–530. doi: 10.1152/jn.1952.15.5.421. [DOI] [PubMed] [Google Scholar]
- Bittencourt JC. Anatomical organization of the melanin-concentrating hormone peptide family in the mammalian brain. Gen Comp Endocrinol. 2011;172:185–197. doi: 10.1016/j.ygcen.2011.03.028. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Chang SJ, Bowers MS, Thompson JL, Vittoz N, Floresco SB, Chou J, Chen BT, Bonci A. Orexin A/hypocretin-1 selectively promotes motivation for positive reinforcers. J Neurosci. 2009;29:11215–11225. doi: 10.1523/JNEUROSCI.6096-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron. 2006;49:589–601. doi: 10.1016/j.neuron.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Boutrel B, Kenny PJ, Specio SE, Martin-Fardon R, Markou A, Koob GF, de Lecea L. Role for hypocretin in mediating stress-induced reinstatement of cocaine-seeking behavior. Proc Natl Acad Sci U S A. 2005;102:19168–19173. doi: 10.1073/pnas.0507480102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM, Chou JK, Bonci A. Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron. 2008;59:288–297. doi: 10.1016/j.neuron.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S, Hopf FW, Nagasaki H, Li CY, Belluzzi JD, Bonci A, Civelli O. The melanin-concentrating hormone system modulates cocaine reward. Proc Natl Acad Sci U S A. 2009;106:6772–6777. doi: 10.1073/pnas.0811331106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayas CV, McGranahan TM, Martin-Fardon R, Weiss F. Stimuli linked to ethanol availability activate hypothalamic CART and orexin neurons in a reinstatement model of relapse. Biol Psychiatry. 2008;63:152–157. doi: 10.1016/j.biopsych.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Debanne D, Guerineau NC, Gahwiler BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. J Physiol. 1996;491:163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiLeone RJ, Georgescu D, Nestler EJ. Lateral hypothalamic neuropeptides in reward and drug addiction. Life Sci. 2003;73:759–768. doi: 10.1016/s0024-3205(03)00408-9. [DOI] [PubMed] [Google Scholar]
- Elias CF, Lee CE, Kelly JF, Ahima RS, Kuhar M, Saper CB, Elmquist JK. Characterization of CART neurons in the rat and human hypothalamus. J Comp Neurol. 2001;432:1–19. doi: 10.1002/cne.1085. [DOI] [PubMed] [Google Scholar]
- Georgescu D, Sears RM, Hommel JD, Barrot M, Bolaños CA, Marsh DJ, Bednarek MA, Bibb JA, Maratos-Flier E, Nestler EJ, DiLeone RJ. The hypothalamic neuropeptide melanin-concentrating hormone acts in the nucleus accumbens to modulate feeding behavior and forced-swim performance. J Neurosci. 2005;25:2933–2940. doi: 10.1523/JNEUROSCI.1714-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez JA, Jensen LT, Fugger L, Burdakov D. Convergent inputs from electrically and topographically distinct orexin cells to locus coeruleus and ventral tegemental area. Eur J Neurosci. 2012;35:1426–1432. doi: 10.1111/j.1460-9568.2012.08057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlin AS, Clemens KJ, McNally GP. Renewal of extinguished cocaine-seeking. Neuroscience. 2008;151:659–670. doi: 10.1016/j.neuroscience.2007.11.018. [DOI] [PubMed] [Google Scholar]
- Harris GC, Aston-Jones G. Arousal and reward: a dichotomy in orexin function. Trends Neurosci. 2006;29:571–577. doi: 10.1016/j.tins.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Henny P, Jones BE. Innervation of orexin/hypocretin neurons by gabaergic, glutamatergic or cholinergic basal forebrain terminals evidenced by immunostaining for presynaptic vesicular transporter and postsynaptic scaffolding proteins. J Comp Neurol. 2006;499:645–661. doi: 10.1002/cne.21131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander JA, Lu Q, Cameron MD, Kamenecka TM, Kenny PJ. Insular hypocretin transmission regulates nicotine reward. Proc Natl Acad Sci U S A. 2008;105:19480–19485. doi: 10.1073/pnas.0808023105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath TL, Gao XB. Input organization and plasticity of hypocretin neurons: possible clues to obesity's association with insomnia. Cell Metab. 2005;1:279–286. doi: 10.1016/j.cmet.2005.03.003. [DOI] [PubMed] [Google Scholar]
- James MH, Charnley JL, Levi EM, Jones E, Yeoh JW, Smith DW, Dayas CV. Orexin-1 receptor signalling within the ventral tegmental area, but not the paraventricular thalamus, is critical to regulating cue-induced reinstatement of cocaine-seeking. Int J Neuropsychopharmacol. 2011;14:684–690. doi: 10.1017/S1461145711000423. [DOI] [PubMed] [Google Scholar]
- James MH, Yeoh JW, Graham BA, Dayas CV. Insights for developing pharmacological treatments for psychostimulant relapse targeting hypothalamic peptide systems. J Addict Res Ther. 2012;S4:008. [Google Scholar]
- Jupp B, Lawrence AJ. New horizons for therapeutics in drug and alcohol abuse. Pharmacol Ther. 2009;125:138–168. doi: 10.1016/j.pharmthera.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Kauer J, Malenka R. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Kuzmiski JB, Bains JS. Metabotropic glutamate receptors: gatekeepers of homeostasis. J Neuroendocrinol. 2010;22:785–792. doi: 10.1111/j.1365-2826.2010.02020.x. [DOI] [PubMed] [Google Scholar]
- Lawrence AJ, Cowen MS, Yang HJ, Chen F, Oldfield B. The orexin system regulates alcohol-seeking in rats. Br J Pharmacol. 2006;148:752–759. doi: 10.1038/sj.bjp.0706789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahler SV, Smith RJ, Aston-Jones G. Interactions between VTA orexin and glutamate in cue-induced reinstatement of cocaine seeking in rats. Psychopharmacology (Berl) 2012 doi: 10.1007/s00213-012-2681-5. DOI: 10.1007/s00213-012-2681-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant NJ, Hamlin AS, McNally GP. Lateral Hypothalamus is required for context-induced reinstatement of extinguished reward seeking. J Neurosci. 2009;29:1331–1342. doi: 10.1523/JNEUROSCI.5194-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant NJ, Furlong TM, McNally GP. Medial dorsal hypothalamus mediates the inhibition of reward seeking after extinction. J Neurosci. 2010;30:14102–14115. doi: 10.1523/JNEUROSCI.4079-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR, Spanagel R, Luscher C. Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci. 2009;12:1036–1041. doi: 10.1038/nn.2367. [DOI] [PubMed] [Google Scholar]
- Millan EZ, Furlong TM, McNally GP. Accumbens shell-hypothalamus interactions mediate extinction of alcohol seeking. J Neurosci. 2010;30:4626–4635. doi: 10.1523/JNEUROSCI.4933-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morshedi MM, Meredith GE. Repeated amphetamine administration induces Fos in prefrontal cortical neurons that project to the lateral hypothalamus but not the nucleus accumbens or basolateral amygdala. Psychopharmacology (Berl) 2008;197:179–189. doi: 10.1007/s00213-007-1021-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussawi K, Zhou W, Shen H, Reichel CM, See RE, Carr DB, Kalivas PW. Reversing cocaine-induced synaptic potentiation provides enduring protection from relapse. Proc Natl Acad Sci U S A. 2011;108:385–390. doi: 10.1073/pnas.1011265108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olds J, Milner P. Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol. 1954;47:419–427. doi: 10.1037/h0058775. [DOI] [PubMed] [Google Scholar]
- Petrovich GD, Holland PC, Gallagher M. Amygdalar and prefrontal pathways to the lateral hypothalamus are activated by a learned cue that stimulate eating. J Neurosci. 2005;25:8295–8302. doi: 10.1523/JNEUROSCI.2480-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovich GD, Setlow B, Holland PC, Gallagher M. Amygdalo-hypothalamic circuit allows learned cues to override satiety and promote eating. J Neurosci. 2002;22:8748–8753. doi: 10.1523/JNEUROSCI.22-19-08748.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Y, Liu ZW, Borok E, Rabenstein RL, Shanabrough M, Lu M, Picciotto MR, Horvath TL, Gao XB. Prolonged wakefulness induces experience-dependent synaptic plasticity in mouse hypocretin/orexin neurons. J Clin Invest. 2007;117:4022–4033. doi: 10.1172/JCI32829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, You ZB, Wise RA. Reinstatement of cocaine seeking by hypocretin (orexin) in the ventral tegmental area: independence from the local corticotropin-releasing factor network. Biol Psychiatry. 2009;65:857–62. doi: 10.1016/j.biopsych.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]